 Wenzheng Xia1†
Wenzheng Xia1† Meng Hou
Meng Hou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 27 May 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00821
This article is part of the Research TopicCell Signaling Mediating Critical Radiation ResponsesView all 15 articles
Glioblastoma (GBM) is the most common and lethal brain tumor in adults. Ionizing radiation (IR) is a standard treatment for GBM patients and results in DNA damage. However, the clinical efficacy of IR is limited due to therapeutic resistance. The programmed death ligand 1 (PD-L1) blockade has a shown the potential to increase the efficacy of radiotherapy by inhibiting DNA damage and repair responses. The miR-33a-5p is an essential microRNA that promotes GBM growth and self-renewal. In this study, we investigated whether a PD-L1 inhibitor (a small molecule inhibitor) exerted radio-sensitive effects to impart an anti-tumor function in GBM cells by modulating miR-33a-5p. U87 MG cells and U251 cells were pretreated with PD-L1 inhibitor. The PD-L1 inhibitor-induced radio-sensitivity in these cells was assessed by assaying cellular apoptosis, clonogenic survival assays, and migration. TargetScan and luciferase assay showed that miR-33a-5p targeted the phosphatase and tensin homolog (PTEN) 3′ untranslated region. The expression level of PTEN was measured by western blotting, and was also silenced using small interfering RNAs. The levels of DNA damage following radiation was measured by the presence of γ-H2AX foci, cell cycle, and the mRNA of the DNA damage-related genes, BRCA1, NBS1, RAD50, and MRE11. Our results demonstrated that the PD-L1 inhibitor significantly decreased the expression of the target gene, miR-33a-5p. In addition, pretreatment of U87 MG and U251 cells with the PD-L1 inhibitor increased radio-sensitivity, as indicated by increased apoptosis, while decreased survival and migration of GBM cells. Mir-33a-5p overexpression or silencing PTEN in U87 MG and U251 cells significantly attenuated PD-L1 radiosensitive effect. Additionally, PD-L1 inhibitor treatment suppressed the expression of the DNA damage response-related genes, BRCA1, NBS1, RAD50, and MRE11. Our results demonstrated a novel role for the PD-L1 inhibitor in inducing radio- sensitivity in GBM cells, where inhibiting miR-33a-5p, leading to PTEN activated, and inducing DNA damage was crucial for antitumor immunotherapies to treat GBM.
Glioblastomas (GBM) are one of the most treatment-resistant tumors, often recurring after chemotherapy and radiation treatment (1). Amount of effort has been taken to identify therapeutics that radio-sensitize GBMs because most patients will receive radiation treatment (2). However, it's difficult to identify such radiosensitive chemotherapeutic agents because GBMs exhibit redundant pro-growth and pro-survival pathways, leading to chemotherapy resistance (3). To overcome such resistance, it's needed to devise therapeutic strategies targeting such redundant treatment-resistant pathways to increase the radiosensitivity of GBMs.
Programmed death ligand 1 (PD-L1)regulates the immune system by binding the programmed cell death protein 1 (PD-1) receptor as an immune checkpoint protein (4, 5). By combining with PD-1 on immune cells, PD-L1 helps tumor cells escape from the immune system and survive (6). Therefore, abrogation of the PD-1 and PD-L1 interaction has acted as an effective therapeutic strategy to enhance antitumor immunity across multiple malignancies. The immune checkpoint blockade has shown a therapeutic effect in immunosuppressive GBMs (7). With respect to radiosensitization in colorectal carcinoma and breast cancer cell lines, knockdown of PD-L1 sensitizes cells to radiotherapy (8). Although the impact of the PD-L1 blockade on radiosensitization has been suggested in GBM, its role has yet to be fully elucidated.
MicroRNAs (miRNAs) are a series of small, noncoding RNA molecules, typically about 18–22 nucleotides in length in the mature form (9). miRNAs negatively regulate gene expression at the post-transcriptional level by inhibiting mRNA translation and/or promoting mRNA degradation (10). In recent years, abundant miRNAs have been found to be deregulated in many types of cancer: some function as tumor suppressors and others as tumor promoters (11). The miR-33a-5p is located on chromosome 22, and its high expression is related with the poor prognosis of GBM patients (12). miR-33a-5p influences the radiation resistance-associated pathway protein, STAT3, leading to radiation resistance in GBM cells. Importantly, miR-33a-5p is also an essential component of the PTEN regulatory network (13).
PTEN is a critical inhibitor of cell proliferation, viability, and migration in GBMs (14, 15). Following the treatment of cells with PD-1 inhibitors (e.g., pembrolizumab), genomic and transcriptomic analyses revealed a significant accumulation of PTEN mutations, leading to an immunotherapeutic response in GBMs (16). Thus, in the present study, we sought to reveal whether the PD-L1 inhibitor could influence the radiosensitivity of GBM cells by modulating the miR-33a-5p/PTEN pathway.
Genetic alterations involved in GBM progression or recurrence have close relationships with DNA damage response (DDR) (17). DDR contributes to malignancy by regulating diverse cellular functions, including cell metabolism, proliferation and programmed cell death (18). Importantly, the DDR induces chemo- or radio-resistance in GBM (19). Thus, targeting the DDR could promote the growth-suppressive effects of radiation (20). Immune checkpoint blockade using a PD-L1 antagonist targeted the DDR and induced radiosensitization in tumor cells (8). Thus, direct pharmacological targeting of PD-L1 is an attractive approach for sensitizing GBMs to radiation.
Here, we suggested that immune checkpoint blockade using the PD-L1 inhibitor was a potent therapy for GBM radio-sensitization. Furthermore, we showed that the PD-L1 inhibitor induced radiosensitization by modulating the miR-33a-5p/PTEN pathway. Thus, we propose that the immune checkpoint blockade is a promising treatment strategy for GBM radiosensitization.
The U87 MG human glioblastoma cell line and U251 cells were obtained from the American Type Culture Collection (ATCC). They were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic solution at 37°C with 5% CO2. Experiments were performed with cells grown to 70% confluency.
For the PD-L1 blockade, cells were fed media containing 4 mg/mL PD-L1 inhibitor (Abcam, ab230369) and incubated at 37°C, as previously described (21).
Cells were placed in a cesium-137 source irradiator. Cells were irradiated using a single dose of 10 Gy, as previously reported (22).
