 Christophe Blanquart
Christophe Blanquart Marie-Claude Jaurand
Marie-Claude Jaurand Didier Jean
Didier Jean
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 25 March 2020
Sec. Thoracic Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00388
This article is part of the Research Topic Emerging Therapies for Malignant Mesothelioma View all 11 articles
Malignant mesothelioma (MM), especially its more frequent form, malignant pleural mesothelioma (MPM), is a devastating thoracic cancer with limited therapeutic options. Recently, clinical trials that used immunotherapy strategies have yielded promising results, but the benefits are restricted to a limited number of patients. To develop new therapeutic strategies and define predictors of treatment response to existing therapy, better knowledge of the cellular and molecular mechanisms of MM tumors and sound preclinical models are needed. This review aims to provide an overview of our present knowledge and issues on both subjects. MM shows a complex pattern of molecular changes, including genetic, chromosomic, and epigenetic alterations. MM is also a heterogeneous cancer. The recently described molecular classifications for MPM could better consider inter-tumor heterogeneity, while histo-molecular gradients are an interesting way to consider both intra- and inter-tumor heterogeneities. Classical preclinical models are based on use of MM cell lines in culture or implanted in rodents, i.e., xenografts in immunosuppressed mice or isografts in syngeneic rodents to assess the anti-tumor immune response. Recent developments are tumoroids, patient-derived xenografts (PDX), xenografts in humanized mice, and genetically modified mice (GEM) that carry mutations identified in human MM tumor cells. Multicellular tumor spheroids are an interesting in vitro model to reduce animal experimentation; they are more accessible than tumoroids. They could be relevant, especially if they are co-cultured with stromal and immune cells to partially reproduce the human microenvironment. Even if preclinical models have allowed for major advances, they show several limitations: (i) the anatomical and biological tumor microenvironments are incompletely reproduced; (ii) the intra-tumor heterogeneity and immunological contexts are not fully reconstructed; and (iii) the inter-tumor heterogeneity is insufficiently considered. Given that these limitations vary according to the models, preclinical models must be carefully selected depending on the objectives of the experiments. New approaches, such as organ-on-a-chip technologies or in silico biological systems, should be explored in MM research. More pertinent cell models, based on our knowledge on mesothelial carcinogenesis and considering MM heterogeneity, need to be developed. These endeavors are mandatory to implement efficient precision medicine for MM.
The therapeutic options for malignant mesothelioma (MM) are limited, especially for the most common form of mesothelioma, malignant pleural mesothelioma (MPM). Current MPM chemotherapy is based on intravenous injections of pemetrexed (PMTX) and cisplatin or carboplatin. Recently, this basic treatment has been improved by the addition of vascular endothelial growth factor (VEGF) antibodies (bevacizumab), where the overall survival of patients receiving PMTX, cisplatin, and bevacizumab was significantly enhanced (MAPS study) (1). Furthermore, immunotherapy-based strategies are currently becoming attractive therapeutic options, and several clinical trials have recently been performed. A phase II study using monoclonal antibodies against cytotoxic T-lymphocyte antigen 4 (CTLA4; tremelimumab) in patients showing progression of the disease after first-line treatment yielded encouraging results, but it was performed in a small number of patients (2). Another phase II study (DETERMINE) investigated the effect of tremelimumab in patients whose disease had progressed after one or two systemic treatments. There were no benefits, but the safety profile was acceptable (3). A more recent phase 2 study (IFCT-1501 MAPS2) reported the use of immune control checkpoint inhibitors, programmed cell death protein 1 (PD-1; nivolumab) and cytotoxic T-lymphocyte-associated protein 4 (CTLA4; ipilimumab), alone or in combination. The results showed objective anti-tumor responses and a significant increase in survival without progression and global survival (4). Clinical trials for cell-based immunotherapy using dendritic cells or chimeric antigen receptor T (CAR-T) cells have also yielded promising results (5–10).
In the future, new clinical trials will be developed that utilize novel anti-cancer compounds or immunological modulators in association with chemotherapies or in combination with immunological approaches. The efficiency of current treatments are dependent on the integrity of metabolic pathways and DNA repair mechanisms that account for resistance mechanisms. Overall, therapy improvements require better knowledge of the state of the cell regulatory pathways. In addition, immunotherapies need sound knowledge about the immunological status of the tumor. To date, molecular data are not ordinarily used to assist therapeutic decisions, and thus there is an urgent need for their use in translational medicine. To reach these goals, two different fields must be investigated: (i) the cellular and molecular status of MM tumors, regarding mutations, alterations in regulatory pathways, and the microenvironment landscape, and (ii) the methodology of preclinical assays to soundly test specific anti-tumor agents. The aim of this review is to provide an update on our present knowledge and issues on these subjects and to provide perspectives for advancements in MM treatment.
Malignant mesothelioma are heterogeneous tumors that show a complex pattern of molecular changes, including genetic, chromosomic, and epigenetic alterations, all of which should be considered to model this pathology. Of all the MM types defined by tumor location, MPM has the best described molecular alterations and heterogeneity, and thus we will focus on it. Notably, recent integrative multi-omics analysis as well as next generation sequencing (NGS) studies on malignant peritoneal mesothelioma (MPeM) showed similarities to MPM in terms of molecular alterations, even though some alterations, such as ALK rearrangement, are only found in MPeM (11–13).
Recent NGS studies identified a low mutation burden in MPM compared to other adult solid tumors (14). However, this mutation burden could be underestimated by classical NGS analyses, which focus on the detection of changes at the nucleotide level. Early karyotyping analyses and molecular cytogenetic techniques, such as comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) arrays, showed that MPM is characterized by numerous chromosomal abnormalities, including abundant numeric and structural chromosome changes and recurrent alterations in specific chromosome regions (15). More recently, a combination of high-density array-CGH with targeted NGS demonstrated the presence of chromothripsis in the 3p21 region, which includes the BAP1 gene (16). Chromothripsis and also chromoplexy were confirmed on several other chromosome regions in MPM using mate-pair sequencing (17). These numerous inter- or intra-chromosomal rearrangements may result in the disruption of tumor suppressor genes (TSG) as well as the amplification of oncogenes or fusion genes that can drive carcinogenesis.
The mutated genes in MPM are essentially TSG that are inactivated by several mechanisms, including single nucleotide variants, copy number losses, gene fusions, and splicing alterations (14, 18). The only recurrent oncogenic mutation was identified in the promoter of TERT, which encodes telomerase, the essential enzyme that maintains the length of the telomeres (19). The most frequently altered TSG are CDKN2A, BAP1, and NF2, and to a lesser extent TP53, SETD2, and LATS2. All of the other mutated genes show <3% somatic mutation (14, 18). Germline mutations that predispose to MPM were first identified in BAP1, but two recent studies also highlighted germline mutations in several other genes that are less common than in BAP1. They are mainly involved in cell-cycle, chromatin regulation and DNA repair (20–24). Up to 7% of MPM patients may have germline mutations, but experimental validations are needed to confirm that some of these genes are MPM susceptibility genes (like BAP1).
