 María Florencia Mercogliano1
María Florencia Mercogliano1 Roxana Schillaci
Roxana Schillaci- 1Laboratorio de Biofisicoquímica de Proteínas, Instituto de Química Biológica de la Facultad de Ciencias Exactas y Naturales-Consejo Nacional de Investigaciones Científicas y Técnicas (IQUIBICEN-CONICET), Buenos Aires, Argentina
- 2Laboratory of Molecular Mechanisms of Carcinogenesis, Instituto de Biología y Medicina Experimental (IBYME-CONICET), Buenos Aires, Argentina
Breast cancer is the most frequently diagnosed cancer and the principal cause of mortality by malignancy in women and represents a main problem for public health worldwide. Tumor necrosis factor α (TNFα) is a pro-inflammatory cytokine whose expression is increased in a variety of cancers. In particular, in breast cancer it correlates with augmented tumor cell proliferation, higher malignancy grade, increased occurrence of metastasis and general poor prognosis for the patient. These characteristics highlight TNFα as an attractive therapeutic target, and consequently, the study of soluble and transmembrane TNFα effects and its receptors in breast cancer is an area of active research. In this review we summarize the recent findings on TNFα participation in luminal, HER2-positive and triple negative breast cancer progression and metastasis. Also, we describe TNFα role in immune response against tumors and in chemotherapy, hormone therapy, HER2-targeted therapy and anti-immune checkpoint therapy resistance in breast cancer. Furthermore, we discuss the use of TNFα blocking strategies as potential therapies and their clinical relevance for breast cancer. These TNFα blocking agents have long been used in the clinical setting to treat inflammatory and autoimmune diseases. TNFα blockade can be achieved by monoclonal antibodies (such as infliximab, adalimumab, etc.), fusion proteins (etanercept) and dominant negative proteins (INB03). Here we address the different effects of each compound and also analyze the use of potential biomarkers in the selection of patients who would benefit from a combination of TNFα blocking agents with HER2-targeted treatments to prevent or overcome therapy resistance in breast cancer.
Introduction
Breast cancer comprises 24.2% of total cancers and is the leading cause of cancer mortality in women worldwide (15.0%), constituting a complex problem for public health. Breast cancer is a heterogeneous disease comprised by different subtypes which have distinct characteristics, therapies, and survival rates. TNFα, on the other hand, is a pleiotropic cytokine that participates in a wide spectrum of processes with contradictory effects which range from cell survival, proliferation, immunostimulation to cell death, and immunosuppression. The final effect depends on the concentration and context in which it is found. TNFα, as a central inflammation mediator, has been linked to breast cancer initiation, progression, and metastasis [reviewed in (1)]. Given these facts, understanding TNFα's pivotal role in breast cancer is of great importance, and evidence highlights this cytokine as a novel target to develop therapies that could be combined with the current breast cancer treatments to overcome or avoid resistance and achieve a better clinical outcome, as well as to reduce the social cost of the disease.
TNFα and its Receptors: an Overview
TNFα was named due to original research that in 1975 determined that TNFα causes hemorrhagic necrosis of tumors when found in high concentration (2–4). Later, it was discovered that TNFα was involved in a plethora of cellular processes and, more importantly, that it had paradoxical effects. It was initially described that TNFα was mainly produced by activated macrophages, monocytes, NK cells, T lymphocytes, neutrophils and mast cells, but afterwards it was discovered that it was also expressed by non-immune cells like fibroblasts, endothelial cells, cardiac myocytes and neurons, among others (5, 6). TNFα expression is induced transcriptionally by nuclear factor κB (NF-κB) (7), c-Jun, activator protein 1 (AP1) and nuclear factor associated with activated T lymphocytes (NFAT) (6).
TNFα is encoded by a single copy gene located in human chromosome 6 and is closely located to the genes of the major histocompatibility complex (8). It has four exons and three introns, but most of the protein is encoded in the last exon, while the first ones contain the leader peptide sequence. TNFα is synthesized as a type II membrane protein of 26 kDa that forms a homotrimer and is known as transmembrane TNFα (tmTNFα). Interestingly, tmTNFα can act both as a ligand by binding mainly to TNFα Receptor 2 (TNFR2) or can function as a receptor itself (9). tmTNFα on the cell surface can bind to its receptors on the target cell and modulate either physiological or pathological responses which are not only related to the immune system. For example, tmTNFα is involved in host defense (10), regulation of cytotoxic activity against cells and tumors (11), immunoglobulin production by B cells (12, 13), activation of T lymphocytes (9) and NK cells (14), stimulation of monocytes to generate cytokine production (15), activation of endothelium (9, 13), adipocyte differentiation (16) and cardiac hypertrophy (17), among others. When acting as a receptor, tmTNFα is said to “reverse signal” (18, 19), as other TNFα family members, like CD30L (20), CD40L (21), CD137L (22), meaning that it signals outside-to-inside back to the tmTNFα expressing cell (23). Although reverse signaling has been studied mostly in activation and modulation of the immune system, this mechanism has not been completely characterized (19). tmTNFα has an external C-terminus and a cytoplasmic N-terminus that is liberated by proteolytic cleavage, rendering an active soluble mature TNFα (sTNF) of 17 kDa by TNFα converting enzyme (TACE/ADAM17) (24, 25). This TNFα monomer forms homotrimers of a total molecular weight of 52 kDa with a very potent autocrine, paracrine, and endocrine effect (26). However, the sequence and mechanisms that lead this selective cleavage process remain unclear (27).
Both tmTNFα and sTNFα can bind to two membrane receptors: TNFα Receptor 1 (TNFR1/CD120a, 55 kDa) and TNFR2/CD120b (75 kDa) (28) causing their trimerization, a process required to recruit signaling proteins to its cytoplasmic domain to form an adaptor scaffold that will trigger different pathways depending on which receptor is activated, the cell type and the overall cellular context.
TNFR1 and TNFR2 are members of the TNFα receptor superfamily, which is characterized by the presence of cysteine clusters and are classified as type I membrane proteins. The extracellular ligand binding domain of each receptor shares only 28% homology, which explains the broad spectrum of processes that this superfamily can control. The intracellular domains of the receptors have no sequence homology and are void of enzymatic activity, therefore triggering signaling through recruitment of cytosolic adaptor proteins (29). To add yet another layer of complexity to the TNFα pathway, both receptors can be found in soluble forms and are proteolytically cleaved by TACE, as is the sTNFα form. These soluble receptors regulate availability of TNFα by sequestering it, or they can protect it from degradation, enabling it to achieve a sustained signal (30, 31). TNFR1 is expressed at low levels in the majority of nucleated cell types [reviewed in (32)] and can be activated by both sTNFα and tmTNFα. TNFR2 is mostly expressed in restricted cell subtypes like neurons, oligodendrocytes, astrocytes, endothelial cells, and subpopulations of T lymphocytes, among others (33, 34), and tmTNFα is required for its full activation.
Signal transduction through TNFR1 can induce opposite effects: it can either stimulate cell survival and proliferation, or apoptosis and cell death, depending on signal strength, the signaling molecules recruited to the scaffold and on the crosstalk with other pathways (35). TNFR1 has a cytoplasmic death domain that recruits TNFR1-Associated Death Domain protein (TRADD) and TNF Receptor Associated Factor 2 (TRAF2). It can form two different complexes, depending on the engaged scaffold: complex I culminates in the activation of survival pathways stimulating signaling mediated by JNK, NF-κB, AP-1, MAPK pathways (36). On the other hand, the formation of complex II recruits Fas-Associated protein with Death Domain (FADD) and pro-caspases (37) which in turn create a death-inducing signaling complex that concludes in apoptosis (38).
On the contrary, TNFR2 primarily stimulates cell activation, migration, and proliferation (39). Even though TNFR2 lacks the death domain, it can also bind TRAF2 through TNFα Receptor Associated Factor 1 (TRAF1) and can activate the canonical and non-canonical NF-κB pathway just like TNFR1, but the process is slower (40) and the activation is more sustained (41). There have been some other descriptions of TNFR2 activating TNFR1 pathways, such as NF-κB, AP-1, JNK and MAPK (34, 42), by recruiting scaffold proteins such as Receptor Interacting Protein (RIP-1) and TRADD via TRAF2, resulting in apoptosis as well as in TNFR1 activation. Given that TNFR2 has lower affinity for TNFα than TNFR1, it can augment TNFα concentration near TNFR1 and constitute the so-called ligand passing process, thereby stimulating TNFR1 signaling (43).
In summary, the intricate pathway of TNFα accounts for two receptors that can be found in a transmembrane or soluble form and can activate unique signaling cascades, but can also converge, depending on the adaptor proteins recruited. In addition, TNFα also exists in a transmembrane and soluble form, adding further layers of complexity to the TNFα pathway which explain the various and opposite cellular processes in which TNFα is involved.
The Role of TNFα in the Normal Mammary Gland
In the normal mammary gland, TNFα elicits proliferation, morphogenetic branching, and differentiation of said tissue (44, 45). Varela and Ip reported that mammary epithelial rat cells express tmTNFα and sTNFα as well as both TNFRs, and that their expression is independently regulated during mammary gland development. TNFR1 is the receptor that mediates proliferation of normal epithelial cells, while TNFR2 regulates casein accumulation (46). TNFα effect in the normal mammary gland is predominantly exerted by NF-κB in normal rat mammary epithelial cells in culture (47). Interestingly, Varela et al. showed that p50/50 NF-κB homodimers were the ones to mediate TNFα effects, unlike the classical heterodimer of p50/p65 (47).
During the different stages of mammary gland differentiation, TNFα expression is tightly regulated and its concentration varies. Through puberty, TNFα is present to stimulate branching morphogenesis, increasing expression of MMP9 to allow tissue remodeling (45). In the course of pregnancy, TNFα stimulates proliferation and morphological differentiation but inhibits casein accumulation via TNFR1 until lactation (46). This effect is mediated by TNFα-induced NF-κB pathway activation and by TNFα repression of STAT5 phosphorylation in mammary epithelial cell lines in vitro (48), but it has also been reported that NF-κB could be activated by other factors such as EGFR (49). During lactation, sTNFα decreases, while tmTNFα is expressed at high levels like both TNFRs. Therefore, NF-κB pathway activation is reduced due to diminished nuclear p50 and p65 (48). Finally, during involution of the mouse mammary gland in vivo, sTNFα is the main form found. It acts via TNFR1, activating TWEAK (50, 51), LIF, p42/p44 MAPK and STAT3 (52) to induce apoptosis (53, 54).
The Relevance of TNFα in Cancer
The presence of inflammatory mediators in the tumor microenvironment (TME), generated either by the tumor cells or the tumor infiltrating cells, has been widely recognized as one of the hallmarks of cancer [reviewed in (55, 56)]. There is evidence that relates pro-inflammatory cytokines to cancer as functional polymorphisms in the cytokines genes have been associated with cancer severity. In addition cell populations that secrete inflammatory mediators corresponding to either tumor cells or host immune cells, inflammatory cytokines have been detected in cancer patients and associated to poor prognosis, as reviewed by Mantovani and Balkwill (57, 58). As mentioned before, TNFα is a major mediator of inflammation (59) with ambiguous effects and has been detected in human ovarian (60–62), breast (62, 63), endometrial (62), oral (64), pancreatic (65), gastric (66), liver (67), prostate, bladder and colorectal (68) cancer as well as in lymphomas (69) and leukemias. It has been reported that knockout mice for TNFα are resistant to chemical-induced carcinogenesis (70–72). There has been quite a big controversy regarding TNFα expression as a parameter to predict clinical outcome in breast cancer patients (73–76). In this sense, recent meta-analysis showed that the TNFα−308 polymorphic site (77) but not−238 (78) associates with breast cancer patient survival. The information here presented postulates TNFα as a central player in cancer initiation, progression, invasion and metastasis, and proposes it as an attractive therapeutic target for the benefit of cancer patients.
TNFα in Breast Cancer: Signaling, Progression, and Metastasis
Low grade inflammation has a key role in cancer pathogenesis and progression [reviewed in (56, 57, 79)]. It has been reported that TNFα treatment in certain breast cancer cell lines inhibits proliferation and induces apoptosis (80). On the other hand, it has also been shown that most breast cancer cell lines are resistant to TNFα-induced apoptosis, and that their proliferation, survival, and progression are mediated by said cytokine (81). This difference could be due to the differential expression of TNFRs, of the Bcl-2 family, of caspases activation or of ceramide expression (80).
It has been previously reported that inflammation is associated with poor prognosis and a higher recurrence risk in breast cancer patients (82, 83). TNFα mRNA and protein were present in malignant and stromal cells in biopsies of breast cancer patients, and especially in those with worse prognosis (74). Lately, several studies have shown the prometastatic role of TNFα and its participation in the epithelial-to-mesenchymal (EMT) process necessary for migration of tumor cells to establish metastasis (84). In particular, prolonged exposure to TNFα of breast cancer cell lines induces the upregulation of the transcriptional repressor Twist1 via activation of IKKβ and NF-κB and induces EMT and cancer stemness properties (85). In a study of a cohort of patients with inflammatory breast cancer it was found that there was a direct correlation between TNF-α production by peripheral blood T lymphocytes and the detection of circulating tumor cells expressing EMT markers (86).
Breast cancer diagnosis is carried out by determination by immunohistochemistry of the HER2 receptor, of hormonal receptors (estrogen and progesterone receptor, ER and PR, respectively) and of the proliferation marker Ki67. According to the expression of these receptors, breast cancer is classified into the following subtypes: luminal A or B (ER-positive, PR-positive and high or low Ki67, respectively), luminal B HER2-positive (ER-positive, PR-positive, HER2-positive), HER2-positive and triple negative breast cancer (TNBC, ER-negative, PR-negative, HER2-negative) (87).
In the following sections we will address the role of TNFα in each of these subtypes and its effect on signaling, proliferation, progression, and metastasis.
Luminal Breast Cancer Subtype
This breast cancer subtype is characterized by the presence of ER and PR and the incidence for luminal A and luminal B subtype is 30–40 and 20–30% (88), respectively. Regarding survival rate luminal A and B has the best overall survival with 92.5% at 4-years followed by luminal B/HER2-positive with 90.3% (89). Since PR expression is driven by ER, PR is said to parallel ER expression. We will therefore focus on the relationship between ER and TNFα [reviewed in (90)].
About two thirds of breast tumors express ER enabling estrogens to drive their growth. Estrogen production takes place in ovaries, muscle, liver, breast, and adipose tissue. The main sources of plasmatic estrogens in pre-menopausal women are the ovaries, and in postmenopausal women, the sources are the adipose tissue and muscle (91). In breast cancer, estradiol is considered to have the strongest estrogenic effects and also has a higher concentration than in normal breast tissue. Indeed, the levels of estradiol in breast tumors of postmenopausal women were reported to be 10–100 times higher than its serum concentration, and values were similar to those found in pre-menopausal women (92, 93). This fact was supported by the finding that the expression level of aromatase mRNA is enhanced in breast cancer, compared with normal breast tissue. Breast tumors, therefore, proved to be autonomous in retaining a constant estradiol concentration, even against a steep plasma to tumor concentration gradient in postmenopausal patients. Briefly, estradiol can be derived from testosterone, estrone or estradiol sulfate due to the catalytic activity of aromatase, 17b-hydroxysteroid dehydrogenase (17-OHSD) or estrone sulphatase, respectively. It was demonstrated by immunohistochemistry and by in situ hybridization that aromatase is expressed mainly in malignant human breast epithelial cells (94).
Many cytokines, such as TNFα, IL-6 and PGE2, stimulate aromatase activity in primary cultured human mammary adipose tissue. In this regard, it was reported that aromatase mRNA levels positively correlate with TNFα, IL-6, and COX2 mRNA levels (95). Moreover, it was shown that TNFα induces aromatase gene expression through c-fos and c-jun binding on the AP-1 element present on exon 1.4 together with the glucocorticoid receptor (91). Considering that aromatase is only expressed in undifferentiated adipose fibroblasts but not in the mature adipocytes, it is also possible that TNFα and IL-6 contribute to augment aromatase mRNA expression by increasing this population in breast cancer, also given that both cytokines are inhibitors of adipogenic differentiation (96). On the other hand, IL-10 through inhibition of TNFα-induced p42/p44 MAPK activation can suppress aromatase mRNA expression in human adipose tissue (97) (Figure 1).
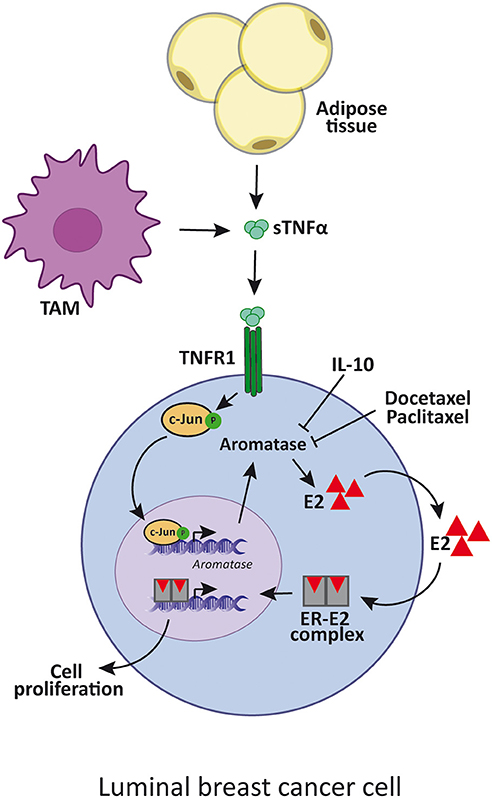
Figure 1. TNFα enhances luminal breast cancer cell proliferation by aromatase upregulation. TNFα is produced by adipose cells, TAM or tumor cells itself, and induces the expression of aromatase. This enzyme increases estradiol synthesis which binds to ER that, in turn, promotes luminal cancer cell proliferation. IL-10 and docetaxel and paclitaxel inhibit aromatase synthesis by reducing TNFα signaling. sTNFα, soluble TNFα; TAM, tumor-associated macrophages; E2, estradiol; ER, estrogen receptor.
Reports in favor of the anti-proliferative and apoptotic effect of TNFα on luminal breast cancer have only been executed on the MCF-7 cell line. However, controversial results have been found since a study showed that MCF-7 lines from different laboratories had different expression levels of the anti-apoptotic protein Bcl-2, which consequently modified the sensitivity of the cells to TNFα-induced apoptosis (80).
For instance, it was reported that TNFα induces a cytotoxic effect in luminal breast cancer cell lines in absence of ubiquitin editing enzyme TNFα-induced protein 3 (TNFAIP3 also called A20) (98), but this protein has a wide range of effects in different tissues (99, 100). Not only does A20 protects cells from TNFα cytotoxic effects but it also contributes to a more aggressive phenotype in response to TNFα stimulation.