Cells were harvested using RIPA buffer (Sigma-Aldrich). Proteins were separated on 10% SDS-PAGE gels and transferred to polyvinylidene difluoride membranes. The membranes were incubated overnight with the following primary antibodies: anti-PD-L1 (ab205921,1:500), anti-PTEN (ab32199, 1:750), anti-gamma H2AX (ab2893, 1:500), and anti-β-actin (ab8227, 1:1000). Following incubation, membranes were washed, incubated for 1 h with appropriate secondary antibodies conjugated to horseradish peroxidase, and developed using chemiluminescent substrates. The stained protein bands were visualized using a Bio-Rad ChemiDoc XRS instrument, and quantified and analyzed using the Quantity One software.
The extent of apoptotic cell death was assayed using the Annexin V-FITC Apoptosis Detection Kit, according to the manufacturer's instructions. Briefly, cells were harvested and washed in ice-cold phosphate-buffered saline (PBS), resuspended in 300 μL of binding buffer and incubated with 5 μL of Annexin V-FITC solution for 30 min at 4°C in the dark. This was followed by incubation with 5 μL of propidium iodide (PI) for 5 min. The samples were immediately analyzed by bivariate flow cytometry on the BD FACSCanto II instrument, equipped with Cell Quest software (BD). Approximately 1-5 × 105 cells were analyzed in each sample (23).
A colony formation (clonogenic) assay was used in order to determine cell survival. Briefly, remaining U87 MG cells and U251 cells after indicated treatments were trypsinized and plated into 6-well plates. The density of per well is 1,500 cells. Colonies were allowed to grow (~9–10 days). Fixed cells were then stained with a 0.5% (v/v) crystal violet (Sigma-Aldrich) solution. Only colonies consisting of ≥50 cells were counted as previously described (24).
DAPI labeled U87 MG cells and U251 cells were plated in the upper compartment of 0.8 μm Transwell chambers. After 6–8 h migration to the underside of the top chamber, a fluorescence microscope was applied to evaluate the migration of the fluorescently-labeled U87 MG cells and U251 cells. Each experiment was performed in triplicate.
Cells were immediately lysed in 500 μl TRIzol reagent (ThermoFisher Scientific) and stored at −80°C before purification using a standard phenol-chloroform extraction protocol with the RNAqueous Micro Kit (ThermoFisher Scientific). The transcriptome was analyzed using an Affymetrix human microarray (ThermoFisher Scientific) and normalized based on quantiles.
Total RNA from clonal cells was isolated using RNeasy spin columns (Qiagen), per the manufacturer's protocol. For reverse-transcription reactions, first-strand cDNA was synthesized using Superscript reverse transcriptase (Invitrogen), per the manufacturer's protocol. TaqMan probes (Applied Biosystems) were used to estimate the level of gene expression of miR-33a-5p, PTEN, BRCA1, NBS1, RAD50, and MRE11. GAPDH and U6 were used as housekeeping genes (25). The primer sets (Invitrogen) used are listed in Table 1.

Table 1. Primer sequences.
The 3′-UTR of PTEN was synthesized, annealed, and inserted into the SacI and HindIII sites of the pMIR-reporter luciferase vector (Ambion), downstream of the luciferase stop codon. To induce mutagenesis, the sequences complementary to the binding sites of miR-33a-5p in the 3′-UTR of PTEN (gguuuUGCUACUCUAAUGCAu) were replaced by gguuuUGCUACUCUUUACGUu. The constructs were validated by sequencing. U87 MG cells and U251 cells were seeded into a 24-well plate to perform the luciferase assay. After overnight culture, cells were co-transfected with the wild-type or mutated plasmid, and equal amounts of the miR-33a-5p mimic or the miR- negative control mimic (miR-NC mimic). Luciferase assays were performed using the Dual Luciferase Reporter Assay System (Promega) 24 h after transfection.
Before transfection with the miR-33a-5p mimic and negative control (NC) mimic, U87 MG cells and U251 cells were seeded into 6-well plates at a density of 1 × 105 cells per well, and incubated for 12 h. For the overexpression of miR-33a-5p, cells were transfected with the miR-33a-5p mimic or the NC mimic (Invitrogen, Carlsbad, CA, USA) using the X-treme transfection reagent (Roche Applied Science, Penzberg, Germany), according to the manufacturer's protocol. Forty-eight hours after transfection, cells were harvested for further analysis. The transfection efficiency was analyzed by qRT-PCR.
The siRNAs were applied to knock down PTEN expression in U87 MG cells and U251 cells. A non-targeting siRNA was used as a negative control (Invitrogen). The target sequences were as follows: PTEN: 5′-AAAGAGATCGTTAGCAGAA-3′; and Control: 5′-ACACGTCCGAACATACTAC-3′. Transfection efficiency was detected by qRT-PCR and western blotting.
Briefly, cells were fixed by 3% paraformaldehyde and then permeabilized with 0.5% Triton X-100. Cells were then blocked by 5% goat serum, followed by incubation with primary antibodies. Cells were then incubated with fluorescent secondary antibodies and DAPI to stain the targeted proteins and the nucleus, respectively (8). The cells were then analyzed by fluorescence microscopy.
Further, 70% cold anhydrous ethanol was applied to fix the cells. Then, the cells were treated with propidium iodide (PI) (Sigma, St. Louise, MO, USA) and RNase A. A flow cytometer equipped with Cell Quest software was used to detect the cell cycle distribution.
Data were expressed as the mean ± standard deviation (SD). Differences among groups were tested by one-way analysis of variance, and comparisons between two groups were evaluated by the Student's t-test, using the SPSS package v19.0 (SPSS Inc., Chicago, IL, USA). A P-value of < 0.05 was considered statistically significant.
To determine whether the PD-L1 inhibitor would radiosensitize U87 MG cells and U251 cells, we first examined the expression of PD-L1 in the U87 MG cells and U251 cells under radiotherapy conditions. The results suggested that radiation induced PD-L1 expression (Figures 1A,B). We then administered the PD-L1 inhibitor two hours before radiation treatment and found that U87 MG cells and U251 cells were sensitized, as indicated by increased apoptosis (Figures 1C–E) and decreased cellular survival (Figures 1F,G). Cellular migration of the U87 MG cells and U251 cells was also inhibited by PD-L1 inhibitor (Figures 1H–J).
