
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Oncol. , 21 May 2020
Sec. Head and Neck Cancer
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00822
 Jean-Philippe Foy1,2,3
Jean-Philippe Foy1,2,3 Chloé Bertolus3David Boutolleau4,5Henri Agut4,5Antoine Gessain6Zdenko Herceg7
Chloé Bertolus3David Boutolleau4,5Henri Agut4,5Antoine Gessain6Zdenko Herceg7 Pierre Saintigny1,2,8*
Pierre Saintigny1,2,8*In some western countries, an increasing incidence of oral squamous cell carcinoma (OSCC) has been observed in non-smoker non-drinker patients (NSND), mostly in women with HPV-negative OSCC. In the context of the unknown etiology and mechanisms of tumorigenesis of OSCC in NSND, we discuss data supporting the hypothesis of a viral origin not related to HPV. OSCC from NSND are characterized by an antiviral DNA methylation and gene expression signature. Based on the similar increasing incidence of oral tongue SCC (OTSCC) and oropharyngeal SCC (OPSCC) in young women and men respectively, we hypothesize that changes in sexual behaviors may lead to an increasing incidence of herpesvirus in the oral cavity, especially HSV-2, similarly to what has already been described in HPV-positive OPSCC. Because viral genome integration has not been detected in OSCC from NSND, a “hit and run” viral mechanism involving epigenome deregulation could therefore play a key role at early steps of oral carcinogenesis in this population of patients. In conclusion, epidemiological, clinical and molecular data supports a “hit and run” viral origin of OSCC from NSND.
Head and neck cancer (HNC) is ranked as the seventh most frequent cancer worldwide and is a significant cause of cancer-associated morbidity and mortality (1). A squamous origin is more common in HNC which is strongly associated with consumption of tobacco and alcohol. However, HNC is a heterogeneous disease which can be caused by alternative etiological factors, especially viral infection, and which includes different anatomical subsites: oral cavity, oropharynx, nasopharynx, hypopharynx and larynx. Squamous cell carcinomas (SCC) of the larynx and hypopharynx are strongly associated with smoking and drinking habits while nasopharyngeal SCC as well as some oropharyngeal SCC are caused by a viral infection involving Epstein - Barr virus (EBV) and Human Papillomavirus (HPV), respectively. Although alcohol and tobacco still remain associated with oral cavity and oropharyngeal cancers, the incidence of oropharyngeal and oral SCC is increasing in young patients, suggesting alternative etiological factors (2). Notably, an increasing incidence of HNSCC has been observed in young to middle-aged men with oropharyngeal SCC (OPSCC) (3) and has been associated with HPV infection (4, 5). HNSCC can also affect the oral cavity of non-smoker non-drinker (NSND) patients, especially in young and elderly women with oral tongue SCC (OTSCC) and gingival SCC respectively (6–8). Indeed, although some epidemiological studies may provide inconsistent data on the incidence of OSCC worldwide, a previous study using data from 22 international cancer registries showed that the increasing incidence of oral tongue SCC was reported among subjects <45 years old in some countries (9). However, as opposed to HPV-related OPSCC, a direct oncogenic role of HPV during oral carcinogenesis has not been demonstrated (10–12) and therefore, has not been associated with the similar increasing incidence of OTSCC in young women (2, 11). This contrasts with recent reports by us and others showing that OSCC affecting NSND are characterized by gene expression profiles compatible with an antiviral response (13, 14). Herein, we review and discuss the arguments to support the hypothesis of a viral origin of OSCC in NSND alternative to HPV, based on epidemiological, clinical as well as molecular data.
Constitutional genetic abnormalities may also potentially explain the occurrence of OSCC in young NSND (2). Previously published genetic variations, such as gene polymorphisms (15, 16), have been associated with OSCC but further investigations are required in order to provide conclusive evidence of their relation with smoking and/or drinking habits or not. Moreover, the recent increase of OSCC in young NSND is unlikely to be due to changes in the genetic makeup considering the timescale needed for the acquisitions of new genetic polymorphisms.
Notably, further studies are needed in order to investigate the role of predisposing genetic disorders such as Fanconi and dyskeratosis congenita, into oral carcinogenesis not related to tobacco/alcohol consumption.
Extrinsic environmental risk factors contribute significantly to most common cancers (17). In particular, the International Agency for Research on Cancer (IARC) estimated that ~12% of cancers are caused by one of seven known oncogenic viruses: hepatitis B and C viruses (HBV, HCV); human T-lymphotropic virus 1 (HTLV-1); HPV; Kaposi's sarcoma-associated herpes virus (HHV-8); Merkel cell polyomavirus; and Epstein-Barr virus (EBV). In the field of HNSCC, EBV and HPV are involved in nasopharyngeal SCC as well as in OPSCC (4, 5) respectively. Although a comprehensive RNA-sequencing for viral pathogen discovery failed to reveal any potentially causative integrated viruses in OSCC from NSND, the involvement of a non-integrated virus in the initiation or promotion of OSCC cannot be excluded (18). Moreover, additional risk factors may potentially contribute to oral carcinogenesis in NSND. In particular, chronic local inflammation, as observed in periodontal diseases, may induce changes in oral microbiota that would increase the risk of oral cancer (19–21). Interestingly, the reported relationship between HPV status and oral microbiota (22) suggests that an increasing incidence of viral infection in the oral cavity may also interfere with the oral microbial flora and therefore, may contribute to oral cancer.
Moreover, alternative etiological factors, such as drug consumption (23), household air pollution (24), occupation (25), have been proposed to explain oral cancer not related to tobacco/alcohol habits, but require validation in large prospective cohorts of patients. In contrast, some factors, such as the regular consumption of fruits and vegetables could play a role in reducing oral cancer risk (26). While the link between these factors and oral cancer development is not definitive, they may interact with oncogenic viruses and therefore play a possible role in NSND developing OSCC.
In conclusion, a comprehensive epidemiological analysis of the geographical distribution of OSCC in NSND, which is still poorly known worldwide, could help to provide a better understanding of the potential role of genetic predisposition factors as well as the involvement of specific infectious agents in this disease.
