 Robert A. Belderbos
Robert A. Belderbos Heleen Vroman
Heleen Vroman Joachim G. J. V. Aerts
Joachim G. J. V. Aerts- 1Department of Pulmonary Medicine, Erasmus MC Rotterdam, Rotterdam, Netherlands
- 2Erasmus MC Cancer Institute, Erasmus MC Rotterdam, Rotterdam, Netherlands
Malignant pleural mesothelioma (MPM) is a treatment recalcitrant tumor with a poor overall survival (OS). Current approved treatment consists of first line chemotherapy that only modestly increases OS, illustrating the desperate need for other treatment options in MPM. Unfortunately, clinical studies that investigate the effectivity of checkpoint inhibitor (CI) treatment failed to improve clinical outcome over current applied therapies. In general, MPM is characterized as an immunological cold tumor with low T-cell infiltration, which could explain the disappointing results of clinical trials investigating CI treatment in MPM. Currently, many other therapeutic approaches, such as cellular therapies and cancer vaccines are investigated that could induce a tumor-specific immune response and increase of the number of tumor-infiltrating lymphocytes. In this review we will discuss these novel treatment approaches for MPM.
Introduction
Malignant pleural mesothelioma (MPM) is a lethal cancer with limited treatment options (1–3). Current first-line treatment, consisting of platinum/antifolate combination therapy, leads to a median overall survival (OS) of 9–2 months (4). The addition of Bevacizumab to first-line treatment increased OS by 2.7 months and is now the accepted standard therapy in France (5, 6). Since then, no new treatments that could improve the outcome for MPM were reported. Immunotherapies, aiming at the activation of the immune system by blocking inhibitory checkpoint receptors, called checkpoint inhibitor (CI) treatment have drastically improved OS for non-small cell lung cancer and melanoma patients (7). So far, CI treatment has been promising for a small group of MPM patients in phase I/II trials, with response rates between 9 and 29% (8–17). However, unfortunately the DETERMINE phase IIb trial failed to show superiority of anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (Tremelimumab) over placebo in a second or third-line setting for MPM (18). Moreover, in the PROMISE-meso trial, blockade of programmed cell death protein 1 (PD1) failed to prolong progression free survival (PFS) or OS compared to second-line chemotherapy (gemcitabine/vinerolbine) treatment (19). Combination treatment of monoclonal antibodies (mAbs) targeting PD1 or PD1 ligand (PD-L1) with anti-CTLA4 mAb seems to be more effective than CI monotherapy in MPM (10, 20, 21). The results of the ongoing Checkmate 753 phase III trial are awaited (NCT02899299), where Nivolumab (PD1 blockade) and Ipilimumab (CTLA-4 blockade) are combined as first line therapy in unresectable MPM and compared to first-line chemotherapy consisting of pemetrexed and cisplatin or carboplatin (22). As CI treatment, especially anti-PD(L)1 mAb, reinvigorates T-cells, the low number of tumor-infiltrating T-cells (TILs) in MPM might explain the relatively low response rates found in clinical trials investigating anti-PD1/PD-L1 treatment (23). Tumors with high numbers of TILs respond better to CIs (24). In MPM, dendritic cells (DCs) are reduced in both their numbers and their functionality, which could explain the low numbers of TILs (25). Induction of tumor-specific T-cells that infiltrate tumor and kill tumor cells upon antigen recognition by secretion of perforins, granzymes and death ligands, such as Fas and TRAIL could improve clinical outcomes (26, 27). Cancer vaccines and DC-therapy can induce activation and proliferation of tumor specific T-cells. Additionally, chimeric antigen receptor (CAR) T-cells, specific for a tumor antigen, can be used to target specific tumor antigens directly. Recent developments in therapies initiating a tumor directed immune response, such as cancer vaccines, DC-therapy and CAR T-cell therapy in a clinical setting in MPM will be discussed in this review (Figure 1).
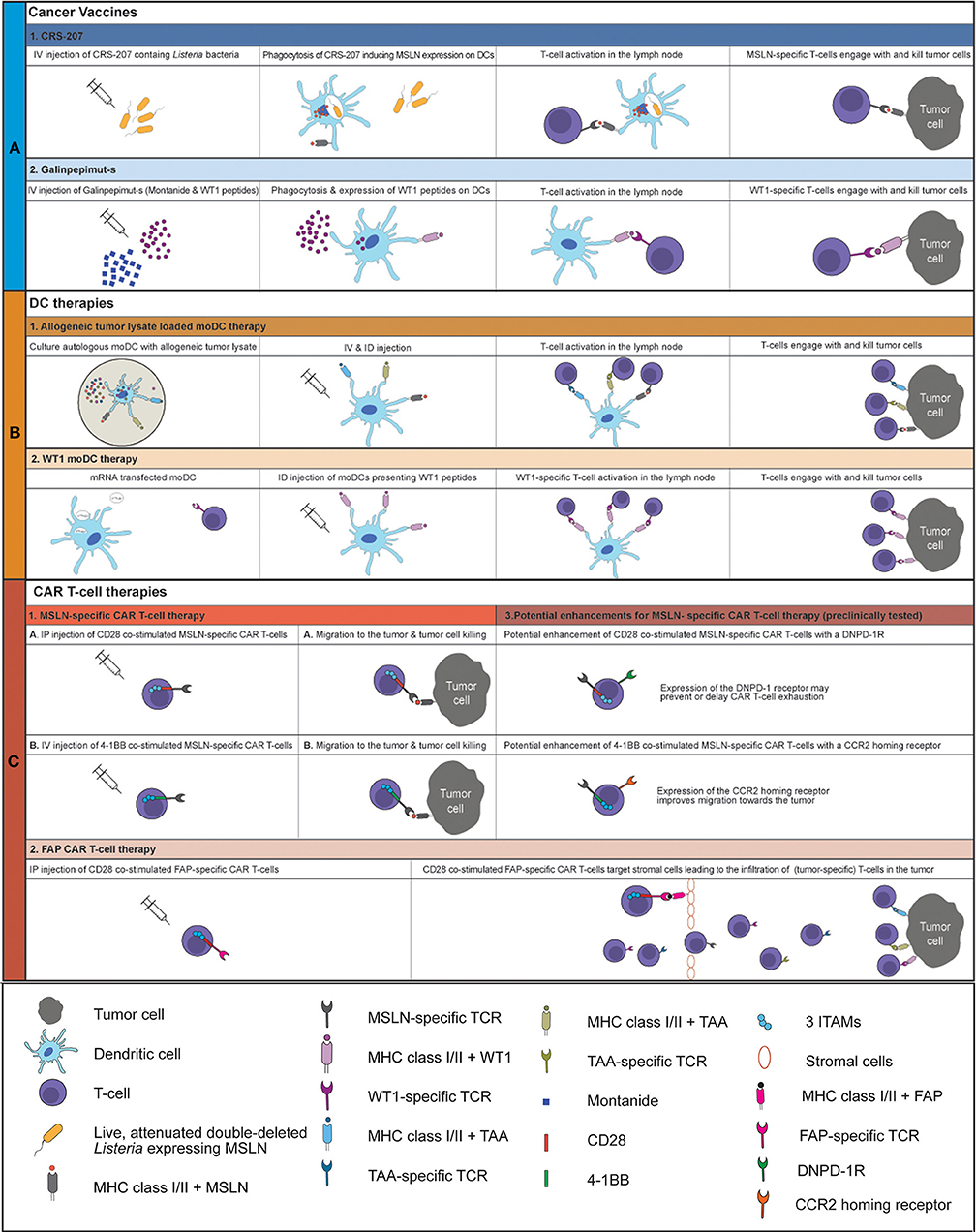
Figure 1. Overview of current clinically tested cancer vaccines and cellular therapies for MPM An overview of the working mechanism of CRS-207 (A1), Galinpepimut-s (A2), allogeneic tumor lysate loaded moDC therapy (B1), WT1 moDC therapy (B2), MSLN-specific CD28 co-stimulated CAR T-cell therapy (C1A), MSLN-specific 4-1BB co-stimulated CAR T-cell therapy (C1B) and FAP CAR T-cell therapy (C2). The potential enhancements of MSLN-specific CAR T-cell therapy are displayed in C3. IV, intravenous; ID, intradermal; IP, intrapleural; MSLN, mesothelin; moDC, monocyte-derived dendritic cell; MHC, major histocompatibility complex; TCR, T-cell receptor; WT1, Wilms Tumor 1 protein; TAA, tumor-associated antigen; ITAM, Immunoreceptor tyrosine-based activation motif; DNPD-1R, dominant negative PD1 receptor; CCR2, CC chemokine receptor 2; FAP, fibroblast activation protein.