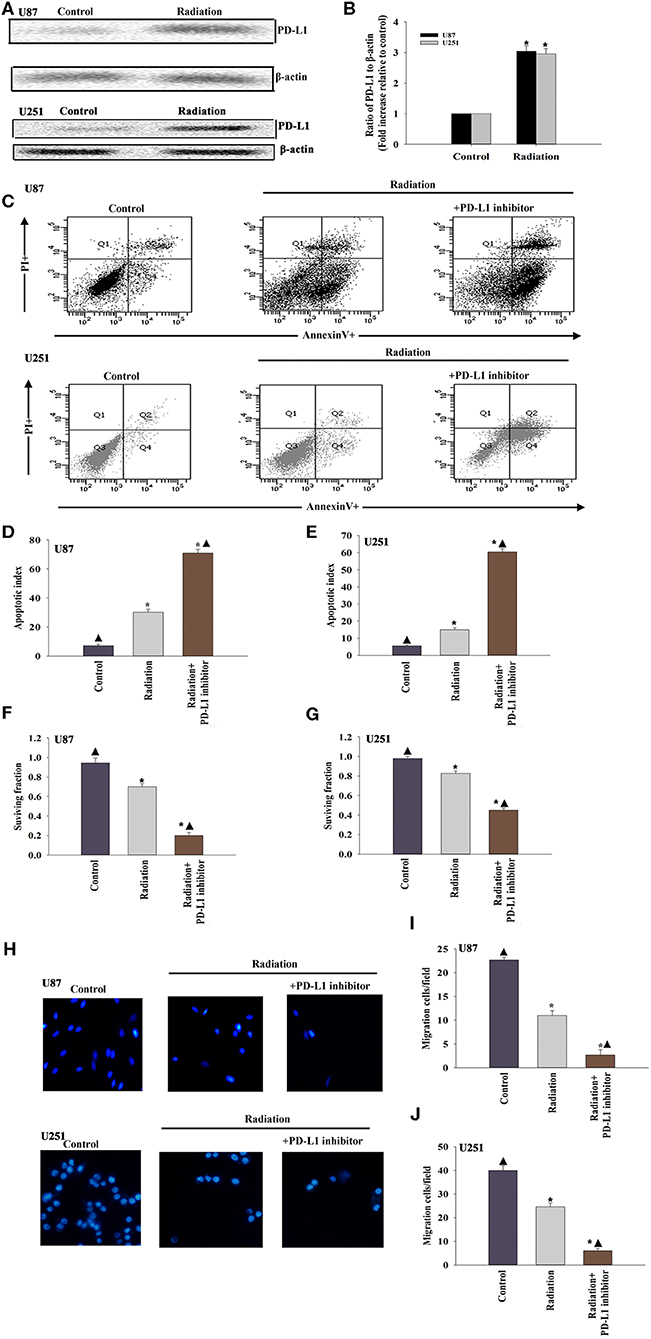
Figure 1. The immune checkpoint inhibitor sensitizes GBM to radiotherapy. (A,B) Representative images of western blots of PD-L1 and β-actin in radiated or untreated U87 MG cells and U251 cells. Fold changes were normalized to β-actin. Each column represents the mean ± SD from three independent experiments; *P < 0.05, vs. Control. U87 MG cells and U251 cells were subjected to radiation, with or without PD-L1-inhibitor treatment. Untreated U87 MG cells and U251 cells were used as the control separately. (C) Representative distributions of PI and Annexin V staining from FACScan flow cytometric analyses of apoptotic cells. (D,E) Percentage of apoptotic cells in above conditions. (F,G) Colony formation was presented as a bar graph in the U87 MG cells and U251 cells. (H) Fluorescence microscope images of the migrated U87 MG cells and U251 cells. (I,J) Data are presented as the number of migrated cells. Each column represents the mean ± SD from three independent experiments; *P < 0.05, vs. Control; ▴P < 0.05, vs. Radiation.
To examine the effect of miRNAs in the PD-L1 inhibitor-induced sensitization to radiotherapy, miRNA microarray probes were used. We found that the expression of specific miRNAs in–PD-L1 inhibitor treated before radiated U87 MG cells was significantly altered when compared with that in only radiated cells. Among them, miR-33a-5p was significantly downregulated in the PD-L1 inhibitor group, and therefore, we selected the down-regulated miR-33a-5p and verified the expression level using real-time PCR. The results showed that radiation induced increasing expression of miR-33a-5p, compared to the untreated cells. While, PD-L1 inhibitor decreased the expression of the miR-33a-5p (Figures 2A,B).
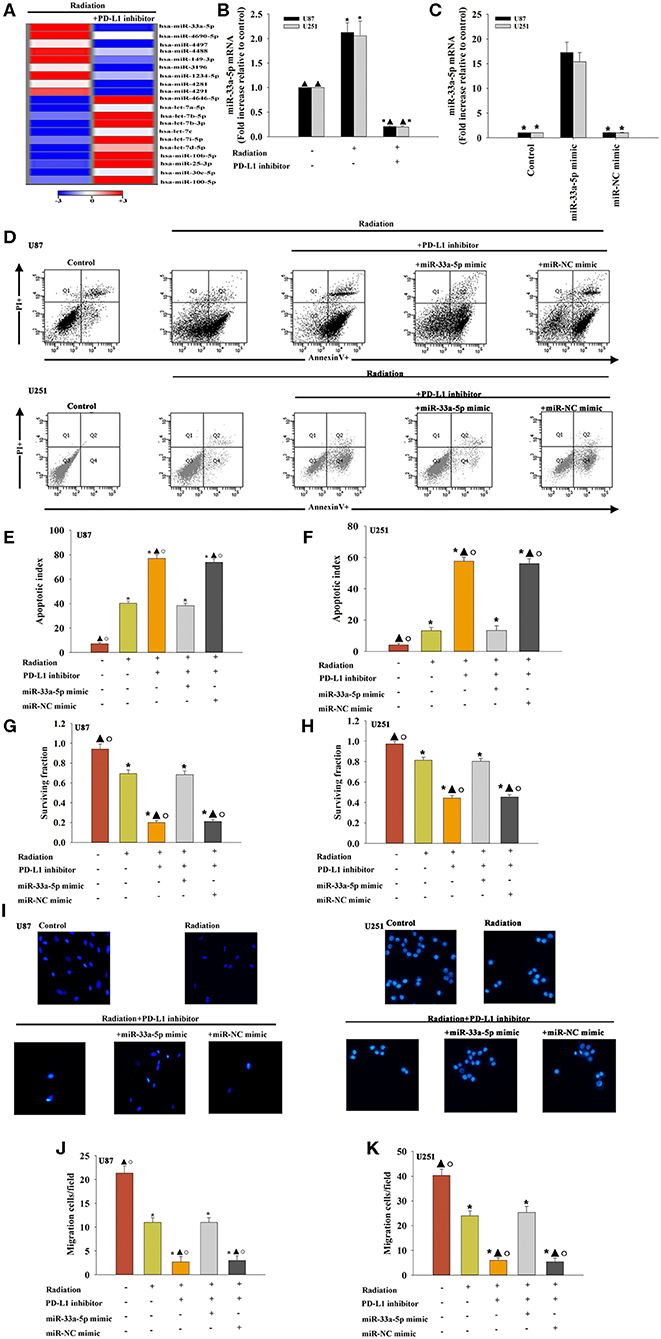
Figure 2. Effect of the PD-L1 inhibitor on miRNA expression in glioma cells. (A) Heat map of miRNAs differentially regulated by the PD-L1 inhibitor in radiated U87 MG cells. “Red” indicates up-regulation, and “blue” indicates down-regulation. (B) RT-qPCR validation of the differentially regulated miRNAs in U87 MG cells and U251 cells treated with radiation, with or without PD-L1 inhibitor pre-treatment. Untreated U87 MG cells and U251 cells were used as the control separately. *P < 0.05, vs. Control; ▴P < 0.05, vs. Radiation. U87 MG cells and U251 cells were transfected with a mimic control or the miR-33a-5p mimic, treated with the PD-L1 inhibitor, and subjected to radiation. In parallel, U87 MG cells and U251 cells, treated or untreated with the PD-L1 inhibitor were radiated. Un-treated U87 MG cells and U251 cells were used as the control separately. (C) The transfection efficiency was analyzed by qRT-PCR; *P < 0.05, vs. the miR-33a-5p mimic. (D) Representative distributions of PI and Annexin V staining from FACScan flow cytometric analyses of