
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 03 June 2020
Sec. Cancer Epidemiology and Prevention
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00710
 Xueting Li1
Xueting Li1 Yuanli Liu1
Yuanli Liu1 Yan Wang1
Yan Wang1 Xue Li1
Xue Li1 Xinran Liu1
Xinran Liu1 Mengru Guo1
Mengru Guo1 Yiwei Tan1Xiaofa Qin2
Yiwei Tan1Xiaofa Qin2 Xiuhong Wang1*Mingshan Jiang3*
Xiuhong Wang1*Mingshan Jiang3*Sucralose is a calorie-free high-intensity artificial sweetener that is widely used in thousands of foods and beverages all over the world. Although it was initially regarded as a safe, inert food additive, its adverse effect on gut microbiota and health has drawn more and more attention as evidence accumulates. Studies by us and others revealed that sucralose exacerbated gut damage and inflammation in animal models for inflammatory bowel disease (IBD), including those for both ulcerative colitis, and Crohn's disease. Our study demonstrated that sucralose greatly aggravated dextran sulfate sodium (DSS)-induced colitis along with causing changes in gut microbiota, the gut barrier and impaired inactivation of digestive proteases mediated by deconjugated bilirubin. It is well-documented that IBD greatly increases the risk of colorectal cancer (CRC), the globally third-most-common cancer, which, like IBD, has a high rate in the developed countries. Azoxymethane (AOM)/DSS has been the most commonly used animal model for CRC. In this study, we further explored the effect of sucralose on tumorigenesis and the possible mechanism involved using the AOM/DSS mouse model. First, 1.5 mg/ml sucralose was included in the drinking water for 6 weeks to reach a relatively stable phase of impact on gut microbiota. Then, 10 mg/kg AOM was administered through intraperitoneal injection. Seven days later, 2.5% DSS was put in the drinking water for 5 days, followed by 2 weeks without DSS. The 5 days of DSS was then repeated, and the mice were sacrificed 6 weeks after AOM injection. The results showed that sucralose caused significant increases in the number and size of AOM/DSS-induced colorectal tumors along with changes in other parameters such as body and spleen weight, pathological scores, mortality, fecal β-glucuronidase and digestive proteases, gut barrier molecules, gut microbiota, inflammatory cytokines and pathways (TNFα, IL-1β, IL-6, IL-10, and TLR4/Myd88/NF-κB signaling), and STAT3/VEGF tumor-associated signaling pathway molecules. These results suggest that sucralose may increase tumorigenesis along with dysbiosis of gut microbiota, impaired inactivation of digestive protease, damage to the gut barrier, and exacerbated inflammation.
Colorectal cancer (CRC) is the third most commonly diagnosed cancer worldwide, with an incidence ranking third in men, only after lung and prostate cancer, and second in women, only after breast cancer (1). CRC is high in developed countries, and there is a trend of increase in multiple developing countries (2). Although an increase in screening is leading to a decline in CRC incidence in the elderly population, multiple studies in countries such as the USA, Canada, UK, Germany, Norway, Denmark, Sweden, Slovenia, Australia, and New Zealand have shown increases in CRC in those younger than 50 (2–4). A study using data on 143.7 million people aged 20–49 years from 20 European countries revealed an increase of CRC incidence from 2004/2005 to 2016 at 7.9% per year among subjects aged 20–29 years, 4.9% per year in the 30–39-years age group, and 1.6% per year in the 40–49-years age group (4). However, the cause for this remains a mystery.
Studies have found that CRC may be affected by multiple factors such as race and ethnicity, heredity, smoking, and alcohol (5). The high level of CRC in developed countries in North America, Europe, Australia, New Zealand, Japan, and South Korea suggested a close association between CRC and the modern lifestyle (2, 5). Intriguingly, inflammatory bowel disease (IBD), a devastating inflammatory disease of the gut, is also very high in developed countries (6, 7), and IBD greatly increased the risk of CRC with a hazard ratio as high as 33.3 (5), suggesting a likely intimate link between CRC and IBD (8–10). Recently, dysbiosis of microbiota in the development of IBD and CRC has drawn more and more attention (11–15). Accumulating evidence suggests that dysbiosis of gut microbiota, gut inflammation, and CRC may have intimate interaction and connection. There are reports that IBD and CRC may involve decreased abundance and diversity of gut bacteria along with a changed ratio of Firmicutes, Bacteroidetes, and Proteobacteria (14–16). Stool from patients with CRC had changes in the abundances of Fusobacterium nucleatum, Peptostreptococcus anaerobius, Peptostreptococcus stomatis, Parvimonas micra, Solobacterium moorei, and Gemella morbillorum (16, 17). An important question is what causes this dysbiosis of gut microbiota in modern society.
Not surprisingly, diet, not only in terms of nutrients but also of dietary chemicals, may have a huge impact on gut microbiota (11, 14, 15). In fact, it has been long proposed that food additives such as saccharin and sucralose may have played an important causative role in IBD due to their inhibition of gut bacteria and the resultant impaired inactivation of digestive proteases by deconjugated bilirubin through the action of bacterial β-glucuronidase (18, 19). Papers published in recent years in Nature showed that food additives such as artificial sweeteners and emulsifiers increased the risk of diabetes, obesity, and colitis through their adverse impact on gut bacteria (20, 21). Studies by us and others revealed that sucralose, the widely used artificial sweetener, increased the risk of IBD in animal models for both ulcerative colitis and Crohn's disease (22, 23). Our study showed that sucralose exacerbated dextran sulfate sodium (DSS)-induced colitis along with dysbiosis of gut microbiota, decrease in bacterial β-glucuronidase, increase in fecal digestive protease, and aggravated damage of the gut barrier and gut tissue. As azoxymethane (AOM)/DSS is the most commonly used animal model for CRC (24), and microbiota dysbiosis and barrier dysfunction may be a common ground for IBD and CRC (25, 26), we investigated the effect of sucralose on AOM/DSS-induced tumorigenesis in mice.
C57BL/6 mice (4 weeks old, from the Laboratory Animal Center of the Second Affiliated Hospital of Harbin Medical University) were adapted to the environment for 2 weeks before the experiment and were raised under specific pathogen-free conditions in the Animal Experimental Center of Harbin Medical University (24–25°C, humidity 70–75%, 12 h light/dark) with a standard diet and drinking water. All animal experiments strictly followed the requirements of animal feeding regulations of Harbin Medical University and met the ethical requirements for animal experiments.