There have been various reports of NF-κB repression by ER accounted for different mechanisms (101), such as prevention of NF-κB binding to DNA (102), recruitment of co-repressors (103), competition for co-activators (104), and prevention of NF-κB translocation to the nucleus (105), among others. Even though clinical data reported that ER-positive breast tumors with constitutively active NF-κB are more aggressive and less responsive to treatment (106), very few studies indicated that a positive transcriptional crosstalk could exist (107, 108). It was Frasor et al. who showed that treatment with TNFα and estradiol regulated a set of genes that are clinically relevant because they can distinguish patients with poor response to endocrine treatment (109). In fact, both molecules act together to promote survival of breast cancer cells and progression onto a more aggressive phenotype (110). In this regard, by using global run-on coupled with deep sequencing (GRO-seq) in MCF-7 breast cancer cells, it was demonstrated that TNFα was responsible for exposing latent estrogen receptor binding sites to which estradiol could bind to regulate gene expression. The availability of these enhancers was dependent on TNFα induction of the NF-κB pathway and the pioneer factor FOXA1. The proliferative effects of TNFα in human breast cancer cell lines in vitro were shown to be mediated by TNFR1, which activates JNK and PI3K/AKT which stimulates NF-κB. The latter, in turn, induces cyclin D1 expression which concludes in cellular proliferation (Figure 2) (108, 111). Interestingly the same effects were reported for estradiol treatment (108). TNFα can also stimulate cancer cells proliferation through the p42/p44 MAPK pathway mediated by TNFR1 and TNFR2 independently of NF-κB, establishing an alternative pathway to breast cancer proliferation (111) (Figure 2). MCF-7 cell line is sensitive to TNFα-induced apoptosis through JNK pathway activation, while p42/p44 MAPK is weakly activated (112). Instead, in T-47D cell line, TNFα has proliferative effects and causes potent activation of p42/p44 MAPK, while JNK is only transiently activated (111). Moreover, when NF-κB pathway activation is insufficient, JNK stimulates apoptosis (36, 113).
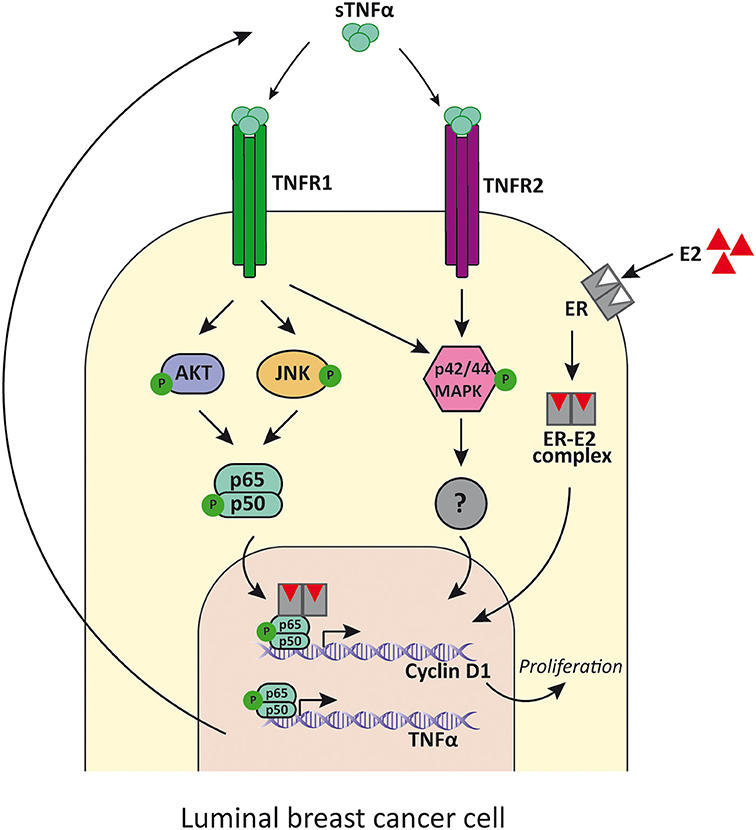
Figure 2. TNFα signaling in luminal breast cancer cells. TNFα binds to TNFR1 and induces the activation of PI3K/Akt, p42/p44MAPK and JNK, while its binding to TNFR2 mainly induces p42/p44MAPK activation. PI3K/Akt and JNK then activates NF-κB that promotes the transcription of cyclin D1 and the consequent cell proliferation. In addition, NF-κB also induces the expression of TNFα, establishing a positive feed-back. E2 binds to ER and also promotes luminal cancer cell proliferation through cyclin D1 upregulation by tethering NF-κB and enhances its activity. sTNFα, soluble TNFα; TNFR1, TNFα receptor 1; TNFR2, TNFα receptor 2; E2, estradiol; ER, estrogen receptor.
Additionally, TNFα elicited metastatic properties in the originally non-metastatic MCF-7 cell line, such as cell spreading and formation of FAK/paxillin protrusions through Src activation, elevated expression of CD44, VLA6 and β1, cell migration and initiation of the EMT process, resistance to chemotherapy and secretion of pro-tumoral factors such as chemokines and cytokines (114, 115). Moreover, TNFα in combination with estradiol and EGF had a stronger effect than each molecule alone (114). EMT has been found to be accountable for the change to a fibroblast-like morphology, loss of adhesion and junction molecules, and greater invasion and cell motility (116). EMT has also been associated with the development and maintenance of breast cancer stem cells. All the above-mentioned characteristics have been linked not only to maintaining the tumor but also to the acquisition of metastatic potential, therapy resistance, and relapse (117). All these effects were proven to be exerted by TNFα in MCF-7 and T-47D cell lines (118, 119). TNFα also modulates MMPs expression, in particular MMP9, in MCF-7 cells (120). This protein family is involved in cell migration and invasion of the extracellular matrix (120, 121). All these facts show that TNFα plays a critical role in luminal breast cancer, inducing tumor promotion, proliferation, survival, progression and metastasis, and that it can be an attractive target for potential new therapies to enhance patients' successful outcome.
TNBC Subtype
As stated before, TNBC is characterized by not expressing clinically significant levels of ER, PR and HER2 receptors. This breast cancer subtype accounts for 15–20% of breast cancers and is associated with younger women (<35 years) (122) in Hispanic and/or African American populations (123). It is usually diagnosed in an advanced stage, has a high risk of metastasis development and exhibits poor response to therapy (124, 125). Consequently, presence of TNBC has a survival rate of 77%, and together with the HER2-positive subtype are the ones that have shorter 4-year survival (89).
Regarding TNFα, there have been paradoxical results of its implication in TNBC proliferation and survival. It has been stated that TNFα can induce apoptosis (126) and promote tumor growth in TNBC, depending on the specific cell line and the cellular context in which it is found (127–129). Qiao et al. demonstrated that TNFα induces apoptosis in BT549 cells, activating c-Jun which, in turn, downregulates anti-apoptotic genes, and upregulates pro-apoptotic cascades. In the same study, they showed that TNFα can also have a pro-malignant effect, stimulating c-Jun and inducing cell invasion (126). Other groups have proved that treatment with exogenous TNFα cannot trigger apoptosis in MDA-MB-468 (127) and MDA-MB-231 cell lines (130). On the contrary, when TNFα was silenced, cell proliferation and motility were abolished and apoptosis was induced (128).
When promoting tumor growth, TNFα activates NF-κB and p38/MAPK pathways which stimulate signal transducer and activator of transcription 3 (STAT3) (127), a known transcription factor classified as an oncogene (131). This cascade generates a positive feedback loop because activated STAT3 increases the expression of HBXIP (Hepatitis B Virus X-Interacting Protein) which augments TNFR1 that will bind TNFα and continue the activation of NF-κB and p38/MAPK. MUC4, a membrane glycoprotein, has also been shown to promote cell proliferation mediated by β-catenin and an increase in cyclin D1 expression in TNBC cell lines (132).
As stated before, TNFα has been associated with cancer cell motility, invasion and EMT. TNBC is not an exception since there have been several reports of different proteins that can mediate said processes. Important players related to growth, migration and invasion in breast cancer are the MMPs (120, 121), in particular the expression of MMP9 exerted by TNFα-induced AP-1 activation (133) or CDKNA1/p21(134), which can act as an oncogene (135). It has been described that p42/p44MAPK (136) and PI3K/Akt (135) are critical for TNFα-induced MMP9 expression in cancer. Using specific pathway inhibitors, it was clear in the MDA-MB-231 cell line that the p42/p44MAPK pathway was the one responsible for TNFα-induced MMP9 expression. TNFα activates AP-1 through PI3K/Akt and p42/p44 MAPK pathways and elicits the expression of ZEB2, an EMT regulator in TNBC cell lines (137). In this regard, it has been found that PI3K/Akt and p42/p44MAPK pathways are enhanced in human TNBC samples and have a central role in EMT promotion and cell invasion in breast tumors (138) as well as in cancers in general (Figure 3).
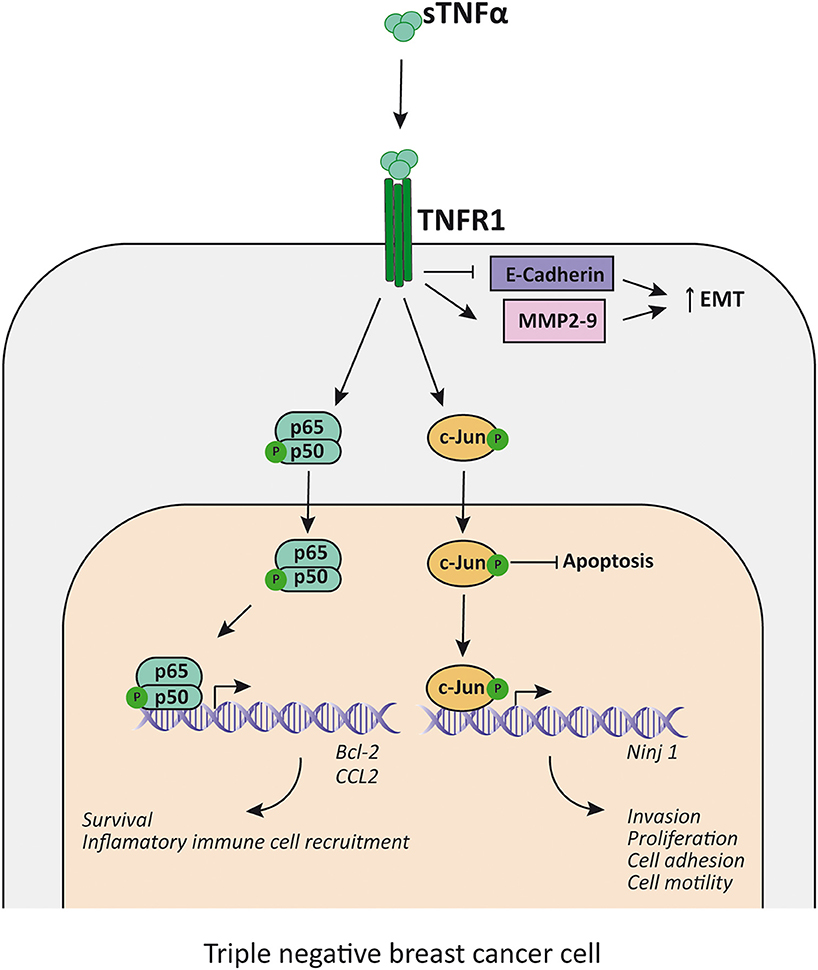
Figure 3. TNFα promotes proliferation and EMT in TNBC cells. TNFα, acting mainly through TNFR1, induces NF-κB and c-Jun activation, which inhibits apoptosis and enhance the transcription of survival factors (i.e., Bcl-2), the expression of inflammatory cell recruitment chemokines (i.e., CCL2) and promotes transcription of factors related to EMT (i.e., increases Ninj1, MMP2, and MMP9 transcription and decreases E-cadherin transcription). sTNFα, soluble TNFα; TNFR1, TNFα receptor 1; MMP, metalloproteinase; EMT, epithelial-mesenchymal transition.
Overexpression of A20 protein, mentioned in the luminal subtype section, is also involved in the aggressiveness of TNBC and is effectively highly expressed in this subtype according to TCGA data analysis (100). It has been suggested that A20 may induce expression of HSP70, which protects TNBC cells from TNFα cytotoxicity in vivo in mice models (139). It has also been demonstrated that A20 protein activates the inflammatory IL-6/phospho-STAT3 pathway, downregulates SOC3 and upregulates HSP70 and the expression of inflammatory cytokines like IL-6, IL-8, CCL5, and TGF-β upon TNFα stimulus. These events conclude in the expression of EMT markers and contribute to a more aggressive phenotype, a higher metastatic potential and the generation of cancer stem cells in TNBC cell lines (99).
We previously discussed the role of MUC4 in TNBC proliferation, but it was also found to have a pivotal position in invasion and metastasis. In this sense, MUC4 (132) and FAK (140) are overexpressed in invasive TNBC but not in normal breast tissues. MUC4 not only influences proliferation but also regulates the metastatic potential of TNBC suppressing F-actin formation and regulating FAK phosphorylation, thus increasing cell motility in vitro (141).
HER2-Positive Subtype
Expression of the HER2 receptor is found in 13–20% of breast cancers (142), contributing to oncogenic transformation, a more aggressive phenotype and the worst 4-year survival (82.7%) together with the TNBC subtype (89), as already stated.
HER2 is an orphan tyrosine kinase receptor located in the cytoplasmic membrane with no known ligand, that belongs to the HER family (143). The three other members of the HER family are the EGFR/HER1, HER3, and HER4 [reviewed in (144)]. In presence of their specific ligands, the receptors can homo- or heterodimerize, upon which transphosphorylation of tyrosine residues occurs [reviewed in (145)]. This leads to recruitment of adaptor proteins or enzymes capable of activating different signaling cascades that conclude in tumor cell proliferation and survival [reviewed in (146)]. HER2/HER3 heterodimer is the duo with higher oncogenic activity (147). It stimulates primarily the PI3K/Akt and p42/p44MAPK pathways (148), but it can also activate NF-κB anti-apoptotic signaling (149). We have previously reported that in HER2-positive breast cancer cell lines, TNFα transactivates HER2, inducing phosphorylation of Tyr877 through c-Src, in a PKC-dependent manner. This elicits autophosphorylation and leads to the formation of the HER2/HER3 heterodimer which in turn activates the PI3K/Akt pathway and concludes in proliferation mediated by NF-κB-induced cyclin D1 expression (150) (Figure 4). Our group demonstrated that stable overexpression of TNFα in BT-474 cells, a human HER2-positive breast cancer cell line, led to high activation levels of Akt and NF-κB pathways (151). This overactivated signaling conferred higher survival capacity, a major colony forming potential and an increased proliferation in strict culture conditions with respect to the parental cell line (151). All these events are of major significance in the processes of tumor establishment and of an increased metastatic potential. We demonstrated that TNFα has a central role in mediating proliferative effects and tumor progression in HER2-positive breast cancer in vitro and in human xenograft models (150, 151).
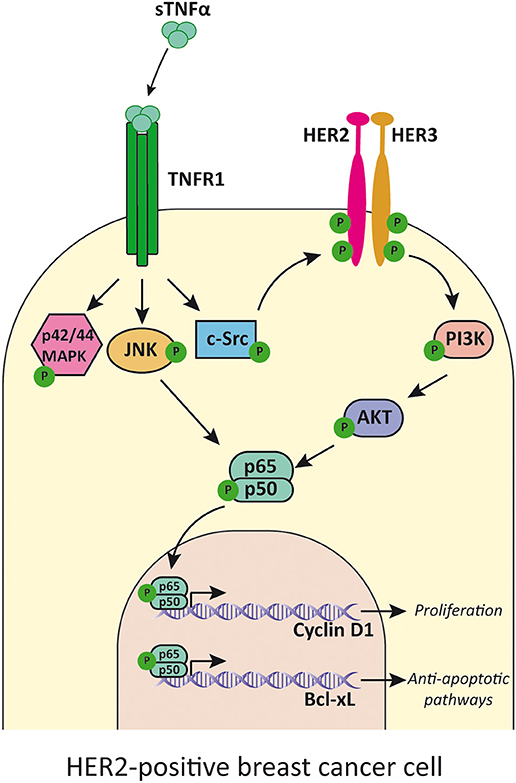
Figure 4. TNFα transactivates HER2 receptor and induces cell proliferation. TNFα, acting mainly through TNFR1, activates p42/p44MAPK, JNK, and c-Src. This last kinase phosphorylates HER2 and induces HER2/HER3 heterodimer formation and activation of PI3K/Akt pathway. JNK and Akt activates NF-κB that induces transcription of cyclin D1 and Bcl-xL. TNFα, soluble TNFα; TNFR1, TNFα receptor 1.
The mucin family of proteins has been shown to be another important player in HER2-positive breast cancer. Li et al. described that heregulin stimulation, the preferred HER3 ligand, induces phosphorylation of mucin 1 (MUC1) which interacts with γ-catenin and p120 catenin (152). This complex is then translocated to the nucleus where it interacts with different transcription factors like STAT1, ERα, β-catenin, and p53 to regulate transcription of genes involved in tumor promotion (153). MUC4, another mucin family member, has been demonstrated to interact with HER2 and to promote HER2 and HER3 translocation to the cytoplasmic membrane, augmenting the number of receptors available on the cell surface and maintaining them anchored to the membrane during longer periods of time (154). Consequently, there is an increase in the signaling cascade of the HER2/HER3 heterodimer through activation of PI3K (155), causing an increase in cell proliferation and survival in HER2-positive breast cancer (156). MUC4 is aberrantly expressed in a variety of cancers, including breast tumors, and in this setting it can also induce phosphorylation of HER2, which results in inactivation of the pro-apoptotic pathway and in stimulation of pro-survival proteins (157). To our knowledge, we are the first group to report that TNFα increases MUC4 expression through activation of the NF-κB pathway in HER2-positive breast cancer (151).
TNFα and the Immune Response Against Cancer
TNFα is known to play a dual role in oncoimmunology [reviewed in (158)] as it can behave as an immunostimulant or immunosuppresor cytokine. TNFα is crucial not only to mediate killing of several tumor types, but also for proper proliferation of T lymphocytes, B cells, NK cells, macrophages, and dendritic cells (DC). In spite of this, evidence throughout time has shown that TNFα has a critical role in the inflammatory environment that favors cancer development, acting as a tumor-promoting factor [reviewed in (159)].