apoptotic cells. (E,F) Apoptotic cells in the above conditions. (G,H) Colony formation was presented as a bar graph in the U87 MG cells and U251 cells. (I) Fluorescence microscope images of the migrated U87 MG cells and U251 cells. (J,K) Data are presented as the number of migrated cells. Each column represents the mean ± SD from three independent experiments; *P < 0.05, vs. Control; ▴P < 0.05, vs. Radiation; °P < 0.05, vs. Radiation + PD-L1 inhibitor + miR-33a-5p mimic.
To determine the role of miR-33a-5p in PD-L1 inhibitor-induced radio-sensitization, U87 MG cells and U251 cells were transfected with the miR-33a-5p mimic (Figure 2C), and the negative control before treatment with the PD-L1 inhibitor. Compared to the radiation only group, treatment with the PD-L1 inhibitor induced more cellular apoptosis, while the overexpression of miR-33a-5p significantly inhibited apoptosis (Figures 2D–F). PD-L1 inhibitor treatment reduced the U87 MG cells and U251 cells surviving fraction compared to the radiation-only group; however, the overexpression of miR-33a-5p promoted the survival of U87 MG cells and U251 cells (Figures 2G,H). Transfection with the miR-33a-5p mimic dramatically promoted cell migration as well (Figures 2I–K). No apparent changes in the control miR-mimic were detected.
To further explore the molecular action of miR-33a-5p in GBM tissues, we searched for potential miR-33a-5p targets using TargetScan. Bioinformatics databases were used to check the potential targets. PTEN is considered to be a putative target of miR-33a-5p (Figure 3A). Thus, GBM cells were co-transfected with a wild-type PTEN-luciferase reporter vector, together with the miR-33a-5p mimic or the miR-NC mimic, and tested luciferase activity. miR-33a-5p-transfected cells showed a remarkable reduction of luciferase activities from the PTEN reporter in U87 MG cells and U251 cells (Figures 3B,C). At meanwhile, compared with the mutated 3′-UTR, the luciferase activities of the cells transfected with the wild-type 3′-UTR showed significant reductions (Figures 3B,C). Next, western blot analyses were performed to evaluate PTEN protein expression. We found that the expression of PTEN was downregulated in the radiation group, while it was increased following treatment with the PD-L1 inhibitor. Overexpression of miR-33a-5p reversed the inducement of PD-L1 inhibitor (Figures 3D–F).
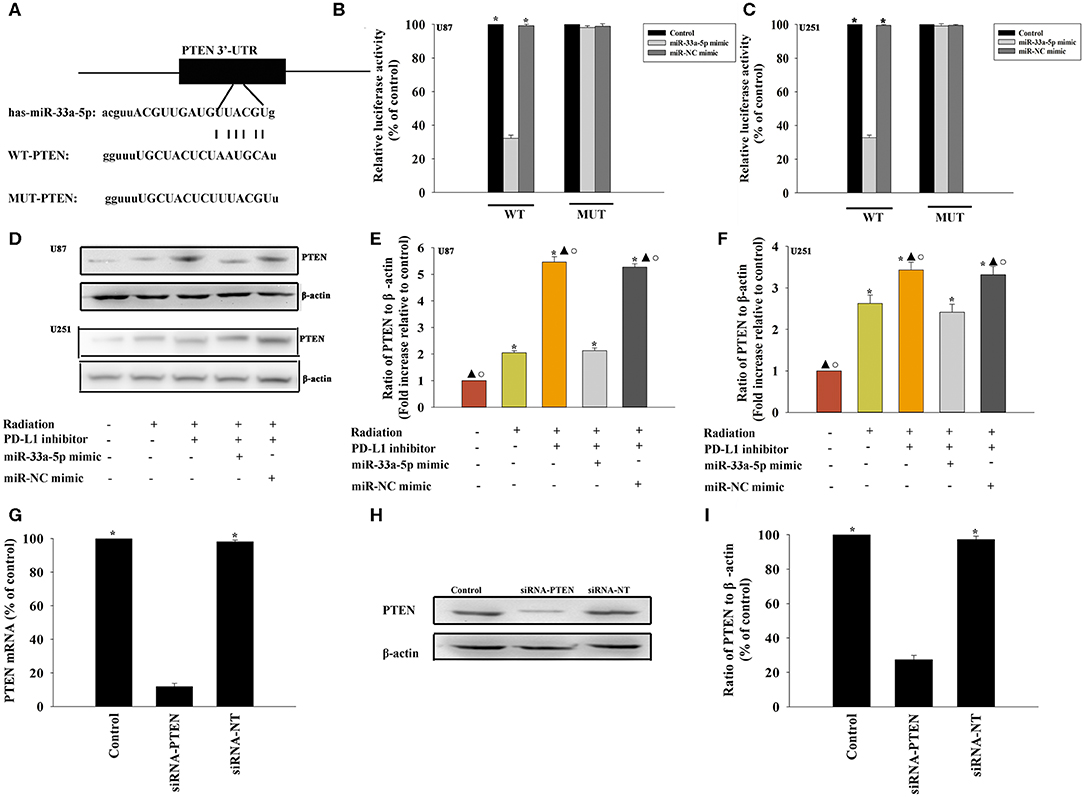
Figure 3. PTEN is a direct target of miR-33a-5p. (A) The predicted binding sites between miR-33a-5p and the PTEN 3′-UTR. (B,C) A dual luciferase assay was performed in U87 MG cells and U251 cells after co-transfection with PTEN 3′-UTR wild type (WT) or mutant (MUT) plasmids, miR-33a-5p mimics, and miR-NC mimic. *P < 0.05, vs. the miR-33a-5p mimic in the WT group; (D–F) western blot analysis of PTEN and β-actin protein levels in follow cells; U87 