
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 09 April 2020
Sec. Pharmacology of Anti-Cancer Drugs
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00484
This article is part of the Research Topic Novel Drugs Targeting the Microenvironment and the Epigenetic Changes in Hematopoietic Malignancies View all 6 articles
 Andra Marcu1,2†
Andra Marcu1,2† Andrei Colita3,4†Letitia Elena Radu1,2Cristina Georgiana Jercan1,2Ana Maria Bica1Minodora Asan1
Andrei Colita3,4†Letitia Elena Radu1,2Cristina Georgiana Jercan1,2Ana Maria Bica1Minodora Asan1 Daniel Coriu1,4
Daniel Coriu1,4 Alina Daniela Tanase1,5Carmen C. Diaconu6Cristina Mambet6Anca Botezatu7
Alina Daniela Tanase1,5Carmen C. Diaconu6Cristina Mambet6Anca Botezatu7 Sergiu Pasca8Patric Teodorescu9,10Gabriela Anton7Petruta Gurban11
Sergiu Pasca8Patric Teodorescu9,10Gabriela Anton7Petruta Gurban11 Anca Colita1,2*
Anca Colita1,2*Background: Juvenile myelomonocytic leukemia (JMML) is a rare myelodysplastic/myeloproliferative neoplasm diagnosed in young children, characterized by somatic or germline mutations that lead to hyperactive RAS signaling. The only curative option is hematopoietic stem cell transplantation (HSCT). Recent data showing that aberrant DNA methylation plays a significant role in pathogenesis and correlates with clinical risk suggest a possible benefit of hypomethylating agents (HMA) in JMML treatment.
Aim: The aim is to report the results of HMA-based therapy with 5-azacytidine (AZA) in three JMML patients treated in a single center, non-participating in EWOG-MDS study.
Methods: The diagnosis and treatment response were evaluated according to international consensus criteria. AZA 75 mg/m2 intravenous (i.v.) was administered once daily on days 1–7 of each 28-day cycle. All patients were monitored for hematologic response, spleen size, and evolution of extramedullary disease. Targeted next generation sequencing (NGS) were performed after the 3rd AZA cycle and before SCT to evaluate the molecular alterations and genetic response.
Results: Three patients diagnosed with JMML were treated with AZA (off-label indication) in Pediatric Department of Fundeni Clinical Institute, Bucharest, Romania between 2017 and 2019. There were two females and one male with median age 11 months, range 2–16 months. The cytogenetic analysis showed normal karyotype in all patients. Molecular analysis confirmed KRAS G13D mutation in two patients and NRAS G12D mutation in one patient. The clinical evaluation showed important splenomegaly and hepatomegaly in all 3 pts. One patient received AZA for early relapse after haploidentical HSCT and the other two patients received upfront AZA, as bridging therapy before HSCT. After HMA therapy, 2/3 patients achieved clinical partial response (cPR), 1/3 had clinical stable disease (cSD) and all had genetic stable disease (gSD) after 3 cycles and were able to receive the planned HSTC. One patient achieved clinical and genetic complete response before HSCT. During 22 cycles of AZA there were only four adverse events but only one determined dose reduction and treatment delay.
Conclusion: Our data show that AZA monotherapy is safe and effective in controlling disease both in upfront and relapsed patients in order to proceed to HSCT.
Juvenile myelomonocytic leukemia (JMML) is a rare myeloproliferative/myelodysplastic neoplasm of early infancy and childhood defined by an excessive production of mature and immature myeloid cells, predominantly of monocytic and granulocytic lineages (1, 2). Therapeutic approaches range from watchful monitoring to allogeneic hematopoietic stem cell transplantation (HSCT) performed in early stages (3). Clinical symptoms result from hematopoietic insufficiency and leukemic infiltration of various organs, such as spleen, liver, skin, lung, and gastrointestinal tract (4). Conventional cytogenetics studies indicate monosomy 7 in up to 25% of JMML patients and other abnormalities in 10% of cases. However, a normal karyotype is diagnosed in about two-thirds of patients (5–7). Strikingly, many JMML children with a normal karyotype exhibit an elevated level of fetal hemoglobin (HbF) (8).