Cancer Vaccines
Cancer vaccines can be made of tumor lysate, single or multiple peptides, viruses, or attenuated bacteria. The purpose of vaccinating cancer patients is to elicit a tumor-specific type 1-polarized T-cell response, leading to clinical benefit for the patient. Immunostimulatory adjuvants, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and toll-like receptor (TLR) ligands are often combined with cancer vaccines, to attract and activate antigen presenting cells (APC) that will take up the cancer vaccines (28). Certain adjuvants, such as Montanide, protect the peptides in the cancer vaccine and create a depot for slow antigen release that attracts lymphocytes and DCs, therefore called depot adjuvants (29). For MPM, Wilms Tumor 1 (WT-1) peptide-based vaccine, Galinpepimut-S and CRS-207 are the most thoroughly evaluated cancer vaccines and will be discussed in more detail.
WT-1 Cancer Vaccines
WT-1 is a protein expressed on almost all (97%) MPM cells with a variable distribution and intensity and serves as an immunohistochemical marker for MPM diagnosis, making WT-1 an appropriate target for immunotherapy (30). The cancer vaccine, Galinpepimut-S consist of four WT-1 peptides of different lengths that can be presented in both MHC class I and II molecules, permitting the activation of both CD4+ and CD8+ T-cells (31). Treatment with Galinpepimut-S was investigated in a randomized phase II study in MPM patients with positive (> 10%) WT-1 expression. Herein, Galinpepimut-S was administrated with adjuvants (GM-CSF and Montanide) and compared to placebo, in which only the adjuvants were administered. Unfortunately, the study was closed after inclusion of 41 patients due to futility of the placebo treatment and a non-significant increase in median OS (4, 5 months) and median PFS (2, 8 months) for Galinpepimut-S treated patients, as compared to the placebo arm (31). In July 2019, a clinical trial which investigates the combined treatment of Galinpepimut-S with nivolumab in patients with WT-1 expressing MPM (NCT04040231) has started.
CRS-207
CRS-207 is a live-attenuated listeria-encoding human mesothelin (MSLN) vaccine. APCs will phagocytose the Listeria bacteria in CRS-207, leading to release of MSLN, that is subsequently presented by APCs to T-cells in the lymph nodes, thereby inducing an MSLN-specific immune response. MSLN is expressed in 90% of epithelioid MPM patients, which comprises up to 80% of all MPM patients (32). MSLN is not expressed in most sarcomatoid MPMs and only minimally in biphasic MPM. MSLN has low expression on non-malignant cells, making it an attractive target for immunotherapy (32, 33). In a phase Ib trial, treatment-naïve MPM patients received 2 CRS-207 doses, followed by 6 cycles of pemetrexed/cisplatin and CRS-207 booster infusions (34). The disease control rate was 89%, with 1 complete response (CR) and 19 partial responses (PR) in 35 evaluable patients. Unfortunate, the median OS was 14.7 months, which is comparable to OS observed after standard chemotherapy treatment (34, 35). Additional trials were initiated with CRS-207 in combination with pembrolizumab (Keytruda), chemotherapy and GM-CSF transfected tumor cell vaccine (GVAX) (NCT 01675765, NCT03175172, NCT02243371), and results are awaited (36). Unfortunately, the Keytruda trial has been halted because of insufficient clinical activity (NCT03175172).
In conclusion, despite careful selection of adjuvants and antigenic targets of cancer vaccines applied in MPM, therapeutic success or induction of a clinically detectable cytolytic immune response has not yet been shown (37). Combining cancer vaccines specifically with agents that target the immunosuppressive tumor microenvironment (TME) might improve clinical outcome. Clinical trials investigating these combination therapies are currently investigated and results are awaited.
DC-Therapy
DCs are low in numbers and are impaired in functionality in MPM patients (25). Moreover, the TME in MPM causes immunosuppression through secretion of immunosuppressive cytokines and expression of inhibitory molecules by tumor cells and immune cells again affecting DC mediated T-cell activation (38–41). To circumvent the immunosuppressive TME, DCs can be activated and loaded with selected tumor associated antigens (TAAs) or whole tumor lysate in vitro. DC-therapy has been developed in three generations. In first generation DC-therapy, monocytes isolated from peripheral blood were cultured with GM-CSF and interleukin (IL) 4, leading to the differentiation into immature monocyte-derived DCs (moDC) (42). These immature moDCs were loaded with TAAs or tumor lysate and reinjected without any further activating stimulation into the patient. Second-generation DC-therapy, additionally stimulated the generated moDCs in vitro with a maturation/activation cocktail, consisting of cytokines and immune stimulants, such as poly IC, TLR ligands and prostaglandin E2 (40–42). Second generation DC-therapy is currently used in various clinical trials. Response rates for second-generation DC-therapy in melanoma, prostate cancer, malignant glioma and renal cell carcinoma vary from 8 to 15% with an increase in OS of ~20% (42, 43). In contrary, an overall response rate of 7.1% was found in studies investigating first-generation DC-therapy in various malignancies, but mainly melanoma (44). Next-generation DC-therapy, aims at using naturally occurring DCs (nDC) that are purified directly from peripheral blood, in vitro loaded TAAs or tumor lysate and activated, and used for DC-therapy. The benefits of using nDCs are a shortened culture-time and lower manufacturing costs. It is also thought that DC-therapy containing nDCs will improve response rates, however this still has to be confirmed in clinical trials (42, 45, 46). DCs can be classically loaded with proteins during culture but TAAs can also be presented via RNA transfection methods or cancer cell-DC fusion (45, 47). The type of antigen source can vary from specific TAAs to complete tumor lysates. Analysis of 173 clinical trials in a wide variety of tumors showed that active immunotherapy using tumor-lysate (ORR 8.1%) was clinically more effective than peptide-based therapies (ORR 3.6%) (48), indicating that vaccinating with a broad range of tumor-associated proteins prohibits escape by the tumor and supports the hypothesis of immunoediting (Box 1).