The epigenomic landscape of MPM has also been investigated, albeit to a lesser extent. A microarray-based methylome analysis demonstrated that MPM has specific patterns of gene methylation compared to normal pleura or other tumors (25, 26). The contribution of DNA methylation to mesothelial carcinogenesis has been clearly established, notably by the downregulation of TSG expression (27). The mechanisms for epigenetic regulation in MPM were principally studied in the context of BAP1 inactivation; they highlighted the role of polycomb repressive complex 2 (PRC2) and histone methyltransferase (28). Other studies also emphasized the involvement of non-coding RNA such as micro-RNA (miRNA) or long non-coding RNA (lncRNA), both of which are deregulated in MPM, in carcinogenesis (29–31).
Altogether, these molecular alterations lead to changes in gene expression and deregulation of several biomolecular pathways, including signaling pathways such as Hippo or the PI3K/AKT/mTOR pathways, the cell cycle and apoptosis, among others (32). The implication for therapy from all these molecular changes has been recently reviewed (33).
Like most adult solid tumors, MM is a heterogeneous cancer with high variability among patients. Hence, the development of experimental models must consider this heterogeneity. Histology defines three major types of MM: epithelioid, the most frequent histological subtype; sarcomatoid, with the worst prognosis; and biphasic, which is a mixture of the two previous morphologies. Histological subtypes within these three types have been defined (34). The histological classification only partially captures the tumor heterogeneity observed at both the molecular and clinical levels (35). Large-scale omics and NGS studies have demonstrated MPM heterogeneity at the molecular level that goes beyond the histological classification (14, 18, 36). The first MPM molecular classification, related to histological types and survival, proposed two tumor subtypes by clustering transcriptomic data (36). A new subtype with a poor prognosis and characterized by a double mutation in the TSG NF2 and LATS2, both of which are involved in the Hippo signaling pathway, was identified by coupling genetic and transcriptomic analysis (37). Other studies have proposed classifications into four subtypes that are also related to prognosis and partially to genetic alterations (14, 18, 38). Interestingly, a meta-analysis that compared the subtypes obtained by clustering from several transcriptomic data sets showed that only the most extreme subtypes, which represent the “pure” epithelioid and sarcomatoid phenotypes, are found in all datasets. These findings suggest that intermediate subtypes might only reflect divisions of a continuum (38, 39). Based on these results, histo-molecular gradients obtained by a signal deconvolution method on transcriptomic data were proposed to consider MPM inter-tumor heterogeneity as well as intra-tumor heterogeneity. These histo-molecular gradients determine the variable proportion of epithelioid and sarcomatoid tumor cell contingents in tumor samples. They also have a strong prognostic value and may be of interest for guiding therapeutic strategies (38, 39). Another recent publication further sustained that MPM heterogeneity is better described by a continuum (40).
Intra-tumor heterogeneity is still partially described in MPM, in part due to the use of omics approaches only in bulk tumor samples. MPM is likely a polyclonal tumor that comprises multiple subclones with variable cellular prevalence (41, 42). To better define the polyclonal tumor origin and understand the tumor evolution of mesothelioma, further studies are required in a larger number of tumor samples. Several studies also highlighted the presence of cancer stem cells in MPM (35). In MPM, heterogeneity is not limited to tumor cells; the tumor microenvironment is also distinct from one patient to another in terms of type and number of stromal and immune cells that infiltrate the tumors (43). Immune signatures are linked to the patients' outcome (44). Spatial heterogeneity of the somatic mutations of cancer cells, as well as the immune microenvironment, was highlighted by studying tumor samples at different anatomic sites (45). In this complex context, the use of the emergent “single cell” approaches will be helpful in providing an accurate characterization of tumor and stromal cell heterogeneity and should contribute to a breakthrough in knowledge about intra-tumor heterogeneity. Besides the inter- and intra-tumor heterogeneity of tumor cells, the evolutionary features of tumors need to be considered to establish a classification that is clinically relevant (46).
In this section, we will focus on preclinical models that are useful for chemotherapy, targeted therapy, or immunotherapy rather than for surgery or radiotherapy, even though those therapies have a place in the treatment of patients. The efficiency of anti-cancer compounds to treat MM patients has been tested using large variety of so-called preclinical MM models. These systems are based on use of human or mammalian MM samples, i.e., xenografts in immunosuppressed mice or isografts in syngeneic rodents. Multiple combinations have been developed based on the nature of the malignant sample (cells or tumor tissue), the recipient (rats or mice), the anti-cancer agent (anti-cancer drug, lytic virus, therapeutic cells sur as dendritic or CAR-T cells, etc.), the agent vector (if any), the method to implant tumor cells, and the analytical method. These models do not exactly reproduce human MM, but they are surrogates for a proof of concept. Preclinical model options are synthetized in Figures 1, 2, and the main points are described below:
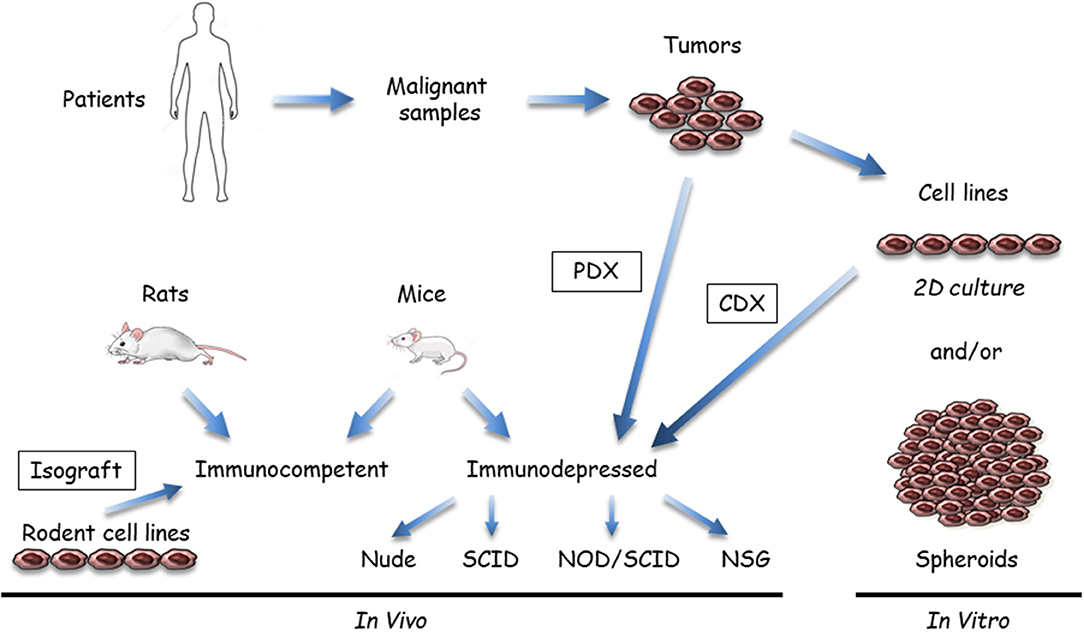
Figure 1. Available malignant mesothelioma (MM) preclinical models that use tumor samples or cell lines. Malignant samples are tumor fragments or more often MM cells obtained after tissue dissociation; these samples are used in vitro or transplanted/inoculated in immunosuppressed or immuno-compatible rodents, almost exclusively mice. In vitro, two-dimensional (2D) cultures or three dimensional (3D) spheroids can be grown from tumor tissue samples. In vivo, MM tumor cells in culture are inoculated either subcutaneously or orthotopically (intracavitary, in the pleura or peritoneum) to generate cell-derived xenograft (CDX) or isograft. Tumor fragments can be also engrafted into immunosuppressed mice (patient-derived xenograft [PDX]). Immunocompetent models mainly comprise syngeneic rodent models. Human immunocompetent models can be obtained using NOD-scid IL2Rγnull (NSG) mice.