The genetic landscape of JMML is dominated by somatic or germline mutations that lead to hyperactive RAS signaling (9). Five molecular alterations of RAS pathway were described in association with five JMML genetic subtypes, that have distinct clinical and hematological features (1). Three subtypes, covering 55–60% of patients, involve heterozygous somatic activating mutations in PTPN11, NRAS, and KRAS genes (1, 9). The other two subtypes (20–30% of JMML cases) occur in patients with underlying constitutional diseases caused by germline mutations in the RAS pathways (RASopathies): neurofibromatosis type 1 (NF1) and Noonan-like “CBL syndrome,” respectively. Hematological disorders develop due to the acquired loss of heterozygosity of the constitutionally affected NF1 or CBL tumor suppressor genes in the hematopoietic progenitors (4, 10, 11). In rare cases of JMML that lack the above-mentioned mutations, heterozygous somatic RRAS mutations have been reported as disease drivers (12). In addition to RAS pathway mutations, whole-exome sequencing has identified secondary molecular events in ~50% of patients. These alterations, including also mutations in epigenetic regulation genes (EZH2, ASXL1, DNMT3A), might impact clinical outcome and therapeutic decisions (1). Interestingly, JMML displays a unique linear pattern of disease progression. As shown previously, all mutations present at diagnosis are acquired by a single dominant clone that is consistently detected at relapse. Also, Stieglietz et al. have reported that the number of somatic mutations identified at diagnosis influences the survival rate, while the type of RAS pathway mutations that does not represent an independent prognostic factor (13).
Aberrant DNA methylation is another factor related to adverse clinical outcome in JMML patients. European Working Group on MDS in Childhood (EWOG-MDS) published in 2011 a study on 127 JMML patients, evaluating DNA methylation at 14 loci through quantitative mass spectrometry, describing CpG island hypermethylation in the BMP4, CALCA, CDKN2B, or RARB promoter regions as being the best predictor of relapse after HSCT (14). Similarly, different other studies showed that DNA hypermethylation is connected to clinical risk (15–18). Recently, three JMML epigenetic subgroups based on DNA methylation profiling were identified. The low methylation cluster is defined by the presence of Noonan syndrome, CBL mutations and the majority of NRAS mutations, having a favorable outcome. The intermediate methylation cluster consists of patients with numeric aberration of chromosome seven (monosomy) and somatic KRAS mutations. The high methylation subgroup includes patients displaying somatic PTPN11 mutation, low platelet count, elevated HbF, and diagnosis established after the age of two. This subgroup was specifically associated with a higher rate of disease relapse and dismal prognosis (2).
The aberrant DNA methylation patterns described in JMML create a premise for the clinical use of hypomethylating agents, such as 5-azacytosine (azacytidine) and 2′-deoxy-5-azacytidine (decitabine). Once entering the cell, azacytidine is activated through consecutive ATP-dependent phosphorylation steps. Eighty to ninety percentage of azacytidine is incorporated into RNA leading to apoptosis (19) 10–20% of azacytidine is integrated into DNA, permanently inhibiting DNA methyltransferase, suppressing its function, and causing its degradation. Methylation marks are lost during DNA replication, reversing silenced tumor suppressor genes and recovering proliferation, and apoptosis control (20–22). Azacytidine induces specific immune responses through azacytidine-induced immune genes and inhibition of regulatory T cells (23–25).
Two girls and one boy, median age 11 months, range 2–16 months, were diagnosed and treated with azacytidine (off label indication) in the Pediatric Department of Fundeni Clinical Institute, Bucharest, Romania, between 2017 and 2019. On admission, mean hemoglobin (Hb) was 8,8 g/dl (range 7,1–10,2 g/dl), mean white blood cell (WBC) count was 22,576 × 109/L (range 19,75–28,21 × 109/L), mean monocyte count was 7,1 × 109/L (range 6,2–8,4 × 109/L) and mean platelet count was 65 × 109/L (range 46–77 × 109/L). All patients presented with blasts in the bone marrow 4–15% (mean 10%), while only 1 patient presented with 5% myeloid blasts in peripheral blood. The cytogenetic analysis showed normal karyotype, while targeted next-generation sequencing (NGS) revealed KRAS G13D mutation in two patients and NRAS G12D mutation in one patient. All patients had important splenomegaly, with mean spleen size of 7 cm (range 3–10 cm) under the costal margin and hepatomegaly with mean liver size of 5 cm (range 4–8 cm) under costal margin. One patient received azacytidine for early relapse after hematopoietic stem cell transplantation (HSCT) and the other two received upfront azacytidine, as bridging therapy before HSCT (Table 1).