Box 1. Immunoediting.
Immunoediting is a term that describes the balance between the prevention of tumor establishment through surveillance by the immune system and tumor cell growth when tumor cells escape from immunosurveillance (49–51).
Immunoediting by malignant cells contains three phases: elimination, equilibrium, and escape:
Elimination: cancer cells are eliminated by the innate and adaptive immune system.
Equilibrium: mutations and adaptations occur in certain cancer cells, leading to escape from the immune system of these cancer cells. During this phase, these mutated/adapted cancer cells will decrease antigen expression and become resistant to the immune system, whereas non-mutated cancer cells will be eliminated by the immune system, thereby increasing the frequency of mutated/adapted cancer cells. This process can take several years (52).
Escape: mutated/adapted cancer cells will proliferate and cause tumor outgrowth that can no longer be hampered or controlled by the immune system (53).
DC-Therapy in MPM
Two types of second-generation DC-therapy have been tested in clinical trials in MPM patients. Autologous moDCs transfected with messenger RNA (mRNA) encoding for WT1 and autologous moDCs loaded with autologous/allogeneic tumor lysate.
WT1-Targeted DC-Therapy
MoDCs transfected with WT1 encoding mRNA have resulted in promising clinical responses in MPM patients, but also in other malignancies. Prolonged stabilization of disease was noted in MPM patients, with OS (from start of chemotherapy) of 35.7 months (54, 55). This study was followed up by a phase I/II trial (MESODEC) in which treatment-naïve patients received WT1-targeting DC-therapy during chemotherapy, followed by pleurectomy/decortication (P/D) in the case of a resectable tumor (NCT02649829). The primary objective of this trial (recruiting since 2017 and enrolling 20 patients) is to assess the feasibility of WT1-targeting DC-therapy in combination with chemotherapy.
Tumor Lysate Loaded DC-Therapy
Two clinical trials that applied DC-therapy that consists of autologous moDCs loaded with autologous tumor lysate have been reported in MPM (56, 57). In the first Phase I clinical trial, ten MPM patients were treated with at least 3 biweekly DC vaccinations. Tumor lysate was prepared from single cell suspensions of tumor cell lines generated from tumor tissue and/or pleural effusions. Three patients had a PR, one had stable disease (SD) and six had progressive disease (PD). Median OS from time of diagnosis was 19 months (57). To improve the efficacy of DC-therapy in a sequential trial, ten MPM patients were treated with a combination of moDCs loaded with autologous tumor lysate and low-dose cyclophosphamide treatment, a chemotherapy that at low concentration specifically targets regulatory T-cells (Tregs) that favor anti-tumor immune responses (40, 58–60). At first radiological evaluation after treatment, one patient had a CR, four had SD and two had PD. Radiological response assessment was impossible in three patients as they had received additional P/D (56). Grade III/IV toxicities did not occur. Moreover, cyclophosphamide treatment indeed selectively depleted Tregs and the frequency of naïve Tregs prior to treatment was positively correlated to OS (61). Two patients were still alive 6 years after diagnosis.
Unfortunately, using autologous tumor material as a source for tumor lysate is not feasible for a large number of patients in a phase II trial, because of the varying quality and/or lack of tumor material. Loading moDCs with allogeneic tumor lysate, serving as an “of-the-shelf” source for antigen-loading material, was compared to autologous tumor lysate-loaded moDC-therapy in mice, and induced similar protection against tumor outgrowth (62). To create allogeneic tumor lysate for clinical trials, cell lines were generated of pleural fluid of 5 MPM patients with different histological subtypes and varying antigen expression. An allogeneic tumor lysate was derived from these cell lines that contained a broad spectrum of TAAs. Two out of nine MPM patients treated with allogeneic tumor lysate-loaded moDCs (MesoPher) in a phase I dose-escalation trial had a PR and two patients are still alive 4 years after start of treatment. Grade III/IV toxicities were not reported (63). This phase I clinical study is followed up by an international, randomized, open-label, multicenter phase III trial (DENIM-trial), that will evaluate the efficacy of autologous moDCs loaded with allogeneic tumor lysate in MPM patients. Recruitment started in June 2018 and the first results are expected in 2021 (64). An overview of finished and ongoing clinical trials investigating DC-therapy in MPM is provided in Table 1.
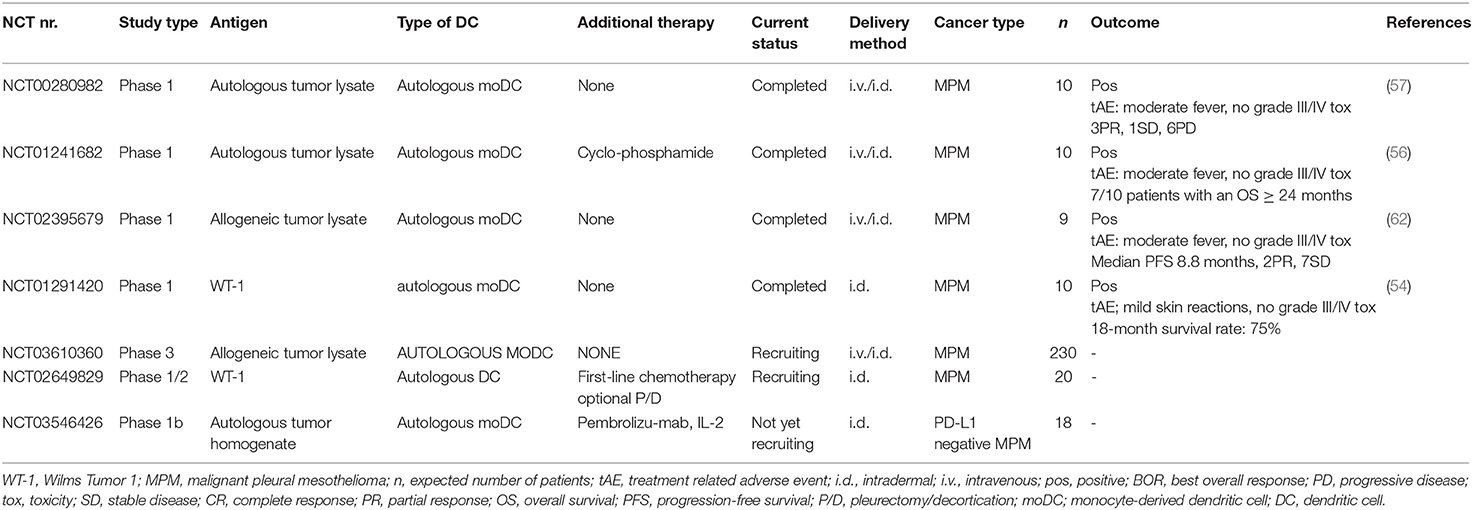
Table 1. Ongoing and completed trials for dendritic cell therapy in mesothelioma.