N-benzoyl-L-tyrosine ethyl ester (BTEE), Na-Benzoyl-Larginine 4-nitroanilide hydrochloride (BAPNA), sucralose (≥98.0% HPLC), and Azoxymethane (AOM) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 4-Nitrophenyl b-D-glucopyranoside was purchased from BBI Life Sciences. DSS (MW: 36–50 kDa) was obtained from MP Biomedical (Solon, OH, USA). The antibodies used in this study were anti-TLR4 (19811-1-AP, Proteintech), anti-VEGF (19003-1-AP, Proteintech), anti-STAT3 (10253-2-AP, Proteintech), anti-Phospho-Stat3 (Tyr705) (#9145, Cell Signaling Technology), anti-MyD88 (#4283, Cell Signaling Technology), anti-TRAF6 (YT4720, Immunoway), anti-inhibitor of NF-κB alpha (IκBα) (#4814, Cell Signaling Technology), and anti-Occludin (13409-1-AP, Proteintech), as well as anti-GAPDH, anti-β-Actin, goat anti-rabbit IgG, and goat anti-mouse IgG from ZSGB-BIO Co. Ltd. (Beijing, China). All other reagents used were of analytical grade.
Before the induction of CRC by AOM, sucralose was added to the drinking water of mice at 1.5 mg/ml for 6 weeks to induce explicit change in the gut microbiota (21, 27). Then, AOM was administered through intraperitoneal injection at 10 mg/kg. Seven days later, 2.5 % DSS was put in the drinking water for 5 days, followed by 2 weeks without DSS. The 5-days DSS was then repeated (28). Sucralose was given for the whole period, and the animals were sacrificed on the 36th day after AOM injection. An illustrated protocol can be seen in Figure 1A. Feces were collected just before the start of experiment and on day 1, 6, 25, and 36 after AOM injection. Gut, spleen, and blood were harvested at the end of the experiment. Samples were stored frozen prior to analysis.
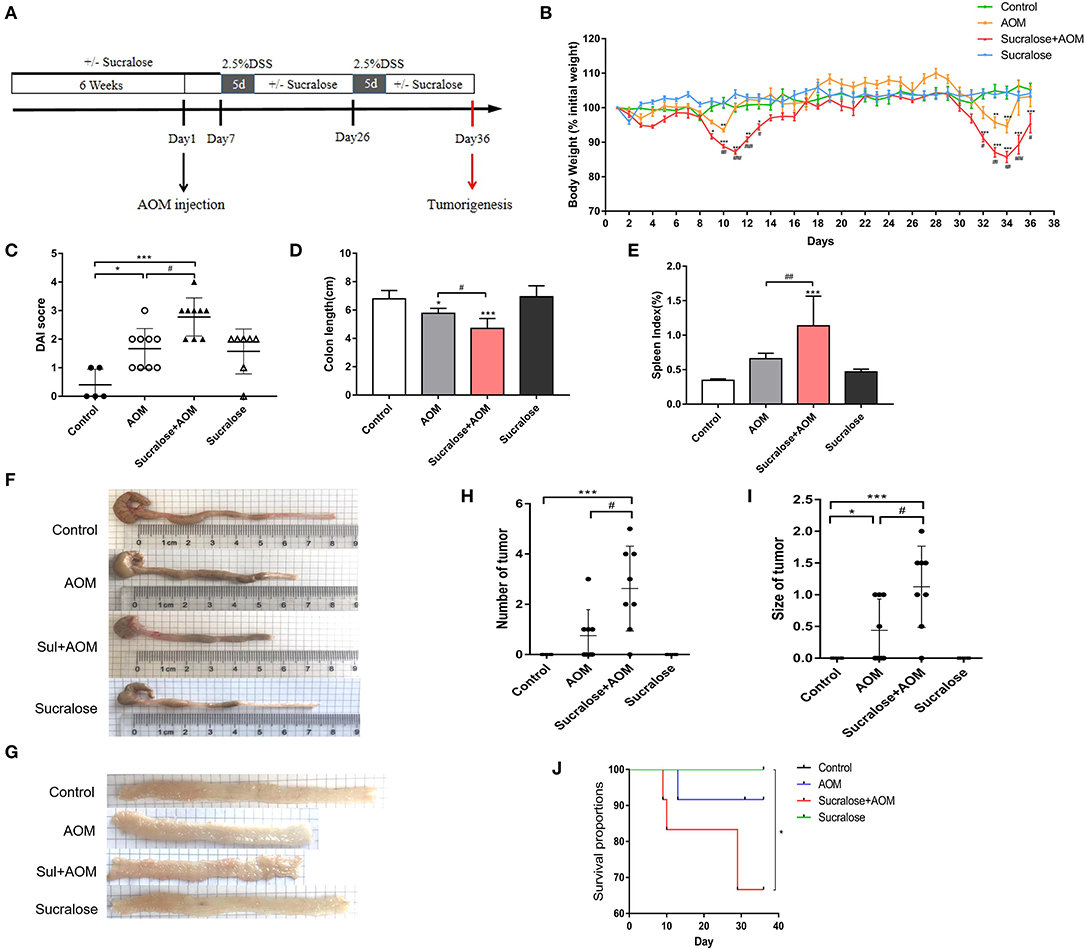
Figure 1. Sucralose significantly increases the severity of disease of AOM/DSS colon cancer mice. (A) Protocol of sucralose and AOM/DSS treatments used in this study. (B) Change in body weight, (C) disease activity index (DAI) score, (D) colon length, (E) spleen weight index, (F) photoraphs showing colon length, (G) photograph of colon tumor, (H) tumor number, (I) tumor size, and (J) survival rate. Compared with the control group: *P < 0.05, **P < 0.01, ***P < 0.001; compared with the AOM group: #P < 0.05, ##P < 0.01, ###P < 0.001, n = 8.
The disease activity index (DAI) was calculated for each animal by adding together the scores for body weight, stool consistency, and stool blood as listed in Supplementary Table 1 and described in our previous studies (29).
Tumor size was measured ex vivo according to the area against a standard background grid. Colonic tissues and tumors were fixed in 4% (w/v) paraformaldehyde and then paraffin-embedded, sectioned, and stained with hematoxylin and eosin (HE). Pathological changes were observed and scored. Colonic damage was graded in a blinded manner, as described in Supplementary Table 2 and in our previous studies (27).
Feces were collected, added to PBS containing PMSF, and homogenized. After centrifuging, the supernatant was collected for the assay. β-glucuronidase, trypsin, and chymotrypsin (amidase) activities were measured by spectrophotometry using 4-Nitrophenyl b-D-glucopyranoside, Na-Benzoyl-L-arginine 4-nitroanilide hydrochloride (BAPNA), and N-benzoyl-l-tyrosine ethyl ester (BTEE) as the substrate, respectively, according to the methods described in detail in our previous studies (27, 30).
Total RNA was isolated from colon tissues using the UNlQ-10 Column Trizol Total RNA Isolation Kit (Sangon Biotech, Shanghai, China). The RNA concentration and OD260/280 absorbance ratio were then measured using a Nanodrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The RNA was transcribed into cDNA using 5X All-In-One RT MasterMix (abm, Jiangsu, China; cat. no. G492). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed in volumes of 20 μl containing 1 μl of each primer (Supplementary Table 3) in the FastStart Universal SYBR Green Master (Roche, Basel, Switzerland; cat.no.04913850001) according to the manufacturer's instructions. PCR amplification was performed with the following conditions: 30 s at 95°C, followed by 40 cycles of 5 s at 95°C and 31 s at 60°C. After that, a melting curve analysis was performed to confirm the specificity of the qRT-PCR. All samples were analyzed in triplicate, and the results were normalized to the expression of GAPDH. The results were calculated by the 2−ΔΔCt equation.