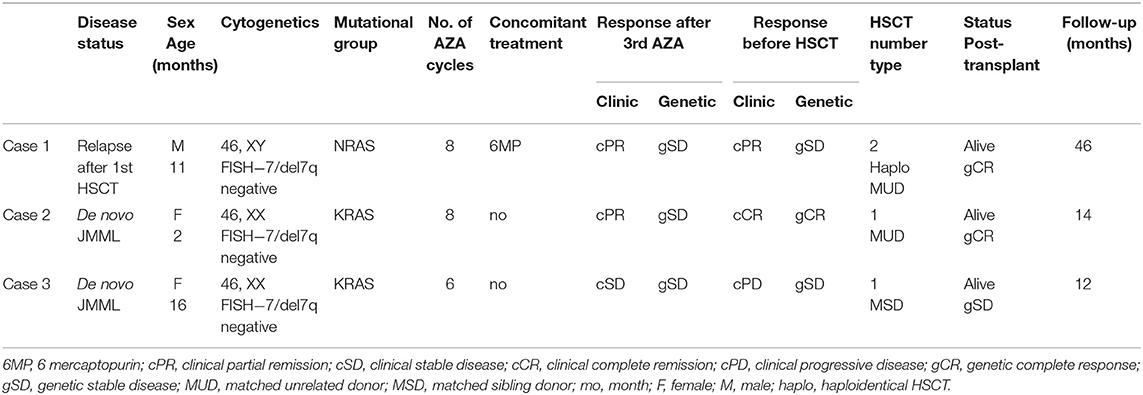
Table 1. Response to azacytidine in children with JMML.
JMML diagnosis and therapy response were evaluated according to international consensus criteria (Table 2) (26). Azacytidine 75 mg/m2 i.v. was administered once daily, on days 1–7 of each 28-day cycle. All patients were monitored for hematologic response, spleen size, and evolution of extramedullary disease. NGS at diagnosis and real-time PCR were performed in order to identify the molecular alterations and assess the genetic response after first three cycles and pre-HSCT. Patients were monitored for adverse events (AEs).
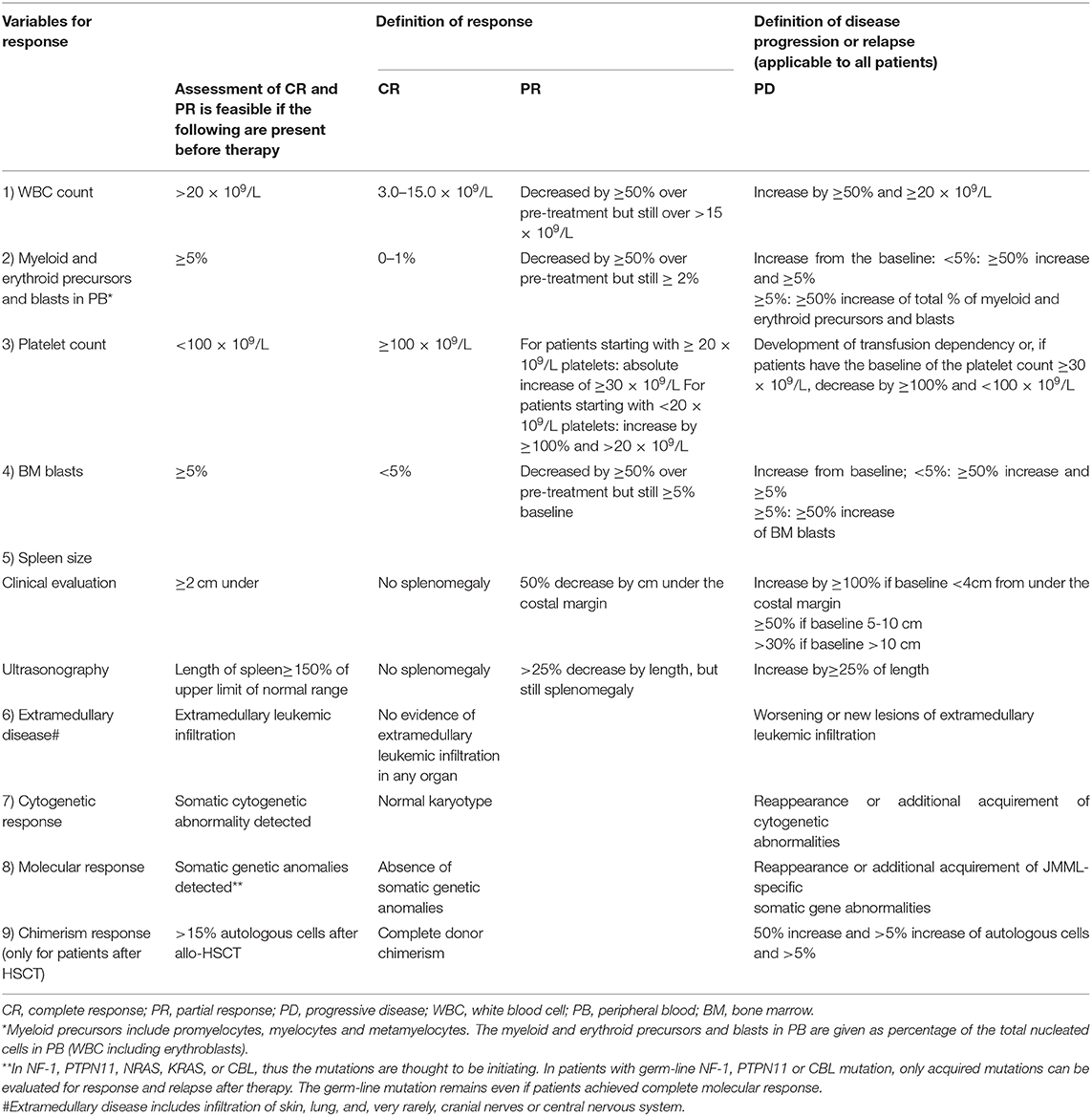
Table 2. Variables for evaluation of response to therapy in JMML (26).
The first case is of an 11-month-old boy diagnosed with NRAS-JMML, treated with 6-mercaptopurine and low-dose cytarabine, while searching for an HLA compatible donor for an allogeneic HSCT. Since the donor was not available at clearance timepoint and considering the emergency for transplantation, he underwent a haploidentical HSCT (haplo HSCT) from his father, with melphalan-fludarabine conditioning regimen, post-transplant cyclophosphamide, and immunosuppressive therapy for graft-vs. -host disease (GvHD) prophylaxis. After engraftment, the chimerism analysis performed on day +19 showed mixed results (53% chimerism from donor). Despite stopping immunosuppression and infusion of donor lymphocytes (DLI), followed by skin and gastrointestinal