Regarding its role as an immunosuppressive cytokine, emerging evidence indicates that, in multiple cancer types, TNFα stimulates and favors not only the accumulation but also the activity of certain immune cell populations, like regulatory T lymphocytes (Tregs) (160), regulatory B lymphocytes (Bregs) (161) and myeloid-derived suppressor cells (MDSCs) (162), in different cancer types. All of the previous mentioned cell types are crucial negative modulators of tumor immune surveillance. A recent study has demonstrated that inflammatory molecules such as PGE2 (163) interleukin-1β (IL-1β) (164) and IL-6 (165) produced by tumor cells induce the stem cells present in the bone marrow of tumor-bearing mice to differentiate into Gr-1+CD11b+ MDSCs and to accumulate in the tumor tissue. This, in turn, allows tumor progression as it impairs the activation of T lymphocytes due to the already known potential of this cell population to suppress not only the innate but also the adaptive immune response (166). In addition, the accumulation of MDSCs in the tumor bed of ovarian cancer was shown to be regulated by the production of CXCL12 and CXCR4, induced by PGE2 (167). In line with this, it has been proved that TNFα is able to affect the leukocyte extravasation by regulating chemokine expression (168) [reviewed in (169)]. For example, in ovarian cancer cell lines, TNFα can behave as a signaling molecule within the producing cell or among the surrounding ones, and can induce CXCR4 expression through NF-κB, enhancing in this way cell invasion (170, 171). Moreover, it has been proved that TNFα secretion by MDSCs in the tumor bed acted as an autocrine cytokine that enhances their T lymphocyte suppressive activity by upregulating different genes associated with immunosuppression, like Nos2 (172).
With respect to the role of TNFα and its signaling pathways in regulating the immune system in cancer, it has been reported that the TNFR2-mediated signaling has a critical role not only in the survival (162) but also in the activity (173) of the MDSCs. In line with this, it has been recently demonstrated that deficiency in either of the two TNFRs caused a diminished accumulation of MDSCs in the tumor bed and the cause underlying this effect was the downregulation of CXCR4 expression on the surface of MDCSs in a breast cancer model (166). This supports the idea that TNFα is involved in differentiation, migration, activation, and survival of the MDSCs in the TME. As mentioned above, TNFR1 has been reported to be the main mediator of TNFα-induced apoptosis (36). However, it was not the scenario that emerged in the majority of cancer cells, because they lost sensitivity to TNFα secreted by CD8+ T lymphocytes and NK cells in melanoma (174). There are several mechanisms involved in the resistance to TNFα-induced cell death that include the relatively high proportion of TNFα receptors associated with anti-apoptotic proteins, such as cFLIP, cIAPs, TRAF2, and the loss of caspase-8 and of 2-aminoethanethiol dioxygenase (Ado), a metabolic protein required for TNFα cytotoxicity (175) [reviewed in (176–178)]. Supporting this evidence, it has been proved that TNFR1 gene-deficient mice showed reduced chemical-induced colon inflammation and tumor incidence (179), indicating a putative role of TNFR1 in inflammation and tumor progression. In addition, posttranslational modifications of TNFR1 are known to affect its activity. It was shown that addition of α2-6 sialic acid to TNFR1 by the Golgi enzyme ST6Gal-l sialyltransferase blocks apoptosis induced by TNFα in human monocytic cells as well as in monocytes from mice that overexpressed the enzyme (180). In a recent study, Holdbrooks et al. showed that through this sialylation-dependent mechanism, TNFR1 internalization induced by TNFα was inhibited. Interestingly, this process led to the blockade of the apoptotic pathway guided by TNFR1 signaling, promoting sustained activation of the NFκB- and Akt-mediated survival pathway (181). These results indicate that in aberrant glycosylation conditions, TNFR1 can switch from the classical apoptotic signaling pathway activated by sTNFα to the one commonly attributed to TNFR2, which favors tumor progression by promoting cell proliferation.
As stated before, TNFR2 has been related to immunosuppression, tumor progression, and metastasis. Studies demonstrated that development and expansion of Treg cells in the thymus is facilitated by TNFR2 (182, 183) and that it mediates the stimulatory effects of TNFα on Treg cells (184, 185). Chen et al. proved that no other T lymphocyte expressed higher levels of TNFR2 than Treg cells, which correlated with a more suppressive population (186). Generally, the TNFα-TNFR2 axis plays a very important role in the enhancement of tumor immune escape (173). In addition, recent preclinical studies showed that TNFR2 blockade was enough to reduce the progression of breast cancer cell lines treated with TNFα (111) and that it promoted TNFα-associated lung cancer cell death (187). Nie et al. obtained inhibition of breast and colon cancer progression by combining TNFR2-blocking antibody with an anti-CD25 antibody (188).
Studies show that TNFα promotes activation-induced cell death (AICD) of CD8+ T lymphocytes (189) through TNFR2 signaling and that it prevents them from infiltrating the tumor bed. TNFR2 expression can be upregulated on CD8+ T lymphocytes (190) and can induce the expression of the inhibitory receptor Tim3 (191). Another evidence of the ability of TNFα to impair the function of antitumor immune cells is that, in melanoma, the cytotoxic response against the tumor that is carried out by CD8+ T lymphocyte is inhibited by this cytokine, which is produced by CD4+ lymphocytes infiltrating the tumor (192). In addition, in an adoptive CD8+ T cell transfer protocol, TNFα induced the dedifferentiation of melanoma cells, facilitating immune escape and melanoma relapse (193). More recently, TNFα has been shown to promote the expression of PD-L1 in cancer cells (194), leading to immunosuppression. Torrey et al. proved that targeting TNFR2 with antagonistic antibodies inhibits not only proliferation of cancer cells but also proliferation of Tregs infiltrating the tumor while simultaneously inducing the expansion of effector T lymphocytes (195).
It has been shown that the antitumor immunity promoted by the crosstalk between DC and NK cells both in human and mouse cancer models is a result of tmTNFα-TNFR2 interaction that triggers the secretion of Th1 cytokines (14, 196) [reviewed in (197)]. Impairing sTNFα-TNFR1 interaction by different strategies (sTNFα blockade, gene deletion or antibody blockade of TNFR1) can prevent autoimmunity, shedding light on the specific role of the sTNF-TNFR1 axis in inflammation (198) [reviewed in (199)]. sTNFα has been associated with all stages of cancer progression, including tumorigenesis, proliferation, angiogenesis, metastasis, and immune escape [reviewed in (158)].
Sobo-Vujanovic et al. have shown that sTNFα blockade with a dominant negative protein specific for sTNFα, INB03, or TNFR1 deletion not only prevented carcinogenesis but also decreased tumor growth and increased survival of methylcholanthrene-injected mice (71). Moreover, they proposed a new role for sTNFα during carcinogenesis in driving MDSCs accumulation in the TME, in acquiring and maintaining their immunosuppressive functions (200), in survival extension (162), and growth stimulated by IL1α, VEGF and GM-CSF [reviewed in (201, 202)]. The authors suggested that this occurred by activation of TNFR1 signaling on MDSCs progenitors, leading to MDSCs generation but also to arrested differentiation (71).
Results from our own group support the idea that TNFα is engaged in mounting an immunosuppressive TME during carcinogenesis, particularly through the sTNFα isoform. In a suitable breast cancer preclinical model, we demonstrated that HER2-positive tumors that are de novo resistant to trastuzumab, an anti-HER2 monoclonal antibody used as a first line treatment for patients with this type of cancer, are sensitized when sTNFα is blocked with INB03 in combination with trastuzumab (203) (Figure 5). Together with tumor growth inhibition, we found that sTNFα blockade with INB03 in combination with trastuzumab increases tumor infiltrating NK cell activation and degranulation, increases macrophage recruitment to the tumor bed, and favors their differentiation to the M1 subtype. In addition, these tumors exhibited a decrease in myeloid cell infiltration and in the percentage of the monocytic subtype of the MDSCs population (203) (Figure 5). These results indicate that sTNFα is responsible for generating an immunosuppressive TME and favoring tumor immune evasion.
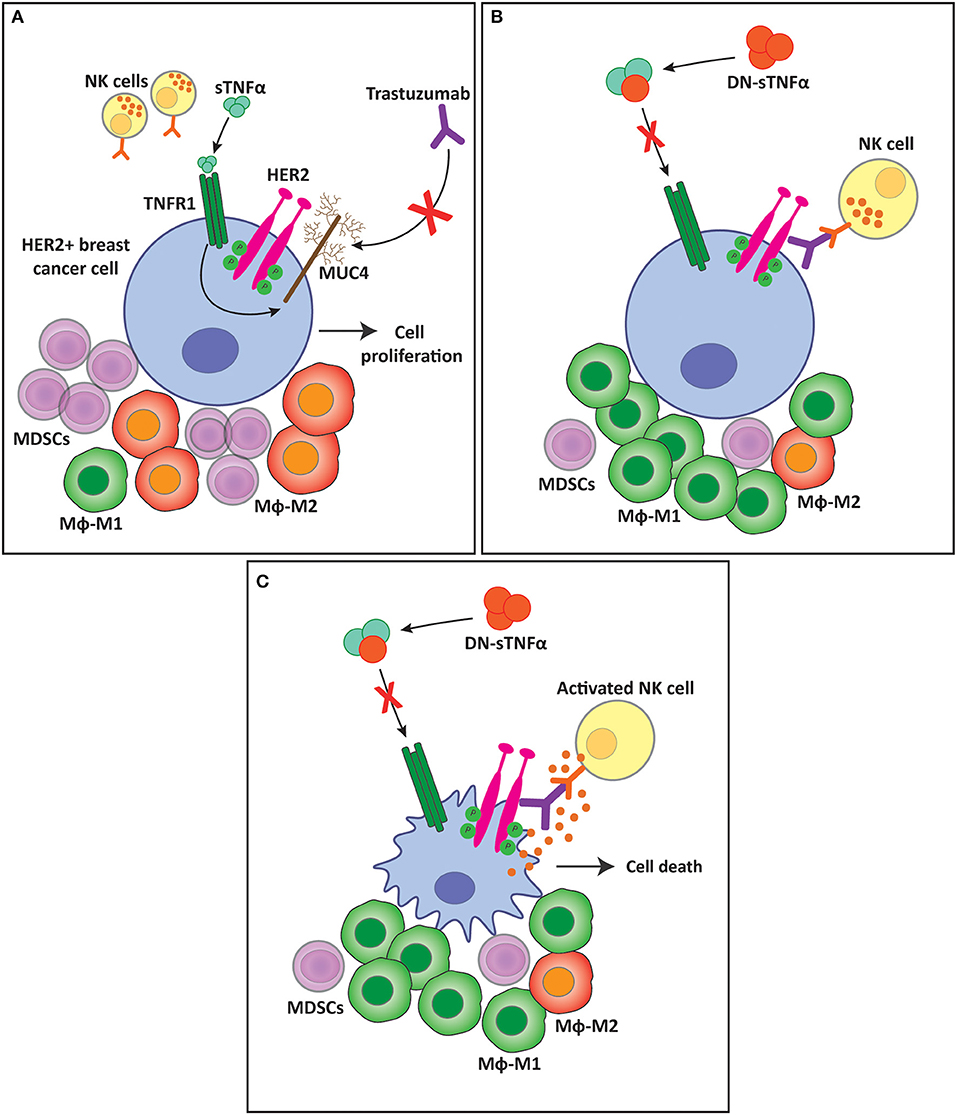
Figure 5. sTNFα blockade overcomes trastuzumab resistance and favors a less immunosuppressive tumor microenvironment. (A) sTNFα induces the upregulation of the membrane glycoprotein MUC4 in HER2-positive breast tumors. MUC4 masks trastuzumab epitope on the HER2 molecule, impairing its binding and ADCC exerted by NK cells, generating resistance to the antibody. This resistance is accompanied by an immunosuppressive tumor microenvironment with an increased infiltration of MDSCs and macrophage polarization to the M2 subtype. (B) sTNFα blockade with a dominant negative protein (DN-sTNFα) downregulates MUC4 expression, enabling trastuzumab to induce NK cell ADCC. This antitumor innate immune response generates a less immunosuppressive tumor microenvironment, decreasing MDSCs infiltration, and increasing macrophage polarization to the M1 subtype. (C) NK cell activation and degranulation induced by trastuzumab treatment kills tumor cells through ADCC. sTNFα, soluble TNFα; TNFR1, TNFα receptor 1; MUC4, mucin 4; MDSC, myeloid-derived suppressor cells; Mϕ, macrophages.
In line with our results, a recent study has demonstrated that HER2-positive breast cancer patients exhibited a specific tumor transcriptome that conditioned their response to adjuvant trastuzumab (204). The study shows that HER2-positive tumors that were sensitive to trastuzumab treatment had a higher expression of certain chemokines related to an antitumor immune response, to migration of T and B lymphocytes to the tumor bed and monocyte recruitment. In particular, patients bearing trastuzumab sensitive tumors showed an upregulation of CCL2 expression in contrast to the HER2-positive tumors that were resistant. The expression profile of the tumors that responded to therapy correlated with an increased CD8+ T lymphocyte infiltration of the tumor and an enrichment of the macrophage M1 subtype genes, suggesting macrophage polarization. In particular, it is proposed that CCL2 recruits mainly monocytes to the TME and that its chemokine action is regulated by the PI3K/NF-κB pathway, activated by the HER2 receptor in the plasma membrane. Triulzi et al. claim that there is a strong relationship between the expression of immune chemokines like CCL2 together with the infiltration of immune cells in the TME and with the efficacy of trastuzumab treatment in HER2-positive patients (204).
Involvement of TNFα and Nf-κB in Breast Cancer Therapy Resistance
Breast cancer treatment with radiotherapy, endocrine treatment, chemotherapy, targeted therapies or immune checkpoint drugs induces, in the majority of cases, an excellent initial tumor response. However, survival of cancer cells after the first line of treatment induces cancer recurrence or their lack of response leads to resistance events that are life-threatening. As described previously in this review, TNFα is not only closely involved in breast cancer onset, progression and in metastasis formation, but it also participates by favoring therapy resistance. As NF-κB is one of the main transcription factors activated by TNFα, we will here also highlight the primary data pointing out to the relationship between NF-κB and resistance to breast cancer therapies. Table 1 shows the reported mechanisms by which TNFα induces resistance to chemotherapy, radiotherapy, tyrosine kinase inhibitors, PARP inhibitors, trastuzumab and anti-immune checkpoint therapies in breast cancer.
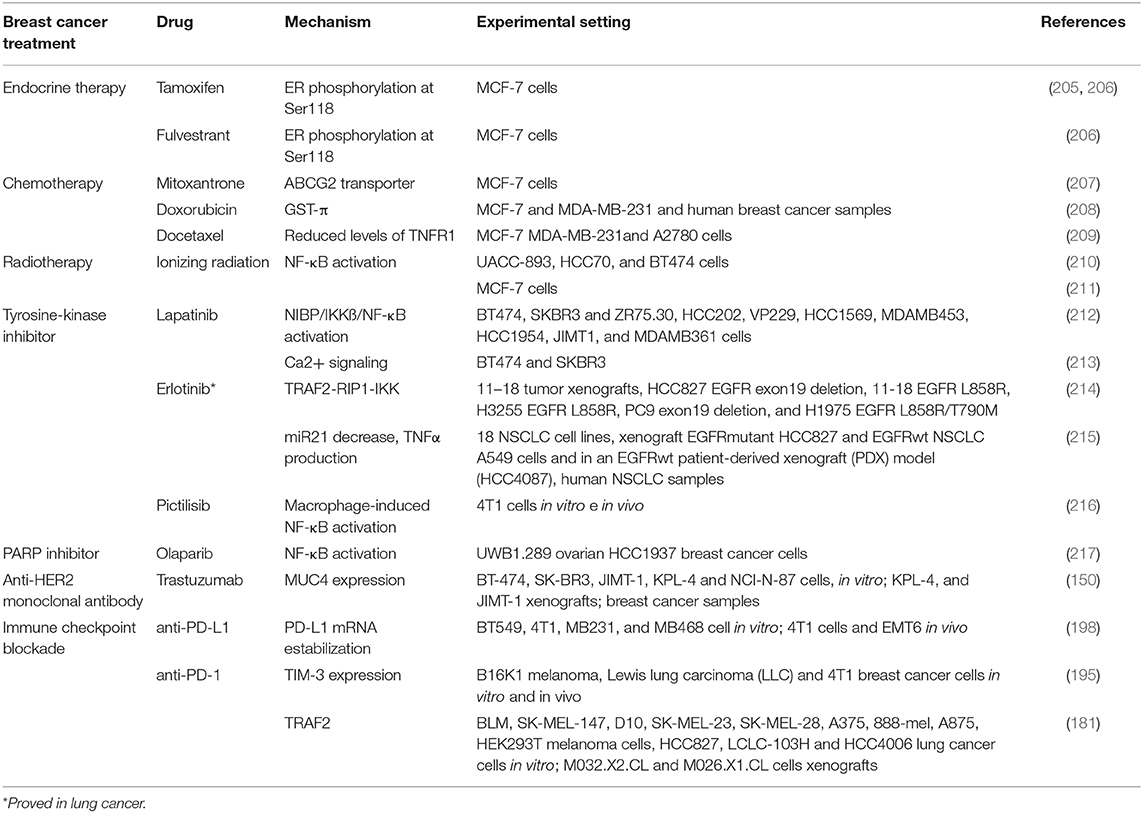
Table 1. Mechanisms of TNFα-induced therapy resistance in breast cancer.
Chemotherapy and Radiotherapy
Radiation therapy or chemotherapy along with radiation treatment combined with breast-conserving surgery (or mastectomy), are the therapeutic approaches used for all breast cancers subtypes. Although these treatments provide a striking increase in survival, tumor recurrence due to chemotherapy and radiotherapy resistance remains a major clinical problem to the successful treatment of breast cancer.
One of the known mechanisms of chemotherapy resistance relies on the ATP binding cassette transporter G2 (ABCG2, also known as breast cancer resistance protein), that causes efflux of several endogenous and exogenous agents, such as chemotherapy drugs, thus conferring a drug resistant phenotype [reviewed in (218)]. In particular, ABCG2 is able to exclude drugs such as mitoxantrone, captothecin, indolocarbazole, topoisomerase I inhibitors, EGFR1 inhibitors, methotrexate, flavopiridol, and quinazoline from the cells (219). Pradhan et al. demonstrated that TNFα synergized with the estradiol effect on ABCG2 expression. This effect was mediated by exposure of a latent NF-κB response element in the ABCG2 promoter induced by estradiol treatment, which allowed NF-κB binding near ER binding site and led to an increase in ABCG2 mRNA and protein expression (220). In the MCF-7 cell line resistant to mitoxantrone, a type II topoisomerase inhibitor which disrupts DNA synthesis, TNFα induced an increase in ABCG2 protein levels (221). These findings are in line with a previous report exploring the positive crosstalk between ER and NF-κB that revealed ABCG2 as a common target of both transcription factors that induced aggressiveness of breast cancer (109). Doxorubicin treatment of chemotherapy-resistant cell lines, such as T-47D and MBCDF, induced NF-κB activation which persisted longer in time than in the sensitive cell lines. This effect was due to a non-canonical phosphorylation of IκBα through c-Abl kinase (222).