Combination Treatment DC-Therapy
Multiple reviews have discussed strategies to combine DC-therapy with other therapeutic agents, such as low-dose chemotherapy to deplete specific immune cell subsets, radiotherapy to induce an abscopal effect or therapies that target specific immune cell subtypes or enzymes (40, 41). CI-treatment is thought to not only complement DC-therapy but work synergistically with DC-therapy. Mice treated with DC-therapy had more tumor-specific CD8+ TILs than mice treated with placebo (65). Moreover, most of these TILs expressed high levels of PD1 on the cell surface, indicating their susceptibility for reinvigoration by CI treatment (65). The increase of TILs induced by DC-therapy may improve the current response rates of CI-treatment in MPM. Moreover, TILs induced by DC-therapy, that are hampered by inhibitory signaling may be reinvigorated. Based on this rationale, nine MPM patients who received autologous DC therapy in our center were sequentially treated with CIs. Three patients had a PR, five had SD and the median OS was 17.5 months from start of CI treatment (66). This data suggests a synergistic effect between DC-therapy and CIs in MPM that warrants further research.
CAR T-cell Therapy
The hypothesis for adoptive T-cell therapy is to introduce tumor-specific T-cells that directly target the tumor cells. The first step toward CAR T-cell therapy was the use of autologous TILs that were expanded in vitro and reinjected after one dose of cyclophosphamide and in combination with IL-2 to treat metastatic melanoma. Objective regression was observed in 11 out of 20 patients with a mean response duration of 5.6 months (2–13 months) (67). Unfortunately, the reproducibility and quality of these TILs could not be guaranteed due to interpatient differences of TILs (68). To avoid the need of TILs, T-cells can be genetically modified to express a T-cell receptor (TCR) that targets tumor-specific antigens. Although promising radiological responses were observed using these transgenic TCR T-cells, clinical use was still restricted to (Human Leukocyte Antigen A2) HLA-A2 patients (69). In an effort to enhance the efficacy of transgenic TCR T-cells and make target-antigen recognition independent of (Major Histocompatibility Complex) MHC, a CAR instead of a TCR was developed (70). A CAR classically consists of an extracellular part with an antigen-recognition domain, a transmembrane domain and an intracellular domain that contains three immune receptor tyrosine-based activation motifs (ITAMs). CAR constructs are transfected into (autologous) T-cells via mRNA or viral transduction (71). Historically, five generations of CAR T-cell therapy are distinguished. The most crucial adjustments that separate different generations concern the characteristics of the intracellular domain, which can contain, apart from the three ITAMs, one or two co-stimulatory molecules, such as CD28 or 4-1BB, and an inducible expression cassette for a protein, as IL-12 or a cytokine receptor, such as IL-2R (72, 73). Currently, two second generation CAR T-cell therapies targeting CD19 have been approved for the treatment of hematological malignancies (74). Although the clinical outcomes for CAR T-cell therapy in treatment-resistant hematological malignancies are impressive with complete response rates varying from 40 to 60%, these responses are not found for solid tumors. Also, CAR T-cell therapy induces severe treatment-related toxicities varying from 49 to 73% (75–77). Cytokine release syndrome (CRS) and neurological events are the most frequent severe treatment-related adverse events. CRS results from an immense release of cytokines from immunotherapy-targeted immune cells and cancer cells. The severity of CRS is dependent on the dosage of CAR T-cells, amount of tumor burden and level of IL-6. Blocking the IL-6 receptor with tocilizumab or neutralizing IL-6 through binding with a mAb siltuximab reduces CRS severity (74). The mechanism driving neurotoxicity, CAR T-cell Related Encephalopathy Syndrome (CRES), is still unknown. Locoregional admission of CAR T-cell therapy could reduce toxicity, however for hematological malignancies this is not an option.
Challenges for CAR T-Cell Therapy in Solid Tumors
CAR T-cell therapy encounters many challenges in solid tumors, such as migration of the CAR T-cells to the tumor, infiltration into the tumor, survival within the immunosuppressive TME as well as the lack of specific targetable tumor-specific antigens (78, 79). In B-cell driven malignancies, CD19 is a perfect target because it is expressed on all tumor cells (80, 81). Finding the perfect tumor-specific antigen to target in solid tumors is challenging due to heterogeneous expression of these tumor antigens. The lack of specific tumor antigens can also lead to severe “on target, off tumor” toxicity, caused by destruction of non-malignant cells expressing the antigen CAR T-cells are directed against (79). To migrate to and infiltrate the TME, CAR T-cells need to be equipped with appropriate tumor homing chemokine receptors and tumor endothelium degrading enzymes. Additionally, chemokines can be injected into the tumor that attract CAR T-cells. Another possibility to circumvent migration difficulties and even avoid development of systemic toxicities is locoregional administration of CAR T-cell therapy, but this is technically not achievable for all solid tumors. The stromal cells that are associated with nearly all epithelioid solid tumors form a physical barrier and severely hamper immune cell infiltration (79). A promising approach to attack the stromal component of the TME, is the development of CAR T-cells targeting (fibroblast activation protein) FAP which is expressed on various stromal cell types (82). Targeting the stromal cells by the FAP-specific CAR T-cells will allow and lead to infiltration of the tumor by TILs. Furthermore, as the target is expressed on non-malignant cells and not the malignant cells, this also reduces the risk of immunoediting and tumor escape. The immunosuppressive environment generated by the TME also affects the cytolytic activity of CAR T-cells and leads to CAR T-cell exhaustion. Secretion of inflammatory cytokines by CAR T-cells could counteract this immunosuppressive environment. Another possibility to directly circumvent exhaustion is to combine CAR T-cell therapy with CI treatment. Recently, CAR T-cells have been genetically modified with silenced PD-(L)1 coinhibitory signaling by the expression of a dominant negative PD1 receptor (DNPD-1R) that lacks an intracellular signaling domain. Although many challenges remain in the treatment of solid tumors with CAR T-cell therapy, current understanding and recent developments show great potential. Many of these new approaches are currently investigated in MPM.
Systemic and Locoregional CAR-T Cell Therapy in MPM
The choice of targetable tumor-antigen is crucial in the development of CAR-T cell therapy for MPM. Several tumor-antigen targets, such as MSLN, WT-1, FAP and the antigens of the ErbB family are evaluated for their applicability for CAR T-cell therapy in MPM. CAR T-cells targeting MSLN, FAP or WT-1 are already investigated in clinical trials, summarized in Table 2. Second generation CD28 FAP CAR T-cells have been evaluated in a phase I trial. Patients with metastatic MPM treated with these CAR T-cells developed no treatment related toxicities. Radiological responses were not reported, but 2 out of 3 patients were still alive with a median follow up of 18 months. Recently, Haas et al. showed that treatment with second generation, 4-1BB MSLN CAR T-cells as monotherapy or in combination with low-dose cyclophosphamide was well-tolerated in patients with MPM, ovarian carcinoma and pancreatic ductal carcinoma (84). One case of dose limiting toxicity (grade 4 sepsis) was reported without the use of cyclophosphamide. No radiological responses were seen and 11 out of 15 patients had SD as best overall response. Moreover, the persistence of CAR T-cells in the peripheral blood was <28 days after injection. Apart from the known hurdles for CAR T-cell therapy in solid tumors, a potential reason for the minimal persistence and clinical efficacy might be a consequence of the murine-derived CAR that was used. A new phase 1 trial has started evaluating a fully human CAR T-cell (Table 1, NCT03054298). CAR T-cells targeting the ErbB family antigens, T1E28z CAR T-cells showed promising results both in vitro and in mouse models, which needs to be validated in a clinical studies (86–88).