grade 4 GvHD and severe lung disease, there was a progressive loss of donor cells. Therapy with azacytidine was initiated while a new work-up for second HSCT was started. The infant achieved clinical partial remission (cPR) after three courses of azacytidine and maintained the same status after eight courses. After 1 year, a 9/10 unrelated donor HSCT was performed, with busulfan-cyclophosphamide-melphalan-ATG conditioning. Chimerism analysis on day +24 showed 100% donor cells. He is now at 2 years after 2nd transplant, with very good clinical condition, no chronic GvHD, normal blood count, full donor chimerism, and full immune recovery.
The second case is of a 2-month-old girl diagnosed with KRAS-JMML, who was started in first line therapy with azacytidine. She tolerated azacytidine courses very well, without hematological toxicities. After three courses, she obtained cPR (normal WBC, absent myeloid/erythroid precursors or blasts in PB, normal PLT count, no blasts in BM, more than 50% reduction in spleen and liver size) and complete clinical remission (cCR) after eight courses. Targeted next-generation sequencing (NGS) revealed the presence of KRAS G13D mutation with 21% variant allele frequency (VAF) at diagnosis. Molecular monitoring by real-time PCR indicated a decrease of mutational load after three courses and genetic complete remission (gCR) after eight courses. 10/10 MUD HSCT, with thiotepa-treosulfan-fludarabine-ATG conditioning has been performed.