Feces on day 36 were used to detect changes in the intestinal flora of the mice. Bacterial DNA in 0.15 g of feces collected by the end of the experiment was extracted by TIANamp Stool DNA Kit (TIANGEN, China) then amplified by qPCR (Applied Biosystems 7500 PCR) using the primers listed in Supplementary Table 4. The abundance of total bacteria in feces was assessed as the relative ratio of 16S RNA (16S) to DNA, while the abundance of certain bacteria was calculated as the relative ratio of the specific sequence to 16S.
Segments of colon were homogenized using RIPA buffer and protein inhibitor cocktail (1:10) (PhosSTOP ESAYpack, Roche). The homogenates were kept on ice for 30 min and centrifuged at 12,000 g for 5 min at 4°C. The protein concentration was determined using the BCA Protein Assay Kit (Beyotime, Shanghai, China). For ELISA assays, colon tissue protein was collected and assayed for IL-1β, IL-10, IL-6, and MPO, blood was collected, and D-Lac in serum was measured according to the manufacturer's instructions. The results for IL-1β, IL-10, and IL-6 are expressed in pg/μg, MPO is expressed in U/mg, and D-Lac is expressed in nmol/L. For Western blot analysis, 20–80 μg of protein was electroblotted onto a PVDF membrane after separation by 10% SDS-polyacrylamide gel electrophoresis. Immunoblots were incubated with primary antibodies against Occludin, STAT3, p-STAT3, VEGF, TNFα, TLR4, MyD88, TRAF6, NF-κB inhibitor alpha (IκBα), and GAPDH or β-Actin. Chemiluminescent signals were analyzed using the Quantity One (version 4.5.2) program (Bio-Rad Laboratories) and Image J software.
The experimental data were plotted and analyzed with Graphpad Prism version 7.0, and the experimental results were expressed as mean ± standard error (n = 5–10 in each group). The results of the experiments were analyzed using repeated-measures ANOVA with Greenhouse-Geisser correction and Tukey's multiple comparisons test or t-test. P < 0.05 being regarded as significant difference. Pearson correlation coefficient was used for correlation analysis, with P < 0.05 being considered statistically significant.
Figure 1A illustrates the protocol of treatment in this study. We can see some fluctuations in body weight in the AOM/DSS groups (Figure 1B), which would be mainly due to the repeated treatment and withdraw of DSS. DSS will result in colitis along with diarrhea, bloody stool, rapid weight loss, inflammation and shorting of the colon, which may gradually recover after withdrawal. Compared with mice treated with AOM/DSS alone, the AOM/DSS + sucralose group showed more severe weight loss, more blood in stool, and a significantly lower DAI score, more shortening of the colon, and higher spleen weight (Figures 1B–F). Sucralose also increased the number and size of tumors induced by AOM/DSS, with a positive rate of 87.5% in the AOM/DSS + sucralose group vs. 50% in the AOM/DSS-alone group (Figures 1G–I). In addition, the AOM/DSS + sucralose group also showed higher mortality (Figure 1J).
We measured the digestive proteases trypsin and chymotrypsin and β-glucuronidase in feces, MUC2 and tight junction molecules (Occludin, ZO-1, claudin-1, and claudin-4) in the mucosa, and D-Lac in serum (a bacterial product of the gut that infiltrates into the blood) as a parameter for gut permeability.
The results showed that mice treated with AOM/DSS alone demonstrated significantly higher levels of fecal trypsin and chymotrypsin but a lower level of fecal β-glucuronidase, as well as exhibiting reduced occludin and increased claudin-1 and claudin-4 in muscosa and elevated serum D-Lac (Figure 2), suggesting damage to the gut barrier and increased gut permeability. AOM/DSS + sucralose caused further increase in fecal trypsin and chymotrypsin and decrease in fecal β-glucuronidase, as well as increased mucosal occludin, claudin-1, and claudin-4, suggesting further damage to the gut barrier (Figure 2). β-glucuronidase showed a significant negative correlation with trypsin and chymotrypsin (Table 1). Surprisingly, the serum D-Lac level of the AOM/DSS + sucralose group, although significantly higher than control, is significantly lower than that of the AOM/DSS group, which will be discussed in detail in the next section.
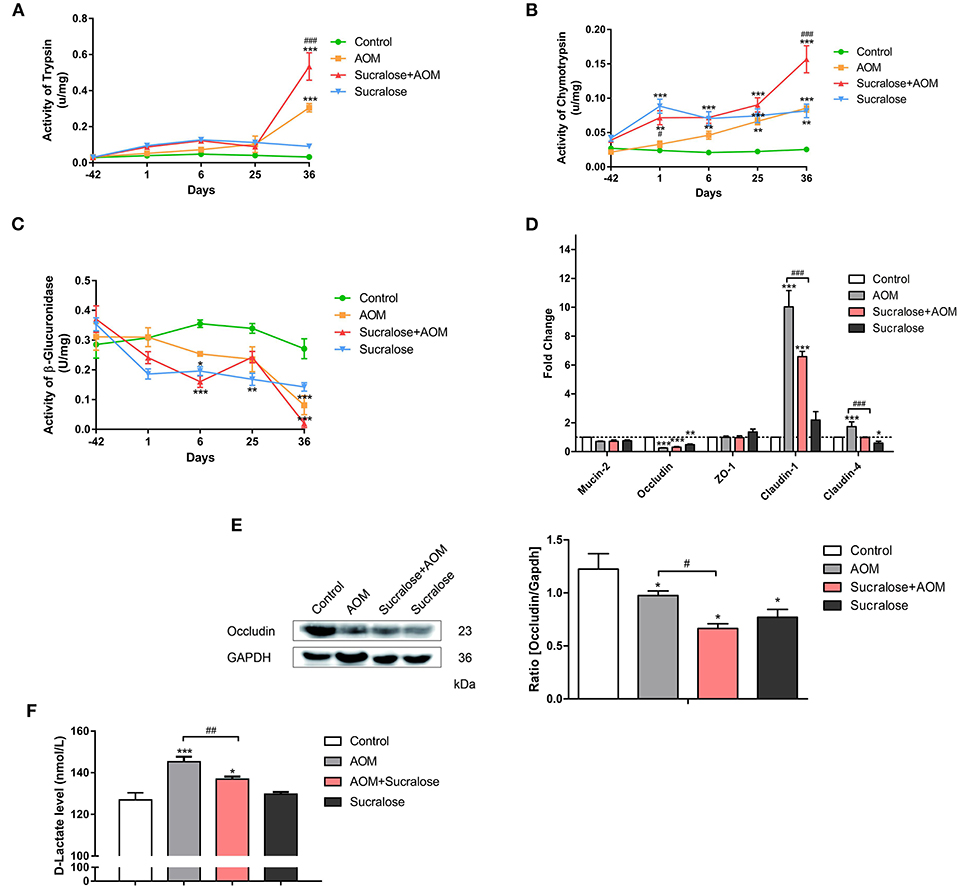
Figure 2. Sucralose aggravates damage to the colonic mucosal barrier in AOM/DSS colon cancer mice. (A) Trypsin activity in feces, (B) chymotrypsin activity in feces, (C) β-glucuronidase activity in feces, (D) changes in mucosal barrier molecule mRNA levels, (E) occluding (tight junction protein) expression, and (F) serum D-Lac levels. Compared with the control group: *P < 0.05, **P < 0.01, ***P < 0.001; compared with the AOM group: #P < 0.05, ##P < 0.01, ###P < 0.001, n = 8.