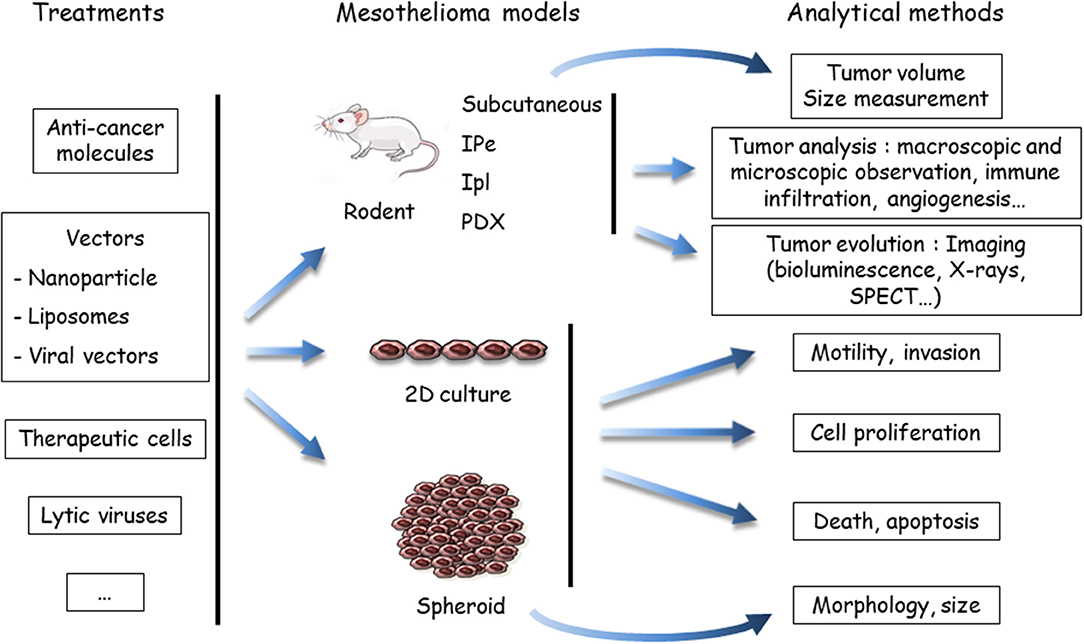
Figure 2. Evaluation of treatments using preclinical models of malignant mesothelioma (MM). Anti-cancer agents are delivered to the MM models, either in a native, or vectorized form (in nanoparticles, liposomes, or viruses). MM cells may be labeled or engineered to allow one to determine tumor growth by imaging methods, mostly bioluminescence. To determine the host response, the analytical method depends on the MM model. In vivo endpoints comprise measuring the tumor volume, quantifying and identifying tumor cells in the pleural effusion or ascites (if produced), or measuring others parameters (angiogenesis, immune infiltration, etc.). In vitro, the endpoints are cell viability, type of death, proliferation, motility, invasion, morphology, and volume (spheroids). Both cell and mouse models have been applied to test the efficiency of drugs in mono- or multimodality chemotherapies, immunotherapies and target therapies, oncovirotherapies, or cell and gene therapies. SPECT, single photon emission computed tomography; IPe, intra-pleural; Ipl: intra-peritoneal; PDX, patient-derived xenograft.
(i) Samples and recipients (Figure 1): Several MM samples are used for preclinical studies. Tumor fragments, pleural liquid, and ascites can be collected from patients. Commercial MM cell lines are available from different companies, but primary MM cell lines are a better model, as extensively discussed in the in vitro models section (see below). Cell models in culture mostly comprise two dimensional (2D) MM cells or three dimensional (3D) multicellular tumor spheroids (MCTS). These cell models are generally monoculture, but new developments include the introduction of stromal and immune cells to better recapitulate the tumor microenvironment. In vivo models are based on the injection of MM cells in subcutaneous (SC) or orthotopic (intrapleural [IPl] or intraperitoneal [IPe]) sites in relevant rodents, mainly mice. Fresh MM tissue samples can be also grown as tumoroids (tumor-derived organoids) in culture or xenografted in immunosuppressed mice as patient-derived xenografts (PDX). Regarding the heterogeneity of human MM, it is important to work with well-characterized MM, particularly when drugs have been designed to target a single protein or a specific pathway. With our developing knowledge of the MM biology, it appears that multiple samples would have to be used. Furthermore, MM classification according to data arising from multi-omic studies (12, 14, 18, 36, 38) might help to define key alterations representative of molecular subtypes of MM and limit the studies on representative samples.
(ii) Anti-cancer compounds (Figure 2): These compounds are intended for chemotherapy, target therapy, immunotherapy, gene therapy, or oncovirotherapy because MM is a compartmentalized tumor with accessibility for in vivo local delivery (47, 48). They are used alone or in combination with other compounds, and as a single molecule or vectorized. Preclinical studies on the chemotherapeutic agent PMTX illustrated this diversity. The effects of PMTX have been investigated in association with several anti-tumor agents (anti-tubulin, gemcitabine, cisplatin, anti-thymidylate synthase, RNA interference [RNAi] embedded in liposomes, miRNA expressed in adenovirus vector, etc.) to determine a potential synergistic effect (49–55). Liposomal PMTX formulations have been tested in an orthotopic mouse model (56). Due to the diversity of the assays, it is difficult to compare their predictability.