Because NSND suffering from OSCC are epidemiologically different from SD, their molecular profiles have been compared in different studies (7, 14, 18, 27–29), as summarized in Table 1. Few actionable genomic alterations were different between OSCC from NSND and SD. We compared genome wide expression profiles of HPV-negative OSCC from NSND and SD and found that the main biological difference lies in the differential expression of the genes involved in the immune microenvironment (14). Notably, OSCC from NSND were characterized by an enrichment of immune-related pathways involving T-cell activation and differentiation, which was consistent with a higher CD8+ T-cell infiltrate in NSND compared to SD. Moreover, we observed an activation of the interferon-γ response in OSCC from NSND, as previously reported in all HNSCC from NSND (29). Our data highlighted the importance of the microenvironment, and also suggested a higher clinical benefit of indoleamine 2, 3-dioxygénase (IDO1) and programmed death-ligand 1 (PD-L1) inhibition in the subgroup of NSND patients with OSCC. Notably, we identified a set of ~850 genes that were differentially expressed in NSND vs. SD (named NSND gene set), that were validated in four independent datasets: including one cohort of patients treated at our institution (Centre Léon Bérard, CLB, Lyon, France). Using the same methodology (single sample gene set enrichment analysis) and as shown in Figure 1, we have computed the enrichment score of the NSND gene set in OSCC surgically resected in patients from United Kingdom or from Sri Lanka (GSE51010) (30). In line with our previous results, OSCC from NSND had a higher score compared to OSCC from SD in patients from the United Kingdom. No significant difference was found between NSND and SD patients from Sri Lanka who are betel consumers (Figure 1). This observation provides some evidence that the differences in the immune microenvironment between NSND and SD Caucasian patients could be explained by an extrinsic stimulation of the immune response in NSND, similarly to what has been previously described in OSCC from Sri Lankan betel consumers (30), rather than a negative effect of smoking and drinking on the immune response. This is also in line with the observation that no difference in the enrichment score of the NSND gene set in normal oral mucosa from smokers vs. non-smokers was observed, as well as in normal oral keratinocytes treated with ethanol and/or nicotine vs. untreated ones (14). Overall, these results provide some evidence that the difference in OSCC affecting NSND vs. SD patients are not related to the effect of smoking and/or alcohol on normal mucosa.
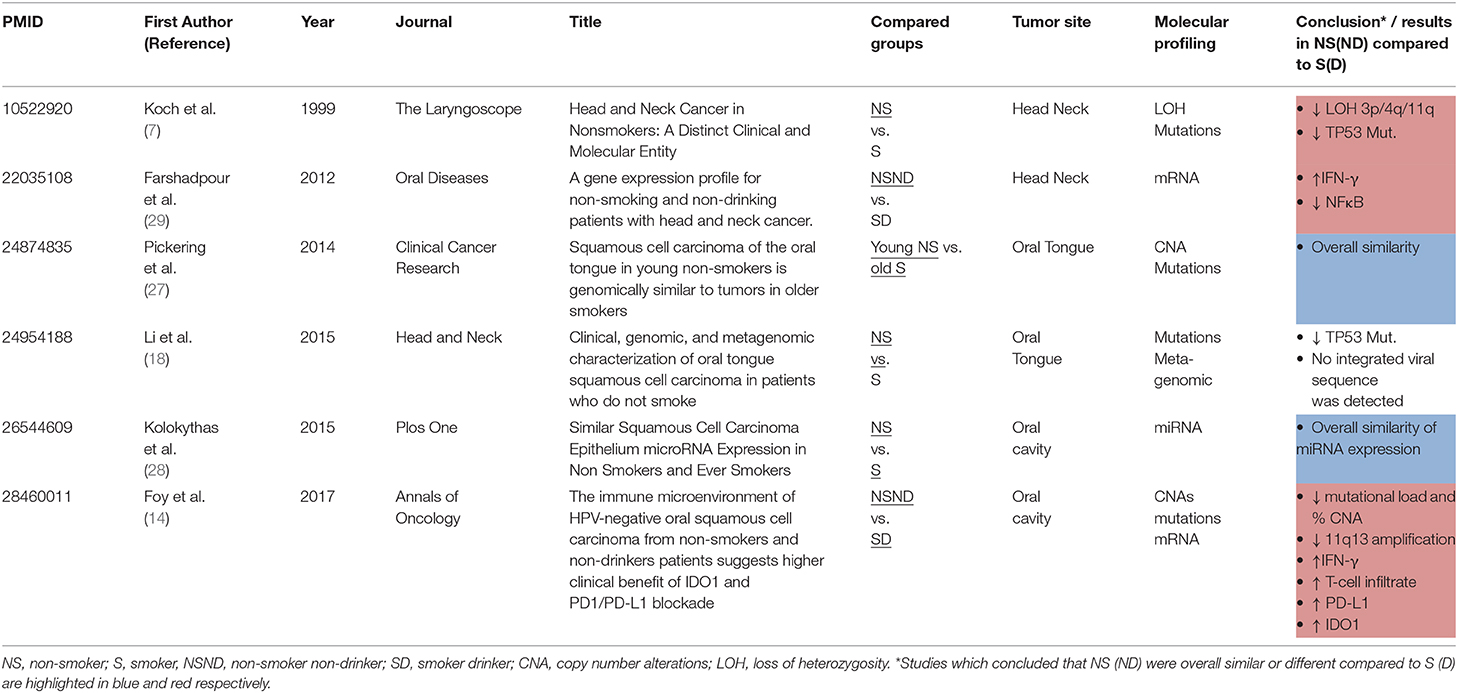
Table 1. Overview of studies investigating differences in molecular profiles of oral/head and neck squamous cell carcinomas in non-smoker (non-drinker) compared to smoker (drinker) patient.