Table 1. Correlation between β-glucuronidase, trypsin and chymotrypsin.
AOM/DSS caused marked epithelial destruction, infiltration of inflammatory cells, crypt deformation, increased abundance of mitotic cells, and a high degree of dysplasia, indicating the existence of inflammation and highly differentiated adenocarcinoma of the colon (Figure 3A), along with increases in microscopic lesion scores (Figure 3B) and MPO activity (Figure 3C). Treatment with sucralose caused even more pronounced changes than did AOM/DSS alone.
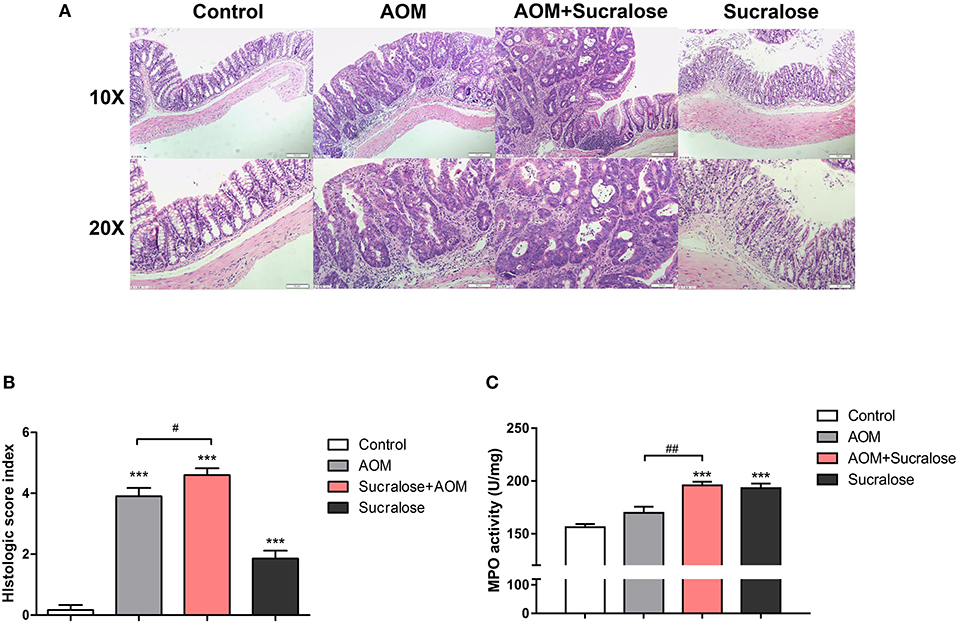
Figure 3. Sucralose significantly increases colonic tissue damage in AOM/DSS colon cancer mice. (A) Light microscopy evaluation of hematoxylin and eosin (H&E)-stained sections and (B) histological score (n = 5, scale bar = 50 μm). (C) MPO activity of colon tissue (n = 5). Compared with the control group: ***P < 0.001; compared with the AOM group: #P < 0.05, ##P < 0.01, n = 8.
We assessed the abundances of total gut bacteria and of the dominant phyla, as well as of some of the species that have been reported to be associated with CRC. Compared to control, all the three AOM/DSS and sucralose treated groups showed significant decreases in bacterial abundance as measured by 16S/DNA (Figure 4A). Treatment with AOM/DSS alone caused a significant increase in Peptostreptococcus anaerobius, while the sucralose-only group showed significant increases in Firmicures, Clostridium symbiosum, and Peptostreptococcus anaerobius and decreases in Solobacterium moorei and Bifidobacteria as compared to control. Compared to the AOM/DSS only group, AOM/DSS + sucralose caused significant increases in Ficmicures, Actinomycetes, Peptostreptococcus stomatis, Clostridium symbiosum, and Peptostreptococcus anaerobius and a decrease in proteobacteria (Figures 4B,C). Table 2 shows the correlations among the changes in gut microbiota and β-glucuronidase, trypsin, and chymotrypsin. β-glucuronidase showed significant positive correlations with 16S/DNA, Proteobacteria, Gemella taiwanensis, Parvimonas micra, and Bifidobacterium and significant negative correlations with Firmicutes, Lactobacillus, and Peptostreptococcus anaerobius. Trypsin showed significant positive correlations with Firmicutes, Actinobacteria, Clostridium symbiosum, Peptostreptococcus anaerobius, Peptostreptococcus stomatis, and Solobacterium moorel and significant negative correlations with Proteobacteria. Gemella taiwanensis, and Parvimonas micra. Chymotrypsin showed significant positive correlations with Firmicutes, Actinobacteria, Clostridium symbiosum, Lactobacillus, Peptostreptococcus anaerobius, and Peptostreptococcus stomatis and significant negative correlations with 16s/DNA and Proteobacteri.
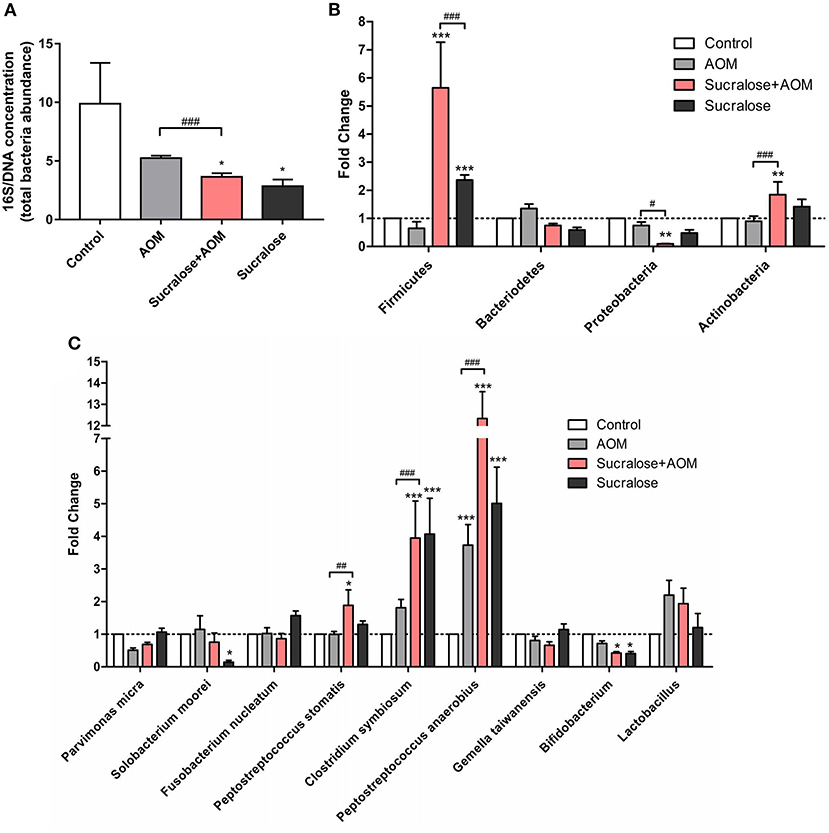
Figure 4. Sucralose alters mouse fecal microbiota composition. (A) Changes in 16S/DNA concentration. (B) Changes in Firmicutes, bacteriodetes, proteobacteria, and actinobacteria. (C) Changes in Parvimonas micra, Solobacterium moorei, Fusobacterium nucleatum, Peptostreptococcus stomatis, Clostridium symbiosum, Gemella, and Peptostreptococcus anaerobius. Compared with the control group: *P < 0.05, **P < 0.01, ***P < 0.001; compared with the AOM group: #P < 0.05, ##P < 0.01, ###P < 0.001, n = 8.