Gemcitabine, a nucleoside analog that acts as an antimetabolite by arresting cells in the S-phase of cell cycle, has been widely used in the treatment of several solid tumors, including breast cancer (207). It was demonstrated in MDA-MB-231 cells that gemcitabine treatment increased TNFα production and the transcriptional activation of NF-κB through IκBα phosphorylation. Phosphorylation blockade with BAY11-7082 increased the cytotoxic activity of gemcitabine, suggesting that NF-κB is promoting breast cancer cell survival in this model. A similar behavior was obtained in MCF-7 cells treated with docetaxel. TNFα levels increased upon docetaxel treatment and were associated with the induction of docetaxel resistance, cell survival dependent on TNFR2/NF-κB signaling, and degradation of TNFR1. tmTNFα has also been linked to chemotherapy resistance. In this case, tmTNFα expressed in breast cancer cells, transmit outside-to-inside signals in the cells promoting activation of NF-κB through the p42/p44 MAPK signaling pathway to promote glutathione S-transferase- π (GST-π) expression, an enzyme that reduces intracellular doxorubicin levels. In addition, inhibition of tmTNFα expression improved sensitivity to docetaxel in breast cancer cells in vivo and in vitro. In breast cancer patient samples, tmTNFα expression was associated with tumor size, incidence of metastasis, and HER2 expression, while it was absent in peritumoral breast tissue (223).
Radiotherapy is frequently applied as a standard protocol after surgery. There are reports in chemotherapy resistance that indicate that NF-κB is highly activated in irradiated cells. The irradiation of MCF-7 cells with low dose of ionizing radiation (2 or 10 Gy of 137Cs γ-rays) proved to induce two phases of NF-κB activation (209). The first was triggered by the irradiation itself and induced an increase in TNFα transcription which led to a second phase of sustained NF-κB activation. TNFα from irradiated cells activated NF-κB in the non-targeted unirradiated bystander cells as well, establishing a feedforward effect. This data suggested that the TNFα/NF-κB pathway induced by irradiated cells could support the survival and progression of tumor cells. In addition, TNFα pretreatment of breast cancer cells protected them from radiation insults (208, 211).
In the clinical setting, the evaluation of NF-κB in pre- and post-chemotherapy breast cancer samples from patients subjected to neoadjuvant chemotherapy with anthracycline- and/or taxane, showed that activation of NF-κB was associated with chemoresistance. Furthermore, nuclear NF-κB staining was more frequently present in breast cancer cells after chemotherapy (210). As reported for endocrine-resistant breast cancer, NF-κB activation promoted the expression of the inhibitors of apoptosis (IAPs) and Bcl-xL, contributing to a chemotherapy and radiotherapy resistant phenotype which was also associated with treatment response [reviewed in (224, 225)].
Endocrine Therapy
As stated previously, the proliferative effects of estradiol in breast cancer are mediated by ER. Several therapeutic strategies were developed to target this axis. Tamoxifen is a pioneer selective estrogen receptor modulator that competitively inhibits estrogen binding to ER in breast tissue and has been widely used to treat ER-positive breast cancers in pre- and post-menopausal women (226). Fulvestrant is a pure antagonist that blocks ER, induces its downregulation, and promotes interactions with corepressors (227). Another option to tackle the estradiol/ER pathway is to inhibit estradiol production by aromatase inhibitors. Currently, the third generation of aromatase inhibitors, such as anastrozole and letrozole, is used in post-menopausal women with breast cancer (228). Despite these therapies, over 30% of patients with ER-positive breast cancer experience relapse, a fact that highlights the study of the resistance mechanisms to tamoxifen, fulvestrant and aromatase inhibitors, as an important field of research [reviewed in (229)]. With reference to TNFα participation in hormone therapy resistance, Castellaro et al. showed that MCF-7 cells become resistant to tamoxifen or fulvestrant treatment when cultured with macrophages previously exposed to TNFα. This acquired resistance is dependent on ER phosphorylation at Ser118 induced by p42 MAPK and STAT3. This leads to the formation of an NF-κB/STAT3/phospho-ER complex that binds to the cyclin D1 gene promoter which, in turn, promotes cell proliferation independently of ER ligand status surpassing the efficacy of ER antagonists (230).
There is a strong body of evidence that shows that NF-κB modulates ER activity, thus modifying the response of ER-positive breast cancer cells to endocrine therapy. This is supported by the fact that NF-κB trans-represses ER functions that can proceed through protein-protein interaction or by recruitment of co-repressors. The lack of ER activation resulting from the administration of aromatase or ER inhibitors drives NF-κB-mediated tumor progression by releasing NF-κB from the inhibitory control of ER (231). Constitutive NF-κB activity and hyperactive p42/p44 MAPK are correlated with hormone resistance and emergence of ER-negative breast cancer cell populations (231). NF-κB activation also contributes to the aggressiveness of the tumor leading to the expression of several pro-survival genes, such as survivine and the multidrug transporter protein ABCG2 (206). The complexity of ER effects from liganded and unliganded ER activation and NF-κB activation through its canonical and non-canonical pathways, envision a broader network of interactions that would participate in hormone resistance which remains to be further explored.
In anti-estrogen-resistant LCC9 and MCF-7 cell lines, the small molecule inhibitor of the NF-κB pathway, parthenolide, was able to restore the sensitivity to fulvestrant in vitro (232). The LCC9 cells also exhibited a higher expression of p65 NF-κB than the parental cells. In line with these results, Zhou et al. using BT474 and MCF7/HER2 cell lines resistant to tamoxifen, demonstrated that NF-κB inhibition by parthenolide and bortezomib was able to overcome tamoxifen resistance. This effect was accompanied by the binding increase of the co-repressor NCOR1 to ER, which is associated with histone deacetylase activity and transcriptional repression of ER (233).
Clinical data support the in vitro findings. Several studies performed in ER breast cancer showed that the activation of NF-κB correlated with tamoxifen resistance, early relapse, a more aggressive tumor phenotype and a reduced overall survival (106, 234). It was demonstrated that the levels of NF-κB bound to DNA in breast cancer patients inversely correlate with ER expression in tissue samples (106). Taken as a whole, all these reports support the fact that crosstalk between ER and NF-κB pathways should be involved in the progression of ER-positive tumors, but do not take into account TNFα signaling. These findings propose a potential clinical use of NF-κB inhibitors to treat hormone-resistant breast cancer. Trinh et al. showed that the administration of bortezomib was able to delay disease progression in 22% of the patients with metastatic cancer resistant to endocrine therapy (235).
Although there are several reports showing TNFα participation in aromatase induction (described in the luminal breast cancer section), few studies have addressed its impact on aromatase inhibitors resistance. A report using genome wide expression profiling of 81 ER-positive breast carcinomas before and during treatment with an anastrozole in the neoadjuvant setting, identified an inflammatory signature that was correlated with poor response. The expressions of SLAMF8 and TNFα as well as lymphocytic infiltration were associated with lower effect on the antiproliferative response to the aromatase inhibitor. The data obtained in this study were validated in an independent cohort pointing to a clear involvement of TNFα in resistance to aromatase inhibitors.
Tyrosine Kinases Inhibitors
Tyrosine kinases inhibitors (TKIs) are small molecules designed to compete with ATP at its binding site in the tyrosine kinase domain in the receptors to reduce phosphorylation. Their targets are proto-oncogene and oncogene products that have phospho-tyrosine kinase activities, thus inhibiting cancer cell proliferation. Several TKIs are used to treat breast cancer and are directed against EGFR (gefitinib, lapatinib, erlotinib), HER2 (lapatinib, neratinib), CDK4/6 (ribociclib, palbociclib, abemaciclib), and the PI3K/Akt/mTOR pathway (alpelisib, pictisilib, everolimus), among others [reviewed in (236)].
EGFR/HER2 Inhibitors
Lapatinib is a dual EGFR/HER2 tyrosine kinase inhibitor. It is used in metastatic breast cancer with capecitabine and with letozole in hormone positive HER2-positive breast cancer previously treated with trastuzumab (237), and as a second line of treatment in HER2-positive breast cancer after trastuzumab failure (238, 239). Wetterskog et al. identified NIK and IKKβ binding protein (NIBP) as a mediator of lapatinib resistance by studying HER2-amplified primary tumors and cell lines using a high throughput lapatinib small interfering RNA sensitivity screen, with a library targeting 369 genes recurrently amplified and overexpressed in HER2-positive breast cancer (240). It has been previously demonstrated that NIBP activated NF-κB signaling through IKKβ kinase (241). Therefore, the authors proposed the need of a simultaneous blockade of HER2 and NIBP that activates NF-κB by IKKβ and IKKβ activation, respectively, to overcome resistance to the TKI. Acquired resistance to lapatinib has been studied by several groups in HER2-positive breast cancer cell lines adapted to grow under lapatinib treatment. As constitutive activation of NF-κB of lapatinib-resistant cells is a common hallmark, inhibition of cell growth and apoptosis in vitro and in vivo, could be achieved by lapatinib administration together with an IKK inhibitor (242). Another work demonstrated that lapatinib resistance induced by NF-κB activation could be prevented by a sublethal concentration of an intracellular calcium chelator (212). In the case of hormone-positive HER2-positive breast cancer cell lines, the increased ER signaling and its transcriptional activity in response to lapatinib were described as a source of lapatinib resistance enhancing FOXO3a and caveolin-1 expression (212). On the other hand, lapatinib was also studied to treat TNBCs and was found to activate NF-κB via the Src family kinase and IκBα phosphorylation. This effect was not mediated by EGFR and only lapatinib, but no other EGFR inhibitor was able to synergize the anti-tumor activity of proteasome inhibitors in vitro and in vivo (243).
EGFR is frequently overexpressed in sporadic TNBC, and TKIs targeting EGFR are being used in clinical trials (213) [reviewed in (244)]. However, to the best of our knowledge, there are no reports showing the participation of TNFα or NF-κB in resistance to EGFR TKIs in breast cancer. In contrast, seminal findings by Bivona et al. have demonstrated in EGFR-mutant lung cancer cells that inhibition of NF-κB enhanced erlotinib-induced decrease in cell proliferation in erlotinib-sensitive and resistant cells (245). Then, the TRAF2-RIP1-IKK complex was identified as the signaling mediator of the NF-κB transcriptional activation induced by erlotinib (246). It has been recently demonstrated that EGFR inhibition by erlotinib in non-small cells lung cancer, independent of the EGFR mutation status, induced downregulation of miR-21, which in turn increased TNFα mRNA stability and its protein synthesis. TNFα then induced NF-κB activation and enhanced TNFα production itself in a feedforward loop. In the same experimental setting, blockade of TNFα enhanced EGFR TKIs antitumor effect in mutated EGFR tumors and overcame resistance to EGFR-targeted therapies. A similar mechanism of erlotinib resistance was found to take place in glioma cells (247).
PI3K Inhibitors
Alpelisib, a PI3K inhibitor, has been recently approved by the FDA for advanced or metastatic HER2-negative, hormone receptor positive with PIK3CA-mutation, that exhibits progression to endocrine therapy in breast cancer. However, up to today there have been no reports of resistance to alpeslisib linked to NF-κB or TNFα. However, in vivo findings using murine 4T1 TNBC cells co-cultured with macrophages, showed that pictisilib, another PI3K inhibitor, induced NF-κB activation in 4T1 cells and turned them resistant to this TKI. Addition of aspirin to the culture reinstated the apoptotic effect of pictisilib. Experiments performed in vivo proved that pictisilib administration induced the expression of macrophage-associated cytokines and chemokines, macrophage recruitment to the tumor bed, and poor antitumor effect. However, treatment with aspirin and pictisilib decreased macrophage infiltration, tumor growth and pulmonary metastasis (214), highlighting the importance of macrophage-induced NF-κB activation in tumor cells resistant to pictisilib. In line with these findings, TNFα derived from macrophages, turned melanoma cells resistant to MEK inhibition through activation of NF-κB (215).
CDK4/6 Inhibitors
Ribociclib, palbociclib and abemaciclib bind to CDK4 and 6 ATP pocket, which induces CDK4/6 inhibition. These drugs are used in metastatic hormone receptor positive, HER2-negative breast cancer together with aromatase inhibitors in first line or with fulvestrant as second line (216). The resistance mechanisms where NF-κB was involved were obtained from data in glioblastoma cell lines where it was found that abemaciclib treatment was ineffective to inhibit cell proliferation. This effect was produced by abemaciclib activation of NF-κB that, in turn, induced the production of hepatocyte growth factor, brain-derived neurotrophic factor, and nerve growth factor, activating c-Met and TrkA-B. Then, dual inhibition of c-Met/Trk and CDK4/6 was proposed for a clinical trial (248). In addition, prolonged exposure to pablociclib was reported to induce fibroblast senescence in an NF-κB dependent manner, and these cells were able to enhance melanoma growth in mice and recruitment of MDCS to the tumor bed.
Poly-Adenosine Diphosphate (ADP) Ribose Polymerase (PARP) Inhibitors
The enzyme poly ADP ribose polymerase (PARP) repairs single strand breaks in DNA. Then, during DNA replication under treatment with PARP inhibitors, the single strand breaks result in double strand breaks when the DNA helix unwinds. Patients with hereditary breast and ovarian cancer syndrome have mutations in the homologous recombination repair enzymes BRCA1 and BRCA2, which are unable to repair these double strand breaks under PARP inhibitors treatment. The affected cells then enter apoptosis. PARP inhibitor-resistant breast and ovarian cancer cell lines harboring BRCA1 mutation were used to study gene expression by RNA sequencing (249). Pathway analysis showed that ~50% of the differentially expressed genes were NF-κB-regulated genes and that TNFα was the upstream regulator of several of them. It was also demonstrated that treatment of PARP inhibitor-resistant cells with bortezomib induced cell death to a greater extent than the parental cell line, confirming the involvement of the NF-κB pathway in the resistant phenotype. Bortezomib treatment increases PARP inhibitor sensitivity of the resistant cell lines, suggesting a possible therapeutic combination in the clinical setting (249).
Trastuzumab
Trastuzumab, a humanized monoclonal antibody against HER2, has changed the poor outcome of patients with HER-positive breast cancer since its approval in 1998. Its therapeutic effect includes blockade of HER2 signaling through p42/p44 MAPK and PI3K/Akt pathways, inhibition of HER2 shedding, downregulation of HER2 availability, inhibition of tumor angiogenesis, and ADCC (217, 250–254). However, de novo and acquired resistance to trastuzumab reaches 27 and 42% in the adjuvant and neoadjuvant settings, respectively, hampering its clinical benefit (255, 256).
As mentioned before (HER2-positive section), NF-κB activation was found to be associated with ER-negative, HER2-positive breast cancer (257). Bortezomib treatment demonstrated to induce apoptosis and to act synergistically with trastuzumab in this breast cancer subtype (258, 259). In the case of HER2-positive ER-positive breast cancer cell lines resistant to trastuzumab, the NF-κB pathway showed to be constitutively activated and its blockade improved the tumor response to trastuzumab (260).
Our findings identified TNFα as an important player in trastuzumab resistance in HER2-positive breast and gastric cancer. We proved that TNFα overexpression turned trastuzumab-sensitive cells and tumors into resistant ones. On the other hand, de novo trastuzumab-resistant breast cancer cell lines exhibited greater TNFα levels than the sensitive ones (151). In vivo blockade of TNFα in mice bearing trastuzumab-resistant breast tumors inhibited tumor growth when trastuzumab was simultaneously administrated (151, 203). The antitumor effect was similar either when both tmTNF and sTNF were neutralized with etanercept, a fusion protein of TNFR2 and human IgG, or when only sTNF was blocked with INB03. Our study revealed that the mechanism underlying TNFα-induced trastuzumab resistance was mediated by MUC4 expression. MUC4 is masks the trastuzumab binding epitope on the HER2 molecule (261). Furthermore, MUC4 confers antiadhesive properties to tumor cells allowing their systemic dissemination. Our findings demonstrated that TNFα was able to upregulate MUC4 expression through NF-κB signaling pathway, impairing trastuzumab binding and ADCC. In addition, we proved that MUC4 expression in breast cancer specimens is an independent prognostic biomarker of poor response to adjuvant trastuzumab. These results suggest that MUC4 expression in HER2-positive breast cancer samples can be useful to select patients that can benefit from trastuzumab and TNFα-blocking therapies to avoid or overcome trastuzumab resistance (151). Our team also contributed to the characterization of MUC4 expression in different histological breast cancer subtypes. We identified that the invasive micropapillary carcinoma of the breast (IMPC), a low-incidence entity of breast cancer, was the one that had higher MUC4 expression in contrast to the infiltrating ductal carcinoma, the infiltrating lobular carcinoma or the mucinous carcinoma (262). In the HER2-positive breast cancers, IMPC was present in 18% of the samples in either its pure or mixed phenotype and was associated with hormone receptor expression. Despite the fact that HER2-positive/hormone receptor positive tumors have nowadays a prognosis similar to that of luminal B breast cancer (89), patients bearing IMPC exhibited a poor response to trastuzumab in the adjuvant setting (262).
Immune Check-Point Inhibitors
In the past years, the impressive advances in immunotherapy irrupted into the cancer arena after the improvement of outcome in melanoma patients treated with the antibodies against the so-called immune checkpoints (263). After being triggered by an antigen, the physiological immune response goes through an effector phase that must be shut down, when the pathogen has been eliminated, to maintain homeostasis and prevent autoimmune symptoms or even autoimmune diseases. One of these self-control mechanisms relies on molecules that are expressed in the late effector phase of the immune response. CTLA-4 and PD-1 are molecules upregulated in activated T lymphocytes. They interact with CD80/CD86 or PD-L1 and PD-L2, respectively, which are present in dendritic cells, triggering negative signals in T lymphocytes. Tumor cells have the ability to express PD-L1 and/or PD-L2 mimicking cells from the host, thus impeding T lymphocytes attack. Therefore, the immunotherapeutic strategy that emerged is based on blocking CTL4/CD80/CD86 and PD-1/PD-L1 interaction with specific antibodies to avoid antitumor immune response shutdown (264). The FDA approved antibodies are: ipilimumab (anti-CTLA-4), pembrolizumab, nivolumab, cemiplimab-rwlc (anti-PD-1) and atezolimumab, avelumab, durvalumab (anti-PD-L1). They are used to treat bladder, cervical, colon, head and neck, liver, lung, renal, stomach cancers, and Hodgking lymphoma (265). The most promising results with immune checkpoint blockade (ICB) were obtained with nivolumab in refractory Hodgkin's lymphoma where the objective response rate was 87% (266). Thus, the reinvigoration of the antitumor immune response pursues the promotion of cancer cell cytotoxicity. The immunotherapy-based on atezolimumab has also recently reached FDA approval to treat TNBC, the breast cancer subtype with poorest survival, whose current treatment relies only on aggressive chemotherapy. In locally advanced or metastatic TNBC atezolizumab was found to be effective only in those tumors which express PD-L1 in their tumor-infiltrating lymphocytes (TILs) (267). However, the overall survival of these patients at 24 months was 50.7% in the atezolizumab combined with nab-paclitaxel group vs. 36.9% in the group treated with chemotherapy alone (267). In general, only a small number of the patients subjected to anti-immune checkpoint therapy experienced benefit. In addition, severe immune-related adverse events (irAEs) are frequently reported, and they stem from blocking the immune system control letting local and systemic autoimmune responses arise.