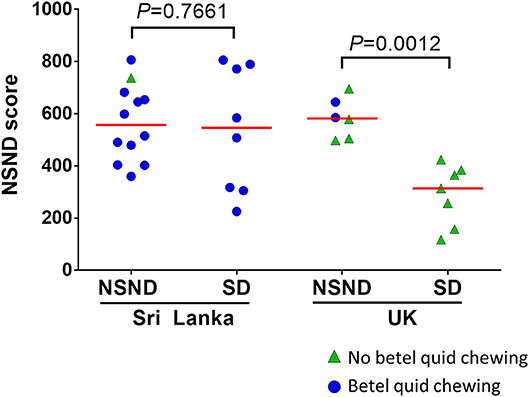
Figure 1. Enrichment scores for the non-smoker non-drinker (NSND) gene set in patients with OSCC from Sri Lankan and United Kingdom (UK). We previously identified a set of genes that were differentially expressed between NSND and smoker drinker patients (14). Using the single sample Gene Set Enrichment Analysis, we computed a score of this gene set (NSND score) in a publically available gene expression dataset of OSCC from Sri Lankan and UK patients (GSE51010) (30).
Interestingly, JAK2 was among the genes overexpressed in NSND compared to SD, in multiple cohorts (Figure 2A). Moreover, JAK2 copy number gain was observed in some tumors from NSND (Figure 2B), and was correlated with its gene expression level (Figure 2C). JAK2 is involved in the gamma (or type II) interferon activation in response to intracellular pathogens, including viruses. Intriguingly, concurrent overexpression and amplification of JAK2 has recently been found in three other cancers: a subgroup of gastric cancers (31), Hodgkin lymphoma, and a subgroup of triple negative breast cancer (32, 33), all being related to EBV infection. It is tempting to hypothesize that gene and expression level alterations of JAK2 reflect the chronic inflammatory response induced by an infectious agent such a viral infection. Moreover, the lower mutational load in OSCC from NSND compared to SD suggests that the potent IFN-γ-mediated immune response is independent from the mutation load and linked to a dominant tumor antigen, as observed in the case of viral-induced carcinogenesis such as in HPV-related OPSCC (14). Overall, these observations suggest the potential role of chronic-viral stimulation during oral carcinogenesis in NSND. However, caution should be made regarding these data, because of the lack of specific signature of viral infection.
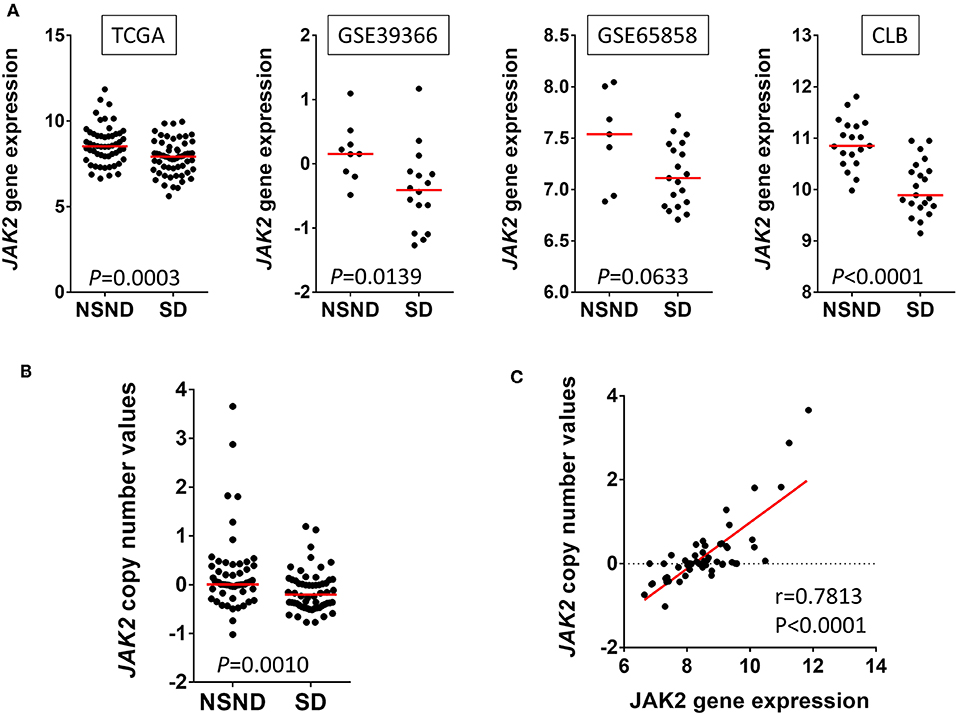
Figure 2. Janus kinase 2 (JAK2) alterations in human papillomavirus (HPV)-negative oral squamous cell carcinoma (OSCC) from non-smokers non-drinkers (NSND) and smokers drinkers (SD). (A) We extracted JAK2 gene expression data from four independent cohorts of HPV-negative OSCC, as previously defined (14): The Cancer Genome Atlas (TCGA), GEO1 and GEO2 from GSE39366 and GSE65858 respectively, and Centre Léon Bérard (CLB, Lyon, France). Overexpression of JAK2 in NSND as compared with SD was observed in all four cohorts (Whitney test). In TCGA, we compared copy number linear value in OSCC from NSND and SD (Mann Whitney test) (B) and tested its correlation with gene expression in OSCC from NSND (Pearson correlation) (C).
A recent study has investigated DNA methylation subtypes of HNSCC, and has identified a CpG Island Methylator Phenotype (CIMP)-atypical subtype of HNSCC more commonly affecting the oral cavity of non-smoker and female patients. Consistently with our results, this subtype was characterized by an antiviral gene expression profile associated with pro-inflammatory M1 macrophages and CD8+ T cell infiltration (13). Viral-associated epigenetic changes are known to be associated with cancer development (32, 33), and CIMP has already been associated with EBV and HPV associated carcinogenesis (34, 35). Upregulation of DNA-methyltransferases in virus-infected cells may lead to aberrant DNA methylation (36) as well as miRNA deregulation such as downregulation of the tumor suppressor miR-200 as observed in EBV-associated gastric adenocarcinomas (37). Thus, DNA methylation profile of OSCC in NSND strengthens the hypothesis of their viral origin.