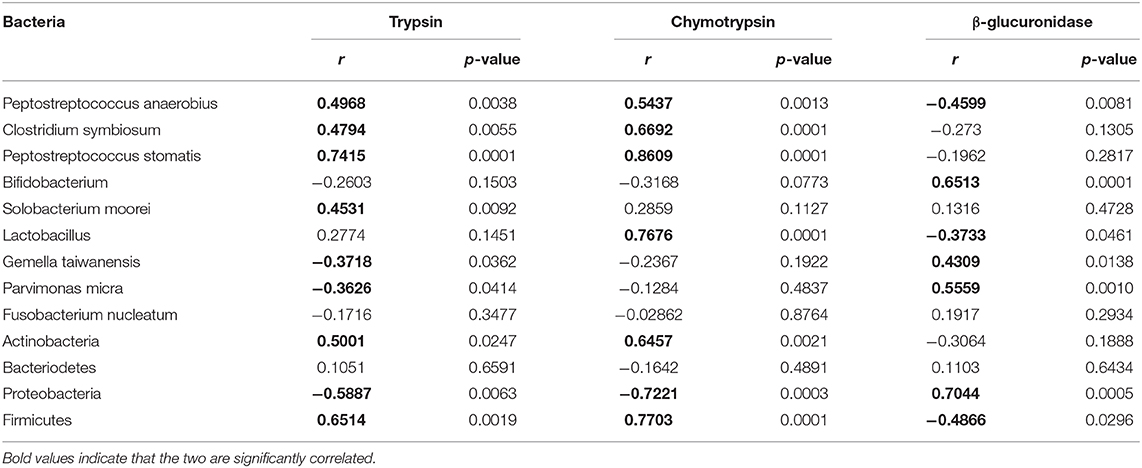
Table 2. Correlation between CRC-associated bacterial abundances and trypsin, chymotrypsin, and β-glucuronidase.
We measured a variety of inflammatory cytokines as well as key molecules of the TLR4/MyD88/TRAF6/NF-κB pathway associated with gut inflammation by increased infiltration of components from gut bacteria.
In terms of mRNA expression (Figure 5A), the AOM/DSS group showed significant increases in IL-1β, IL-10, and TRAF6 vs. control. Sucralose alone also resulted in significantly increased expressions of TNFα and TLR4. Compared to the AOM/DSS-alone group, the AOM/DSS + sucralose group showed significantly higher expressions of TNFα and IL-1β and lower levels of IL-10 and TRAF6.
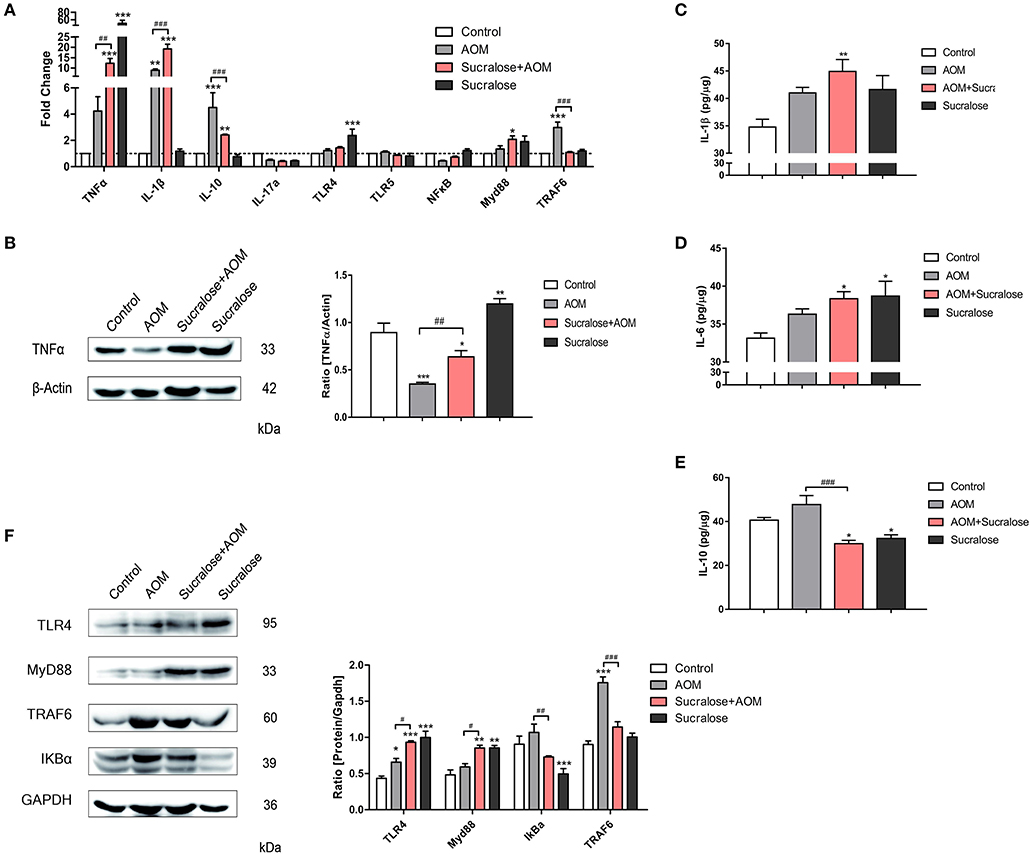
Figure 5. Sucralose promotes expression of pro-inflammatory factors. (A) Changes in mRNA levels of cytokines and TLR4/MyD88/TRAF6/NF-κB signaling pathways (n = 8). (B) Protein expression of TNFα. (C) Proteins of IL-1β in colon tissues. (D) Protein levels of IL-6 in colon tissue. (E) Protein levels of IL-10 in colon tissues. (F) Changes in protein levels of TLR4/MyD88/TRAF6/NF-κB signaling pathways. Compared with the control group: *P < 0.05, **P < 0.01, ***P < 0.001; compared with the AOM group: #P < 0.05, ##P < 0.01, ###P < 0.001, n = 5.