(iii) Analytical methods (Figure 2): The endpoints for in vitro assays comprise the determination of cell proliferation, cell death, motility and invasive properties, and spheroid state (morphology and volume). In vivo tumor analyses involve macroscopic and microscopic observations and evaluation of immune infiltration and angiogenesis. The key point is to monitor tumor evolution, especially for orthotropic tumor grafts. Different analytical methods have been developed for in situ tumor visualization. Firefly luciferase (luc)-engineered cells can be detected by a non-invasive bioluminescence imaging method, as in rats injected with luc-MM cells in the pleural cavity (57). However, data have shown that magnetic resonance imaging (MRI) is a more reliable method for MPM tumor burden measurement compared to bioluminescence (57). Computed tomography scanning may be also of interest, as shown with a lung cancer cell line in mice (58). Tumor lesions and the localization of epidermal growth factor receptor (HER) were visualized with single photon emission computed tomography (SPECT) and MRI in an orthotopic MM model with radiolabeled specific antibodies (59). Bioluminescence of luc-expressed MM remains the most common strategy to monitor tumor development in orthotopic models. These in vivo imaging methods require specific equipment and facilities and, for bioluminescence detection, the genetic modification of tumor cells with the luc gene. The introduction of an exogenous gene might have an impact on cell mechanisms and immune response.
In the following subsections, we detail the present and ongoing models, with a focus on their interest, limitations, and impacts to assess emerging therapies. The advantages and limitations of the mesothelioma preclinical models are presented in Table 1.
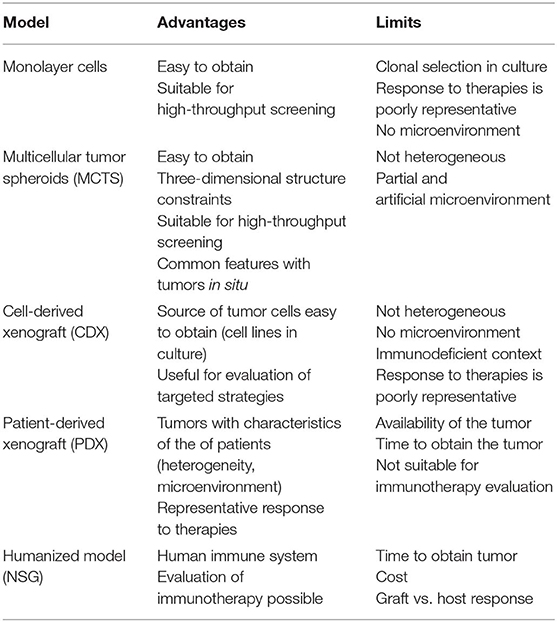
Table 1. Advantages and limitations of the different models of mesothelioma.
MM cell lines have been widely used to study MM pathogenesis and evaluate the activity of numerous anti-cancer agents. The first MM cell lines were established in 1982 from the abdominal fluid of a patient (60). In 1987, a MM cell line was established from a surgical sample of malignant pleura, namely H-Meso-1 (61). Since that study, numerous cell lines have been established from samples of patients by different groups to constitute local biocollection. Some of these collections have been extensively characterized (19, 36, 37, 62–66), as well as 21 cell lines in the Genomics of Drug Sensitivity in Cancer (GDSC) database (https://www.cancerrxgene.org/celllines). MPM cell lines present common characteristics with regard to tumors and might lead to the identification of new biomarkers (62, 67, 68). One study discussed the limits of these cell models. The authors found strong molecular differences between primary and commercial cell lines (67), mainly due to a high number of divisions after their establishment, and thus an increased risk of new karyotypic changes. These models remain interesting for screening and preliminary investigations. Primary tumor cells represent an intriguing alternative because they share similar molecular characteristic with the primary tumor, even though they show a reduction of subclonal diversity (15, 42, 67). However, the necessity to perform studies before 6–10 passages limits the number of experiments. The most appropriate strategy would probably be to conduct large screening studies on cancer cell lines and then confirm the findings with primary cancer cells. The results obtained with cell lines should be confirmed on samples from patients (if applicable).
The in vivo models that use wild type rodents and genetically modified mice (GEM) are summarized in Figure 3. GEM have been generated to obtain “spontaneous” MM, without exposure to asbestos fibers, by heterozygous (htz) or homozygous (hom) conditional mutation of Ink4a and/or Nf2 and/or Trp53, or by IPl/IPe injection of AdCre to mimic the human condition (69). In this system, the rate of MPM is dependent on the type of inactivated genes; high rates occur with at least two hom genes, including Trp53. Survival generally exceeds 30–50 weeks, although shorter survivals occur in a few situations with hom/hom combinations (70). Similar results were recently reported by inactivating Ink4a, Nf2, and Bap1 (71). The generated MM express a similar morphology to human MM, with a proportion of each histological type depending on the modified genes.
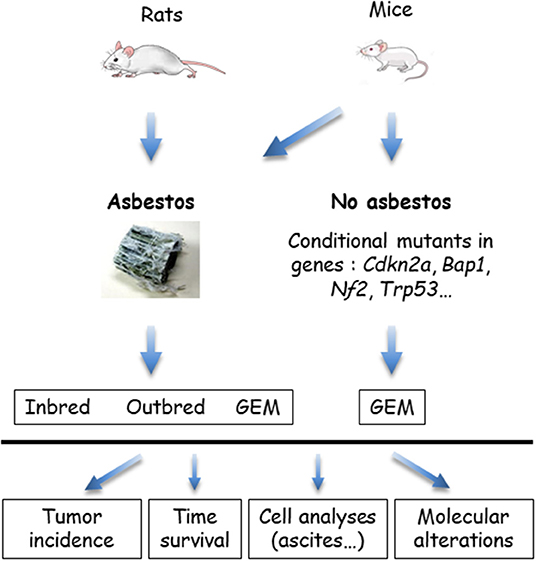
Figure 3. In vivo models of malignant mesothelioma (MM) in asbestos-treated rodents and genetically modified mice (GEM). MM can be obtained from wild type (WT) rats or mice, exposed to asbestos, or GEM. Asbestos-induced MM models are generated by the injection of asbestos fibers intracavitary, in the pleura or peritoneum, mostly peritoneal in mice. Conditional mutant mice are obtained by engineering the major genes altered in MM. Asbestos-recipient animals and GEM may be investigated for tumor incidence, survival, quantitative and qualitative analyses of cells in ascites or pleural fluid, and molecular alterations in tumors.
MM have also been generated in several types of GEM exposed to asbestos fibers IPe injections. Experimental cancers induced in animals by the responsible carcinogen better reflect the natural history of these cancers. They are particularly relevant for the coupled asbestos-MM condition, given that the large majority of human MM cases are linked to asbestos exposure. With regard to conditional mutant mice, it takes several months to more than 1 year before the development of a MM. While the morphological features are reproduced, the sarcomatoid MM subtype most frequently forms, contrary to what is observed in humans (70).