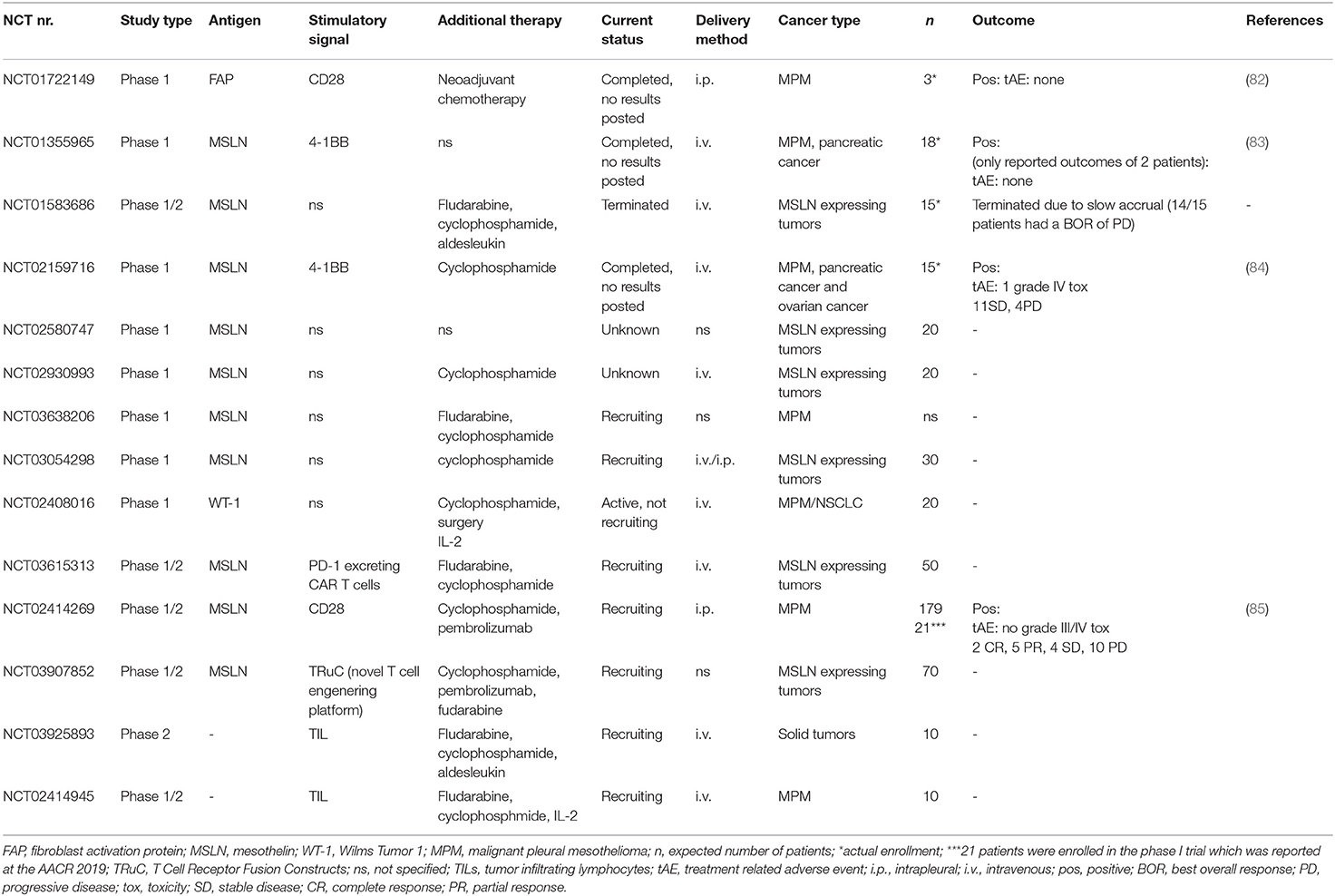
Table 2. Ongoing and completed trials for T-cell therapy in mesothelioma.
Currently methods to improve migration to the tumor site are heavily studied in mouse models. Herein, MSLN CAR T-cells that expressed a tumor homing chemokine receptor CCR2 showed improved tumor infiltration (89). Moreover, in an orthotopic mouse model of MPM, migration toward the tumor was circumvented by intra-pleural administration of second generation, CD28-costimulated MSLN CAR T-cells and led to a larger reduction of pleural an metastatic tumor load as compared to intravenous administration (90). Moreover, the intra-pleural treatment dose was 30-fold lower than the intravenous administered dose and elicited no grade III/IV toxicities.
In a clinical setting, no'on-target, off-tumor' effects were seen when 21 patients with malignant pleural disease were treated with CD28-costimulated MSLN CAR-T cells intrapleurally (85, 90, 91). In this study 19 out of 21 patients had MPM, of whom 13 were subsequently treated with pembrolizumab (anti-PD1). In total two patients had a CR, five had PR and four had SD as best overall response (85). Just as for DC therapy, Combining CAR T-cell therapy with anti-PD1 treatment showed promising clinical results. In a MPM mouse model, combined treatment of anti-PD1 mAb with CAR T-cell therapy improved treatment efficacy. CAR T-cell exhaustion can also be prevented by genetically modifying the CAR T-cells to express a dominant negative PD1 receptor (DNPD-1R) that lacks an intracellular signaling domain, avoiding the need for CI treatment and their related toxicities (92). A trial with CAR T-cells with a DNPD1R is expected to start in 2020 (93).
Conclusions
MPM remains a treatment-recalcitrant tumor with few registered treatment options. CI treatment failed to improve clinical outcome which might correlate with the low number of TILs in MPM. Cancer vaccines, DC-therapy and CAR T-cell therapy all induce a tumor directed immune response and increase the number of tumor-specific T-cells. Both cellular therapies and cancer vaccines face many challenges such as, migration of therapy-induced T-cells to the tumor, infiltration into the tumor, survival within the immunosuppressive TME and finding an optimal targeting approach. Improvement of cancer vaccines and cellular therapies and multimodal approaches that circumvent and overcome these difficulties should be investigated thoroughly. As both cancer vaccines and cellular therapies aim to induce infiltration of tumor-specific T cells into the TME, CI treatment serves as an ideal therapeutic option to block inhibitory signaling and reinvigorate TILs leading to enhancement of both treatments. In conclusion, additional research is needed to investigate and compare effectivity of cancer vaccines and cellular therapies for a cold tumor like MPM. Evaluating and influencing characteristics of the TME in MPM that withhold T-cell infiltration or impair cytotoxic T-cell function, is warranted to create a holistic treatment approach.