From an epidemiological perspective, an intriguing observation over the past three decades has been the simultaneous increasing incidence of OPSCC in young men as well as OSCC in young women (11, 38–40). In order to illustrate these observations, we mined the SEER database and compared the incidence of OT cancer in women and of OP cancer in men less than 50 years old (41). As shown in Figure 3A, a parallel increase of the incidence of OT cancer in female and of OP cancer in males less that 50 was observed between 1980 and 2013. We contrasted this increase with the decrease in cigarette consumption during the same period of time based on data provided by Ng et al. (42). Consistently, Figure 3B shows a Pearson correlation of 0.57 (P = 0.0006) between the incidence of OT cancer in women and of OP cancer in men <50 years old. This observation is consistent with a previous study showing that the increasing incidence of both OTSCC and OPSCC had simultaneously started in the 1980s (43).
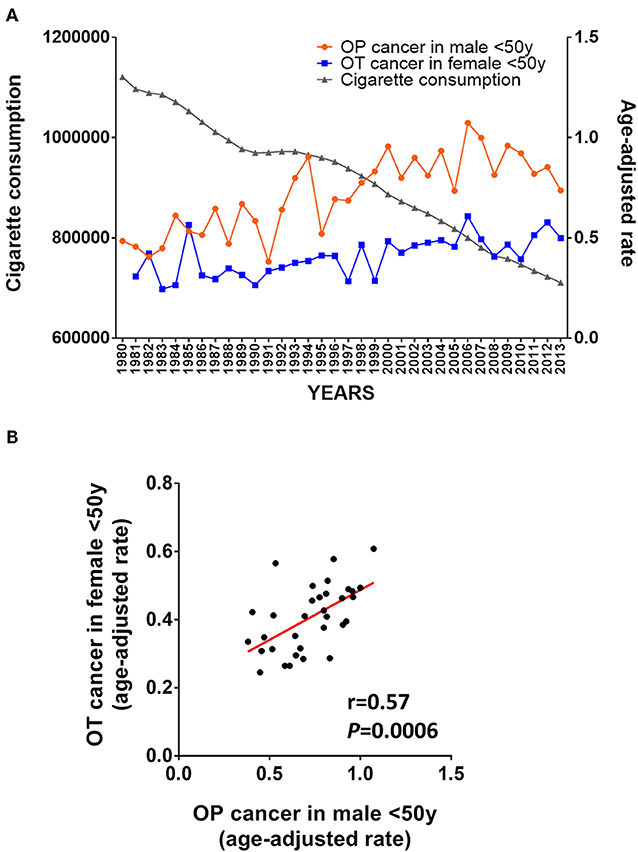
Figure 3. Epidemiological argument supporting a viral origin of OSCC in non-smoker non-drinker (NSND). (A) Evolution of age-adjusted cancer incidence in young patients (<50 years-old) from the SEER database (41) and global cigarette consumption between 1980 and 2013 from the Regional Office for the Americas (AMRO) as previously reported (42); (B) Pearson's correlation of the age-adjusted incidence of OT and OP cancer in young female and male between 1980 and 2013.
The increasing incidence of OPSCC in young NSND men has been associated with an increasing rate of oral HPV infection due to oral sexual behaviors (4, 44). Markedly, the decrease in age of sexual debut as well as the increased number of sexual partners may have contributed to a rise in oral/oropharyngeal HPV infection (44, 45), and a higher risk of HPV transmission from women to men has been proposed to explain the higher incidence of OPSCC in men compared to women (46).
Based on these observations, we propose that the increasing incidence of OTSCC in young NSND may also be linked to one or several sexual transmissible virus which could be transmitted by oral sexual behaviors and which could promote oral carcinogenesis. If this is true, because the increasing incidence of OSCC from NSND is mainly observed in women, transmission of the potential oncogenic virus(es) should be higher from men to women rather than from women to men.
Molecular profile and epidemiological data on OSCC from NSND support the hypothesis of a viral origin possibly due to changes in sexual behaviors (44, 45). As previously described, viral infections are common among humans, but rarely lead to cancer (47). Epidemiological criteria for causality, proposed by Austin Bradford Hill, have been used in order to provide some evidence for a causal relationship between viral infection and cancer. A total of nine criteria were described, including the strength and consistency of association as well as experimental verification and analogy (47). In order to show the strength and consistency of the association between virus(es) and OSCC in NSND, one of them and/or its viral sequence should be detected in all tumor cells, in different studies by independent investigators. However, using three separate transcriptomic analyses, no viral sequence was found to be integrated in the tumor genome of 20 OSCC from non-smokers (18). In the TCGA cohort of our previous study (14), information on the presence of integrated viral sequences was retrieved for 22 OSCC in NSND, from a previous publication (48). DNA virus transcript was detected in only one OSCC which harbored a human herpes virus type 1. These results do not support the hypothesis of a viral integration-driven oral carcinogenesis in NSND, although it cannot be definitively excluded with regard to the small number of samples as well as the methodology of these studies. A mechanism that has been proposed by which carcinogenesis can be promoted by a viral infection without host genome integration is the “hit and run” phenomenon (18). It has been defined by the International Agency for Research on Cancer as the involvement of a virus in the initiation or promotion of cancer without being required for the maintenance of the transformed phenotype (49). It has been originally described in order to explain the oncogenic potential of herpesviruses, especially herpes simplex virus 2 (HSV-2), in the late 1970s (50) and the early 1980s (51). Further evidence of the role of the “hit and run” mechanism in oncogenesis was provided for adenovirus in the early 2000s (52), and also discussed for HPV in HNSCC (53) or gamma herpesviruses, especially EBV (54). However, a definitive experimental proof of the hit-and-run phenomenon as a mechanism of carcinogenesis is still lacking.