TNFα is an important player in the effectiveness of ICB. First, seminal findings by Lim et al. demonstrated the important participation of TNFα in PD-L1 biology (194). Using 4T1 cells and human TNBC cell lines, they demonstrated that TNFα induces COP9 signalosome 5 (C5N5), a protein that inhibits PD-L1 ubiquitination, through NF-κB activation. Then, PD-L1 protein increases its stability in tumor and dendritic cells, maintaining tumor immune evasion. In this experimental setting, the inhibition of NF-κB activation by curcumin was able to downregulate PD-L1 expression (194). Second, TNFα has been recognized as a mediator of ICB resistance in several cancer types. Preclinical studies performed in melanoma, lung and breast cancer show that TNFα induces expression of PD-L1. Treatment with anti-PD-1 allows TNFα-induced expression of T lymphocyte Immunoglobulin and TIM-3, another checkpoint molecule, in CD4+ and CD8+ TILs. In addition, TNFα also promotes death of CD8+ TILs (191). The above-mentioned effects were proved to be TNFR1-dependent, highlighting sTNFα's participation in immune surveillance evasion. The administration of anti-TNFα reduced CD8+ TILs cell death, TIM-3 and PD-L1 expression, decreasing T lymphocyte exhaustion upon anti-PD-1 therapy. In melanoma, anti-PD-1 therapy induced the regression of 20% of the tumors, while a synergistic antitumor effect was observed when anti-PD-1 was used in combination with anti-TNFα antibodies causing tumor regression of 75% (191).
As it was stated before, TNFR1 signaling can induce apoptosis or promote proliferation and survival depending on the adaptor proteins available in the cytoplasm. In the case of most cancer cells, TRAF2 is abundant, and the final outcome produced by TNFα is tumor growth. Recently, Vredevoogd et al., have described that TRAF2 induces resistance to T lymphocyte cytotoxicity in melanoma and lung cancer (175). Blockade of the TRAF2 pathway increased the susceptibility of these models to ICB. Consistently, patients with tumors harboring inactivating mutations in TRAF2 are more susceptible to T lymphocytes killing. On the other hand, it is known that hypoxia in solid tumors drives the formation of abnormal new vessels that provide an irregular blood flow and deficient drug delivery into the tumors. Concurrently, the endothelial cells of these vessels poorly express leucocyte adhesion molecules, impairing leukocytes extravasation. Fusion protein Cys–Asn–Gly–Arg–Cys–Gly–TNFα (NGR-TNFα), which targets the tumor vasculature delivering low amounts of TNFα, activates endothelial cells and allows CD8+ T lymphocytes infiltration (268). The simultaneous administration of NGR-TNFα with anti-PD-1 or anti-CTLA4 and with adoptive T lymphocyte therapy increased the effectiveness of each therapy alone in prostate and melanoma models (269). Finally, TNFα is induced by ICB and has been shown to be one of the important players in the irAEs. The irAEs, i.g. colitis, are managed by corticosteroid administration (270, 271), but refractory cases are treated with infliximab once ICB is suspended. Considering the aforementioned mechanisms of TNFα-induced ICB resistance, preclinical studies investigated the prophylactic effect of TNFα blockade both to impede irAEs development and to enhance ICB‘s efficacy. Melero and co-workers used etanercept prior to treatment with anti-CTLA4 and anti-PD-1 and found an improved anti-tumor efficacy of the ICB and also a decrease in colitis. These findings were achieved in tumor-transplanted mouse models of fibrosarcoma, melanoma, and colon cancer and in xenografts of colon cancer. This work verified an increase of CD8+ TILs, a decrease in T lymphocytes exhaustion and inactivation-induced cell death of T lymphocytes as described before (191). Based on this evidence, an ongoing clinical trial was launched in melanoma patients, combining nivolumab + Ipilimumab + the anti-TNFα certolizumab (272).
Clinical Relevance of TNFα and its Blockade as Therapies in Cancer
Pioneer findings of Balkwill et al. demonstrated the usefulness of the treatment with anti-TNFα antibodies to prevent chemical-induced skin papilloma and inhibit growth of a transplantable breast cancer (273). These results opened up a new field of application of anti-TNFα drugs, so far devoted just to treat inflammatory and autoimmune diseases for more than 20 years.
The current anti-TNFα FDA approved therapies available are antibodies (infliximab, adalimumab, and golimumab), PEGylated Fab fragment (certolizumab pegol) and the fusion protein of TNFR2 with the Fc of the human IgG1 (etanercept). They all block both tmTNF and sTNF. As detailed above, active TNFα consists of a homotrimer that is continuously exchanging TNFα monomers. The dissociation rate is independent of TNFα concentration, while the association rate is not. The latter is very important in maintaining bioactivity at high concentrations, but not at low concentrations. The use of Förster resonance energy transfer (FRET) assay shed light on the difference of TNFα blockade with the above-mentioned inhibitors. In the case of adalimumab, infliximab, or etanercept treatment, the TNFα exchange between monomers and trimers is blocked but not by administration of certolizumab or golimumab. These findings might explain that under conditions of incomplete blocking, residual TNFα bioactivity can be found in vitro for adalimumab (274). The analysis of all the FDA approved drugs targeting TNFα revealed essentially no differences among them in the treatment of rheumatoid arthritis and spondyloarthritis (275). Regarding etanercept treatment, Liu et al. showed that certain polymorphisms in the TNFα gene promoter could predict a good response to TNFα-blocking therapies, especially for etanercept treatment in patients with spondyloarthritis (276). In this sense, another important contribution was made by Murdaca et al. that reported that the +489 TNFα polymorphism is associated with susceptibility and severity of psoriatic arthritis and this single nucleotide variant is responsible for a higher production of TNFα. Moreover, they informed that patients with +489 polymorphisms are associated with a better response to etanercept and adalimumab. On the contrary, in their cohort −238 and −408 polymorphisms do not influence the clinical outcome of patients with psoriatic arthritis which was related with the outcome of other auto-inflammatory immune diseases in other ethnicities (277). However, the application of these drugs in cancer will require a deeper study to determine which strategy would render the best outcome.
Due to TNFα‘s role in the immune system as a factor that causes tumor necrosis and controls infections, it was thought that the use of TNFα inhibitors could unleash cancer development and/or grant susceptibility to them. In this regard, there has been a good number of studies that addressed this issue in patients with inflammatory autoimmune diseases treated with different TNFα-blocking agents. No increased risk of developing malignancies was found in patients with rheumatoid arthritis and with immune-mediated diseases in general, psoriatic arthritis, ankylosing spondylitis and inflammatory bowel disease (278–283).
There is one study in patients with inflammatory bowel disease that previously had cancer, where the authors found that anti-TNFα treatment had a mild risk of incident cancer (284). These results are conflictive because it is well-known that patients who had a neoplastic disease in the past have an increased risk of developing new cancers, therefore the effect cannot be entirely attributed to anti-TNFα therapies. Current recommendations counterindicate the use of TNFα-blockers in patients with inflammatory bowel disease who had a cancer diagnosis in the last 5 years (285). On the other hand, there was another study of patients with rheumatoid arthritis and breast cancer that showed that TNFα-blockade did not increase recurrence of breast cancer with respect to patients treated otherwise (280).
In addition, two reports have described that anti-TNFα therapies cause an increased risk of non-melanoma skin cancers (279) and hematological malignancies in patients with rheumatoid arthritis. In the latter report it was also stated that rheumatoid arthritis patients have an increased risk of lymphomas due to the disease itself (286), which prevents us from speculating about the role of TNFα inhibitors.
The use of TNFα blocking agents for treating breast cancer has been poorly explored. In this regard there is only one phase II clinical trial that showed that etanercept is safe and well-tolerated in breast cancer patients, but that it did not produce objective disease responses, which could be due to the advanced stage of the patients (287). Taking all this into account, TNFα-blocking agents are a promising tool to treat breast cancer patients in combination with current cancer therapies.
Several findings support the fact that TNFR1 may be responsible for TNFα pro-tumorigenic actions [reviewed in (158, 288)]. If so, sTNFα neutralization would be an important path to explore. Indeed, Sobo-Vujanovic et al. and our own work demonstrated that sTNFα blockade prevents chemical induced skin carcinogenesis and overcomes trastuzumab resistance in HER2-positive breast cancer models, respectively (71, 151). The sTNFα blockade also preserves the cross talk between NK cells and dendritic cells that is mediated by tmTNFα, making less immunosuppressive targeting TNFα (14).
The facts exposed here postulate TNFα as an attractive target potentially useful to treat different breast cancer subtypes. These TNFα-blocking drugs should be studied in the clinical setting to potentiate current therapies and overcome treatment resistance thereby achieving a better outcome for the patients.
Conclusion
TNFα has been proved to play a central role in initiation, promotion, and metastasis in most of cancers, in particular in breast cancer. It is also important at the onset of the immune response and exerts a cytotoxic effect against infected and in tumor cells. However, TNFα has a great variety of biological effects, from cell proliferation, inflammation, pro-survival activity, to cell death. These dramatically different outcomes are due to different TNFRs and the signaling adaptor proteins present in each cell, and to the different forms of TNFα: sTNFα and tmTNFα.
As cancer evolves after immunoedition acquires the ability to evade immunosurveillance. Along this process, tumor cells reduce their immunogenicity, and among other features, they become resistant to the cytotoxic activity of TNFα. The described adaptive mechanisms to avoid the lethal activity of TNFα include downregulation of TNFRs, increase of TNFR adaptor proteins which favors survival activity and glycosylation of TNFRs. In this scenario, TNFα induces survival and even cancer cell proliferation. Indeed, tumor cells themselves are able to produce TNFα and can increase its concentration upon therapy insults such as chemo/radiotherapy, targeted therapies and immunotherapy with ICB, to take advantage of NF-κB signaling to promote survival. In this way, it favors the emergence of therapy-resistant breast cancer cells directly linked to the recurrence of the disease.
The approval of ICB for the treatment of TNBC opens up a long sought new therapeutic possibility for this aggressive breast cancer subtype which is otherwise only handled with chemotherapy. However, irAEs frequently leads to ICB treatment withdrawal. As it is acknowledged that TNFα serum levels parallels the irAEs, the proposal of administration of TNFα blocking agents together with ICB will give chances to increase the number of patients able to complete the treatment. In addition, TNFα neutralization would also exert additional beneficial activities preventing T lymphocytes death and exhaustion.
On the other hand, TNFα also upregulates pro-metastatic factors, such as MMPs and MUC4, and enhances tumor dissemination. In particular, in HER2-positive breast cancer, MUC4 expression impairs trastuzumab efficacy by masking the binding epitope in the HER2 molecule and is an independent biomarker of poor survival in patients treated with adjuvant trastuzumab. All this evidence provides a proof-of-concept for proposing the blockade of TNFα as a promising therapy in breast cancer. Anti-TNFα therapies have been used for more than 20 years to treat autoimmune and inflammatory diseases. This fact envisions a shorter path to use TNFα-blocking agents together with the appropriate oncology treatments. Moreover, MUC4 expression in cancer specimens would help select the subgroup of HER2-positive breast cancer patients that benefit from the combination of anti-TNFα therapies with the standard treatments, as we previously demonstrated in HER2-positive breast cancer. This approach will contribute to precision medicine and will optimize the economic resources devoted to oncology.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from National Cancer Institute (Argentina) 2018, IDB/PICT 2017-1517 from the National Agency of Scientific Promotion of Argentina (ANPCyT), Florencio Fiorini Foundation and Alberto J. Roemmers Foundation awarded to RS; a grant from Oncomed-Reno, CONICET 1819/03, awarded to PE and RS; grants from National Cancer Institute (Argentina) 2018, IDB/PICT 2015-1587, IDB/PICT 2017-1587, from ANPCyT, PID 2012-066 from Consejo Nacional de Investigaciones Científicas y Técnicas and from the National Institute of Cancer from Argentina (Argentina) 2018–2019, all of them awarded to PE. We are grateful to Fundación René Baron and Fundación Williams for their institutional support to IBYME-CONICET.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Waters JP, Pober JS, Bradley JR. Tumour necrosis factor and cancer. J Pathol. (2013) 230:241–8. doi: 10.1002/path.4188
2. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. (1975) 72:3666–70. doi: 10.1073/pnas.72.9.3666
3. Green S, Dobrjansky A, Carswell EA, Kassel RL, Old LJ, Fiore N, et al. Partial purification of a serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. (1976) 73:381–5. doi: 10.1073/pnas.73.2.381
4. Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature. (1984) 312:724–9. doi: 10.1038/312724a0
5. Falvo JV, Tsytsykova AV, Goldfeld AE. Transcriptional control of the TNF gene. Curr Dir Autoimmun. (2010) 11:27–60. doi: 10.1159/000289196
6. Tsai EY, Yie J, Thanos D, Goldfeld AE. Cell-type-specific regulation of the human tumor necrosis factor alpha gene in B cells and T cells by NFATp and ATF-2/JUN. Mol Cell Biol. (1996) 16:5232–44. doi: 10.1128/mcb.16.10.5232
7. Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. (1990) 171:35–47. doi: 10.1084/jem.171.1.35
8. Spriggs DR, Deutsch S, Kufe DW. Genomic structure, induction, and production of TNF-α. Immunol Ser. (1992) 56:3–34.
9. Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. (1995) 83:793–802. doi: 10.1016/0092-8674(95)90192-2
10. Birkland TP, Sypek JP, Wyler DJ. Soluble TNF and membrane TNF expressed on CD4+ T lymphocytes differ in their ability to activate macrophage antileishmanial defense. J Leukoc Biol. (1992) 51:296–9. doi: 10.1002/jlb.51.3.296
11. Decker T, Lohmann-Matthes ML, Gifford GE. Cell-associated tumor necrosis factor (TNF) as a killing mechanism of activated cytotoxic macrophages. J Immunol. (1987) 138:957–62.
12. Macchia D, Parronchi P, Piccinni MP, Simonelli C, Mazzetti M, Ravina A, et al. In vitro infection with HIV enables human CD4+ T cell clones to induce noncognate contact-dependent polyclonal B cell activation. J Immunol. (1991) 146:3413–8.
13. Macchia D, Almerigogna F, Parronchi P, Ravina A, Maggi E, Romagnani S. Membrane tumour necrosis factor-α is involved in the polyclonal B-cell activation induced by HIV-infected human T cells. Nature. (1993) 363:464–6. doi: 10.1038/363464a0
14. Xu J, Chakrabarti AK, Tan JL, Ge L, Gambotto A, Vujanovic NL. Essential role of the TNF-TNFR2 cognate interaction in mouse dendritic cell-natural killer cell crosstalk. Blood. (2007) 109:3333–41. doi: 10.1182/blood-2006-06-026385
15. Rossol M, Meusch U, Pierer M, Kaltenhäuser S, Häntzschel H, Hauschildt S, et al. Interaction between transmembrane TNF and TNFR1/2 mediates the activation of monocytes by contact with T cells. J Immunol. (2007) 179:4239–48. doi: 10.4049/jimmunol.179.6.4239
16. Xu H, Sethi JK, Hotamisligil GS. Transmembrane tumor necrosis factor (TNF)-alpha inhibits adipocyte differentiation by selectively activating TNF receptor 1. J Biol Chem. (1999) 274:26287–95. doi: 10.1074/jbc.274.37.26287
17. Mann DL. Stress-activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol. (2003) 65:81–101. doi: 10.1146/annurev.physiol.65.092101.142249
18. Harashima S, Horiuchi T, Hatta N, Morita C, Higuchi M, Sawabe T, et al. Outside-to-inside signal through the membrane TNF-alpha induces E-selectin (CD62E) expression on activated human CD4+ T cells. J Immunol. (2001) 166:130–6. doi: 10.4049/jimmunol.166.1.130
19. Eissner G, Kirchner S, Lindner H, Kolch W, Janosch P, Grell M, et al. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J Immunol. (2000) 164:6193–8. doi: 10.4049/jimmunol.164.12.6193
20. Wiley SR, Goodwin RG, Smith CA. Reverse signaling via CD30 ligand. J Immunol. (1996) 157:3635–9.
21. Blotta MH, Marshall JD, DeKruyff RH, Umetsu DT. Cross-linking of the CD40 ligand on human CD4+ T lymphocytes generates a costimulatory signal that up-regulates IL-4 synthesis. J Immunol. (1996) 156:3133–40.
22. Schwarz H. Biological activities of reverse signal transduction through CD137 ligand. J Leukoc Biol. (2005) 77:281–6. doi: 10.1189/jlb.0904558
23. Eissner G, Kolch W, Scheurich P. Ligands working as receptors: reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. (2004) 15:353–66. doi: 10.1016/j.cytogfr.2004.03.011
24. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. (1997) 385:729–33. doi: 10.1038/385729a0
25. Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. (2018) 53:45–53. doi: 10.1016/0092-8674(88)90486-2
26. Perez C, Albert I, DeFay K, Zachariades N, Gooding L, Kriegler M. A nonsecretable cell surface mutant of tumor necrosis factor (TNF) kills by cell-to-cell contact. Cell. (1990) 63:251–8. doi: 10.1016/0092-8674(90)90158-b
27. Brakebusch C, Varfolomeev EE, Batkin M, Wallach D. Structural requirements for inducible shedding of the p55 tumor necrosis factor receptor. J Biol Chem. (1994) 269:32488–96.
28. Tartaglia LA, Weber RF, Figari IS, Reynolds C, Palladino MA Jr, Goeddel DV. The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc Natl Acad Sci USA. (1991) 88:9292–6. doi: 10.1073/pnas.88.20.9292
29. Ledgerwood EC, Pober JS, Bradley JR. Recent advances in the molecular basis of TNF signal transduction. Lab Invest. (1999) 79:1041–50.
30. Aderka D, Engelmann H, Maor Y, Brakebusch C, Wallach D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J Exp Med. (1992) 175:323–9. doi: 10.1084/jem.175.2.323
31. Watts AD, Hunt NH, Madigan MC, Chaudhri G. Soluble TNF-alpha receptors bind and neutralize over-expressed transmembrane TNF-alpha on macrophages, but do not inhibit its processing. J Leukoc Biol. (1999) 66:1005–13. doi: 10.1002/jlb.66.6.1005
32. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. (2008) 117:244–79. doi: 10.1016/j.pharmthera.2007.10.001
33. Choi SJ, Lee KH, Park HS, Kim SK, Koh CM, Park JY. Differential expression, shedding, cytokine regulation and function of TNFR1 and TNFR2 in human fetal astrocytes. Yonsei Med J. (2005) 46:818–26. doi: 10.3349/ymj.2005.46.6.818
34. Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov. (2010) 9:482. doi: 10.1038/nrd3030
35. Ihnatko R, Kubes M. TNF signaling: early events and phosphorylation. Gen Physiol Biophys. (2007) 26:159–167.
36. Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. (1996) 87:565–76. doi: 10.1016/s0092-8674(00)81375-6
37. Chinnaiyan AM, Tepper CG, Seldin MF, O'Rourke K, Kischkel FC, Hellbardt S, et al. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. (1996) 271:4961–5. doi: 10.1074/jbc.271.9.4961
38. Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. (1995) 14:5579–88.