As for the protein level by Western blot assay (Figures 5B–F), the AOM/DSS group showed significant increases in IL-6, TLR4, and TRAF6 but a lower level of TNFα vs. control. Sucralose alone also resulted in significantly increased expressions of TNFα, TLR4, and Myd88 but decreases in IL-10 and IκBα. Compared to the AOM/DSS-alone group, the AOM/DSS + sucralose group showed significantly higher levels of TNFα, TLR4, and Myd88 but lower levels of IL-10, IκBα, and TRAF6.
We analyzed the mRNA level of tumor-associated genes such as Cadherin-1, Ki-67, PCNA, β-catenin, STAT3, and COX2 (Figure 6A), as well as the protein levels of tumor marker VEGF, its downstream transcription activator STAT3 and p-STAT3, and the tumor proliferation marker PCNA (Figures 6B–D). AOM/DSS did not show an effect on VEGF but increased the level of PCNA. Compared to AOM/DSS alone, the AOM/DSS + sucralose group showed significant increases in VEGF, STAT3, p-STAT3, and PCNA. Sucralose alone showed a high level of VEGF and PCNA.
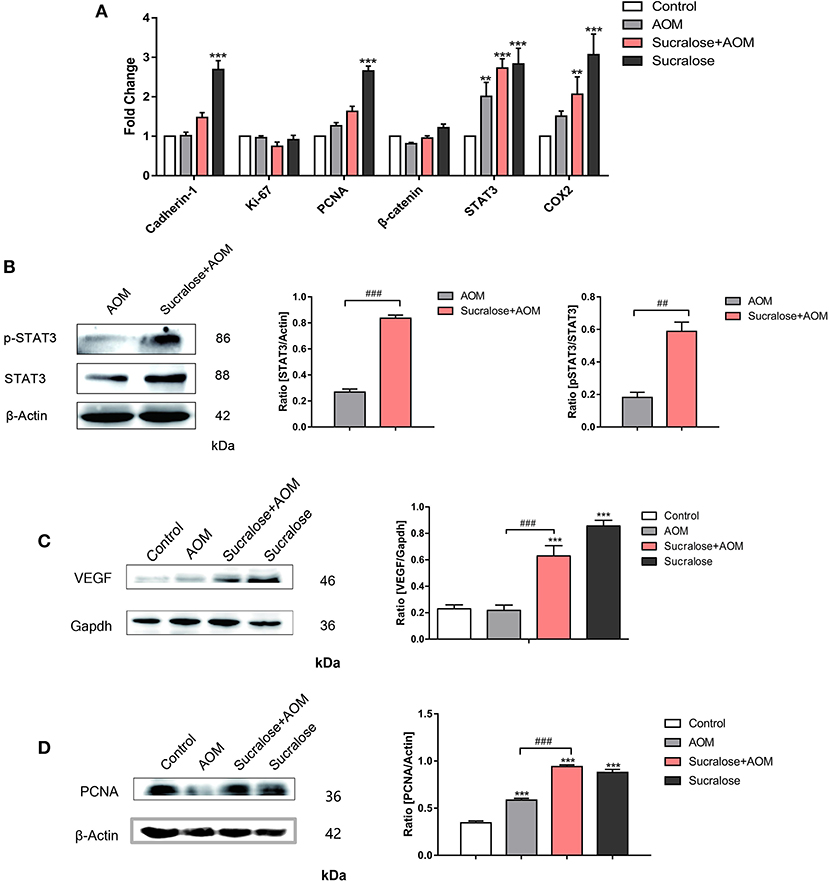
Figure 6. Sucralose promotes tumor-associated gene expression. (A) mRNA levels of tumor-associated genes. (B) Changes in STAT3 and pSTAT3 in tumor tissues. (C) Changes in VEGF protein in colon tissue. (D) Expression of PCNA protein in colon tissues. Compared with the control group: **P < 0.01, ***P < 0.001; compared with the AOM group: ##P < 0.01, ###P < 0.001, n = 5.
From the results above, we can see that sucralose caused significant increases in the number and size of AOM/DSS-induced colorectal tumors. A likely mechanistic explanation would be that inflammation was exacerbated by sucralose, as demonstrated by the higher expression or protein levels of TNFα and IL-1β, decrease in IL-10, and increased weight of spleen in the AOM/DSS + sucralose group compared to the AOM/DSS-alone group. Inflammation serves as a critical link between IBD and CRC (10). Interestingly, despite more damage to the gut barrier, as demonstrated by the reduced mucosal occludin, claudin-1, and claudin-4, the AOM/DSS + sucralose group had a lower level of serum D-lactate, a product of gut bacteria, than the AOM/DSS-alone group. This discrepancy may likely be due to the significant reduction in gut bacteria by sucralose shown in this study (Figure 4A) as well as in a study by others that sucralose can cause general inhibition of both total anaerobes and aerobic bacteria (25). This suggests the exacerbated inflammation in the sucralose group may be caused by factors other than increased infiltration of bacterial components. As AOM/DSS + sucralose caused greater increases in fecal trypsin and chymotrypsin, we propose that more severe damage to the gut barrier and increased infiltration of other toxicants such as DSS are the result of more severe impaired inactivation of digestive proteases and that changes in gut microbiota may have played a critical role.
Under normal conditions, digestive proteases such as trypsin and chymotrypsin are present at very low levels in feces (31–34). This inactivation of digestive proteases depends on gut bacteria, as a large amount of digestive proteases appear in the lower gut when animals are raised under germ-free conditions (35) or are treated with antibiotics (36) or saccharin and sucralose (37, 38). Both our in vitro and in vivo studies showed inhibition of digestive proteases by unconjugated but not by conjugated bilirubin (39, 40). Thus, deconjugation of bilirubin by the bacterial β-glucuronidase enriched in certain kinds of gut bacteria may be the key link between gut bacteria and the inactivation of digestive proteases in gut lumen. This also provided an explanation of how a reduction in gut bacteria can result in more damage and pathogenesis of the gut, which is also shown in other multiple studies. For instance, a study by Zhan et al. found that germ-free mice developed significantly more and larger tumors compared with conventional specific pathogen-free (SPF) mice after AOM and DSS treatment despite the lack of early acute inflammation (41). The germ-free mice showed a delay in intestinal epithelial repair of DSS-induced injury, resulting in a late onset of proinflammatory and protumorigenic responses and increased epithelial proliferation and microadenoma formation. In another study, it was shown that germ-free mice had enhanced hemorrhaging, epithelial injury, and mortality as a consequence of a weakened intestinal barrier despite only minimal inflammation (42). Impairment of the deconjugated bilirubin-mediated inactivation of digestive proteases due to a reduction in or lack of gut bacteria, and thus of the bacterial β-glucuronidase needed for bilirubin deconjugation, may be a shared key mechanism for all these mysteries (43). This notion is strongly supported by the significant negative correlation between β-glucuronidase and trypsin and chymotrypsin shown in Table 1 as well as the significant positive correlation between gut bacteria abundance and fecal β-glucuronidase and significant negative correlation between gut bacteria abundance and digestive proteases such as chymotrypsin shown in Table 2. Despite this, as shown in Table 2, although gut bacteria such as Firmicutes at the phylum level showed significant negative correlations with β-glucuronidase and negative correlations with trypsin and chymotrypsin, the lower level of gut bacteria of the Firmicutes phylum such as Gemella taiwanensis, Lactobacillus, Peptostreptococcus anaerobius, and Parvimonas micra may have an opposite correlation. Studies have shown that even the same species of gut bacteria may have different impacts. For instance, the enterotoxigenic strain of B. fragilis may serve as a trigger for colitis and tumorigenesis, while the non-toxigenic strain of B. fragilis may serve as an anti-inflammatory probiotic and protect against colitis and CRC (11). Thus, it remains a formidable task to elucidate the association between dysbiosis of gut bacteria and diseases.