Herpesviruses are extremely widespread among humans and represent a large family of DNA viruses. More than 90% of adults have been infected with at least one of these, and a latent form of the virus remains in most people. There are 9 herpesviruses types known to infect humans: herpes simplex viruses 1 and 2 (HSV-1 and HSV-2), varicella-zoster virus (VZV), Epstein–Barr virus (EBV), human cytomegalovirus (CMV), human herpesvirus 6A and 6B (HHV-6A and HHV-6B), human herpesvirus 7 (HHV-7), and Kaposi's sarcoma-associated herpesvirus (HHV-8). A majority of them is transmitted by saliva, and is characterized by the latent and recurring infections. It is plausible that specific herpesviruses are involved in oral carcinogenesis, through a hit and run mechanism, when affecting individuals with a specific genetic context. In particular, oral shedding HSV-2 has been previously reported (55, 56) and changes in oral sexual behaviors (44, 45) could lead to an increasing incidence of oral infection by HSV-2 which is usually found in genital areas, but related evidence-based literature is scarce. Interestingly, HSV-2 infection has been described as more easily transmitted from men to women than from women to men (57), that would be in line with the increasing incidence of OSCC mostly observed in women, as discussed in the previous section. Moreover, the predilection for HPV-positive HNSCC to occur in palatine and lingual tonsils has been associated with local features such as anatomy of reticular crypt epithelium as well as the local lymphoid microenvironment of the oropharynx (53, 58). Similarly, in order to explain the predilection for OSCC in NSND to occur in the oral cavity, the neurotropism of herpes simplex viruses such as HSV-2 may be associated with the rich innervation (sensitive, sensory/gustation and motor) of the oral cavity involving different cranial nerves.
Besides the involvement of one specific virus in oral carcinogenesis, changes in sexual behaviors (44, 45) may also cause oral transmission of several different genital viruses as well as other microbial species, resulting in changes in the normal microbial flora of the oral cavity. Indeed, a viral-mediated deregulation of the oral microbiome has been already observed in HPV-positive HNSCC which is characterized by a different taxonomic composition of the microbiota in saliva, compared to the HPV-negative disease (22). Such changes in oral microbiome may be associated with chronic inflammation such as in periodontitis and lead to malignant transformation of the oral mucosa (19–21). A recent study has also suggested an association between periodontal pathogens and OSCC in NSND (59).
Finally, an intriguing observation is the difference in antiviral immune response between male and female in terms of prevalence, intensity and pathogenesis of viral infection. Indeed, higher innate and adaptive immune responses in female may contribute to an increased risk of immunopathology due to aberrant host inflammatory responses (60). Bringing this notion to oncogenesis may also explain the higher incidence of this cancer in female compared to male.
The evidence suggesting a causal link between herpesvirus infection and the development of OSCC is circumstantial. While the support for this relationship may grow with new studies, the molecular mechanism by which herpesviruses may trigger oral carcinogenesis is largely unknown. We propose that herpesviruses promote oral carcinogenesis, especially in NSND, through a direct, yet to be explored, hit and run mechanism involving epigenome deregulation. The epigenome plays the pivotal role in the establishment and stable propagation of gene activity states over cell generations. Epigenetic mechanisms have been implicated in modulating the gene expression programme in response to environmental exposures, including biological agents, namely viruses (61, 62). In addition, epigenetic modifications are known to play important roles in protecting against viral infection, and there is evidence that some viruses may hijack cellular machineries to promote the viral life cycle (61, 63). Therefore, epigenome reconfiguration, potentially involving DNA methylation and non-coding RNAs, and consequently gene expression reprogramming, may be induced by a transient infection by herpesvirus (“hit”) and these changes may be propagated over many cell division even after the clearance of the virus (“run”). Herpesvirus may hijack cellular defense systems by directly interacting with epigenetic (DNA methylation) machinery, thereby deregulating different key host genes and pathways via an epigenetic strategy, a scenario analogous to that suggested for different oncogenic viruses (32, 61, 64). Several factors related to the DNA methylation process, including DNA methyltransferases (DNMTs) and DNA demethylases (TET enzymes), may be involved in this process. An alternative, albeit not mutually exclusive, mechanism by which herpesvirus may deregulate the epigenome states and gene expression program operating in hit and run oncogenesis in oral mucosa, may involve interaction between viral proteins and host proteins involved in regulation of transcriptional programme, such as transcription factors. This may notably concern a subset of transcription factors (known as “pioneer transcription factors”) which have the capacity to modulate epigenetic states, through opening chromatin and inducing nucleosome remodeling, eviction or affecting DNA-nucleosome interaction thereby making an unscheduled DNA accessibility to other transcriptional factors (65). Another possibility is that the proteins encoded by herpesvirus may interact with cellular proteins that protect the genome from aberrant epigenetic modifications (such as aberrant DNA methylation) that may stably silence or upregulate the key cellular genes involved in the control of cell proliferation and genomic stability.
Identifying and characterizing potential epigenome deregulation in the herpesvirus-mediated hit and run oncogenesis should contribute to understanding the mechanism of oral carcinogenesis. This in turn should serve as the basis for development of methylation-based biomarkers that may be used for follow-up of individuals at higher risk and preventive strategies aimed at reversing early key molecular events leading to cancer development.
In conclusion, we propose an overview of epidemiological, clinical and molecular data which are in favor of the viral origin of OSCC in NSND (Figure 4). Markedly, OSCC from NSND are characterized by an antiviral DNA methylation and gene expression signature. Because viral genome integration has not been detected in OSCC from NSND, a “hit and run” viral mechanism involving epigenome deregulation could therefore play a key role at early steps of oral carcinogenesis in this population of patients. Based on the similar increasing incidence of OTSCC and OPSCC in young women and men respectively, we hypothesize that changes in sexual behaviors may lead to an increasing incidence of herpesvirus in the oral cavity, especially HSV-2, similarly to what has already been described in HPV-positive OPSCC.
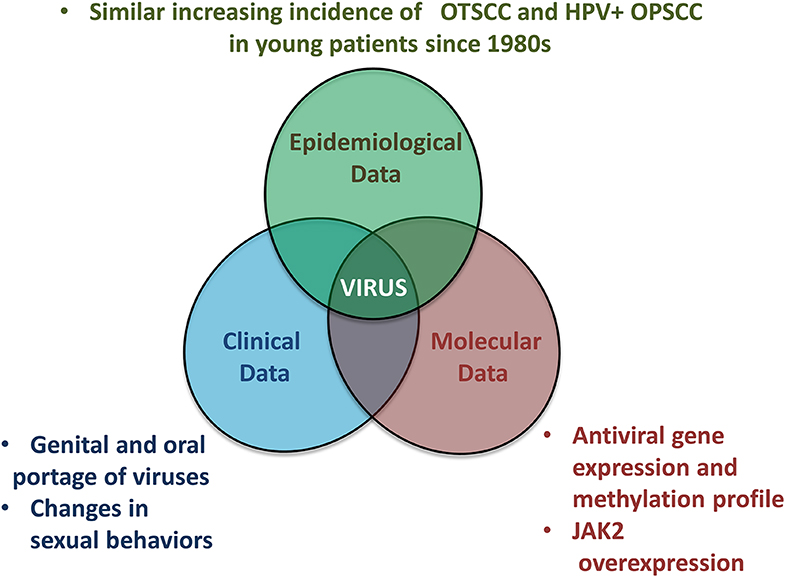
Figure 4. Overview of epidemiological, clinical and molecular evidence linking OSCC affecting non-smokers non-drinkers to a viral origin.