MG cells and U251 cells were transfected with a mimic control or the miR-33a-5p mimic, treated with the PD-L1 inhibitor, and subjected to radiation. In parallel, U87 MG cells and U251 cells, treated or untreated with the PD-L1 inhibitor were radiated. Untreated U87 MG cells and U251 cells were used as the control separately. *P < 0.05, vs. Control; ▴P < 0.05, vs. Radiation; °P < 0.05, vs. Radiation + PD-L1 inhibitor + miR-33a-5p mimic. (G–I) U87 MG cells were transfected with siRNA-PTEN, or with siRNA-NT as a control. siRNA-mediated transfection efficiency was determined by qRT-PCR (G) and western blotting (H,I). Each column represents the mean ± SD from three independent experiments; *P < 0.05, vs. siRNA-PTEN.
A previous study demonstrated that PTEN mediated the DNA damage response to radiosensitize high-grade gliomas (26). To assess whether the downregulation of PTEN reversed the PD-L1 inhibitor-mediated radio-sensitization, we used siRNA-PTEN to down regulate PTEN expression (Figures 3G–I). To test whether the PD-L1 inhibitor and PTEN have a role in cell apoptosis following radiation, we performed FACS analysis to determine the rates of cell apoptosis. The combined treatment of the PD-L1 inhibitor + radiation significantly increased cell apoptosis, compared to that observed following radiation only. As expected, the silencing of PTEN partially abolished the effect caused by PD-L1 inhibitor + radiation treatment (Figures 4A–C). Similarly, we also found that silencing PTEN partially reversed the inhibition of survival that was induced by the PD-L1 inhibitor + radiation treatment (Figures 4D,E). Our results also showed that PTEN silencing reversed the inhibition of U87 MG cells and U251 cells migration that was induced by the PD-L1 inhibitor (Figures 4F–H).
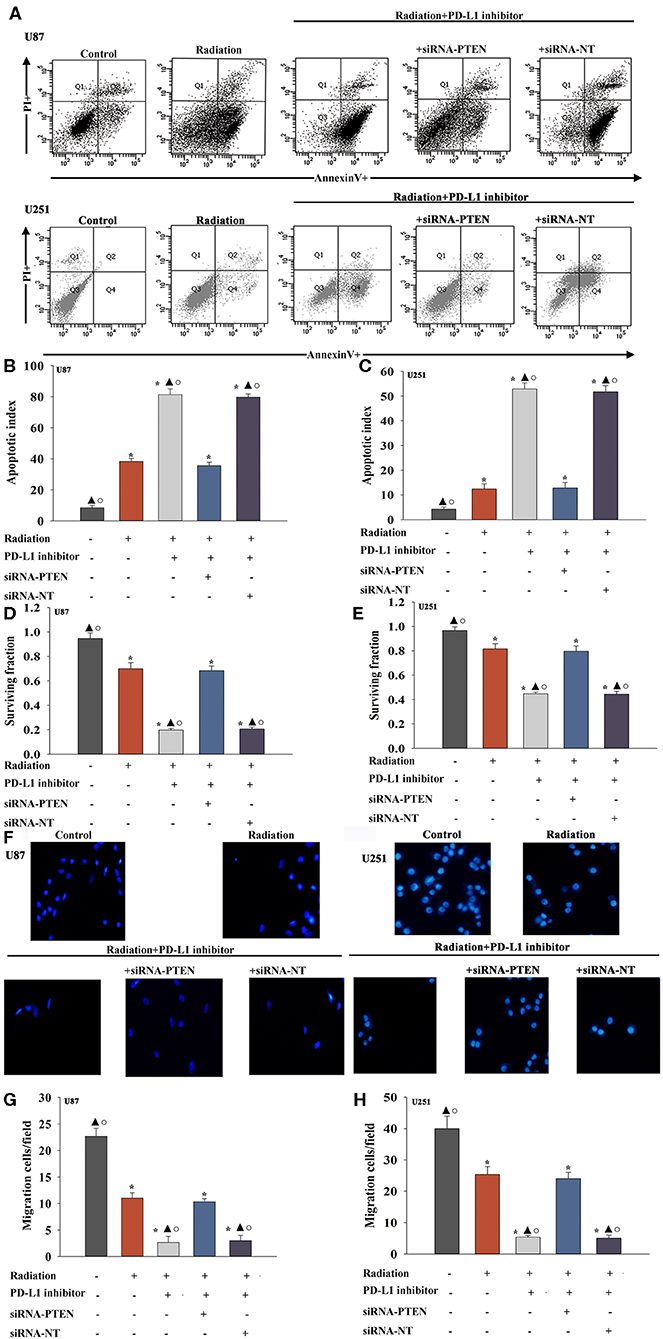
Figure 4. PD-L1 inhibitor confers radio-sensitization by targeting PTEN. U87 MG cells and U251 cells were transfected with siRNA against PTEN, or with siRNA-NT as a control, followed by treatment with the PD-L1 inhibitor and radiation. In parallel experiments, U87 MG and U251 cells were treated with radiation alone, or treated with the PD-L1 inhibitor in the presence of radiation. U87 MG and U251 cells under normal culture conditions were used as the control separately. (A) Representative distributions of PI and Annexin V staining from FACScan flow cytometric analyses of apoptotic cells. (B,C) Apoptotic cells in the above conditions. (D,E) Colony formation was presented as a bar graph in the U87 MG cells and U251 cells. (F) Fluorescence microscope images of the migrated U87 MG cells and U251 cells. (G,H) Data are presented as the number of migrated cells. Each column represents the mean ± SD from three independent experiments; *P < 0.05, vs. Control; ▴P < 0.05, vs. Radiation; °P < 0.05, vs. Radiation + PD-L1 inhibitor + siRNA-PTEN.