The third case is of a 16-month-old girl with KRAS-JMML, who was started on upfront azacytidine. The patient's first clinical and hematological abnormalities were noted at 6 months, but the diagnosis was made 10 months later, when she presented with massive hepato-splenomegaly and respiratory manifestations due to leukemic infiltration. Targeted NGS at diagnosis identified KRAS G13D mutation with a VAF of 38%. She received six courses of azacytidine as bridging therapy for matched sibling donor HSCT. Evaluation after first three courses showed clinical stable disease (cSD) (normal WBC, no blasts in PB or in the BM, but the patient still presented massive hepato-splenomegaly, and thrombocytopenia). She developed pneumonia complicated with lung abscess and received antibiotic treatment for 28 days, with 2 weeks delay in azacytidine administration, and loss of therapeutic response. After six courses of azacytidine we noted clinical progressive disease (cPD) based on the development of platelet transfusion dependency and increase of spleen size after initial reduction. The molecular monitoring confirmed gSD. Matched sibling donor HSCT with thiotepa-treosulfan-fludarabine conditioning has been performed.
We report 4 AEs (fever CTCAE grade 4 – 1 patient, diarrhea CTCAE grade 2 - 2 patients, urticaria and rash grade 2 CTCAE - 1 patient) during 22 cycles of azacytidine, with dose reduction for one course and delay for the next course because of pneumonia. The hematologic monitoring during azacytidine cycles showed normal Hb value, normal WBC, and differential count for all patients, as well as normal PLT count for two of them after the first three courses. No hematologic toxicities were reported in our series (Figure 1). All AEs were managed with standard supportive care and without modifications or delay in azacytidine treatment.
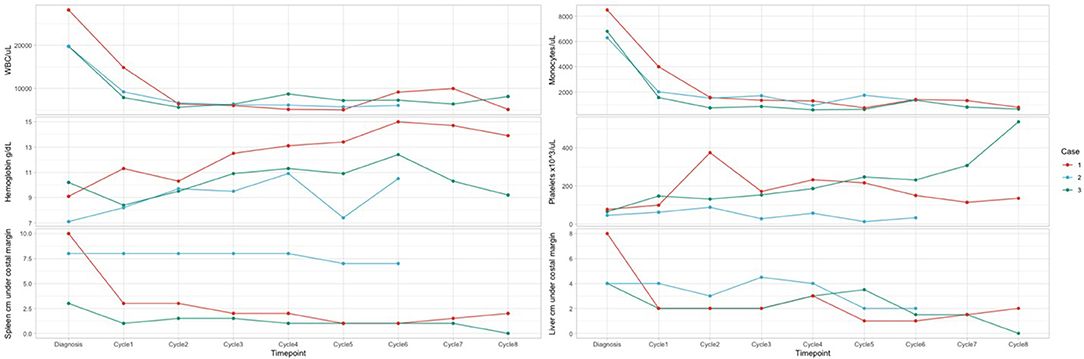
Figure 1. Hematology parameters in dynamics for the patients. Case 2: At diagnosis: KRAS G13D mutation with 21% variant allele frequency (VAF). Molecular monitoring by NGS after 3 courses of therapy indicated a decrease of mutational load to 12%. Molecular monitoring by NGS after 8 courses of therapy indicated complete molecular remission. After HSCT: complete molecular remission. Case 3: At diagnosis: KRAS G13D mutation with a VAF of 38%. Molecular monitoring by NGS after 3 courses of therapy: mutational load 37.5%. Molecular monitoring by NGS at the end of therapy: VAF 35.2%. After HSCT: VAF 2.5%.