Publicly available datasets were analyzed in this study. This data can be found here: The Cancer Genome Atlas; Gene expression Omnibus (GSE51010).
J-PF, ZH, and PS devised the conceptual ideas and drafted the original manuscript. J-PF performed the literature search and draw the figures. CB, DB, HA, and AG contributed to review and revisions of the final draft. All authors approved the final version of the manuscript.
This work was supported by a Priorité Cancers Tabac grant (TABAC-16-025) funded by INCa, Fondation ARC and Ligue nationale contre le cancer; the LYriCAN Grant INCa-DGOS-4664; INCa PLBIO17-338 Projets libres de Recherche Biologie et Sciences du Cancer; JP-F was supported by a fellowship grant soutien pour la formation à la recherche translationnelle en cancérologie funded by l' ITMO cancer from INCa and AVIESAN, and from the Association Française pour le Développement de la Stomatologie (AFDS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. (2015) 136:E359–86. doi: 10.1002/ijc.29210
2. Hussein AA, Helder MN, de Visscher JG, Leemans CR, Braakhuis BJ, de Vet HCW, et al. Global incidence of oral and oropharynx cancer in patients younger than 45 years vs. older patients: A systematic review. Eur J Cancer. (2017) 82:115–27. doi: 10.1016/j.ejca.2017.05.026
3. Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. (2011) 29:4294–301. doi: 10.1200/JCO.2011.36.4596
4. Mork J, Lie AK, Glattre E, Hallmans G, Jellum E, Koskela P, et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med. (2001) 344:1125–31. doi: 10.1056/NEJM200104123441503
5. Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. (2000) 92:709–20. doi: 10.1093/jnci/92.9.709
6. Dahlstrom KR, Little JA, Zafereo ME, Lung M, Wei Q, Sturgis EM. Squamous cell carcinoma of the head and neck in never smoker-never drinkers: a descriptive epidemiologic study. Head Neck. (2008) 30:75–84. doi: 10.1002/hed.20664
7. Koch WM, Lango M, Sewell D, Zahurak M, Sidransky D. Head and neck cancer in nonsmokers: a distinct clinical and molecular entity. Laryngoscope. (1999) 109:1544–51. doi: 10.1097/00005537-199910000-00002
8. DeAngelis A, Breik O, Koo K, Iseli T, Nastri A, Fua T, et al. Non-smoking, non-drinking elderly females, a 5year follow-up of a clinically distinct cohort of oral squamous cell carcinoma patients. Oral Oncol. (2018) 86:113–20. doi: 10.1016/j.oraloncology.2018.09.004
9. Ng JH, Iyer NG, Tan MH, Edgren G. Changing epidemiology of oral squamous cell carcinoma of the tongue: A global study. Head Neck. (2017) 39:297–304. doi: 10.1002/hed.24589
10. Lingen MW, Xiao W, Schmitt A, Jiang B, Pickard R, Kreinbrink P, et al. Low etiologic fraction for high-risk human papillomavirus in oral cavity squamous cell carcinomas. Oral Oncol. (2013) 49:1–8. doi: 10.1016/j.oraloncology.2012.07.002
11. Patel SC, Carpenter WR, Tyree S, Couch ME, Weissler M, Hackman T, et al. Increasing incidence of oral tongue squamous cell carcinoma in young white women, age 18 to 44 years. J Clin Oncol. (2011) 29:1488–94. doi: 10.1200/JCO.2010.31.7883
12. Bertolus C, Goudot P, Gessain A, Berthet N. Clinical relevance of systematic human papillomavirus (HPV) diagnosis in oral squamous cell carcinoma. Infect Agent Cancer. (2012) 7:13. doi: 10.1186/1750-9378-7-13
13. Brennan K, Koenig JL, Gentles AJ, Sunwoo JB, Gevaert O. Identification of an atypical etiological head and neck squamous carcinoma subtype featuring the CpG island methylator phenotype. EBioMedicine. (2017) 17:223–36. doi: 10.1016/j.ebiom.2017.02.025
14. Foy JP, Bertolus C, Michallet MC, Deneuve S, Incitti R, Bendriss-Vermare N, et al. The immune microenvironment of HPV-negative oral squamous cell carcinoma from never-smokers and never-drinkers patients suggests higher clinical benefit of IDO1 and PD1/PD-L1 blockade. Ann Oncol. (2017) 28:1934–41. doi: 10.1093/annonc/mdx210
15. Lau HK, Wu ER, Chen MK, Hsieh MJ, Yang SF, Wang LY, et al. Effect of genetic variation in microRNA binding site in WNT1-inducible signaling pathway protein 1 gene on oral squamous cell carcinoma susceptibility. PLoS ONE. (2017) 12:e0176246. doi: 10.1371/journal.pone.0176246
16. Carneiro NK, Oda JM, Losi Guembarovski R, Ramos G, Oliveira BV, Cavalli IJ, et al. Possible association between TGF-beta1 polymorphism and oral cancer. Int J Immunogenet. (2013) 40:292–8. doi: 10.1111/iji.12037
17. Wu S, Powers S, Zhu W, Hannun YA. Substantial contribution of extrinsic risk factors to cancer development. Nature. (2016) 529:43–7. doi: 10.1038/nature16166
18. Li R, Faden DL, Fakhry C, Langelier C, Jiao Y, Wang Y, et al. Clinical, genomic, and metagenomic characterization of oral tongue squamous cell carcinoma in patients who do not smoke. Head Neck. (2015) 37:1642–9. doi: 10.1002/hed.23807
19. Tezal M, Sullivan MA, Hyland A, Marshall JR, Stoler D, Reid ME, et al. Chronic periodontitis and the incidence of head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. (2009) 18:2406–12. doi: 10.1158/1055-9965.EPI-09-0334
20. Schmidt BL, Kuczynski J, Bhattacharya A, Huey B, Corby PM, Queiroz EL, et al. Changes in abundance of oral microbiota associated with oral cancer. PLoS ONE. (2014) 9:e98741. doi: 10.1371/journal.pone.0098741
21. Gholizadeh P, Eslami H, Yousefi M, Asgharzadeh M, Aghazadeh M, Kafil HS. Role of oral microbiome on oral cancers, a review. Biomed Pharmacother. (2016) 84:552–8. doi: 10.1016/j.biopha.2016.09.082
22. Guerrero-Preston R, Godoy-Vitorino F, Jedlicka A, Rodriguez-Hilario A, Gonzalez H, et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, human papilloma virus infection and surgical treatment. Oncotarget. (2016) 7:51320–34. doi: 10.18632/oncotarget.9710