These sophisticated models have been mainly used for mechanistic purposes, with a focus on the molecular mechanism of MM formation or mechanism of action of asbestos fibers. According to our knowledge, GEM with mutated genes that are relevant to human MM have not been used to test the effects of drugs. However, models of colorectal, non-small-cell lung, and pancreatic cancers have been used to predict therapeutic responses (72, 73). Although these models are physiologically different from humans, GEM mice form tumors that carry relevant gene changes, show histological similarities, and should allow one to perform tests in an immunocompetent environment. However, there are several biological and technical pitfalls. For instance, the tumor evolution can differ among mice, with the possible occurrence of metastases, other types of tumors may be generated, and the physiological differences between mice and human may bias the predictive value of the assays. Otherwise, the complexity of these models makes it difficult to produce homogeneous data from a rather small number of mice, to detect the tumor without autopsy, to follow its evolution, and to determine the right time of its development to establish the planed protocol.
Recent successes were obtained with the use of immune checkpoint inhibitors in clinical trials (2, 4, 74–76). However, the response rate remains limited and, therefore, the objectives are now to extend the benefit of these approaches to a large number of patients. Preclinical studies performed in appropriate in vivo models are mandatory to obtain relevant results and achieve this objective. The first criterion is the presence of a completely functional immune system, a factor that excludes xenograft models that use human tumor cells. Several immunocompetent models of MM have been developed in rodents using cell lines obtained after inoculation of asbestos fibers in the peritoneal cavity (Table 2).

Table 2. The main immunocompetent rodent models of mesothelioma.
C57Bl/6 mice models with murine MM cell lines have been extensively used to evaluate immunotherapeutic approaches. The most utilized cell lines are AE17 and AK7 (77, 78). These cells have been modified, including exogenous expression of Ova as a neo-antigen, to increase their immunogenicity and evaluate a strategy to improve the anti-tumor immune response (78). Models of MM on the Balb/C genetic background, using AB1 and AB12 cells lines, are also available. CBA/J mice injected with AC29 cells can also be used as an immunocompetent model of MM; however, they have been exploited less than the other previously cited models (79). The main injection route to induce MM is IPe. Immunocompetent IPl models of MM have also been developed, but they are not frequently used due to the procedure required to access to pleural space. In both cases, tumor development is monitored by the previously mentioned imaging methods. SC injections are also used to overcome practical concerns, such as measurement of the tumor size and intra-tumor injection of therapeutics, but SC location is far from the pathophysiological context.
A MM model in Fischer F344 rats following IPl injection of IL45 cell line has also been described (80). With regard to rodent models, IL45 cells expressing luc was used to improve the monitoring of tumor development (83). Recently, models of MPeM in Fischer F344 rats have been developed (81). This effort generated several MM cell lines with distinct aggressiveness. Depending on the cell lines used, the immune infiltrate was different (with or without lymphocyte and/or macrophage infiltration of the tumors) (82). Therefore, the efficacy of immunotherapy approaches could be evaluated using this model in the appropriate immune context.
The previously mentioned rodent models allow one to evaluate therapeutic strategies in a living organism with several constraints: elimination, diffusion in the tissue, bio-distribution, and toxicity. These models were particularly used for the evaluation of target therapies using inhibitors of histone deacetylases or signal pathways such as Hippo or focal adhesion kinase (FAK) (84–86), anti-mesothelin or anti-podoplanin antibodies (87–92), or CAR-T cells (5, 93, 94). However, they showed major limitations: (i) differences compared to humans in the immune system and metabolism of chemotherapeutic agents for syngeneic rodent models; and (ii) the use of a human cell line to induce tumors, which does not reflect human intra-tumor heterogeneity, in the context of immunodeficient mice (xenografts). In order to improve the relevance of rodent models, PDX models were developed and they implies implanting a tumor fragment from a patient into an immunodeficient mouse. For MM, the implantation was only heterotopic. Mouse strains with different levels of immunodeficiency can be used depending on the objective of the experiment (Nude, SCID, NOD-SCID, or NOD-scid IL2Rγnull [NSG]) (95). The first description of PDX models of MM was in 1980 (96). Tumors from three patients were transplanted into nude mice but only two tumors grew. Recently, SC implantation of tumors from 50 patients with MPM was evaluated in nude mice (97). This methodology maintains the heterogeneity of the tumor and its microenvironment at least during the first generation. However, the limitations include (i) a high proportion (60%) of MPM do not grow as PDX; (ii) the tumor microenvironment is replaced by murine cells over generations; and (iii) the immune context is modified, which is not suitable for evaluation of immunotherapeutic strategies (95, 97). PDX also requires access to tumor samples, which is not easy in the case of a relatively rare cancer as MM.
The use of a humanized mouse model of MM might be a good alternative to study the anti-tumor immune response. In these models, the mouse immune system is replaced by a human immune system. NSG mice are used for this research; they are deficient in the interleukin 2 receptor gamma subunit (IL-2Rγ) that is involved in differentiation and function of many hematopoietic cells (98). This feature confers a great advantage to study immunotherapy strategies in an environment that closely resembles human patients. However, these models present some limits, including the cost, the time to obtain NSG mice reconstituted with a human immune system, the risk of an incomplete differentiation of haematopoietic stem cells, and the graft vs. host reaction, which could limit the duration of the experiments.
In order to overcome some defaults of the existing in vivo models, 3D tumor spheroids, positioned between 2D cell culture and animal models, have been developed (99). MCTS involves culturing tumor cells in non-adherent conditions to obtain well-rounded cellular structures after 48–72 h. This culture mode has been applied to MM cell lines. The 3D organization of cells induces major changes in gene expression compared to 2D culture (100, 101). Indeed, some pathways involved in resistance to cell death are differentially regulated in monolayer and MCTS, and thus these models better mimic resistance to treatment compared with monolayer cells (64, 102–106). These models also reproduce the diffusion constraint of therapeutic molecules, such as antibodies or nanovectors (106–109). The 3D structures also share common features with tumors from patients (101). This aspect has been notably demonstrated in the field of autophagy (110–112). Indeed, resistance to treatment is associated with autophagy in MCTS and tumors in situ.