Treatment with HMA aims for clinical and hematological response, transfusion independency, and prolonged survival after HSCT, azacytidine being one of the most used agents. Acknowledging its tolerable toxicity and cytoreductive activity, azacytidine becomes a suitable choice for bridging treatment before HSCT, as well as strategy for second HSCT or palliation (27). JMML is challenging and difficult to treat, the only curative option being HSCT. In its absence, the median survival time from diagnosis is <1year (28).
The published data on HMA therapy in relapsed patients after transplantation is limited to three cases. Cseh et al. (27) reported that azacytidine is correlated with partial response during three cycles in one patient, but all three patients eventually progressed and died. Still, we report a NRAS-JMML patient who relapsed after haploidentical HSCT and didn't respond to DLI, despite severe, grade 4 GvHD. He received eight cycles of azacytidine with clinical partial response. After 2nd unrelated HSCT, the patient obtained complete remission, with full donor chimerism. He is alive, fully recovered at 2 years after the 2nd transplant and, in our knowledge, is the first patient showing a favorable response to azacytidine in relapse after HSCT.
Furlan et al. (29) reported a JMML patient with monosomy 7 and KRAS mutation who received upfront azacytidine. Good clinical response was documented, with regression of splenomegaly, and monocyte count after the first course of treatment and disappearance of molecular alterations after cycle five (for monosomy 7) and seven (for KRAS mutation). The eight courses of azacytidine were followed by allogeneic HSCT with complete remission and disease-free survival at 5 years follow-up. Consequently, further trials were open to evaluate remission response at three cycles of therapy and to establish the remission persistence until transplantation. Cseh et al. (27) mentioned three complete clinical, cytogenetic and/or molecular remissions out of nine patients with JMML who received azacytidine before HSCT. Two of the patients had somatic PTPN11 mutation and one had KRAS mutation, thus showing that certain patients respond to this treatment. Although azacytidine may induce complete clinical, cytogenetic and/or molecular remission before allogeneic HSCT, complete remission has not been maintained without transplant.
Interim analysis of the prospective AZA-JMML-001 study evaluating upfront azacytidine in JMML (30) reported 18 patients with JMML (13 PTPN11-, 3 NRAS-, 1 KRAS-, 1 NF1-mutated), classified in DNA methylation classes (MC): high - 11, intermediate (int) – 5 or low for two patients. 11 patients (61%) were in cPR after three cycles of azacytidine, while seven had PD at same treatment stage or prior. All seven patients from the int/low MC and 4/11 from high MC achieved cPR. Seventeen patients received HSCT at a median of 58 days (37–518 days) from last azacytidine dose. Fourteen patients were leukemia-free at a median follow-up of 15.7 months (0.1–31.7 months) after HSCT. Two patients from the high MC relapsed after allograft. 16/18 patients were alive at a median follow-up of 19.8 months (2.6–37.3 months) from diagnosis. One patient discontinued HSCT prior to cycle 3 azacytidine and died from PD. One non-responder patient died from transplant-related causes.
In our case-series we rport cPR at cycle 3 azacytidine in all patients, with gSD. After eight and, respectively, six courses of HMA treatment, we report cCR for one patient, cPR for one patient and cPD for one patient, while genetic response was complete for only one patient before transplantation. Four AEs were reported during 22 cycles of azacytidine, but only one determined dose reduction and treatment delay. Regarding the failure of engraftment for the first transplant for the first patient, it should be mentioned that it was not a failure to engraft, but engraftment with mixed progressive chimerism, followed by complete receptor hematopoiesis, probably in the context of important splenomegaly.
The heterogeneity of disease evolution in different patients cannot be well-explained yet, though the importance of methylation groups and secondary mutations has already been established.
In accordance with international data, our patient series shows that azacytidine monotherapy is well-tolerated in patients with de novo JMML, as well as in patients with relapse after previous treatments, even transplantation. Although the long-term advantage of azacytidine before transplant remains to be fully assessed, responses show it is effective in JMML and provides clinical benefit without severe adverse events.
The study was reviewed and approved by the Ethics Committee of the Fundeni Clinical Institute. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
All authors have read and approved the manuscript and contributed to data gathering. AM, AndC, and AncC wrote the manuscript.