23. Hashibe M, Morgenstern H, Cui Y, Tashkin DP, Zhang ZF, Cozen W, et al. Marijuana use and the risk of lung and upper aerodigestive tract cancers: results of a population-based case-control study. Cancer Epidemiol Biomarkers Prev. (2006) 15:1829–34. doi: 10.1158/1055-9965.EPI-06-0330
24. Raj AT, Patil S, Sarode SC, Sarode GS, Rajkumar C. Evaluating the association between household air pollution and oral cancer. Oral Oncol. (2017) 75:178–9. doi: 10.1016/j.oraloncology.2017.11.012
25. Kjaerheim K, Haldorsen T, Lynge E, Martinsen JI, Pukkala E, Weiderpass E, et al. Variation in nordic work-related cancer risks after adjustment for alcohol and tobacco. Int J Environ Res Public Health. (2018) 15:12. doi: 10.3390/ijerph15122760
26. Cirmi S, Navarra M, Woodside JV, Cantwell MM. Citrus fruits intake and oral cancer risk: A systematic review and meta-analysis. Pharmacol Res. (2018) 133:187–94. doi: 10.1016/j.phrs.2018.05.008
27. Pickering CR, Zhang J, Neskey DM, Zhao M, Jasser SA, Wang J, et al. Squamous cell carcinoma of the oral tongue in young non-smokers is genomically similar to tumors in older smokers. Clin Cancer Res. (2014) 20:3842–8. doi: 10.1158/1078-0432.CCR-14-0565
28. Kolokythas A, Zhou Y, Schwartz JL, Adami GR. Similar squamous cell carcinoma epithelium microRNA expression in never smokers and ever smokers. PLoS ONE. (2015) 10:e0141695. doi: 10.1371/journal.pone.0141695
29. Farshadpour F, Roepman P, Hordijk GJ, Koole R, Slootweg PJ. A gene expression profile for non-smoking and non-drinking patients with head and neck cancer. Oral Dis. (2012) 18:178–83. doi: 10.1111/j.1601-0825.2011.01861.x
30. Saeed AA, Sims AH, Prime SS, Paterson I, Murray PG, Lopes VR. Gene expression profiling reveals biological pathways responsible for phenotypic heterogeneity between UK and Sri Lankan oral squamous cell carcinomas. Oral Oncol. (2015) 51:237–46. doi: 10.1016/j.oraloncology.2014.12.004
31. Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. (2014) 513:202–9. doi: 10.1038/nature13480
32. Niller HH, Wolf H, Minarovits J. Viral hit and run-oncogenesis: genetic and epigenetic scenarios. Cancer Letters. (2011) 305:200–17. doi: 10.1016/j.canlet.2010.08.007
33. Li HP, Leu YW, Chang YS. Epigenetic changes in virus-associated human cancers. Cell Res. (2005) 15:262–71. doi: 10.1038/sj.cr.7290295
34. Lleras RA, Smith RV, Adrien LR, Schlecht NF, Burk RD, Harris TM, et al. Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin Cancer Res. (2013) 19:5444–55. doi: 10.1158/1078-0432.CCR-12-3280
35. Birdwell CE, Queen KJ, Kilgore PC, Rollyson P, Trutschl M, Cvek U, et al. Genome-wide DNA methylation as an epigenetic consequence of Epstein-Barr virus infection of immortalized keratinocytes. J Virol. (2014) 88:11442–58. doi: 10.1128/JVI.00972-14
36. Hino R, Uozaki H, Murakami N, Ushiku T, Shinozaki A, Ishikawa S, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. (2009) 69:2766–74. doi: 10.1158/0008-5472.CAN-08-3070
37. Giudice A, D'Arena G, Crispo A, Tecce MF, Nocerino F, Grimaldi M, et al. Role of viral miRNAs and epigenetic modifications in epstein-barr virus-associated gastric carcinogenesis. Oxid Med Cell Longev. (2016) 2016:6021934. doi: 10.1155/2016/6021934
38. Braakhuis BJ, Leemans CR, Visser O. Incidence and survival trends of head and neck squamous cell carcinoma in the Netherlands between 1989 and 2011. Oral Oncol. (2014) 50:670–5. doi: 10.1016/j.oraloncology.2014.03.008
39. Simard EP, Torre LA, Jemal A. International trends in head and neck cancer incidence rates: differences by country, sex and anatomic site. Oral Oncol. (2014) 50:387–403. doi: 10.1016/j.oraloncology.2014.01.016
40. Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. (2009) 45:309–16. doi: 10.1016/j.oraloncology.2008.06.002
41. Surveillance Epidemiology and End Results (SEER). Program Populations (1969-2015). (2016). Available online at: www.seer.cancer.gov/popdata
42. Ng M, Freeman MK, Fleming TD, Robinson M, Dwyer-Lindgren L, Thomson B, et al. Smoking prevalence and cigarette consumption in 187 countries, 1980-2012. JAMA. (2014) 311:183–92. doi: 10.1001/jama.2013.284692
43. Shiboski CH, Schmidt BL, Jordan RC. Tongue and tonsil carcinoma: increasing trends in the US population ages 20-44 years. Cancer. (2005) 103:1843–9. doi: 10.1002/cncr.20998
44. D'Souza G, Agrawal Y, Halpern J, Bodison S, Gillison ML. Oral sexual behaviors associated with prevalent oral human papillomavirus infection. J Infect Dis. (2009) 199:1263–9. doi: 10.1086/597755
45. Bajos N, Bozon M, Beltzer N, Laborde C, Andro A, Ferrand M, et al. Changes in sexual behaviours: from secular trends to public health policies. AIDS. (2010) 24:1185–91. doi: 10.1097/QAD.0b013e328336ad52
46. Giuliano AR, Nyitray AG, Kreimer AR, Pierce Campbell CM, Goodman MT, Sudenga SL, et al. EUROGIN 2014 roadmap: differences in human papillomavirus infection natural history, transmission and human papillomavirus-related cancer incidence by gender and anatomic site of infection. Int J Cancer. (2015) 136:2752–60. doi: 10.1002/ijc.29082
47. Sarid R, Gao SJ. Viruses and human cancer: from detection to causality. Cancer Lett. (2011) 305:218–27. doi: 10.1016/j.canlet.2010.09.011
48. Khoury JD, Tannir NM, Williams MD, Chen Y, Yao H, Zhang J, et al. Landscape of DNA virus associations across human malignant cancers: analysis of 3,775 cases using RNA-Seq. J Virol. (2013) 87:8916–26. doi: 10.1128/JVI.00340-13
49. Humans IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Human Papillomaviruses. (Lyon: International Agency for Research on Cancer) (2007).