Author Contributions
RB drafted and wrote the paper and contributed to the conception of the work. JA and HV contributed to the conception of the work and substantively revised the manuscript. All authors approved the submitted version.
Conflict of Interest
JA reports receiving commercial research grants from Amphera and Roche, holds ownership interest (including patents) in Amphera BV, and is a consultant/advisory board member for Amphera, Boehringer Ingelheim, Bristol-Myers Squibb, Eli-Lilly, MSD, and Roche.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Baas P, Fennell D, Kerr KM, Schil PE, Haas RL, Peters S. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2015) 26:199. doi: 10.1093/annonc/mdv199
2. Yap TA, Aerts JG, Popat S, Fennell DA. Novel insights into mesothelioma biology and implications for therapy. Nat Rev Cancer. (2017) 17:475–88. doi: 10.1038/nrc.2017.42
3. Carbone M, Adusumilli PS, Alexander HR Jr, Baas P, Bardelli F, et al. Mesothelioma: Scientific clues for prevention, diagnosis, and therapy. CA Cancer J Clin. (2019) 69:402–29. doi: 10.3322/caac.21572
4. Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. (2003) 21:2636–44. doi: 10.1200/JCO.2003.11.136
5. Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, et al. French cooperative thoracic, bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet. (2016) 387:1405–14. doi: 10.1016/S0140-6736(15)01238-6
6. Brosseau S, Assoun S, Naltet C, Steinmetz C, Gounant V, Zalcman G. A review of bevacizumab in the treatment of malignant pleural mesothelioma. Future Oncol. (2017) 13:2537–46. doi: 10.2217/fon-2017-0307
7. Ribas, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359:1350–5. doi: 10.1126/science.aar4060
8. Calabro L, Maio M. Immune checkpoint blockade in malignant mesothelioma. Semin Oncol. (2015) 42:418–22. doi: 10.1053/j.seminoncol.2015.02.001
9. Calabro L, Morra A, Fonsatti E, Cutaia O, Fazio C, Annesi D, et al. Efficacy and safety of an intensified schedule of tremelimumab for chemotherapy-resistant malignant mesothelioma: an open-label, single-arm, phase 2 study. Lancet Respir Med. (2015) 3:301–9. doi: 10.1016/S2213-2600(15)00092-2
10. Disselhorst MJ, Quispel-Janssen J, Lalezari F, Monkhorst K, de Vries JF, van der Noort V, et al. Ipilimumab and nivolumab in the treatment of recurrent malignant pleural mesothelioma (INITIATE): results of a prospective, single-arm, phase 2 trial. Lancet Respir Med. (2019) 7:260–70. doi: 10.1016/S2213-2600(18)30420-X
11. Hassan R, Thomas A, Nemunaitis JJ, Patel MR, Bennouna J, Chen FL, et al. Efficacy and safety of avelumab treatment in patients with advanced unresectable mesothelioma: phase 1b results from the JAVELIN Solid Tumor Trial. JAMA Oncol. (2019) 5:351–7. doi: 10.1001/jamaoncol.2018.5428
12. Lievense LA, Sterman DH, Cornelissen R, Aerts JG. Checkpoint blockade in lung cancer and mesothelioma. Am J Respir Crit Care Med. (2017) 196:274–82. doi: 10.1164/rccm.201608-1755CI
13. Okada, Kijima T, Aoe K, Kato T, Fujimoto N, Nakagawa K, et al. Clinical efficacy and safety of nivolumab: results of a multicenter, open-label, single-arm, Japanese Phase II study in Malignant Pleural Mesothelioma (MERIT). Clin Cancer Res. (2019) 25:5485–92. doi: 10.1158/1078-0432.CCR-19-0103
14. Quispel-Janssen J, van der Noort V, de Vries JF, Zimmerman M, Lalezari F, Thunnissen E, et al. Programmed death 1 blockade with nivolumab in patients with recurrent malignant pleural mesothelioma. J Thorac Oncol. (2018) 13:1569–76. doi: 10.1016/j.jtho.2018.05.038
15. Metaxas Y, Rivalland G, Mauti LA, Klingbiel D, Kao S, Schmid S, et al. Pembrolizumab as palliative immunotherapy in malignant pleural mesothelioma. J Thorac Oncol. (2018) 13:1784–91. doi: 10.1016/j.jtho.2018.08.007
16. Nowak, Kok P, Lesterhuis W, Hughes B, Brown C, Kao S, et al. OA08.02 DREAM - A Phase 2 trial of durvalumab with first line chemotherapy in mesothelioma: final result. J Thorac Oncol. (2018) 13:S338–9. doi: 10.1016/j.jtho.2018.08.276
17. Nowak AK, McDonnell A, Cook A. Immune checkpoint inhibition for the treatment of mesothelioma. Expert Opin Biol Ther. (2019) 19:697–706. doi: 10.1080/14712598.2019.1606209
18. Maio M, Scherpereel A, Calabro L, Aerts J, Cedres Perez S, Bearz A, et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): a multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. (2017) 18:1261–73. doi: 10.1016/S1470-2045(17)30446-1
19. Popat S. A multicentre randomized phase III trial comparing pembrolizumab (P) vs single agent chemotherapy (CT) for advanced pre-treated malignant pleural mesothelioma (MPM) – results from the European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso trial. Ann Oncol. (2019) 30 (suppl_5): v851–934. 2019:394. doi: 10.1093/annonc/mdz394.091
20. Scherpereel, Mazieres J, Greillier L, Lantuejoul S, Do P, Bylicki O, et al. French cooperative thoracic, nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): a multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol. (2019) 20:239–53. doi: 10.1016/S1470-2045(18)30765-4
21. Calabro L, Morra A, Giannarelli D, Amato G, D'Incecco A, Covre A, et al. Tremelimumab combined with durvalumab in patients with mesothelioma (NIBIT-MESO-1): an open-label, non-randomised, phase 2 study. Lancet Respir Med. (2018) 6:451–60. doi: 10.1016/S2213-2600(18)30151-6
22. Gerard Z, Solange P, Aaron Scott M, Thierry Marie J, Sanjay P, Arnaud S, et al. Checkmate 743: A phase 3, randomized, open-label trial of nivolumab (nivo) plus ipilimumab (ipi) vs pemetrexed plus cisplatin or carboplatin as first-line therapy in unresectable pleural mesothelioma. J Clin Oncol. (2017) 35:TPS8581. doi: 10.1200/JCO.2017.35.15_suppl.TPS8581
23. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. (2013) 339:286–91. doi: 10.1126/science.1232227
24. Aerts JG, Hegmans JP. Tumor-specific cytotoxic T cells are crucial for efficacy of immunomodulatory antibodies in patients with lung cancer. Cancer Res. (2013) 73:2381–8. doi: 10.1158/0008-5472.CAN-12-3932
25. Cornwall SM, Wikstrom M, Musk AW, Alvarez J, Nowak AK, Nelson DJ. Human mesothelioma induces defects in dendritic cell numbers and antigen-processing function which predict survival outcomes. Oncoimmunology. (2016) 5:e1082028. doi: 10.1080/2162402X.2015.1082028