We gratefully acknowledge the funding from the project Competitiveness Operational Programme (COP) A1.1.4. ID: P_37_798 MyeloAL-EDiaProT, Contract 149/26.10.2016 (SMIS: 106774), MyeloAL Project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer A-AZ declared a shared affiliation, with no collaboration, with several of the authors, PT and SP, to the handling editor at the time of review.
1. Niemayer CM. JMML genomics and decisions. Hematol Am Soc Hematol Educ Program. (2018) 2018:307–12. doi: 10.1182/asheducation-2018.1.307
2. Lipka DB, Witte T, Toth R, Yang J, Wiesenfarth M, N?llke P, et al. RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat. Commun. (2017) 8:2126. doi: 10.1038/s41467–017-02177-w
3. Niemeyer CM, Flotho C. Juvenile myelomonocytic leukemia: who's the drive at the wheel? Blood. (2019) 133:1060–70. doi: 10.1182/blood-2018–11-844688
4. Krombholz CF, Aumann K, Kollek M, Bertele D, Fluhr S, Kunze M, et al. Long-term serial xenotransplantation of juvenile myelomonocytic leukemia recapitulates human disease in Rag2-/-γc-/- mice. Haematologica. (2016) 101:597–606. doi: 10.3324/haematol.2015.138545
5. Niemeyer CM, Kratz C. Juvenile myelomonocytic leukemia. Curr Oncol Rep. (2003) 5:510–5. doi: 10.1007/s11912–003-0013-y
6. Loh ML. Childhood myelodysplastic syndrome: focus on the approach to diagnosis and treatment of juvenile myelomonocytic leukemia. Hematol Am Soc Hematol Educ Program. (2010) 2010:357–62. doi: 10.1182/asheducation-2010.1.357
7. Niemeyer CM, Kratz CP. Paediatric myelodysplastic syndrome and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol. (2008) 140:610–24. doi: 10.1111/j.1365–2141.2007.06958.x
8. Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. (2015) 125:1083–90. doi: 10.1182/blood-2014–08-550483
9. Locatelli F, Algeri M, Merli P, Strocchio L. Novel approaches to diagnosis and treatment of Juvenile Myelomonocytic Leukemia. Expert Rev Hematol. (2018) 11:129–43. doi: 10.1080/17474086.2018.1421937
10. Steinemann D, Arning L, Praulich I, Stuhrmann M, Hasle H, Stary J, ey al. Mitotic recombination and compound-heterozygous mutations are predominant NF1-inactivating mechanisms in children with juvenile myelomonocytic leukemia and neurofibromatosis type 1. Haematologica. (2010) 95:320–3. doi: 10.3324/haematol.2009.010355
11. Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. (2009) 114:1859–63. doi: 10.1182/blood-2009–01-198416
12. Flex E, Jaiswal M, Pantaleoni F, Martinelli S, Strullu M, Fansa EK, et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet. (2014) 23:4315–27. doi: 10.1093/hmg/ddu148
13. Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang YD, Mazor T, et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet. (2015) 47:1326–33. doi: 10.1038/ng.3400
14. Olk-Batz C, Poetsch AR, N?llke P, Claus R, Zucknick M, Sandrock I, et al. European Working Group of Myelodysplastic Syndromes in Childhood (EWOG-MDS). aberrant DNA methylation characterizes juvenile myelomonocytic leukemia poor outcome. Blood. (2011) 117:4871–80. doi: 10.1182/blood-2010–08-298968
15. Poetsch AR, Lipka DB, Witte T, Claus R, N?llke P, Zucknick M, et al. RASA4 undergoes DNA hypermethylation in resistant juvenile myelomonocytic leukemia. Epigenetics. (2014) 9:1252–60. doi: 10.4161/epi.29941
16. Sakaguchi H, Muramatsu H, Okuno Y, Makishima H, Xu Y, Furukuwa-Hibi Y, et al. Aberrant DNA methylation is associated with a poor outcome in juvenile myelomonocytic leukemia. PLoS ONE. (2015) 10:e0145394. doi: 10.1371/journal.pone.0145394