The main weakness of the current 3D models is the absence of cells from the microenvironment. An alternative to MCTS is the use of tumoroids, which include tumor cells and infiltrated cells (99, 113). However, these models require access to fresh surgical MM samples. Co-cultures serve as an alternative. MCTS of non-small-cell lung cancer, pancreatic, and breast cancer tumor cells supplemented with fibroblasts and/or macrophages have been described (114–116). The tumor-associated macrophages (TAM) obtained in these models present similar characteristics to those observed in tumors, namely increased resistance to treatment and an improved cytokine environment. These aspects are crucial for immunology studies. MCTS that include different cell types might constitute interesting tools for preliminary studies. They are achieved by combining stromal and immune cells issued from cell lines or isolated from different donors. However, although their relevance needs to be confirmed, MCTS reproduce a partial human microenvironment that is completely absent from cell-derived xenograft.
Multiple classical preclinical models of cancer have been applied, and new ones are under development, to test the potential effect of anti-cancer drugs on human MM. Each available model has benefits and limits (Table 1), and they must be selected depending on the objectives of the experiment. Overall, these models incompletely reproduce human MM, given that they do not consider the anatomical or biological tumor microenvironment or the intra-tumor heterogeneity of the tumors. The immunological context is not fully reconstructed, even with humanized mice. Three-dimensional spheroid cultures that have been developed as in vitro systems, and co-cultures with immune and stromal cells should be considered to improve the relevance of these models. Inter-tumor heterogeneity is also insufficiently studied because most models proceed with MM cell lines or tumors not always characterized at the molecular level, especially concerning the mutation burden and chromosomal abnormalities of the tumor cells. These models remain surrogates; however, they are of paramount importance in translational research and this encourage new developments to improve their predictability. Among recent developments are PDX models and the generation of GEM that carry mutations identified in human MM tumor cells. These approaches may be useful, but PDX and GEM models in general are complex and have limitations in the immune environment and animal cost. Their application in the context of MM heterogeneity will require the use of multiple cell lines according to their molecular profile. To achieve sound results with significant statistical value, including kinetics and dose-effect relationships, a large number of animals would also be needed, unless solid ancillary results are available. Besides economic issues, the 3R rules (Replacement, Reduction, and Refinemen) on the use of animals in scientific procedures are recommended. The identification of biomarkers to follow tumor evolution in response to anti-cancer drugs is of importance to limit the number of animals (117). Lower attrition rates for oncology drugs would be obtained with more predictive models (72, 104, 117, 118). Consequently, it is necessary to develop alternatives for replacement, robust and reproducible bases for reduction, and the use of advanced technologies for refinement (119).
Appropriately designed and analyzed preclinical assays are required (72), with the aim to identify new anti-cancer compounds for MM and novel biomarkers for sensitivity or resistance, which are essential to predict the tumor response. Although animal models are considered to be the most relevant, the development of sophisticated in vitro multicellular models should be encouraged. The continuing increase in the knowledge about mesothelial carcinogenesis will permit the use of more pertinent cell models that represent the MM tumor. New approaches not yet used in MM should be explored, including organ-on-a-chip technologies or in silico biological systems using computational modeling and machine learning (120, 121). Powerful technological tools should allow researchers to establish models with MM cells that grow in a more accurate tumor microenvironment, and possible in situ molecular analyses of tumor cells. The use of well-characterized tumor cells, classified in subgroups of molecular classifications or characterized by histo-molecular gradients, is particularly important regarding the molecular heterogeneity of human MM. This endeavor should allow researchers to obtain representative results of a given type of tumors.
The ongoing preclinical models should be improved with regard to precision, reproducibility, and predictivity, and the results should be supported by different approaches. Some standardization might be helpful. The use of existing consortia and/or the development of new consortia will allow the inclusion of more tumor samples in studies and increase the number of relevant cell models. These factors will enable researchers to adequately cover mesothelioma heterogeneity and be able to afford the high costs of new technologies. Some authors have recommended improving the reliability of preclinical cancer studies by using detailed information on the experimental methodology, different approaches, the publication of negative data, and better dialogue between physicians and scientists (122, 123). These factors are particularly important within the actual context of precision medicine, which implements complex methodologies and multidisciplinary investigations and has a high cost.
M-CJ and CB prepared the figures. CB, M-CJ, and DJ wrote the review.
This work was supported by Inserm, CNRS, Hadassah France, the National Research Agency under the Programme d'Investissements d'Avenir (ANR-16-IDEX-0007), the Pays de la Loire Region research program by a financial support from the Institut de Recherche en Santé Respiratoire des Pays de la Loire, the Chancellerie des Universités de Paris (Legs POIX), the Ligue Contre le Cancer (Ile de France committee), and ARSMeso44. The authors thank the cluster LUNG innOvatiOn (LUNG O2) for logistic support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2D, two dimensional; 3D, three dimensional; CDX, cell-derived xenograft; CGH, comparative genomic hybridization; GEM, genetically modified mice; hom, homozygous; htz, heterozygous; IPl, intra-pleural; IPe, intra-peritoneal; luc, firefly luciferase; MCTS, multicellular tumor spheroids; MM, malignant mesothelioma; MPM, malignant pleural mesothelioma; MPeM, malignant peritoneal mesothelioma; MRI, magnetic resonance imaging; NGS, next generation sequencing; NSG, NOD-scid IL2Rγnull; PDX, patient-derived xenografts; PMTX, pemetrexed; SC, subcutaneous; SNP, single nucleotide polymorphism; SPECT, single photon emission computed tomography; TSG, tumor suppressor genes.