We evaluated whether DNA repair could be impaired in cells treated with the PD-L1 inhibitor. Immunofluorescence was used to examine the kinetics of radiation-induced γ-H2AX foci following PD-L1 inhibitor treatment, which are indication of DNA double-strand breaks (Figure 5A). After PD-L1 inhibitor treatment, cells had higher basal levels of γ-H2AX, trapping in S-phase (Figures 5B,C), which was likely the result of the decreased repair of spontaneous DNA damage. PD-L1 inhibitor +radiation also increased the expression of γ-H2AX, compared with radiation alone (Figures 5D,E). However, either miR-33a-5p overexpression or silencing PTEN, could abolished the inducement of DNA damage by PD-L1 inhibitor (Figures 5A–E).
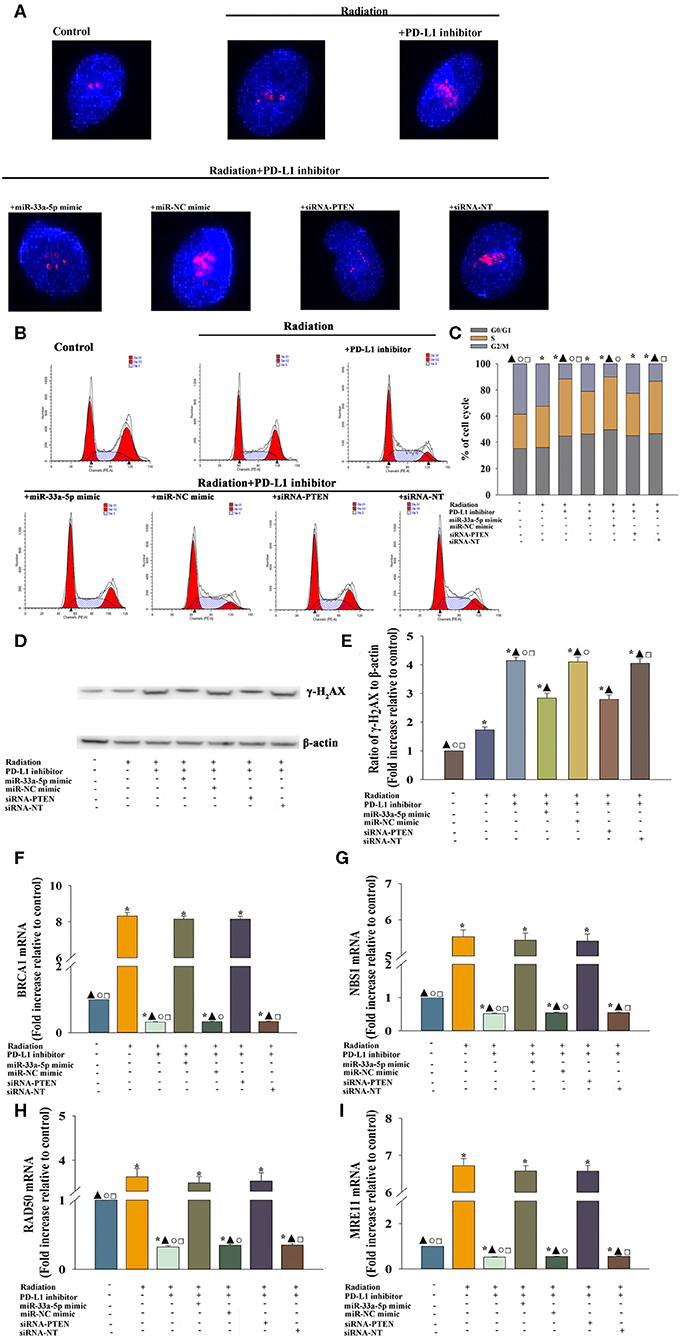
Figure 5. PD-L1 inhibitor-induced DNA damage and the reduced DNA damage response to take radiosensitization effect. U87 MG cells were transfected with siRNA against PTEN, or with siRNA-NT as a control, the miR-33a-5p mimic, or the miR-mimic control. Following transfection, cells were treated with the PD-L1 inhibitor in the presence of radiation. In parallel experiments, U87 MG were treated with radiation, or the PD-L1 inhibitor in the presence of radiation. U87 MG cells under normal culture conditions were used as the control. (A) The formation and resolution of γ-H2AX foci were assessed using immunofluorescence. (B,C) Cell cycle distribution was analyzed. (D,E) Western blot analysis of γ-H2AX and β-actin protein levels. (F–I) qRT-PCR analysis of BRCA1 (F); NBS1 (G); RAD50 (H); and MRE11 (I). *P < 0.05, vs. Control; ▴P < 0.05, vs. Radiation; °P < 0.05, vs. Radiation + PD-L1 inhibitor + miR-33a-5p mimic. □P < 0.05, vs. Radiation + PD-L1 inhibitor + siRNA-PTEN.
Then we searched to explore mechanistic insight into how the PD-L1 inhibitor affected the DDR. Thus, we assessed whether the important DDR pathway genes was affected by the PD-L1 inhibitor in U87 MG cells. We found that the expression of the DDR-related genes, BRCA1 (Figure 5F), NBS1 (Figure 5G), RAD50 (Figure 5H), and MRE11 (Figure 5I) were decreased following treatment with the PD-L1 inhibitor + radiation, compared to the levels of expression observed following radiation only. Additionally, there was a particularly reversed effect of DDR in the miR-33a-5p-overexpression or PTEN-silencing groups (Figures 5F–I).