27. Martínez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res. (2015) 21:5047–56. doi: 10.1158/1078-0432.CCR-15-0685
28. Gouttefangeas C, Rammensee HG. Personalized cancer vaccines: adjuvants are important, too. Cancer Immunol Immunother. (2018) 67:1911–8. doi: 10.1007/s00262-018-2158-4
29. van Doorn E, Liu H, Huckriede A, Hak E. Safety and tolerability evaluation of the use of Montanide ISA51 as vaccine adjuvant: a systematic review. Hum Vaccin Immunother. (2016) 12:159–69. doi: 10.1080/21645515.2015.1071455
30. Eguchi T, Kadota K, Mayor M, Zauderer MG, Rimner A, Rusch VW, et al. Cancer antigen profiling for malignant pleural mesothelioma immunotherapy: expression and coexpression of mesothelin, cancer antigen 125, and Wilms tumor 1. Oncotarget. (2017) 8:77872–82. doi: 10.18632/oncotarget.20845
31. Zauderer MG, Tsao AS, Dao T, Panageas K, Lai WV, Rimner A, et al. A Randomized phase II trial of adjuvant galinpepimut-S, WT-1 analogue peptide vaccine, after multimodality therapy for patients with malignant pleural mesothelioma. Clin Cancer Res. (2017) 23:7483–9. doi: 10.1158/1078-0432.CCR-17-2169
32. Servais EL, Colovos C, Rodriguez L, Bograd AJ, Nitadori J, Sima C, et al. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin Cancer Res. (2012) 18:2478–89. doi: 10.1158/1078-0432.CCR-11-2614
33. Ordonez NG. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod Pathol. (2003) 16:192–7. doi: 10.1097/01.MP.0000056981.16578.C3
34. Hassan R, Alley E, Kindler H, Antonia S, Jahan T, Honarmand S, et al. Live-attenuated, listeria monocytogenes expressing mesothelin (CRS-207) with chemotherapy for treatment of malignant pleural mesothelioma. Clin Cancer Res. (2019) 25:5787–98. doi: 10.1158/1078-0432.CCR-19-0070
35. Jahan T, Hassan R, Alley E, Kindler H, Antonia S, Whiting C, et al. 208O_PR: CRS-207 with chemotherapy (chemo) in malignant pleural mesothelioma (MPM): results from a phase 1b trial. J Thorac Oncol. (2016) 11:S156. doi: 10.1016/S1556-0864(16)30330-6
36. Evan WA, Tawee T, Thierry Marie J, Leena G, Tobias P, James S, et al. A phase II single-arm study of CRS-207 with pembrolizumab (pembro) in previously treated malignant pleural mesothelioma (MPM). J Clin Oncol. (2019) 37:29. doi: 10.1200/JCO.2019.37.8_suppl.29
37. Vermaelen K. Vaccine strategies to improve anti-cancer cellular immune responses. Front Immunol. (2019) 10:8. doi: 10.3389/fimmu.2019.00008
38. Devaud C, John LB, Westwood JA, Darcy PK, Kershaw MH. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology. (2013) 2:25961. doi: 10.4161/onci.25961
39. Gkretsi V, Stylianou A, Papageorgis P, Polydorou C, Stylianopoulos T. Remodeling components of the tumor microenvironment to enhance cancer therapy. Front Oncol. (2015) 5:214. doi: 10.3389/fonc.2015.00214
40. Belderbos RA, Aerts J, Vroman H. Enhancing dendritic cell therapy in solid tumors with immunomodulating conventional treatment. Mol Ther Oncolytics. (2019) 13:67–81. doi: 10.1016/j.omto.2019.03.007
41. van Gulijk M, Dammeijer F, Aerts J, Vroman H. Combination strategies to optimize efficacy of dendritic cell-based immunotherapy. Front Immunol. (2018) 9:2759. doi: 10.3389/fimmu.2018.02759
42. Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. (2017) 38:577–93. doi: 10.1016/j.it.2017.05.006
43. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. (2014) 15:e257–67. doi: 10.1016/S1470-2045(13)70585-0
44. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. (2004) 10:909–15. doi: 10.1038/nm1100
45. Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. (2016) 22:1897–906. doi: 10.1158/1078-0432.CCR-15-1399
46. Bol KF, Schreibelt G, Rabold K, Wculek SK, Schwarze JK, Dzionek A, et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J Immunother Cancer. (2019) 7:109. doi: 10.1186/s40425-019-0580-6
47. Wilgenhof S, Corthals J, Heirman C, van Baren N, Lucas S, Kvistborg P, et al. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J Clin Oncol. (2016) 34:1330–8. doi: 10.1200/JCO.2015.63.4121
48. Neller MA, Lopez JA, Schmidt CW. Antigens for cancer immunotherapy. Semin Immunol. (2008) 20:286–95. doi: 10.1016/j.smim.2008.09.006
49. Claesson MH. Why current peptide-based cancer vaccines fail: lessons from the three Es. Immunotherapy. (2009) 1:513–6. doi: 10.2217/imt.09.35
50. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. (2007) 121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x
51. Belderbos RA, Cornelissen R, Aerts JV. Immunotherapy of mesothelioma: vaccines and cell therapy. In: Ceresoli GL, Bombardieri E, D'Incalci M, editors. Mesothelioma: From Research to Clinical Practice. Cham: Springer International Publishing (2019). p. 271–80. doi: 10.1007/978-3-030-16884-1_19
52. O'Donnell JS, M.Teng WL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
53. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
54. Berneman Z, Germonpré P, Huizing M, Velde A, Nijs G, Stein B, et al. Dendritic cell vaccination in malignant pleural mesothelioma: A phase I/II study. J Clin Oncol. (2014) 32:7583. doi: 10.1200/jco.2014.32.15_suppl.7583
55. Berneman Z, A. Van de Velde, Anguille S, Willemen Y, Huizing M, Germonpré P, et al. Vaccination with Wilms' Tumor Antigen (WT1) mRNA-electroporated dendritic cells as an adjuvant treatment in 60 cancer patients: report of clinical effects and increased survival in acute myeloid leukemia, metastatic breast cancer, glioblastoma and mesothelioma. Cytotherapy. (2016) 18:S13–4. doi: 10.1016/j.jcyt.2016.03.036
56. Cornelissen R, Hegmans JJ, Maat WM, Kaijen-Lambers MEH, Bezemer K, Hendriks RW, et al. Extended tumor control after dendritic cell vaccination with low-dose cyclophosphamide as adjuvant treatment in patients with malignant pleural mesothelioma. Am J Respir Critic Care Med. (2016) 193:1023–31. doi: 10.1164/rccm.201508-1573OC
57. Hegmans JP, Veltman JD, Lambers ME, de Vries IM, Figdor CG, Hendriks RW, et al. Consolidative dendritic cell-based immunotherapy elicits cytotoxicity against malignant mesothelioma. Am J Respir Critic Care Med. (2010) 181:1383–90. doi: 10.1164/rccm.200909-1465OC
58. Chen G, Emens LA. Chemoimmunotherapy: reengineering tumor immunity. Cancer Immunol Immunother. (2013) 62:203–16. doi: 10.1007/s00262-012-1388-0
59. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
60. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. (2007) 56:641–8. doi: 10.1007/s00262-006-0225-8
61. Noordam L, M.Kaijen EH, Bezemer K, Cornelissen R, Maat L, Hoogsteden HC, et al. Low-dose cyclophosphamide depletes circulating naive and activated regulatory T cells in malignant pleural mesothelioma patients synergistically treated with dendritic cell-based immunotherapy. Oncoimmunology. (2018) 7:e1474318. doi: 10.1080/2162402X.2018.1474318
62. Aerts JG, Goeje P, Cornelissen R, Kaijen-Lambers M, Bezemer K, Leest C, et al. Autologous dendritic cells pulsed with allogeneic tumor cell lysate in mesothelioma: from mouse to human. Clin Cancer Res. (2017) 24:2522. doi: 10.1158/1078-0432.CCR-17-2522
63. Cornelissen R, Belderbos R, Aerts J. Abstract 2249: checkpoint inhibitor therapy after dendritic cell vaccination elicits tumor response in mesothelioma patients. Cancer Res. (2019) 79:2249. doi: 10.1158/1538-7445.AM2019-2249
64. Belderbos RA, Baas P, Berardi R, Cornelissen R, Fennell DA, van Meerbeeck JP, et al. A multicenter, randomized, phase II/III study of dendritic cells loaded with allogeneic tumor cell lysate (MesoPher) in subjects with mesothelioma as maintenance therapy after chemotherapy: DENdritic cell Immunotherapy for Mesothelioma (DENIM) trial. Transl Lung Cancer Res. (2019) 8:280–5. doi: 10.21037/tlcr.2019.05.05
65. Dammeijer F, Lievense LA, Kaijen-Lambers ME, van Nimwegen M, Bezemer K, Hegmans JP, et al. Depletion of tumor-associated macrophages with a CSF-1R kinase inhibitor enhances antitumor immunity and survival induced by DC immunotherapy. Cancer Immunol Res. (2017) 5:535–46. doi: 10.1158/2326-6066.CIR-16-0309
66. Belderbos DR, Gulijk M, Lukkes M, Dumoulin D, Cornelissen R, Aerts J, et al. 09 Checkpoint Inhibitors Synergize With Dendritic Cell-Therapy in Pre-Clinical Models And Mesothelioma Patients. Abstract WCLC (2019) 2019:598. doi: 10.1016/j.jtho.2019.08.598
67. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
68. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
69. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
70. Fousek K, Ahmed N. The evolution of T-cell therapies for solid malignancies. Clin Cancer Res. (2015) 21:3384–92. doi: 10.1158/1078-0432.CCR-14-2675
71. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics. (2016) 3:16006. doi: 10.1038/mto.2016.6
72. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. (2017) 5:22. doi: 10.1186/s40364-017-0102-y
73. Petersen CT, Krenciute G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front Oncol. (2019) 9:69. doi: 10.3389/fonc.2019.00069
74. Sermer D, Brentjens R. CAR T-cell therapy: full speed ahead. Hematol Oncol. (2019) 37 (Suppl. 1):95–100. doi: 10.1002/hon.2591
75. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. (2019) 20:31–42. doi: 10.1016/S1470-2045(18)30864-7
76. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEsJMoa1709866
77. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
78. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. (2018) 9:282. doi: 10.1038/s41419-018-0278-6
79. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. (2019) 10:128. doi: 10.3389/fimmu.2019.00128
80. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
81. Amos SM, Duong CP, Westwood JA, Ritchie DS, Junghans RP, Darcy PK, et al. Autoimmunity associated with immunotherapy of cancer. Blood. (2011) 118:499–509. doi: 10.1182/blood-2011-01-325266
82. Curioni, Britschgi C, Hiltbrunner S, Bankel L, Gulati P, Weder W, et al. A phase I clinical trial of malignant pleural mesothelioma treated with locally delivered autologous anti-FAP-targeted CAR T-cells. Ann Oncol. (2019) 30:v501. doi: 10.1093/annonc/mdz253.052
83. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
84. Haas AR, Tanyi JL, O'Hara MH, Gladney WL, Lacey SF, Torigian DA, et al. Phase I Study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther. (2019) 27:1919–29. doi: 10.1016/j.ymthe.2019.07.015
85. Adusumilli PS, Zauderer MG, Rusch VW, O'Cearbhaill RE, Zhu A, Ngai DA, et al. Abstract CT036: A phase I clinical trial of malignant pleural disease treated with regionally delivered autologous mesothelin-targeted CAR T cells: safety and efficacy. Cancer Res. (2019) 79:CT036. doi: 10.1158/1538-7445.AM2019-CT036
86. Klampatsa, Haas AR, Moon EK, Albelda SM. Chimeric Antigen Receptor (CAR) T cell therapy for Malignant Pleural Mesothelioma (MPM). Cancers. (2017) 9:115. doi: 10.3390/cancers9090115
87. Klampatsa Achkova DY, Davies DM, Parente-Pereira AC, Woodman N, Rosekilly J, et al. Intracavitary 'T4 immunotherapy' of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. (2017) 393:52–9. doi: 10.1016/j.canlet.2017.02.015
88. Al-Taei S, Salimu J, Lester JF, Linnane S, Goonewardena M, Harrop R, et al. Overexpression and potential targeting of the oncofoetal antigen 5T4 in malignant pleural mesothelioma. Lung Cancer. (2012) 77:312–8. doi: 10.1016/j.lungcan.2012.03.008
89. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
90. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. (2014) 6:261ra151. doi: 10.1126/scitranslmed.3010162
91. Zeltsman M, Dozier J, McGee E, Ngai D, Adusumilli PS. CAR T-cell therapy for lung cancer and malignant pleural mesothelioma. Transl Res. (2017) 187:1–10. doi: 10.1016/j.trsl.2017.04.004
92. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
Keywords: mesothelioma, cancer vaccines, dendritic cell therapy, CAR-T cell therapy, immunotherapy
Citation: Belderbos RA, Vroman H and Aerts JGJV (2020) Cellular Immunotherapy and Locoregional Administration of CAR T-Cells in Malignant Pleural Mesothelioma. Front. Oncol. 10:777. doi: 10.3389/fonc.2020.00777
Received: 15 September 2019; Accepted: 21 April 2020;
Published: 03 June 2020.
Edited by:
Nico van Zandwijk, Sydney Local Health District, AustraliaReviewed by:
Matteo Giaj Levra, Centre Hospitalier Universitaire de Grenoble, FranceDavid Marc Davies, King's College London, United Kingdom
Copyright © 2020 Belderbos, Vroman and Aerts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joachim G. J. V. Aerts, ai5hZXJ0c0BlcmFzbXVzbWMubmw=