1. Zalcman G, Mazieres J, Margery J, Greillier L, Audigier-Valette C, Moro-Sibilot D, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the mesothelioma avastin cisplatin pemetrexed study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet. (2016) 387:1405–14. doi: 10.1016/S0140-6736(15)01238-6
2. Calabro L, Morra A, Fonsatti E, Cutaia O, Amato G, Giannarelli D, et al. Tremelimumab for patients with chemotherapy-resistant advanced malignant mesothelioma: an open-label, single-arm, phase 2 trial. Lancet Oncol. (2013) 14:1104–11. doi: 10.1016/S1470-2045(13)70381-4
3. Maio M, Scherpereel A, Calabro L, Aerts J, Cedres Perez S, Bearz A, et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): a multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. (2017) 18:1261–73. doi: 10.1016/S1470-2045(17)30446-1
4. Scherpereel A, Mazieres J, Greillier L, Lantuejoul S, Do P, Bylicki O, et al. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): a multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol. (2019) 20:239–53. doi: 10.1016/S1470-2045(18)30765-4
5. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. (2014) 6:261ra151. doi: 10.1126/scitranslmed.3010162
6. Aerts J, de Goeje PL, Cornelissen R, Kaijen-Lambers MEH, Bezemer K, van der Leest CH, et al. Autologous dendritic cells pulsed with allogeneic tumor cell lysate in mesothelioma: from mouse to human. Clin Cancer Res. (2018) 24:766–76. doi: 10.1158/1078-0432.CCR-17-2522
7. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
8. Belderbos RA, Baas P, Berardi R, Cornelissen R, Fennell DA, van Meerbeeck JP, et al. A multicenter, randomized, phase II/III study of dendritic cells loaded with allogeneic tumor cell lysate (MesoPher) in subjects with mesothelioma as maintenance therapy after chemotherapy: DENdritic cell immunotherapy for mesothelioma (DENIM) trial. Transl Lung Cancer Res. (2019) 8:280–5. doi: 10.21037/tlcr.2019.05.05
9. Cornelissen R, Hegmans JP, Maat AP, Kaijen-Lambers ME, Bezemer K, Hendriks RW, et al. Extended tumor control after dendritic cell vaccination with low-dose cyclophosphamide as adjuvant treatment in patients with malignant pleural mesothelioma. Am J Respir Crit Care Med. (2016) 193:1023–31. doi: 10.1164/rccm.201508-1573OC
10. Hegmans JP, Veltman JD, Lambers ME, de Vries IJ, Figdor CG, Hendriks RW, et al. Consolidative dendritic cell-based immunotherapy elicits cytotoxicity against malignant mesothelioma. Am J Respir Crit Care Med. (2010) 181:1383–90. doi: 10.1164/rccm.200909-1465OC
11. Hung YP, Dong F, Watkins JC, Nardi V, Bueno R, Dal Cin P, et al. Identification of ALK rearrangements in malignant peritoneal mesothelioma. JAMA Oncol. (2018) 4:235–8. doi: 10.1001/jamaoncol.2017.2918
12. Joseph NM, Chen YY, Nasr A, Yeh I, Talevich E, Onodera C, et al. Genomic profiling of malignant peritoneal mesothelioma reveals recurrent alterations in epigenetic regulatory genes BAP1, SETD2, and DDX3X. Mod Pathol. (2017) 30:246–54. doi: 10.1038/modpathol.2016.188
13. Shrestha R, Nabavi N, Lin YY, Mo F, Anderson S, Volik S, et al. BAP1 haploinsufficiency predicts a distinct immunogenic class of malignant peritoneal mesothelioma. Genome Med. (2019) 11:8. doi: 10.1186/s13073-019-0620-3
14. Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. (2016) 48:407–16. doi: 10.1038/ng.3520
15. Jean D, Daubriac J, Le Pimpec-Barthes F, Galateau-Salle F, Jaurand MC. Molecular changes in mesothelioma with an impact on prognosis and treatment. Arch Pathol Lab Med. (2012) 136:277–93. doi: 10.5858/arpa.2011-0215-RA
16. Yoshikawa Y, Emi M, Hashimoto-Tamaoki T, Ohmuraya M, Sato A, Tsujimura T, et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc Natl Acad Sci USA. (2016) 113:13432–7. doi: 10.1073/pnas.1612074113
17. Mansfield AS, Peikert T, Smadbeck JB, Udell JBM, Garcia-Rivera E, Elsbernd L, et al. Neoantigenic potential of complex chromosomal rearrangements in mesothelioma. J Thorac Oncol. (2019) 14:276–87. doi: 10.1016/j.jtho.2018.10.001
18. Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. (2018) 8:1548–65. doi: 10.1158/2159-8290.CD-18-0804
19. Tallet A, Nault JC, Renier A, Hysi I, Galateau-Salle F, Cazes A, et al. Overexpression and promoter mutation of the TERT gene in malignant pleural mesothelioma. Oncogene. (2014) 33:3748–52. doi: 10.1038/onc.2013.351
20. Betti M, Casalone E, Ferrante D, Aspesi A, Morleo G, Biasi A, et al. Germline mutations in DNA repair genes predispose asbestos-exposed patients to malignant pleural mesothelioma. Cancer Lett. (2017) 405:38–45. doi: 10.1016/j.canlet.2017.06.028
21. Hassan R, Morrow B, Thomas A, Walsh T, Lee MK, Gulsuner S, et al. Inherited predisposition to malignant mesothelioma and overall survival following platinum chemotherapy. Proc Natl Acad Sci USA. (2019) 116:9008–13. doi: 10.1073/pnas.1821510116
22. Panou V, Gadiraju M, Wolin A, Weipert CM, Skarda E, Husain AN, et al. Frequency of germline mutations in cancer susceptibility genes in malignant mesothelioma. J Clin Oncol. (2018) 36:2863–71. doi: 10.1200/JCO.2018.36.15_suppl.8564
23. Pastorino S, Yoshikawa Y, Pass HI, Emi M, Nasu M, Pagano I, et al. A subset of mesotheliomas with improved survival occurring in carriers of BAP1 and other germline mutations. J Clin Oncol. (2018) 36:3485–94. doi: 10.1200/JCO.2018.79.0352
24. Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. (2011) 43:1022–5. doi: 10.1038/ng.912
25. Christensen BC, Houseman EA, Godleski JJ, Marsit CJ, Longacker JL, Roelofs CR, et al. Epigenetic profiles distinguish pleural mesothelioma from normal pleura and predict lung asbestos burden and clinical outcome. Cancer Res. (2009) 69:227–34. doi: 10.1158/0008-5472.CAN-08-2586
26. Goto Y, Shinjo K, Kondo Y, Shen L, Toyota M, Suzuki H, et al. Epigenetic profiles distinguish malignant pleural mesothelioma from lung adenocarcinoma. Cancer Res. (2009) 69:9073–82. doi: 10.1158/0008-5472.CAN-09-1595
27. McLoughlin KC, Kaufman AS, Schrump DS. Targeting the epigenome in malignant pleural mesothelioma. Transl Lung Cancer Res. (2017) 6:350–65. doi: 10.21037/tlcr.2017.06.06
28. LaFave LM, Beguelin W, Koche R, Teater M, Spitzer B, Chramiec A, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. (2015) 21:1344–9. doi: 10.1038/nm.3947
29. Felley-Bosco E, Rehrauer H. Non-coding transcript heterogeneity in mesothelioma: insights from asbestos-exposed mice. Int J Mol Sci. (2018) 19:1163. doi: 10.3390/ijms19041163
30. Reid G. MicroRNAs in mesothelioma: from tumour suppressors and biomarkers to therapeutic targets. J Thorac Dis. (2015) 7:1031–40. doi: 10.3978/j.issn.2072-1439.2015.04.56
31. Singh AS, Heery R, Gray SG. In silico and in vitro analyses of LncRNAs as potential regulators in the transition from the epithelioid to sarcomatoid histotype of malignant pleural mesothelioma (MPM). Int J Mol Sci. (2018) 19:1297. doi: 10.3390/ijms19051297
32. Jaurand MC, Jean D. Biomolecular pathways and malignant pleural mesothelioma. In: Mineo CT, editor. Malignant Pleural Mesothelioma: Present Status and Future Directions. Sharjah: Bentham Science Publishers Ltd. (2016). p. 169–92.