To date, approval of food additives has never taken into consideration their impact on gut microbiota and related health. However, overwhelming evidence has shown the critical role of gut microbiota dysbiosis in the dramatic increases of many diseases in modern society (44, 45). In view of the synergistic effect of bacterial glycosidase and pancreatic digestive proteases on the degradation of the protective mucus layer, the frequent, constant use of food additives such as sucralose and saccharin may be more detrimental to gut microbiota and health than antibiotics (46). We may need to adopt a prudent attitude to claiming the absolute safety of currently approved food additives for gut microbiota and health.
IBD and CRC and many other diseases in modern society are multifactorial. Nevertheless, given the substantial synergistic effect of AOM and DSS in the model used in this study as well as the added effect of sucralose on AOM/DSS, there is a major problem with basing safety evaluation on separate assessments of individual compounds. The many kinds of food additives in processed food will have added effects on gut microbiota (47). This remains a major challenge, and there is a long way to go to achieve a precise evaluation of the many environmental factors associated with modernization and to find out the causes and mechanisms of diseases emerging and dramatically increasing in modern society. We hope that this preliminary early-stage study may spur more research in this area.
All datasets generated for this study are included in the article/Supplementary Material.
The animal study was reviewed and approved by Institutional Research Board of Harbin Medical University.
XuetL, YL, YW, XueL, XiL, MG, and YT conducted the experiments. XuetL, XW, and XQ wrote the manuscript. WX and MJ contributed to study supervision. All authors participated in the design, interpretation of the studies and analysis of the data, and review of the manuscript.
This work was supported by the National Natural Youth Foundation of China (no. 81703232), Heilongjiang Postdoctoral Program (LBH-Z13147), and College Student Innovation and Entrepreneurship in Heilongjiang Province (no. 201810226020).
XQ was employed by the company GI Biopharma Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00710/full#supplementary-material
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Torre LA, Soerjomataram I, Hayes RB, Bray F, Weber TK, et al. Global patterns and trends in colorectal cancer incidence in young adults. Gut. (2019) 68:2179–85. doi: 10.1136/gutjnl-2019-319511
3. Araghi M, Soerjomataram I, Bardot A, Ferlay J, Cabasag CJ, Morrison DS, et al. Changes in colorectal cancer incidence in seven high-income countries: a population-based study. Lancet Gastroenterol Hepatol. (2019) 4:511–8. doi: 10.1016/S2468-1253(19)30147-5
4. Vuik FE, Nieuwenburg SA, Bardou M, Lansdorp-Vogelaar I, Dinis-Ribeiro M, Bento MJ, et al. Increasing incidence of colorectal cancer in young adults in Europe over the last 25 years. Gut. (2019) 68:1820–6. doi: 10.1136/gutjnl-2018-317592
5. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. (2019) 14:89–103. doi: 10.5114/pg.2018.81072
6. Kirsner JB. Historical review: the historical basis of the idiopathic inflammatory bowel disease. Inflamm Bowel Dis. (1995) 1:2–26. doi: 10.1002/ibd.3780010103
7. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. (2018) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
8. Nadeem MS, Kumar V, Al-Abbasi FA, Kamal MA, Anwar F. Risk of colorectal cancer in inflammatory bowel diseases. Semin Cancer Biol. (2019). doi: 10.1016/j.semcancer.2019.05.001
9. Stidham RW, Higgins PDR. Colorectal cancer in inflammatory bowel disease. Clin Colon Rectal Surg. (2018) 31:168–78. doi: 10.1055/s-0037-1602237
10. Svrcek M, Borralho Nunes P, Villanacci V, Beaugerie L, Rogler G, De Hertogh G, et al. Clinicopathological and molecular specificities of inflammatory bowel disease-related colorectal neoplastic lesions: the role of inflammation. J Crohns Colitis. (2018) 12:1486–98. doi: 10.1093/ecco-jcc/jjy132
11. Grazioso TP, Brandt M, Djouder N. Diet, microbiota, and colorectal cancer. iScience. (2019) 21:168–87. doi: 10.1016/j.isci.2019.10.011
12. Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol. (2019) 16:690–704. doi: 10.1038/s41575-019-0209-8
13. Mendonca L, Dos Santos Ferreira R, de Cassia Avellaneda Guimaraes R, de Castro AP, Franco OL, Matias R, et al. The complex puzzle of interactions among functional food, gut microbiota, and colorectal cancer. Front Oncol. (2018) 8:325. doi: 10.3389/fonc.2018.00325
14. Rapozo DC, Bernardazzi C, de Souza HS. Diet and microbiota in inflammatory bowel disease: the gut in disharmony. World J Gastroenterol. (2017) 23:2124–40. doi: 10.3748/wjg.v23.i12.2124
15. Neuman MG, Nanau RM. Inflammatory bowel disease: role of diet, microbiota, life style. Transl Res. (2012) 160:29–44. doi: 10.1016/j.trsl.2011.09.001
16. Wong SH, Zhao L, Zhang X, Nakatsu G, Han J, Xu W, et al. gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology. (2017) 153:1621–33.e6. doi: 10.1053/j.gastro.2017.08.022
17. Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WK, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. (2015) 6:8727. doi: 10.1038/ncomms9727
18. Qin X. Impaired inactivation of digestive proteases by deconjugated bilirubin: the possible mechanism for inflammatory bowel disease. Med Hypotheses. (2002) 59:159–63. doi: 10.1016/S0306-9877(02)00243-8
19. Qin X. Etiology of inflammatory bowel disease: a unified hypothesis. World J Gastroenterol. (2012) 18:1708–22. doi: 10.3748/wjg.v18.i15.1708
20. Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature. (2014) 514:181–6. doi: 10.1038/nature13793
21. Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. (2015) 519:92–6. doi: 10.1038/nature14232
22. Wang X, Guo J, Liu Y, Yu H, Qin X. Sucralose increased susceptibility to colitis in rats. Inflamm Bowel Dis. (2019) 25:e3–e4. doi: 10.1093/ibd/izy196