39. Bradley JR. TNF-mediated inflammatory disease. J Pathol. (2008) 214:149–60. doi: 10.1002/path.2287
40. Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. (1994) 78:681–92. doi: 10.1016/0092-8674(94)90532-0
41. Naudé PJW, den Boer JA, Luiten PGM, Eisel ULM. Tumor necrosis factor receptor cross-talk. FEBS J. (2011) 278:888–98. doi: 10.1111/j.1742-4658.2011.08017.x
42. Haider S, Knöfler M. Human tumour necrosis factor: physiological and pathological roles in placenta and endometrium. Placenta. (2009) 30:111–23. doi: 10.1016/j.placenta.2008.10.012
43. Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem. (1993) 268:18542–8.
44. Varela LM, Ip MM. Tumor necrosis factor-α: a multifunctional regulator of mammary gland development. Endocrinology. (1996) 137:4915–24. doi: 10.1210/endo.137.11.8895364
45. Lee PH, Hwang J, Murphy G, Ip MM, Lee EH, Hwang J, et al. Functional significance of MMP-9 in tumor necrosis factor-induced proliferation and branching morphogenesis of mammary epithelial cells. Endocrinology. (2000) 141:3746–73. doi: 10.1210/endo.141.10.7697
46. Ip MM, Shoemaker SF, Darcy KM. Regulation of rat mammary epithelial cell proliferation and differentiation by tumor necrosis factor-alpha. Endocrinology. (1992) 130:2833–44. doi: 10.1210/endo.130.5.1572296
47. Varela LM, Stangle-Castor NC, Shoemaker SF, Shea-Eaton WK, Ip MM. TNFα induces NFκB/p50 in association with the growth and morphogenesis of normal and transformed rat mammary epithelial cells. J Cell Physiol. (2001) 188:120–31. doi: 10.1002/jcp.1103
48. Geymayer S, Doppler W. Activation of NF-κb p50/p65 is regulated in the developing mammary gland and inhibits STAT5-mediated β-casein gene expression. FASEB J. (2000) 14:1159–70. doi: 10.1096/fasebj.14.9.1159
49. Sun L, Carpenter G. Epidermal growth factor activation of NF-kappaB is mediated through IkappaBalpha degradation and intracellular free calcium. Oncogene. (1998) 16:2095–102. doi: 10.1038/sj.onc.1201731
50. Watson CJ, Kreuzaler PA. Remodeling mechanisms of the mammary gland during involution. Int J Dev Biol. (2011) 55:757–62. doi: 10.1387/ijdb.113414cw
51. Baxter FO, Came PJ, Abell K, Kedjouar B, Huth M, Rajewsky K, et al. IKKβ/2 induces TWEAK and apoptosis in mammary epithelial cells. Development. (2006) 133:3485–94. doi: 10.1242/dev.02502
52. Levy CS, Slomiansky V, Gattelli A, Nahmod K, Pelisch F, Blaustein M, et al. Tumor necrosis factor alpha induces LIF expression through ERK1/2 activation in mammary epithelial cells. J Cell Biochem. (2010) 110:857–65. doi: 10.1002/jcb.22595
53. Hojilla CV, Jackson HW, Khokha R. TIMP3 regulates mammary epithelial apoptosis with immune cell recruitment through differential TNF dependence. PLoS ONE. (2011) 6:e26718. doi: 10.1371/journal.pone.0026718
54. Kojima H, Fukazawa Y, Sato T, Enari M, Tomooka Y, Matsuzawa A, et al. Involvement of the TNF-α system and the Fas system in the induction of apoptosis of mouse mammary glands after weaning. Apoptosis. (1996) 1:201–8. doi: 10.1007/BF01321103
55. Coussens LM, Zitvogel L, Palucka K. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. (2013) 339:286–91. doi: 10.1126/science.1232227
57. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. (2008) 454:436–44. doi: 10.1038/nature07205
58. Balkwill F, Mantovani A. Inflammation and cancer: back to virchow? Lancet. (2018) 357:539–45. doi: 10.1016/S0140-6736(00)04046-0
59. Sethi G, Shanmugam MK, Ramachandran L, Kumar AP, Tergaonkar V. Multifaceted link between cancer and inflammation. Biosci Rep. (2012) 32:1–15. doi: 10.1042/BSR20100136
60. Naylor MS, Malik ST, Stamp GW, Jobling T, Balkwill FR. In situ detection of tumour necrosis factor in human ovarian cancer specimens. Eur J Cancer. (1990) 26:1027–30.
61. Gupta M, Babic A, Beck AH, Terry K. TNF-α expression, risk factors, and inflammatory exposures in ovarian cancer: evidence for an inflammatory pathway of ovarian carcinogenesis? Hum Pathol. (2016) 54:82–91. doi: 10.1016/j.humpath.2016.03.006
62. Morgado M, Sutton MN, Simmons M, Warren CR, Lu Z, Constantinou PE, et al. Tumor necrosis factor-α and Interferon-γ stimulate MUC16 (CA125) expression in breast, endometrial and ovarian cancers through NFκB. Oncotarget. (2016) 7:14871–84. doi: 10.18632/oncotarget.7652
63. Leek RD, Landers R, Fox SB, Ng F, Harris L, Lewis CE. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br J Cancer. (1998) 77:2246–51.
64. Tang D, Tao D, Fang Y, Deng C, Xu Q, Zhou J. TNF-Alpha promotes invasion and metastasis via NF-Kappa B pathway in oral squamous cell carcinoma. Med Sci Monit Basic Res. (2017) 23:141–9. doi: 10.12659/MSMBR.903910
65. Egberts J-H, Cloosters V, Noack A, Schniewind B, Thon L, Klose S, et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. (2008) 68:1443–50. doi: 10.1158/0008-5472.CAN-07-5704
66. Zhao C, Lu X, Bu X, Zhang N, Wang W. Involvement of tumor necrosis factor-a in the upregulation of CXCR4 expression in gastric cancer induced by Helicobacter pylori. BMC Cancer. (2010) 10:419. doi: 10.1186/1471-2407-10-419
67. Roberts RA, Kimber I. Cytokines in non-genotoxic hepatocarcinogenesis. Carcinogenesis. (1999) 20:1397–401. doi: 10.1093/carcin/20.8.1397
68. Li X, Wang S, Ren HJ, Ma J, Sun X, Li N, et al. Molecular correlates and prognostic value of tmTNF-α expression in colorectal cancer of 5-Fluorouracil-based adjuvant therapy. Cancer Biol Ther. (2016) 17:684–92. doi: 10.1080/15384047.2016.1187551
69. Warzocha K, Salles G, Bienvenu J, Bastion Y, Dumontet C, Renard N, et al. Tumor necrosis factor ligand-receptor system can predict treatment outcome in lymphoma patients. J Clin Oncol. (1997) 15:499–508. doi: 10.1200/JCO.1997.15.2.499
70. Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, et al. Mice deficient in tumor necrosis factor-α are resistant to skin carcinogenesis. Nat Med. (1999) 5:828.
71. Andrea SV, Lazar V, Albert BDL, Fernando CB, Robert LF, Yan L, et al. Inhibition of soluble tumor necrosis factor prevents chemically induced carcinogenesis in mice. Cancer Immunol Res. (2016) 4:441–51. doi: 10.1158/2326-6066.CIR-15-0104
72. Suganuma M, Okabe S, Marino MW, Sakai A, Sueoka E, Fujiki H. Essential role of tumor necrosis factor alpha (TNF-alpha) in tumor promotion as revealed by TNF-alpha-deficient mice. Cancer Res. (1999) 59:4516–18.
73. Krajcik RA, Massardo S, Orentreich N. No association between serum levels of tumor necrosis factor-α (TNF-α) or the soluble receptors sTNFR1 and sTNFR2 and breast cancer risk. Cancer Epidemiol Biomarkers Prev. (2003) 12:945–6.
74. García-Tuñón I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. Role of tumor necrosis factor-α and its receptors in human benign breast lesions and tumors (in.situ and infiltrative). Cancer Sci. (2006) 97:1044–49. doi: 10.1111/j.1349-7006.2006.00277.x
75. Azmy IAF, Balasubramanian SP, Wilson AG, Stephenson TJ, Cox A, Brown NJ, et al. Role of tumour necrosis factor gene polymorphisms (-308 and−238) in breast cancer susceptibility and severity. Br Cancer Res. (2004) 6:395–400. doi: 10.1186/bcr802
76. Cui LF, Guo XJ, Wei J, Liu FF, Fan Y, Lang RG, et al. Overexpression of TNF-alpha and TNFRII in invasive micropapillary carcinoma of the breast: clinicopathological correlations. Histopathology. (2008) 53:381–8. doi: 10.1111/j.1365-2559.2008.03128.x
77. Yi F, Shi X, Pei X, Wu X. Tumor necrosis factor-alpha-308 gene promoter polymorphism associates with survival of cancer patients: a meta-analysis. Med (United States). (2018) 97:e13160. doi: 10.1097/MD.0000000000013160
78. Zhang Q, Zhao GS, Yuan XL, Li XH, Yang Z, Cui YF, et al. Tumor necrosis factor alpha-238G/A polymorphism and risk of breast cancer. Med (United States). (2017) 96:e7442. doi: 10.1097/MD.0000000000007442
79. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. (2005) 7:211–17. doi: 10.1016/j.ccr.2005.02.013
80. Burow ME, Weldon CB, Tang Y, Navar GL, Krajewski S, Reed JC, et al. Differences in susceptibility to tumor necrosis factor α-induced apoptosis among MCF-7 breast cancer cell variants. Cancer Res. (1998) 58:4940–6.
81. Zhou BP, Hu MC-T, Miller SA, Yu Z, Xia W, Lin S-Y, et al. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kB pathway. J Biol Chem. (2000) 275:8027–31. doi: 10.1074/jbc.275.11.8027
82. Mantovani A, Marchesi F, Porta C, Sica A, Allavena P. Inflammation and cancer: breast cancer as a prototype. Breast. (2018) 16:27–33. doi: 10.1016/j.breast.2007.07.013
83. Cole SW. Chronic inflammation and breast cancer recurrence. J Clin Oncol. (2009) 27:3418–9. doi: 10.1200/JCO.2009.21.9782
84. Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFβ/TNFα-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. (2011) 71:4707–19. doi: 10.1158/0008-5472.CAN-10-4554
85. Li C-W, Xia W, Huo L, Lim S-O, Wu Y, Hsu JL, et al. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of twist1. Cancer Res. (2012) 72:1290–300. doi: 10.1158/0008-5472.CAN-11-3123
86. Cohen EN, Gao H, Anfossi S, Mego M, Reddy NG, Debeb B, et al. Inflammation mediated metastasis: immune induced epithelial-to-mesenchymal transition in inflammatory breast cancer cells. PLoS ONE. (2015) 10:e0132710. doi: 10.1371/journal.pone.0132710
87. Goldhirsch A, Winer EP, Coates AS, Gelber RD, Piccart-Gebhart M, Thürlimann B, et al. Personalizing the treatment of women with early breast cancer: highlights of the st gallen international expert consensus on the primary therapy of early breast cancer 2013. Ann Oncol. (2013) 24:2206–23. doi: 10.1093/annonc/mdt303
88. Fragomeni SM, Sciallis A, Jeruss JS. Molecular subtypes and local-regional control of breast cancer. Surg Oncol Clin N Am. (2018) 27:95–120. doi: 10.1016/j.soc.2017.08.005
89. Howlader N, Cronin KA, Kurian AW, Andridge R. Differences in breast cancer survival by molecular subtypes in the United States. Cancer Epidemiol Biomarkers Prev. (2018) 27:619–26. doi: 10.1158/1055-9965.EPI-17-0627
90. Deblois G, Giguère V. Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nat Rev Cancer. (2013) 13:27–36. doi: 10.1038/nrc3396
91. Zhao Y, Nichols JE, Valdez R, Mendelson CR, Simpson ER. Tumor necrosis factor-alpha stimulates aromatase gene expression in human adipose stromal cells through use of an activating protein-1 binding site upstream of promoter 1.4. Mol Endocrinol. (1996) 10:1350–7. doi: 10.1210/mend.10.11.8923461
92. Bonney RC, Reed MJ, Davidson K, Beranek PA, James VH. The relationship between 17 β-hydroxysteroid dehydrogenase activity and oestrogen concentrations in human breast tumours and in normal breast tissue. Clin Endocrinol. (1983) 19:727–39. doi: 10.1111/j.1365-2265.1983.tb00051.x
93. Blankenstein MA, Maitimu-Smeele I, Donker GH, Daroszewski J, Milewicz A, Thijssen JH. On the significance of in situ production of oestrogens in human breast cancer tissue. J Steroid Biochem Mol Biol. (1992) 41:891–6. doi: 10.1016/0960-0760(92)90443-m
94. Brodie A, Long B, Lu Q. Aromatase expression in the human breast. Breast Cancer Res Treat. (1998) 49(Suppl. 1):S85–119. doi: 10.1023/a:1006029612990
95. Reed MJ, Topping L, Coldham NG, Purohit A, Ghilchik MW, James VH. Control of aromatase activity in breast cancer cells: the role of cytokines and growth factors. J Steroid Biochem Mol Biol. (1993) 44:589–96. doi: 10.1016/0960-0760(93)90264-w
96. Meng L, Zhou J, Sasano H, Suzuki T, Zeitoun KM, Bulun SE. Tumor necrosis factor alpha and interleukin 11 secreted by malignant breast epithelial cells inhibit adipocyte differentiation by selectively down-regulating CCAAT/enhancer binding protein alpha and peroxisome proliferator-activated receptor γ: mechan. Cancer Res. (2001) 61:2250–5.
97. Martínez-Chacón G, Brown KA, Docanto MM, Kumar H, Salminen S, Saarinen N, et al. IL-10 suppresses TNF-α-induced expression of human aromatase gene in mammary adipose tissue. FASEB J. (2018) 32:3361–70. doi: 10.1096/fj.201700938RRR
98. Opipari AW Jr, Hu HM, Yabkowitz R, Dixit VM. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem. (1992) 267:12424–27.