50. Skinner GR. Transformation of primary hamster embryo fibroblasts by type 2 simplex virus: evidence for a “hit and run” mechanism. Br J Exp Pathol. (1976) 57:361–76.
51. Galloway DA, McDougall JK. The oncogenic potential of herpes simplex viruses: evidence for a 'hit-and-run' mechanism. Nature. (1983) 302:21–4.
52. Nevels M, Tauber B, Spruss T, Wolf H, Dobner T. “Hit-and-run” transformation by adenovirus oncogenes. J Virol. (2001) 75:3089–94. doi: 10.1128/JVI.75.7.3089-3094.2001
53. Rautava J, Syrjanen S. Biology of human papillomavirus infections in head and neck carcinogenesis. Head Neck Pathol. (2012) 6:S3–15. doi: 10.1007/s12105-012-0367-2
54. Ambinder RF. Gammaherpesviruses and “Hit-and-Run” oncogenesis. Am J Pathol. (2000) 156:1–3. doi: 10.1016/S0002-9440(10)64697-4
55. Wald A, Ericsson M, Krantz E, Selke S, Corey L. Oral shedding of herpes simplex virus type 2. Sex Transm Infect. (2004) 80:272–6. doi: 10.1136/sti.2003.007823
56. Aggarwal R, Bansal D, Naru J, Salaria M, Rana A, Minz RW, et al. HSV-1 as well as HSV-2 is frequent in oral mucosal lesions of children on chemotherapy. Support Care Cancer. (2014) 22:1773–9. doi: 10.1007/s00520-014-2152-0
57. Wald A, Langenberg AG, Link K, Izu AE, Ashley R, Warren T, et al. Effect of condoms on reducing the transmission of herpes simplex virus type 2 from men to women. JAMA. (2001) 285:3100–6. doi: 10.1001/jama.285.24.3100
58. Franceschi S, Munoz N, Snijders PJ. How strong and how wide is the link between HPV and oropharyngeal cancer? Lancet. (2000) 356:871–2. doi: 10.1016/S0140-6736(00)02673-8
59. Ganly I, Yang L, Giese RA, Hao Y, Nossa CW, Morris LGT, et al. Periodontal pathogens are a risk factor of oral cavity squamous cell carcinoma, independent of tobacco and alcohol and human papillomavirus. Int J Cancer. (2019) 145:775–784. doi: 10.1002/ijc.32152
60. Klein SL. Sex influences immune responses to viruses, and efficacy of prophylaxis and treatments for viral diseases. Bioessays. (2012) 34:1050–9. doi: 10.1002/bies.201200099
61. Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: how environmental factors influence the epigenome. Mutat Res. (2011) 727:55–61. doi: 10.1016/j.mrrev.2011.04.001
62. Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. (2012) 13:97–109. doi: 10.1038/nrg3142
63. van Zuylen WJ, Rawlinson WD, Ford CE. The Wnt pathway: a key network in cell signalling dysregulated by viruses. Rev Med Virol. (2016) 26:340–55. doi: 10.1002/rmv.1892
64. Kaur P, Paliwal A, Durantel D, Hainaut P, Scoazec JY, Zoulim F, et al. DNA methylation of hepatitis B virus (HBV) genome associated with the development of hepatocellular carcinoma and occult HBV infection. J Infect Dis. (2010) 202:700–4. doi: 10.1086/655398
Keywords: herpesvirus, HSV-2, EBV, oral squamous cell carcinoma, non-smoker non-drinker, hit and run
Citation: Foy J-P, Bertolus C, Boutolleau D, Agut H, Gessain A, Herceg Z and Saintigny P (2020) Arguments to Support a Viral Origin of Oral Squamous Cell Carcinoma in Non-Smoker and Non-Drinker Patients. Front. Oncol. 10:822. doi: 10.3389/fonc.2020.00822
Received: 14 July 2019; Accepted: 28 April 2020;
Published: 21 May 2020.
Edited by:
Jan Baptist Vermorken, University of Antwerp, BelgiumReviewed by:
Gemma Gatta, Istituto Nazionale dei Tumori (IRCCS), ItalyCopyright © 2020 Foy, Bertolus, Boutolleau, Agut, Gessain, Herceg and Saintigny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierre Saintigny, cGllcnJlLnNhaW50aWdueUBseW9uLnVuaWNhbmNlci5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.