23. Rodriguez-Palacios A, Harding A, Menghini P, Himmelman C, Retuerto M, Nickerson KP, et al. The artificial sweetener splenda promotes gut proteobacteria, dysbiosis, and myeloperoxidase reactivity in crohn's disease-like ileitis. Inflamm Bowel Dis. (2018) 24:1005–20. doi: 10.1093/ibd/izy060
24. De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, et al. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog. (2011) 10:9. doi: 10.4103/1477-3163.78279
25. Yu LC. Microbiota dysbiosis and barrier dysfunction in inflammatory bowel disease and colorectal cancers: exploring a common ground hypothesis. J Biomed Sci. (2018) 25:79. doi: 10.1186/s12929-018-0483-8
26. Tomasello G, Tralongo P, Damiani P, Sinagra E, Di Trapani B, Zeenny MN, et al. Dismicrobism in inflammatory bowel disease and colorectal cancer: changes in response of colocytes. World J Gastroenterol. (2014) 20:18121–30. doi: 10.3748/wjg.v20.i48.18121
27. Abou-Donia MB, El-Masry EM, Abdel-Rahman AA, McLendon RE, Schiffman SS. Splenda alters gut microflora and increases intestinal p-glycoprotein and cytochrome p-450 in male rats. J Toxicol Environ Health A. (2008) 71:1415–29. doi: 10.1080/15287390802328630
28. Crncec Ilija, Pathria Paulina, Svinka Jasmin et al. Induction of colorectal cancer in mice and histomorphometric evaluation of tumors. Methods Mol Biol. (2015) 1267:145–64. doi: 10.1007/978-1-4939-2297-0_7
29. Zhou JA, Jiang M, Yang X, Liu Y, Guo J, Zheng J, et al. Unconjugated bilirubin ameliorates the inflammation and digestive protease increase in TNBS-induced colitis. Mol Med Rep. (2017) 16:1779–84. doi: 10.3892/mmr.2017.6825
30. Zhou K, Jiang M, Liu Y, Qu Y, Shi G, Yang X, et al. Effect of bile pigments on the compromised gut barrier function in a rat model of bile duct ligation. PLoS ONE. (2014) 9:e98905. doi: 10.1371/journal.pone.0098905
31. Genell S, Gustafsson BE, Ohlsson K. Quantitation of active pancreatic endopeptidases in the intestinal contents of germfree and conventional rats. Scand. J Gastroenterol. (1976) 11:757–762.
32. Gustafsson BE. The physiological importance of the colonic microflora. Scand J Gastroenterol Suppl. (1982) 77:117–31.
33. Midtvedt T, Carlstedt-Duke B, Hoverstad T, Midtvedt AC, Norin KE, Saxerholt H. Establishment of a biochemically active intestinal ecosystem in ex-germfree rats. Appl Environ Microbiol. (1987) 53:2866–71. doi: 10.1128/AEM.53.12.2866-2871.1987
34. Norin KE, Midtvedt T, Gustafsson BE. Influence of intestinal microflora on the tryptic activity during lactation in rats. Lab Anim. (1986) 20:234–7. doi: 10.1258/002367786780865656
35. Norin KE, Gustafsson BE, Midtvedt T. Strain differences in faecal tryptic activity of germ-free and conventional rats. Lab Anim. (1986) 20:67–9. doi: 10.1258/002367786781062188
36. Norin KE. Influence of antibiotics on some intestinal microflora associated characteristics. Anaerobe. (1997) 3:145–8. doi: 10.1006/anae.1997.0091
37. Naim M, Brand JG, Kare MR. Effect of unpalatable diets, food restriction and saccharin-adulterated diet on tryptic, chymotryptic, and amylolytic activity in pancreas, intestine and feces of rats. J Nutr. (1982) 112:2104–15.
38. Li R, Zheng J, Jiang M, Liu Y, Qin X, Wang X. Increased digestive proteases and decreased beta-glucuronidase in feces of rats treated with sucralose and saccharin-another critical evidence that these dietary chemicals may be important causative factors for inflammatory bowel disease. Inflamm Bowel Dis. (2016) 22:E29–30. doi: 10.1097/MIB.0000000000000859
39. Qin X. Inactivation of digestive proteases by deconjugated bilirubin: the possible evolutionary driving force for bilirubin or biliverdin predominance in animals. Gut. (2007) 56:1641–2. doi: 10.1136/gut.2007.132076
40. Zhou K, Jiang M, Qin X, Wang X. Role of bilirubin in digestive proteases inactivation in the lower intestine. Dig Liver Dis. (2015) 47:438–9. doi: 10.1016/j.dld.2015.01.158
41. Zhan Y, Chen PJ, Sadler WD, Wang F, Poe S, Núñez G, et al. Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res. (2013) 73:7199–210. doi: 10.1158/0008-5472.CAN-13-0827
42. Hernández-Chirlaque C, Aranda CJ, Ocón B, Capitán-Cañadas F, Ortega-González M, Carrero JJ, Suárez MD, et al. Germ-free and antibiotic-treated mice are highly susceptible to epithelial injury in DSS colitis. J Crohns Colitis. (2016) 10:1324–35. doi: 10.1093/ecco-jcc/jjw096
43. Qin X. Impaired inactivation of digestive proteases: the possible key factor for the high susceptibility of germ-free and antibiotic-treated animals to gut epithelial injury. World J Gastrointest Pathophysiol. (2017) 8:1–2. doi: 10.4291/wjgp.v8.i1.1
44. Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. (2012) 148:1258–25. doi: 10.1016/j.cell.2012.01.035
45. Ding RX, Goh WR, Wu RN, Yue XQ, Luo X, Khine WWT, et al. Revisit gut microbiota and its impact on human health and disease. J Food Drug Anal. (2019) 27:623–31. doi: 10.1016/j.jfda.2018.12.012
46. Qin X. May dysbiosis caused by dietary chemicals such as sucralose and saccharin be more detrimental to the gut and health than antibiotics? how? Inflamm Bowel Dis. (2019) 25:e20. doi: 10.1093/ibd/izy198
Keywords: sucralose, colitis-associated colorectal cancer, gut microbiota, digestive proteases, gut permeability
Citation: Li X, Liu Y, Wang Y, Li X, Liu X, Guo M, Tan Y, Qin X, Wang X and Jiang M (2020) Sucralose Promotes Colitis-Associated Colorectal Cancer Risk in a Murine Model Along With Changes in Microbiota. Front. Oncol. 10:710. doi: 10.3389/fonc.2020.00710
Received: 27 December 2019; Accepted: 15 April 2020;
Published: 03 June 2020.
Edited by:
Aga Syed Sameer, King Saud bin Abdulaziz University for Health Sciences, Saudi ArabiaReviewed by:
Xiaojie Tan, Second Military Medical University, ChinaCopyright © 2020 Li, Liu, Wang, Li, Liu, Guo, Tan, Qin, Wang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuhong Wang, d2FuZ3hpdWhvbmdAaHJibXUuZWR1LmNu; Mingshan Jiang, am1zaDUwMzFAYWxpeXVuLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.