GBM is the most common malignant primary central nervous system (CNS) tumor in adults, and is resistant to current therapies (27). Current evidence points toward the existence of a small fraction of tumor cells in the bulk tumor that also exhibit radio-resistant properties (28). Glioblastoma has been extensively studied as a paradigm for cancer-associated immunosuppression (29). Mount of immunosuppressive factors were existed on the glioma cell surface (30, 31). Importantly, PD-L1 was upregulated in the GBM microenvironment; thus, protecting GBM from T-cell killing (29). Our results revealed that GBM cells expressed PD-L1, and that radiation induced PD-L1 expression beyond that observed without radiation. H1A is a PD-L1 antibody that destabilizes PD-L1 by disrupting its binding with the PD-L1 stabilizer. Such destabilization results in greater PD-L1 degradation through the lysosome and sensitivity to radiotherapy is increased (8). Our results suggested that the inhibition of PD-L1, using a small molecular inhibitor, increased radio-sensitivity, which was indicated by increased apoptosis, decreased survive, and impaired migration in U87 MG cells and U251 cells.
In this study, our group is the first to report that the PD-L1 inhibitor repressed miR-33a-5p activity. We also demonstrated that the PD-L1 inhibitor was able to induce the expression of the PTEN by inhibiting miR-33a-5p, and further confer radiosensitization in GBM cells. To maintain the primary biological features of those cells, including stemness, self-renewal, and tumor initiation in vivo, the higher level of miR-33a-5p expression in GBM is required (32). Extensive research has been performed to demonstrate the important roles of miR-33a-5p in GBM initiation, progression, and recurrence associated with resistance to radiotherapy (14). Our results showed that radiation induced miR-33a-5p expression, which led to radiation resistance, while reversed by PD-L1 inhibitor. After delivering the PD-L1 inhibitor, we observed an upregulation of the miR-33a-5p target, PTEN. It has been reported that the loss of PTEN promotes gliomagenesis (33) and GBM radiation resistance (34). Similarly, PTEN silencing abolished the PD-L1 inhibitor-induced radiosensitization.
The inhibition of DNA repair was required to overcome radio-resistance (35). Thus, we found that the PD-L1 inhibitor was effective at radiosensitizing U87 MG cells by inhibiting the DDR. Other reports have confirmed the radiosensitizing potential associated with inhibiting the DDR at the pre-clinical level (36, 37). As report, the DDR recently has been confirmed to promote the radiation-induced upregulation of PD-L1 in tumor cells, increased exhaustion of CD8+ T cell induced by radiation, to achieve a greater pro-tumor response (38). Given the well-characterized GBM related immunosuppressive tumor microenvironment, treatment with PD-L1 inhibitors may present important weapons against this disease, such as targeting the DDR in GBM cells following radiation therapy (39, 40). The increasing number of γ-H2AX foci in the S-phase fraction commonly occurred in the DNA damage process (41). Similarly, our results showed that radiation induced the DDR in the U87 MG cells, while the PD-L1 inhibitor impaired the DDR, accompanied with increasing γ-H2AX foci and GBM cells trapping the S-phase; thus, leading to radiation sensitization.
In conclusion, the present study showed that the PD-L1 inhibitor induced radiation sensitization in U87 MG cells and U251 cells by directly targeting miR-33a-5p, activating the PTEN signaling pathway, and inhibiting the DDR process. These findings provide new insights into the understanding of the molecular mechanisms by which PD-L1 inhibitors mediate radiation sensitization in GBM.
The original contributions presented in the study are publicly available. This data can be found here: ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) accession E-MTAB-9007.
WX, JZ, and YT made substantial contributions to the acquisition, analysis and interpretation of data. XWa, XWe, and XZ were the major contributors in writing the manuscript. MH and SL were involved in conception and design, revising it critically for important intellectual content. All authors read and approved the final manuscript.
This study was supported by the national natural science foundation of China (grant no. 81671205 to SL; grant no. 81974186 to SL; grant no.81600278 to WX), the Program from Shanghai Committee of Science and technology (grant no. 18XD1402700 to SL) and the Foundation for Interdisciplinary Research of Shanghai JiaoTong University (grant no. YG2017MS68 to JZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Reifenberger G, Wirsching HG, Knobbe-Thomsen CB, Weller M. Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat Rev. (2017) 14:434–52. doi: 10.1038/nrclinonc.2016.204
2. Brandsma D, Stalpers L, Taal W, Sminia P, van den Bent. MJ Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet. (2008) 9:453–61. doi: 10.1016/S1470-2045(08)70125-6
3. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. Cancer J Clin. (2010) 60:166–93. doi: 10.3322/caac.20069
4. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. (2015) 125:3384–91. doi: 10.1172/JCI80011
5. Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy–inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. (2012) 18:6580–7. doi: 10.1158/1078-0432.CCR-12-1362
6. He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep. (2015) 5:13110. doi: 10.1038/srep13110
7. Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. (2017) 32:253–67.e5. doi: 10.1016/j.ccell.2017.07.006
8. Tu X, Qin B, Zhang Y, Zhang C, Kahila M, Nowsheen S, et al. PD-L1 (B7-H1) competes with the RNA exosome to regulate the dna damage response and can be targeted to sensitize to radiation or chemotherapy. Molecular Cell. (2019) 74:1215–26.e4. doi: 10.1016/j.molcel.2019.04.005
9. Bushati N, Cohen SM. microRNA functions. Ann Rev Cell Dev Biol. (2007) 23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406
10. Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. (2008) 455:64–71. doi: 10.1038/nature07242
11. Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol. (2011) 223:102–15. doi: 10.1002/path.2806
12. Chang M, Qiao L, Li B, Wang J, Zhang G, Shi W, et al. Suppression of SIRT6 by miR-33a facilitates tumor growth of glioma through apoptosis and oxidative stress resistance. Oncol Rep. (2017) 38:1251–58. doi: 10.3892/or.2017.5780
13. Wang H, Sun T, Hu J, Zhang R, Rao Y, Wang S, et al. miR-33a promotes glioma-initiating cell self-renewal via PKA and NOTCH pathways. J Clin Invest. (2014) 124:4489–502. doi: 10.1172/JCI75284
14. Sumazin P, Yang X, Chiu HS, Chung WJ, Iyer A, Llobet-Navas D, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. (2011) 147:370–81. doi: 10.1016/j.cell.2011.09.041
15. Gu Y, Cai R, Zhang C, Xue Y, Pan Y, Wang J, et al. miR-132-3p boosts caveolae-mediated transcellular transport in glioma endothelial cells by targeting PTEN/PI3K/PKB/Src/Cav-1 signaling pathway. FASEB J. (2019) 33:441–54. doi: 10.1096/fj.201800095RR
16. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. (2019) 25:462–9. doi: 10.1038/s41591-019-0349-y
17. Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coskun S, Simon M, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nature Genet. (2016) 48:59–66. doi: 10.1038/ng.3457
19. Goellner EM, Grimme B, Brown AR, Lin YC, Wang XH, Sugrue KF, et al. Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer Res. (2011) 71:2308–17. doi: 10.1158/0008-5472.CAN-10-3213
20. Akamandisa MP, Nie K, Nahta R, Hambardzumyan D, Castellino RC. Inhibition of mutant PPM1D enhances DNA damage response and growth suppressive effects of ionizing radiation in diffuse intrinsic pontine glioma. Neuro-Oncol. (2019) 21:786–99. doi: 10.1093/neuonc/noz053
21. Wang J, Hu C, Wang J, Shen Y, Bao Q, He F, et al. Checkpoint blockade in combination with doxorubicin augments tumor cell apoptosis in osteosarcoma. J Immunother. (2019) 42:321–30. doi: 10.1097/CJI.0000000000000281
22. Lau J, Ilkhanizadeh S, Wang S, Miroshnikova YA, Salvatierra NA, Wong RA, et al. STAT3 Blockade Inhibits Radiation-Induced Malignant Progression in Glioma. Cancer Res. (2015) 75:4302–11. doi: 10.1158/0008-5472.CAN-14-3331
23. Xia W, Zhuang L, Hou M. Role of lincRNAp21 in the protective effect of macrophage inhibition factor against hypoxia/serum deprivationinduced apoptosis in mesenchymal stem cells. Int J Mol Med. (2018) 42:2175–84. doi: 10.3892/ijmm.2018.3767
24. Harder BG, Peng S, Sereduk CP, Sodoma AM, Kitange GJ, Loftus JC, et al. Inhibition of phosphatidylinositol 3-kinase by PX-866 suppresses temozolomide-induced autophagy and promotes apoptosis in glioblastoma cells. Mol Med. (2019) 25:49. doi: 10.1186/s10020-019-0116-z
25. Palanichamy K, Patel D, Jacob JR, Litzenberg KT, Gordon N, Acus K, et al. Lack of constitutively active DNA repair sensitizes glioblastomas to Akt inhibition and induces synthetic lethality with radiation treatment in a p53-dependent manner. Mol Cancer Ther. (2018) 17:336–46. doi: 10.1158/1535-7163.MCT-17-0429
26. Pal S, Kozono D, Yang X, Fendler W, Fitts W, Ni J, et al. Dual HDAC and PI3K inhibition abrogates NFkappaB- and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomas. Cancer Res. (2018) 78:4007–21. doi: 10.1158/0008-5472.CAN-17-3691
27. Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Gen Dev. (2007) 21:2683–710. doi: 10.1101/gad.1596707
28. Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, stem cells. Cell. (2012) 149:36–47. doi: 10.1016/j.cell.2012.03.009
29. Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro-Oncology. (2015) 7:vii9-vii14. doi: 10.1093/neuonc/nov151
30. Badie B, Schartner J, Prabakaran S, Paul J, Vorpahl J. Expression of Fas ligand by microglia: possible role in glioma immune evasion. J Neuroimmunol. (2001) 120:19–24. doi: 10.1016/S0165-5728(01)00361-7
31. Wischhusen J, Jung G, Radovanovic I, Beier C, Steinbach JP, Rimner A, et al. Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer research. (2002) 62:2592–9.
32. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. (2010) 17:362–75. doi: 10.1016/j.ccr.2009.12.049
33. Shin CH, Robinson JP, Sonnen JA, Welker AE, Yu DX, VanBrocklin MW, et al. HBEGF promotes gliomagenesis in the context of Ink4a/Arf and Pten loss. Oncogene. (2017) 36:4610–8. doi: 10.1038/onc.2017.83
34. Miyahara H, Yadavilli S, Natsumeda M, Rubens JA, Rodgers L, Kambhampati M, et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer letters. (2017) 400:110–6. doi: 10.1016/j.canlet.2017.04.019
35. Ahmed SU, Carruthers R, Gilmour L, Yildirim S, Watts C, Chalmers AJ. Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem-like cells. Cancer Res. (2015) 75:4416–28. doi: 10.1158/0008-5472.CAN-14-3790
36. Biddlestone-Thorpe L, Sajjad M, Rosenberg E, Beckta JM, Valerie NC, Tokarz M, et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin Cancer Res. (2013) 19:3189–200. doi: 10.1158/1078-0432.CCR-12-3408
37. Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differe. (2014) 21:258–69. doi: 10.1038/cdd.2013.136
38. Morgan MA, Canman CE. Replication stress: an achilles' heel of glioma cancer stem-like cells. Cancer Rese. (2018) 78:6713–16. doi: 10.1158/0008-5472.CAN-18-2439
39. Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res. (2018) 24:3792–802. doi: 10.1158/1078-0432.CCR-18-0047
40. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. (2018) 24:4175–86. doi: 10.1158/1078-0432.CCR-17-1846
Keywords: glioblastoma, programmed death ligand 1 (PD-L1) blockade, radio-sensitization, miR-33a-5p/PTEN signaling pathway, DNA damage response
Citation: Xia W, Zhu J, Tang Y, Wang X, Wei X, Zheng X, Hou M and Li S (2020) PD-L1 Inhibitor Regulates the miR-33a-5p/PTEN Signaling Pathway and Can Be Targeted to Sensitize Glioblastomas to Radiation. Front. Oncol. 10:821. doi: 10.3389/fonc.2020.00821
Received: 27 November 2019; Accepted: 27 April 2020;
Published: 27 May 2020.
Edited by:
Mary Helen Barcellos-Hoff, University of California, San Francisco, United StatesReviewed by:
Alessandra Cataldo, Istituto Nazionale dei Tumori (IRCCS), ItalyCopyright © 2020 Xia, Zhu, Tang, Wang, Wei, Zheng, Hou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng Hou, MjQ0NTE3ODEzQHFxLmNvbQ==; Shiting Li, bGlzaGl0aW5nQHhpbmh1YW1lZC5jb20uY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.