17. Wilhelm T, Lipka DB, Witte T, Wierzbinska JA, Fluhr S, Helf M, et al. Epigenetic silencing of AKAP12 in juvenile myelomonocytic leukemia. Epigenetics. (2016) 11:110–9. doi: 10.1080/15592294.2016.1145327
18. Fluhr S, Boerries M, Busch H, Symeonidi A, Witte T, Lipka DB, et al. CREBBP is a target of epigenetic, but not genetic, modification in juvenile myelomonocytic leukemia. Clin Epigenetics. (2016) 8:50. doi: 10.1186/s13148–016-0216–3
19. Diesch J, Zwick A, Garz AK, Palau A, Buschbeck M, G?tze KS. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin Epigenetics. (2016) 8:71. doi: 10.1186/s13148–016-0237-y
20. Mund C, Brueckner B, Lyko F. Reactivation of epigenetically silenced genes by DNA methyltransferase inhibitors: basic concepts and clinical applications. Epigenetics. (2006) 1:7–13. doi: 10.4161/epi.1.1.2375
21. Steller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. (2007) 8:286–98. doi: 10.1038/nrg2005
22. Bender CM, Zingg JM, Jones PA. DNA methylation as a target for drug design. Pharm Res. (1998) 15:175–87. doi: 10.1023/A:1011946030404
23. Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. (2014) 5:587–98. doi: 10.18632/oncotarget.1782
24. Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of nonsmall cell lung cancer with azacytidine. Oncotarget. (2013) 4:2067–79. doi: 10.18632/oncotarget.1542
25. Costantini B, Kordasti SY, Kulasekararaj AG, Jiang J, Seidl T, Abellan PP, et al. 5-azacytidine specifically depletes regulatory t cells (Tregs) in myelodysplastic syndrome (MDS) patients. Blood. (2011) 118:787. doi: 10.1182/blood.V118.21.787.787
26. Niemeyer CM, Loh ML, Cseh A, Cooper T, Dvorak CC, Chan R, et al. Criteria for evaluating response and outcome in clinical trials for children with juvenile myelomonocytic leukemia. Haematologica. (2015) 100:17–22. doi: 10.3324/haematol.2014.109892
27. Cseh A, Niemeyer CM, Yoshimi A, Dworzak M, Hasle H, van den Heuvel-Eibrink MM, et al. Bridging to transplant with azacytidine in juvenile myelomonocytic leukemia: a retrospective analysis of the EWOG-MDS study group. Blood. (2015) 125:2311–3. doi: 10.1182/blood-2015–01-619734
28. Dvorak CC, Loh ML. Juvenile myelomonocytic leukemia: molecular pathogenesis informs current approaches to therapy and hematopoietic cell transplantation. Front Pediatrics. (2014) 2:25. doi: 10.3389/fped.2014.00025
29. Furlan I, Batz C, Flotho C, Mohr B, Lübbert M, Suttorp M, Niemeyer CM. Intriguing response to azacytidine in a patient with juvenile myelomonocytic leukemia and monosomy 7. Blood. (2009) 113:2867–8. doi: 10.1182/blood-2008–12-195693
Keywords: juvenile myelomonocytic leukemia, mutation, epigenetics, methylation, azacytidine, hematopoietic stem cell transplantation
Citation: Marcu A, Colita A, Radu LE, Jercan CG, Bica AM, Asan M, Coriu D, Tanase AD, Diaconu CC, Mambet C, Botezatu A, Pasca S, Teodorescu P, Anton G, Gurban P and Colita A (2020) Single-Center Experience With Epigenetic Treatment for Juvenile Myelomonocytic Leukemia. Front. Oncol. 10:484. doi: 10.3389/fonc.2020.00484
Received: 12 February 2020; Accepted: 17 March 2020;
Published: 09 April 2020.
Edited by:
Liren Qian, Sixth Medical Center of PLA General Hospital, ChinaReviewed by:
Alina-Andreea Zimta, Iuliu Haţieganu University of Medicine and Pharmacy, RomaniaCopyright © 2020 Marcu, Colita, Radu, Jercan, Bica, Asan, Coriu, Tanase, Diaconu, Mambet, Botezatu, Pasca, Teodorescu, Anton, Gurban and Colita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anca Colita, YW5jYWNvbGl0YUB5YWhvby5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.