99. Lee E, Ouzounova M, Piranlioglu R, Ma MT, Guzel M, Marasco D, et al. The pleiotropic effects of TNFα in breast cancer subtypes is regulated by TNFAIP3/A20. Oncogene. (2019) 38:469–82. doi: 10.1038/s41388-018-0472-0
100. Lee J-H, Jung SM, Yang K-M, Bae E, Ahn SG, Park JS, et al. A20 promotes metastasis of aggressive basal-like breast cancers through multi-monoubiquitylation of Snail1. Nat Cell Biol. (2017) 19:1260–73. doi: 10.1038/ncb3609
101. Bodine PV, Harris HA, Komm BS. Suppression of ligand-dependent estrogen receptor activity by bone-resorbing cytokines in human osteoblasts. Endocrinology. (1999) 140:2439–51. doi: 10.1210/endo.140.6.6612
102. Galien R, Garcia T. Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res. (1997) 25:2424–9. doi: 10.1093/nar/25.12.2424
103. Cvoro A, Tzagarakis-Foster C, Tatomer D, Paruthiyil S, Fox MS, Leitman DC. Distinct roles of unliganded and liganded estrogen receptors in transcriptional repression. Mol Cell. (2006) 21:555–64. doi: 10.1016/j.molcel.2006.01.014
104. Harnish DC, Scicchitano MS, Adelman SJ, Lyttle CR, Karathanasis SK. The role of CBP in estrogen receptor cross-talk with nuclear factor-kappaB in HepG2 cells. Endocrinology. (2000) 141:3403–11. doi: 10.1210/endo.141.9.7646
105. Ghisletti S, Meda C, Maggi A, Vegeto E. 17β-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol Cell Biol. (2005) 25:2957–68. doi: 10.1128/MCB.25.8.2957-2968.2005
106. Zhou Y, Eppenberger-Castori S, Marx C, Yau C, Scott GK, Eppenberger U, et al. Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. Int J Biochem Cell Biol. (2005) 37:1130–44. doi: 10.1016/j.biocel.2004.09.006
107. Adamson AD, Friedrichsen S, Semprini S, Harper CV, Mullins JJ, White MRH, et al. Human prolactin gene promoter regulation by estrogen: convergence with tumor necrosis factor-alpha signaling. Endocrinology. (2008) 149:687–94. doi: 10.1210/en.2007-1066
108. Rubio MF, Werbajh S, Cafferata EGA, Quaglino A, Coló GP, Nojek IM, et al. TNF-α enhances estrogen-induced cell proliferation of estrogen-dependent breast tumor cells through a complex containing nuclear factor-kappa B. Oncogene. (2006) 25:1367–77. doi: 10.1038/sj.onc.1209176
109. Frasor J, Weaver A, Pradhan M, Dai Y, Miller LD, Lin CY, et al. Positive cross-talk between estrogen receptor and NF-κB in breast cancer. Cancer Res. (2009) 69:8918–25. doi: 10.1158/0008-5472.CAN-09-2608
110. Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M, et al. Structural and molecular mechanisms of cytokine-mediated endocrine resistance in human breast cancer cells. Mol Cell. (2017) 65:1122–35.e5. doi: 10.1016/j.molcel.2017.02.008
111. Rivas MA, Carnevale RP, Proietti CJ, Rosemblit C, Beguelin W, Salatino M, et al. TNF alpha acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-kappa B-dependent pathways. Exp Cell Res. (2008) 314:509–29. doi: 10.1016/j.yexcr.2007.10.005
112. Jupp OJ, McFarlane SM, Anderson HM, Littlejohn AF, Mohamed AA, MacKay RH, et al. Type II tumour necrosis factor-alpha receptor (TNFR2) activates c-Jun N-terminal kinase (JNK) but not mitogen-activated protein kinase (MAPK) or p38 MAPK pathways. Biochem J. (2001) 359:525–35. doi: 10.1042/0264-6021:3590525
113. Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging Cell. (2002) 1:112–6. doi: 10.1046/j.1474-9728.2002.00014.x
114. Weitzenfeld P, Meron N, Leibovich-Rivkin T, Meshel T, Ben-Baruch A. Progression of luminal breast tumors is promoted by ménage à trois between the inflammatory cytokine TNF α and the hormonal and growth-supporting arms of the tumor microenvironment. Mediators Inflamm. (2013) 2013:720536. doi: 10.1155/2013/720536
115. Weitzenfeld P, Kossover O, Körner C, Meshel T, Wiemann S, Seliktar D, et al. Chemokine axes in breast cancer: factors of the tumor microenvironment reshape the CCR7-driven metastatic spread of luminal-A breast tumors. J Leukoc Biol. (2016) 99:1009–25. doi: 10.1189/jlb.3MA0815-373R
116. Wang Y, Zhou BP. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chin J Cancer. (2011) 30:603–11. doi: 10.5732/cjc.011.10226
117. Kotiyal S, Bhattacharya S. Breast cancer stem cells, EMT and therapeutic targets. Biochem Biophys Res Commun. (2014) 453:112–6. doi: 10.1016/j.bbrc.2014.09.069
118. Storci G, Sansone P, Mari S, D'Uva G, Tavolari S, Guarnieri T, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol. (2010) 225:682–91. doi: 10.1002/jcp.22264
119. Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, et al. Inflammatory mediators in breast cancer: coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer. (2011) 11:130. doi: 10.1186/1471-2407-11-130
120. Wolczyk D, Zaremba-Czogalla M, Hryniewicz-Jankowska A, Tabola R, Grabowski K, Sikorski AF, et al. TNF-α promotes breast cancer cell migration and enhances the concentration of membrane-associated proteases in lipid rafts. Cell Oncol. (2016) 39:353–63. doi: 10.1007/s13402-016-0280-x
121. Geng Y, Chandrasekaran S, Hsu J-W, Gidwani M, Hughes AD, King MR. Phenotypic switch in blood: effects of pro-inflammatory cytokines on breast cancer cell aggregation and adhesion. PLoS ONE. (2013) 8:e54959. doi: 10.1371/journal.pone.0054959
122. Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer registry. Cancer. (2007) 109:1721–8. doi: 10.1002/cncr.22618
123. Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. (2009) 9(Suppl. 2):S73–81. doi: 10.3816/CBC.2009.s.008
124. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. (2000) 406:747–52. doi: 10.1038/35021093
125. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. (2004) 10:5367–74. doi: 10.1158/1078-0432.CCR-04-0220
126. Qiao Y, He H, Jonsson P, Sinha I, Zhao C, Dahlman-Wright K. AP-1 is a key regulator of proinflammatory cytokine TNFα-mediated triple-negative breast cancer progression. J Biol Chem. (2016) 291:5068–79. doi: 10.1074/jbc.M115.702571
127. Cai X, Cao C, Li J, Chen F, Zhang S, Liu B, et al. Inflammatory factor TNF-α promotes the growth of breast cancer via the positive feedback loop of TNFR1/NF-κB (and/or p38)/p-STAT3/HBXIP/TNFR1. Oncotarget. (2017) 8:58338–52. doi: 10.18632/oncotarget.16873
128. Pileczki V, Braicu C, Gherman CD, Berindan-Neagoe I. TNF-α gene knockout in triple negative breast cancer cell line induces apoptosis. Int J Mol Sci. (2012) 14:411–20. doi: 10.3390/ijms14010411
129. Yu M, Zhou X, Niu L, Lin G, Huang J, Zhou W, et al. Targeting transmembrane TNF-α suppresses breast cancer growth. Cancer Res. (2013) 73:4061–74. doi: 10.1158/0008-5472.CAN-12-3946
130. Stanculescu A, Bembinster LA, Borgen K, Bergamaschi A, Wiley E, Frasor J. Estrogen promotes breast cancer cell survival in an inhibitor of apoptosis (IAP)-dependent manner. Horm Cancer. (2010) 1:127–35. doi: 10.1007/s12672-010-0018-6
131. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. (1999) 98:295–303. doi: 10.1016/s0092-8674(00)81959-5
132. Mukhopadhyay P, Lakshmanan I, Ponnusamy MP, Chakraborty S, Jain M, Pai P, et al. MUC4 overexpression augments cell migration and metastasis through EGFR family proteins in triple negative breast cancer cells. PLoS ONE. (2013) 8:e54455. doi: 10.1371/journal.pone.0054455
133. Kim S, Choi JH, Bin KJ, Nam SJ, Yang J-H, Kim J-H, et al. Berberine suppresses TNF-alpha-induced MMP-9 and cell invasion through inhibition of AP-1 activity in MDA-MB-231 human breast cancer cells. Molecules. (2008) 13:2975–85. doi: 10.3390/molecules13122975
134. Zaremba-Czogalla M, Hryniewicz-Jankowska A, Tabola R, Nienartowicz M, Stach K, Wierzbicki J, et al. A novel regulatory function of CDKN1A/p21 in TNFα-induced matrix metalloproteinase 9-dependent migration and invasion of triple-negative breast cancer cells. Cell Signal. (2018) 47:27–36. doi: 10.1016/j.cellsig.2018.03.010
135. Lee C-W, Lin C-C, Lin W-N, Liang K-C, Luo S-F, Wu C-B, et al. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-kappaB/p300 binding in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L799–812. doi: 10.1152/ajplung.00311.2006
136. Cohen M, Meisser A, Haenggeli L, Bischof P. Involvement of MAPK pathway in TNF-alpha-induced MMP-9 expression in human trophoblastic cells. Mol Hum Reprod. (2006) 12:225–32. doi: 10.1093/molehr/gal023
137. Qiao Y, Shiue CN, Zhu J, Zhuang T, Jonsson P, Wright APH, et al. AP-1-mediated chromatin looping regulates ZEB2 transcription: new insights into TNFα-induced epithelial-mesenchymal transition in triple-negative breast cancer. Oncotarget. (2015) 6:7804–14. doi: 10.18632/oncotarget.3158
138. Umemura S, Yoshida S, Ohta Y, Naito K, Osamura RY, Tokuda Y. Increased phosphorylation of Akt in triple-negative breast cancers. Cancer Sci. (2007) 98:1889–92. doi: 10.1111/j.1349-7006.2007.00622.x
139. Van Molle W, Wielockx B, Mahieu T, Takada M, Taniguchi T, Sekikawa K, et al. HSP70 protects against TNF-induced lethal inflammatory shock. Immunity. (2002) 16:685–95. doi: 10.1016/s1074-7613(02)00310-2
140. Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, et al. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. (2000) 6:2417–23. doi: 10.1016/S1097-2765(03)00048-0
141. Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial-mesenchymal transition: podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell. (2006) 9:261–72. doi: 10.1016/j.ccr.2006.03.010
142. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. (1987) 235:177–82.
143. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. (2014) 25:282–303. doi: 10.1016/j.ccr.2014.02.025
144. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. (2001) 2:127–37. doi: 10.1038/35052073
145. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
146. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. (2005) 5:341–54. doi: 10.1038/nrc1609
147. Garrett TPJ, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. (2003) 11:495–505. doi: 10.1016/s1097-2765(03)00048-0
148. Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology. (2001) 61(Suppl. 2):1–13. doi: 10.1159/000055396
149. Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS Jr. Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene. (2000) 19:1123–31. doi: 10.1038/sj.onc.1203412
150. Rivas MA, Tkach M, Beguelin W, Proietti CJ, Rosemblit C, Charreau EH, et al. Transactivation of ErbB-2 induced by tumor necrosis factor α promotes NF-κB activation and breast cancer cell proliferation. Breast Cancer Res Treat. (2010) 122:111–24. doi: 10.1007/s10549-009-0546-3
151. Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, et al. TNFα-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clin Cancer Res. (2017) 23:636–48. doi: 10.1158/1078-0432.CCR-16-0970
152. Li Y, Yu W-H, Ren J, Chen W, Huang L, Kharbanda S, et al. Heregulin targets γ-catenin to the nucleolus by a mechanism dependent on the DF3/MUC1 oncoprotein. Mol Cancer Res. (2003) 1:765–75.
153. Leng Y, Cao C, Ren J, Huang L, Chen D, Ito M, et al. Nuclear import of the MUC1-C oncoprotein is mediated by nucleoporin Nup62. J Biol Chem. (2007) 282:19321–30. doi: 10.1074/jbc.M703222200
154. Chaturvedi P, Singh AP, Chakraborty S, Chauhan SC, Bafna S, Meza JL, et al. MUC4 Mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. (2008) 68:2065–70. doi: 10.1158/0008-5472.CAN-07-6041
155. Funes M, Miller JK, Lai C, Carraway KL III, Sweeney C. The mucin Muc4 potentiates neuregulin signaling by increasing the cell-surface populations of ErbB2 and ErbB3*. JBC. (2006) 281:19310–19. doi: 10.1074/jbc.M603225200
156. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. (2003) 100:8933–8. doi: 10.1073/pnas.1537685100
157. Workman HC, Sweeney C, Carraway KL. The membrane mucin Muc4 inhibits apoptosis induced by multiple insults via ErbB2-dependent and ErbB2-independent mechanisms. Cancer Res. (2009) 69:2845–52. doi: 10.1158/0008-5472.CAN-08-2089
158. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. (2009) 9:361–71. doi: 10.1038/nrc2628
159. Wu Y, Zhou BP. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer. (2010) 102:639–44. doi: 10.1038/sj.bjc.6605530
160. Chen X, Bäumel M, Männel DN, Howard OMZ, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol. (2007) 179:154–61. doi: 10.4049/jimmunol.179.1.154
161. Schioppa T, Moore R, Thompson RG, Rosser EC, Kulbe H, Nedospasov S, et al. B regulatory cells and the tumor-promoting actions of TNF-α during squamous carcinogenesis. Proc Natl Acad Sci USA. (2011) 108:10662–67. doi: 10.1073/pnas.1100994108
162. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. (2012) 122:4094–104. doi: 10.1172/JCI64115
163. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. (2007) 67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174
164. Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. (2006) 176:284–90. doi: 10.4049/jimmunol.176.1.284
165. Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. (2007) 67:10019–26. doi: 10.1158/0008-5472.CAN-07-2354
166. Ba H, Li B, Li X, Li C, Feng A, Zhu Y, et al. Transmembrane tumor necrosis factor-α promotes the recruitment of MDSCs to tumor tissue by upregulating CXCR4 expression via TNFR2. Int Immunopharmacol. (2017) 44:143–52. doi: 10.1016/j.intimp.2016.12.028
167. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. (2011) 71:7463–70. doi: 10.1158/0008-5472.CAN-11-2449
168. Ponte AL, Marais E, Gallay N, Langonné A, Delorme B, Hérault O, et al. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. (2007) 25:1737–45. doi: 10.1634/stemcells.2007-0054
169. Sedgwick JD, Riminton DS, Cyster JG, Körner H. Tumor necrosis factor: a master-regulator of leukocyte movement. Immunol Today. (2000) 21:110–13. doi: 10.1016/s0167-5699(99)01573-x
170. Kulbe H, Hagemann T, Szlosarek PW, Balkwill FR, Wilson JL. The inflammatory cytokine tumor necrosis factor-alpha regulates chemokine receptor expression on ovarian cancer cells. Cancer Res. (2005) 65:10355–62. doi: 10.1158/0008-5472.CAN-05-0957
171. Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, et al. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. (2007) 67:585–92. doi: 10.1158/0008-5472.CAN-06-2941
172. Schröder M, Krötschel M, Conrad L, Naumann SK, Bachran C, Rolfe A, et al. Genetic screen in myeloid cells identifies TNF-α autocrine secretion as a factor increasing MDSC suppressive activity via Nos2 up-regulation. Sci Rep. (2018) 8:13399. doi: 10.1038/s41598-018-31674-1
173. Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane TNF- promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J Immunol. (2013) 192:1320–31. doi: 10.4049/jimmunol.1203195
174. Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol. (2018) 3:eaar3451. doi: 10.1126/sciimmunol.aar3451
175. Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell. (2019) 178:585–99.e15. doi: 10.1016/j.cell.2019.06.014
176. Silke J. The regulation of TNF signalling: what a tangled web we weave. Curr Opin Immunol. (2011) 23:620–6. doi: 10.1016/j.coi.2011.08.002
177. Lork M, Verhelst K, Beyaert R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: so similar, yet so different. Cell Death Differ. (2017) 24:1172–83. doi: 10.1038/cdd.2017.46
178. Tsuchiya Y, Nakabayashi O, Nakano H. FLIP the switch: regulation of apoptosis and necroptosis by cFLIP. Int J Mol Sci. (2015) 16:30321–41. doi: 10.3390/ijms161226232
179. Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. (2008) 118:560–70. doi: 10.1172/JCI32453
180. Liu Z, Swindall AF, Kesterson RA, Schoeb TR, Bullard DC, Bellis SL. ST6Gal-I regulates macrophage apoptosis via α2-6 sialylation of the TNFR1 death receptor. J Biol Chem. (2011) 286:39654–62. doi: 10.1074/jbc.M111.276063
181. Holdbrooks AT, Britain CM, Bellis SL. ST6Gal-I sialyltransferase promotes tumor necrosis factor (TNF)-mediated cancer cell survival via sialylation of the TNF receptor 1 (TNFR1) death receptor. J Biol Chem. (2018) 293:1610–22. doi: 10.1074/jbc.M117.801480
182. Mahmud SA, Manlove LS, Schmitz HM, Xing Y, Wang Y, Owen DL, et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat Immunol. (2014) 15:473–81. doi: 10.1038/ni.2849
183. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci Rep. (2013) 3:3153. doi: 10.1038/srep03153
184. Chopra M, Riedel SS, Biehl M, Krieger S, von Krosigk V, Bäuerlein CA, et al. Tumor necrosis factor receptor 2-dependent homeostasis of regulatory T cells as a player in TNF-induced experimental metastasis. Carcinogenesis. (2013) 34:1296–1303. doi: 10.1093/carcin/bgt038
185. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J Exp Med. (2016) 213:1881–900. doi: 10.1084/jem.20151563
186. Chen X, Subleski JJ, Kopf H, Howard OMZ, Männel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol. (2008) 180:6467–71. doi: 10.4049/jimmunol.180.10.6467
187. Sasi SP, Bae S, Song J, Perepletchikov A, Schneider D, Carrozza J, et al. Therapeutic non-toxic doses of TNF induce significant regression in TNFR2-p75 knockdown Lewis lung carcinoma tumor implants. PLoS ONE. (2014) 9:e92373. doi: 10.1371/journal.pone.0092373
188. Nie Y, He J, Shirota H, Trivett AL, Yang D, Klinman DM, et al. Blockade of TNFR2 signaling enhances the immunotherapeutic effect of CpG ODN in a mouse model of colon cancer. Sci Signal. (2018) 11:eaan0790. doi: 10.1126/scisignal.aan0790
189. Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. (1995) 377:348–51. doi: 10.1038/377348a0
190. Kim EY, Teh S-J, Yang J, Chow MT, Teh H-S. TNFR2-deficient memory CD8 T cells provide superior protection against tumor cell growth. J Immunol. (2009) 183:6051–7. doi: 10.4049/jimmunol.0803482
191. Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun. (2017) 8:2256. doi: 10.1038/s41467-017-02358-7
192. Donia M, Andersen R, Kjeldsen JW, Fagone P, Munir S, Nicoletti F, et al. Aberrant expression of MHC Class II in melanoma attracts inflammatory tumor-specific CD4+ T- Cells, which dampen CD8+ T-cell antitumor reactivity. Cancer Res. (2015) 75:3747–59. doi: 10.1158/0008-5472.CAN-14-2956
193. Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. (2012) 490:412–6. doi: 10.1038/nature11538
194. Lim S-O, Li C-W, Xia W, Cha J-H, Chan L-C, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. (2016) 30:925–39. doi: 10.1016/j.ccell.2016.10.010
195. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci Signal. (2017) 10:eaaf8608. doi: 10.1126/scisignal.aaf8608
196. Vujanovic L, Szymkowski DE, Alber S, Watkins SC, Vujanovic NL, Butterfield LH. Virally infected and matured human dendritic cells activate natural killer cells via cooperative activity of plasma membrane-bound TNF and IL-15. Blood. (2010) 116:575–83. doi: 10.1182/blood-2009-08-240325
197. Vujanovic NL. Role of TNF superfamily ligands in innate immunity. Immunol Res. (2011) 50:159–74. doi: 10.1007/s12026-011-8228-8
198. Van Hauwermeiren F, Vandenbroucke RE, Libert C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. (2011) 22:311–19. doi: 10.1016/j.cytogfr.2011.09.004
199. Steed PM, Tansey MG, Zalevsky J, Zhukovsky EA, Desjarlais JR, Szymkowski DE, et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science. (2003) 301:1895–8. doi: 10.1126/science.1081297
200. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-α blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity. (2013) 38:541–54. doi: 10.1016/j.immuni.2013.02.007
201. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
202. Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, et al. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. (2006) 25:387–408. doi: 10.1007/s10555-006-9004-4
203. Schillaci R, Bruni S, De Martino M, Mercogliano MF, Inurrigarro G, Frahm I, et al. Abstract P6-20-14: neutralizing soluble tumor necrosis factor alpha overcomes trastuzumab-resistant breast cancer immune evasion by downregulating mucin 4, improving NK cell function and decreasing myeloid-derived suppressor cells in tumor microenvironmen. Cancer Res. (2019) 79(Suppl. 4):P6–20–14 LP–P6–20–14. doi: 10.1158/1538-7445.SABCS18-P6-20-14
204. Triulzi T, Forte L, Regondi V, Di Modica M, Ghirelli C, Carcangiu ML, et al. HER2 signaling regulates the tumor immune microenvironment and trastuzumab efficacy. Oncoimmunology. (2018) 8:e1512942. doi: 10.1080/2162402X.2018.1512942
205. Sprowl JA, Reed K, Armstrong SR, Lanner C, Guo B, Kalatskaya I, et al. Alterations in tumor necrosis factor signaling pathways are associated with cytotoxicity and resistance to taxanes: a study in isogenic resistant tumor cells. Breast Cancer Res. (2012) 14:R2. doi: 10.1186/bcr3083
206. Wang Q, Wang Y, Wang X, Mo X, Gu J, Lu Z, et al. Survivin up-regulates the expression of breast cancer resistance protein (BCRP) through attenuating the suppression of p53 on NF-κB expression in MCF-7/5-FU cells. Int J Biochem Cell Biol. (2013) 45:2036–44. doi: 10.1016/j.biocel.2013.06.026
207. Hernández-Vargas H, Rodríguez-Pinilla SM, Julián-Tendero M, Sánchez-Rovira P, Cuevas C, Antón A, et al. Gene expression profiling of breast cancer cells in response to gemcitabine: NF-kappaB pathway activation as a potential mechanism of resistance. Breast Cancer Res Treat. (2007) 102:157–72. doi: 10.1007/s10549-006-9322-9
208. Braunstein S, Formenti SC, Schneider RJ. Acquisition of stable inducible up-regulation of nuclear factor-kappaB by tumor necrosis factor exposure confers increased radiation resistance without increased transformation in breast cancer cells. Mol Cancer Res. (2008) 6:78–88. doi: 10.1158/1541-7786.MCR-07-0339
209. Yu H, Aravindan N, Xu J, Natarajan M. Inter- and intra-cellular mechanism of NF-kB-dependent survival advantage and clonal expansion of radio-resistant cancer cells. Cell Signal. (2017) 31:105–11. doi: 10.1016/j.cellsig.2017.01.011
210. Montagut C, Tusquets I, Ferrer B, Corominas JM, Bellosillo B, Campas C, et al. Activation of nuclear factor-kappa B is linked to resistance to neoadjuvant chemotherapy in breast cancer patients. Endocr Relat Cancer. (2006) 13:607–16. doi: 10.1677/erc.1.01171
211. Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc Natl Acad Sci USA. (1989) 86:10104–107. doi: 10.1073/pnas.86.24.10104
212. Xia W, Bacus S, Husain I, Liu L, Zhao S, Liu Z, et al. Resistance to ErbB2 tyrosine kinase inhibitors in breast cancer is mediated by calcium-dependent activation of RelA. Mol Cancer Ther. (2010) 9:292–9. doi: 10.1158/1535-7163.MCT-09-1041
213. Symonds L, Linden H, Gadi V, Korde L, Rodler E, Gralow J, et al. Combined targeted therapies for first-line treatment of metastatic triple negative breast cancer-A phase II trial of weekly nab-paclitaxel and bevacizumab followed by maintenance targeted therapy with bevacizumab and erlotinib. Clin Br Cancer. (2019) 19:e283–96. doi: 10.1016/j.clbc.2018.12.008
214. Usman MW, Gao J, Zheng T, Rui C, Li T, Bian X, et al. Macrophages confer resistance to PI3K inhibitor GDC-0941 in breast cancer through the activation of NF-κB signaling. Cell Death Dis. (2018) 9:809. doi: 10.1038/s41419-018-0849-6
215. Smith MP, Sanchez-Laorden B, O'Brien K, Brunton H, Ferguson J, Young H, et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNF. Cancer Discov. (2014) 4:1214–29. doi: 10.1158/2159-8290.CD-13-1007
216. Schettini F, De Santo I, Rea CG, De Placido P, Formisano L, Giuliano M, et al. CDK 4/6 Inhibitors as single agent in advanced solid tumors. Front Oncol. (2018) 8:608. doi: 10.3389/fonc.2018.00608
217. Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. (2001) 61:4744–49.
218. Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. (1998) 95:15665–70. doi: 10.1073/pnas.95.26.15665
219. Brooks TA, Minderman H, O'Loughlin KL, Pera P, Ojima I, Baer MR, et al. Taxane-based reversal agents modulate drug resistance mediated by P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Mol Cancer Ther. (2003) 2:1195–205.
220. Pradhan M, Bembinster LA, Baumgarten SC, Frasor J. Proinflammatory cytokines enhance estrogen-dependent expression of the multidrug transporter gene ABCG2 through estrogen receptor and NF{kappa}B cooperativity at adjacent response elements. J Biol Chem. (2010) 285:31100–106. doi: 10.1074/jbc.M110.155309
221. Mosaffa F, Lage H, Afshari JT, Behravan J. Interleukin-1 β and tumor necrosis factor-alpha increase ABCG2 expression in MCF-7 breast carcinoma cell line and its mitoxantrone-resistant derivative, MCF-7/MX. Inflamm Res. (2009) 58:669–76. doi: 10.1007/s00011-009-0034-6
222. Esparza-López J, Medina-Franco H, Escobar-Arriaga E, León-Rodríguez E, Zentella-Dehesa A, Ibarra-Sánchez MJ. Doxorubicin induces atypical NF-κB activation through c-Abl kinase activity in breast cancer cells. J Cancer Res Clin Oncol. (2013) 139:1625–35. doi: 10.1007/s00432-013-1476-3
223. Zhang Z, Lin G, Yan Y, Li X, Hu Y, Wang J, et al. Transmembrane TNF-alpha promotes chemoresistance in breast cancer cells. Oncogene. (2018) 37:3456–70. doi: 10.1038/s41388-018-0221-4
224. Li F, Sethi G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta. (2010) 1805:167–80. doi: 10.1016/j.bbcan.2010.01.002
225. Garg AK, Hortobagyi GN, Aggarwal BB, Sahin AA, Buchholz TA. Nuclear factor-kappa B as a predictor of treatment response in breast cancer. Curr Opin Oncol. (2003) 15:405–11. doi: 10.1097/00001622-200311000-00001
226. Francis PA, Pagani O, Fleming GF, Walley BA, Colleoni M, Láng I, et al. Tailoring adjuvant endocrine therapy for premenopausal breast cancer. N Engl J Med. (2018) 379:122–37. doi: 10.1056/NEJMoa1803164
227. Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. (2007) 67:9549–60. doi: 10.1158/0008-5472.CAN-07-1590
228. Nabholtz JM, Buzdar A, Pollak M, Harwin W, Burton G, Mangalik A, et al. Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: results of a North American multicenter randomized trial. Arimidex Study Group. J Clin Oncol. (2000) 18:3758–67. doi: 10.1200/JCO.2000.18.22.3758
229. Murphy CG, Dickler MN. Endocrine resistance in hormone-responsive breast cancer: mechanisms and therapeutic strategies. Endocr Relat Cancer. (2016) 23:R337–52. doi: 10.1530/ERC-16-0121
230. Castellaro AM, Rodriguez-Baili MC, Di Tada CE, Gil GA. Tumor-Associated macrophages induce endocrine therapy resistance in ER+ breast cancer cells. Cancers. (2019) 11:189. doi: 10.3390/cancers11020189
231. Sas L, Lardon F, Vermeulen PB, Hauspy J, Van Dam P, Pauwels P, et al. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res. (2012) 14:212. doi: 10.1186/bcr3196
232. Riggins RB, Zwart A, Nehra R, Clarke R. The nuclear factor kappa B inhibitor parthenolide restores ICI 182,780 (Faslodex; fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol Cancer Ther. (2005) 4:33–41.
233. Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, et al. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer. (2007) 7:59. doi: 10.1186/1471-2407-7-59
234. deGraffenried LA, Chandrasekar B, Friedrichs WE, Donzis E, Silva J, Hidalgo M, et al. NF-kappa B inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann Oncol Off J Eur Soc Med Oncol. (2004) 15:885–90. doi: 10.1093/annonc/mdh232
235. Trinh XB, Sas L, Van Laere SJ, Prové A, Deleu I, Rasschaert M, et al. A phase II study of the combination of endocrine treatment and bortezomib in patients with endocrine-resistant metastatic breast cancer. Oncol Rep. (2012) 27:657–63. doi: 10.3892/or.2011.1562
236. Singla H, Munshi A, Banipal RPS, Kumar V. Recent updates on the therapeutic potential of HER2 tyrosine kinase inhibitors for the treatment of breast cancer. Curr Cancer Drug Targets. (2018) 18:306–27. doi: 10.2174/1568009617666170623122213
237. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-Positive advanced breast cancer. N Engl J Med. (2006) 355:2733–43. doi: 10.1056/NEJMoa064320
238. Cameron D, Casey M, Press M, Lindquist D, Pienkowski T, Romieu CG, et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res Treat. (2008) 112:533–43. doi: 10.1007/s10549-007-9885-0
239. Ryan Q, Ibrahim A, Cohen MH, Johnson J, Ko C, Sridhara R, et al. FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist. (2008) 13:1114–9. doi: 10.1634/theoncologist.2008-0816
240. Wetterskog D, Shiu K-K, Chong I, Meijer T, Mackay A, Lambros M, et al. Identification of novel determinants of resistance to lapatinib in ERBB2-amplified cancers. Oncogene. (2014) 33:966–76. doi: 10.1038/onc.2013.41
241. Hu W-H, Pendergast JS, Mo X-M, Brambilla R, Bracchi-Ricard V, Li F, et al. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J Biol Chem. (2005) 280:29233–41. doi: 10.1074/jbc.M501670200
242. Bailey ST, Miron PL, Choi YJ, Kochupurakkal B, Maulik G, Rodig SJ, et al. NF-kappaB activation-induced anti-apoptosis renders HER2-positive cells drug resistant and accelerates tumor growth. Mol Cancer Res. (2014) 12:408–20. doi: 10.1158/1541-7786.MCR-13-0206-T
243. Chen Y-J, Yeh M-H, Yu M-C, Wei Y-L, Chen W-S, Chen J-Y, et al. Lapatinib-induced NF-kappaB activation sensitizes triple-negative breast cancer cells to proteasome inhibitors. Breast Cancer Res. (2013) 15:R108. doi: 10.1186/bcr3575
244. Nakai K, Hung M-C, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res. (2016) 6:1609–23.
245. Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature. (2011) 471:523–6. doi: 10.1038/nature09870
246. Blakely CM, Pazarentzos E, Olivas V, Asthana S, Yan JJ, Tan I, et al. NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. (2015) 11:98–110. doi: 10.1016/j.celrep.2015.03.012
247. Guo G, Gong K, Ali S, Ali N, Shallwani S, Hatanpaa KJ, et al. A TNF-JNK-Axl-ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma. Nat Neurosci. (2017) 20:1074–84. doi: 10.1038/nn.4584
248. Olmez I, Zhang Y, Manigat L, Benamar M, Brenneman B, Nakano I, et al. Combined c-Met/Trk inhibition overcomes resistance to CDK4/6 inhibitors in glioblastoma. Cancer Res. (2018) 78:4360–69. doi: 10.1158/0008-5472.CAN-17-3124
249. Nakagawa Y, Sedukhina AS, Okamoto N, Nagasawa S, Suzuki N, Ohta T, et al. NF-κB signaling mediates acquired resistance after PARP inhibition. Oncotarget. (2015) 6:3825–39. doi: 10.18632/oncotarget.2868
250. Cuello M, Ettenberg SA, Clark AS, Keane MM, Posner RH, Nau MM, et al. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. (2001) 61:4892–900.
251. Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. (2004) 6:117–27. doi: 10.1016/j.ccr.2004.06.022
252. Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. (2002) 416:279–80. doi: 10.1038/416279b
253. Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. (2006) 94:259–67. doi: 10.1038/sj.bjc.6602930
254. Cooley S, Burns LJ, Repka T, Miller JS. Natural killer cell cytotoxicity of breast cancer targets is enhanced by two distinct mechanisms of antibody-dependent cellular cytotoxicity against LFA-3 and HER2/neu. Exp Hematol. (2018) 27:1533–41. doi: 10.1016/S0301-472X(99)00089-2
255. Perez EA, Romond EH, Suman VJ, Jeong J-H, Sledge G, Geyer CE, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. (2014) 32:3744–52. doi: 10.1200/JCO.2014.55.5730
256. Gianni L, Eiermann W, Semiglazov V, Lluch A, Tjulandin S, Zambetti M, et al. Neoadjuvant and adjuvant trastuzumab in patients with HER2-positive locally advanced breast cancer (NOAH): follow-up of a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet Oncol. (2014) 15:640–7. doi: 10.1016/S1470-2045(14)70080-4
257. Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, et al. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci USA. (2004) 101:10137–42. doi: 10.1073/pnas.0403621101
258. Singh S, Shi Q, Bailey ST, Palczewski MJ, Pardee AB, Iglehart JD, et al. Nuclear factor-kappaB activation: a molecular therapeutic target for estrogen receptor-negative and epidermal growth factor receptor family receptor-positive human breast cancer. Mol Cancer Ther. (2007) 6:1973–82. doi: 10.1158/1535-7163.MCT-07-0063
259. Cardoso F, Durbecq V, Laes J-F, Badran B, Lagneaux L, Bex F, et al. Bortezomib (PS-341, Velcade) increases the efficacy of trastuzumab (Herceptin) in HER-2-positive breast cancer cells in a synergistic manner. Mol Cancer Ther. (2006) 5:3042–51. doi: 10.1158/1535-7163.MCT-06-0104
260. Kanzaki H, Mukhopadhya NK, Cui X, Ramanujan VK, Murali R. Trastuzumab-resistant luminal B breast cancer cells show basal-like cell growth features through NF-κB-activation. Monoclon Antib Immunodiagn Immunother. (2016) 35:1–11. doi: 10.1089/mab.2015.0056
261. Nagy P, Friedlander E, Tanner M, Kapanen AI, Carraway KL, Isola J, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. (2005) 65:473–82.
262. Mercogliano MF, Inurrigarro G, De Martino M, Venturutti L, Rivas MA, Cordo-Russo R, et al. Invasive micropapillary carcinoma of the breast overexpresses MUC4 and is associated with poor outcome to adjuvant trastuzumab in HER2-positive breast cancer. BMC Cancer. (2017) 17:895. doi: 10.1186/s12885-017-3897-x
263. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. (2015) 372:320–30. doi: 10.1056/NEJMoa1412082
265. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. (2018) 62:29–39. doi: 10.1016/j.intimp.2018.06.001
266. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. (2015) 372:311–19. doi: 10.1056/NEJMoa1411087
267. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. (2018) 379:2108–21. doi: 10.1056/NEJMoa1809615
268. Calcinotto A, Grioni M, Jachetti E, Curnis F, Mondino A, Parmiani G, et al. Targeting TNF-α to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J Immunol. (2012) 188:2687–94. doi: 10.4049/jimmunol.1101877
269. Elia AR, Grioni M, Basso V, Curnis F, Freschi M, Corti A, et al. Targeting tumor vasculature with TNF leads effector T cells to the tumor and enhances therapeutic efficacy of immune checkpoint blockers in combination with adoptive cell therapy. Clin Cancer Res. (2018) 24:2171–81. doi: 10.1158/1078-0432.CCR-17-2210
270. Draghi A, Borch TH, Radic HD, Chamberlain CA, Gokuldass A, Svane IM, et al. Differential effects of corticosteroids and anti-TNF on tumor-specific immune responses: implications for the management of irAEs. Int J Cancer. (2019) 145:1408–13. doi: 10.1002/ijc.32080
271. Lesage C, Longvert C, Prey S, Maanaoui S, Dréno B, Machet L, et al. Incidence and clinical impact of anti-TNFα treatment of severe immune checkpoint inhibitor-induced colitis in advanced melanoma: the mecolit survey. J Immunother. (2019) 42:175–9. doi: 10.1097/CJI.0000000000000268
272. Montfort A, Dufau C, Colacios C, Andrieu-Abadie N, Levade T, Filleron T, et al. Anti-TNF, a magic bullet in cancer immunotherapy? J Immunother cancer. (2019) 7:303. doi: 10.1186/s40425-019-0802-y
273. Scott KA, Moore RJ, Arnott CH, East N, Thompson RG, Scallon BJ, et al. An anti-tumor necrosis factor-alpha antibody inhibits the development of experimental skin tumors. Mol Cancer Ther. (2003) 2:445–51.
274. van Schie KA, Ooijevaar-de Heer P, Dijk L, Kruithof S, Wolbink G, Rispens T. Therapeutic TNF inhibitors can differentially stabilize trimeric TNF by inhibiting monomer exchange. Sci Rep. (2016) 6:32747. doi: 10.1038/srep32747
275. Rubbert-Roth A, Atzeni F, Masala IF, Caporali R, Montecucco C, Sarzi-Puttini P. TNF inhibitors in rheumatoid arthritis and spondyloarthritis: are they the same? Autoimmun Rev. (2018) 17:24–28. doi: 10.1016/j.autrev.2017.11.005
276. Liu J, Dong Z, Zhu Q, He D, Ma Y, Du A, et al. TNF-α promoter polymorphisms predict the response to etanercept more powerfully than that to infliximab/adalimumab in spondyloarthritis. Sci Rep. (2016) 6:32202. doi: 10.1038/srep32202
277. Murdaca G, Gulli R, Spanò F, Lantieri F, Burlando M, Parodi A, et al. TNF-α gene polymorphisms: association with disease susceptibility and response to anti-TNF-α treatment in psoriatic arthritis. J Invest Dermatol. (2014) 134:2503–09. doi: 10.1038/jid.2014.123
278. Liu Y, Fan W, Chen H, Yu MX. Risk of breast cancer and total malignancies in rheumatoid arthritis patients undergoing TNF-α antagonist therapy: a meta-analysis of randomized control trials. Asian Pacific J Cancer Prev. (2014) 15:3403–10. doi: 10.7314/APJCP.2014.15.8.3403
279. Askling J, Fahrbach K, Nordstrom B, Ross S, Schmid CH, Symmons D. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. (2011) 20:119–30. doi: 10.1002/pds.2046
280. Raaschou P, Frisell T, Askling J, Baecklund E, Kastbom A, Forsblad H, et al. TNF inhibitor therapy and risk of breast cancer recurrence in patients with rheumatoid arthritis: a nationwide cohort study. Ann Rheum Dis. (2015) 74:2137–43. doi: 10.1136/annrheumdis-2014-205745
281. Bonovas S, Minozzi S, Lytras T, González-Lorenzo M, Pecoraro V, Colombo S, et al. Risk of malignancies using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin Drug Saf. (2016) 15:35–54. doi: 10.1080/14740338.2016.1238458
282. Chen Y, Sun J, Yang Y, Huang Y, Liu G. Malignancy risk of anti-tumor necrosis factor alpha blockers: an overview of systematic reviews and meta-analyses. Clin Rheumatol. (2016) 35:1–18. doi: 10.1007/s10067-015-3115-7
283. Shelton E, Laharie D, Scott FI, Mamtani R, Lewis JD, Colombel JF, et al. Cancer recurrence following immune-suppressive therapies in patients with immune-mediated diseases: a systematic review and meta-analysis. Gastroenterology. (2016) 151:97–109.e4. doi: 10.1053/j.gastro.2016.03.037
284. Poullenot F, Seksik P, Beaugerie L, Amiot A, Nachury M, Abitbol V, et al. Risk of incident cancer in inflammatory bowel disease patients starting anti-TNF therapy while having recent malignancy. Inflamm Bowel Dis. (2016) 22:1362–69. doi: 10.1097/MIB.0000000000000741
285. Dignass A, Van Assche G, Lindsay JO, Lémann M, Söderholm J, Colombel JF, et al. The second european evidence-based consensus on the diagnosis and management of crohn's disease: current management. J Crohns Colitis. (2010) 4:28–62. doi: 10.1016/j.crohns.2009.12.002
286. Askling J, Fored CM, Baecklund E, Brandt L, Backlin C, Ekbom A, et al. Haematopoietic malignancies in rheumatoid arthritis: lymphoma risk and characteristics after exposure to tumour necrosis factor antagonists. Ann Rheum Dis. (2005) 64:1414–20. doi: 10.1136/ard.2004.033241
287. Madhusudan S, Foster M, Muthuramalingam SR, Braybrooke JP, Wilner S, Kaur K, et al. A phase II study of etanercept (Enbrel), a tumor necrosis factor alpha inhibitor in patients with metastatic breast cancer. Clin Cancer Res. (2004) 10:6528–34. doi: 10.1158/1078-0432.CCR-04-0730
Keywords: TNFα, breast cancer, resistance, mucin 4, targeted-therapy
Citation: Mercogliano MF, Bruni S, Elizalde PV and Schillaci R (2020) Tumor Necrosis Factor α Blockade: An Opportunity to Tackle Breast Cancer. Front. Oncol. 10:584. doi: 10.3389/fonc.2020.00584
Received: 18 January 2020; Accepted: 30 March 2020;
Published: 22 April 2020.
Edited by:
Dirk Geerts, University of Amsterdam, NetherlandsReviewed by:
Giuseppe Murdaca, University of Genoa, ItalyGaia Griguolo, University of Padova, Italy
Copyright © 2020 Mercogliano, Bruni, Elizalde and Schillaci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roxana Schillaci, cnNjaGlsbGFjaUBpYnltZS5jb25pY2V0Lmdvdi5hcg==