33. Yap TA, Aerts JG, Popat S, Fennell DA. Novel insights into mesothelioma biology and implications for therapy. Nat Rev Cancer. (2017) 17:475–88. doi: 10.1038/nrc.2017.42
34. Husain AN, Colby T, Ordonez N, Krausz T, Attanoos R, Beasley MB, et al. Guidelines for pathologic diagnosis of malignant mesothelioma: 2012 update of the consensus statement from the international mesothelioma interest group. Arch Pathol Lab Med. (2013) 137:647–67. doi: 10.5858/arpa.2012-0214-OA
35. Oehl K, Vrugt B, Opitz I, Meerang M. Heterogeneity in Malignant Pleural Mesothelioma. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19061603
36. de Reynies A, Jaurand MC, Renier A, Couchy G, Hysi I, Elarouci N, et al. Molecular classification of malignant pleural mesothelioma: identification of a poor prognosis subgroup linked to the epithelial-to-mesenchymal transition. Clin Cancer Res. (2014) 20:1323–34. doi: 10.1158/1078-0432.CCR-13-2429
37. Tranchant R, Quetel L, Tallet A, Meiller C, Renier A, de Koning L, et al. Co-occurring mutations of tumor suppressor genes, LATS2 and NF2, in malignant pleural mesothelioma. Clin Cancer Res. (2017) 23:3191–202. doi: 10.1158/1078-0432.CCR-16-1971
38. Blum Y, Meiller C, Quetel L, Elarouci N, Ayadi M, Tashtanbaeva D, et al. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat Commun. (2019) 10:1333. doi: 10.1038/s41467-019-09307-6
39. Blum Y, Jaurand MC, De Reynies A, Jean D. Unraveling the cellular heterogeneity of malignant pleural mesothelioma through a deconvolution approach. Mol Cell Oncol. (2019) 6:1610322. doi: 10.1080/23723556.2019.1610322
40. Alcala N, Mangiante L, Le-Stang N, Gustafson CE, Boyault S, Damiola F, et al. Redefining malignant pleural mesothelioma types as a continuum uncovers immune-vascular interactions. EBioMedicine. (2019) 48:191–202. doi: 10.1016/j.ebiom.2019.09.003
41. Comertpay S, Pastorino S, Tanji M, Mezzapelle R, Strianese O, Napolitano A, et al. Evaluation of clonal origin of malignant mesothelioma. J Transl Med. (2014) 12:301. doi: 10.1186/s12967-014-0301-3
42. Oey H, Daniels M, Relan V, Chee TM, Davidson MR, Yang IA, et al. Whole-genome sequencing of human malignant mesothelioma tumours and cell lines. Carcinogenesis. (2019) 40:724–34. doi: 10.1093/carcin/bgz066
43. Minnema-Luiting J, Vroman H, Aerts J, Cornelissen R. Heterogeneity in immune cell content in malignant pleural mesothelioma. Int J Mol Sci. (2018) 19:1041. doi: 10.3390/ijms19041041
44. Salaroglio IC, Kopecka J, Napoli F, Pradotto M, Maletta F, Costardi L, et al. Potential diagnostic and prognostic role of microenvironment in malignant pleural mesothelioma. J Thorac Oncol. (2019) 14:1458–71. doi: 10.1016/j.jtho.2019.03.029
45. Kiyotani K, Park JH, Inoue H, Husain A, Olugbile S, Zewde M, et al. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology. (2017) 6:e1278330. doi: 10.1080/2162402X.2016.1278330
46. Maley CC, Aktipis A, Graham TA, Sottoriva A, Boddy AM, Janiszewska M, et al. Classifying the evolutionary and ecological features of neoplasms. Nat Rev Cancer. (2017) 17:605–19. doi: 10.1038/nrc.2017.69
47. Mutti L, Peikert T, Robinson BWS, Scherpereel A, Tsao AS, de Perrot M, et al. Scientific advances and new frontiers in mesothelioma therapeutics. J Thorac Oncol. (2018) 13:1269–83. doi: 10.1016/j.jtho.2018.06.011
48. Tagawa M, Tada Y, Shimada H, Hiroshima K. Gene therapy for malignant mesothelioma: current prospects and challenges. Cancer Gene Ther. (2013) 20:150–6. doi: 10.1038/cgt.2013.1
49. Abu Lila AS, Kato C, Fukushima M, Huang CL, Wada H, Ishida T. Downregulation of thymidylate synthase by RNAi molecules enhances the antitumor effect of pemetrexed in an orthotopic malignant mesothelioma xenograft mouse model. Int J Oncol. (2016) 48:1399–407. doi: 10.3892/ijo.2016.3367
50. Favoni RE, Daga A, Malatesta P, Florio T. Preclinical studies identify novel targeted pharmacological strategies for treatment of human malignant pleural mesothelioma. Br J Pharmacol. (2012) 166:532–53. doi: 10.1111/j.1476-5381.2012.01873.x
51. Giovannetti E, Zucali PA, Assaraf YG, Leon LG, Smid K, Alecci C, et al. Preclinical emergence of vandetanib as a potent antitumour agent in mesothelioma: molecular mechanisms underlying its synergistic interaction with pemetrexed and carboplatin. Br J Cancer. (2011) 105:1542–53. doi: 10.1038/bjc.2011.400
52. Hanauske AR. The role of alimta in the treatment of malignant pleural mesothelioma: an overview of preclinical and clinical trials. Lung Cancer. (2004) 45(Suppl. 1):S121–4. doi: 10.1016/j.lungcan.2004.04.021
53. Iwahori K, Serada S, Fujimoto M, Ripley B, Nomura S, Mizuguchi H, et al. SOCS-1 gene delivery cooperates with cisplatin plus pemetrexed to exhibit preclinical antitumor activity against malignant pleural mesothelioma. Int J Cancer. (2013) 132:459–71. doi: 10.1002/ijc.27611
54. Leon LG, Gemelli M, Sciarrillo R, Avan A, Funel N, Giovannetti E. Synergistic activity of the c-Met and tubulin inhibitor tivantinib (ARQ197) with pemetrexed in mesothelioma cells. Curr Drug Targets. (2014) 15:1331–40. doi: 10.2174/1389450116666141205160924
55. Ueno T, Toyooka S, Fukazawa T, Kubo T, Soh J, Asano H, et al. Preclinical evaluation of microRNA-34b/c delivery for malignant pleural mesothelioma. Acta Med Okayama. (2014) 68:23–6. doi: 10.18926/AMO/52140
56. Ando H, Kobayashi S, Abu Lila AS, Eldin NE, Kato C, Shimizu T, et al. Advanced therapeutic approach for the treatment of malignant pleural mesothelioma via the intrapleural administration of liposomal pemetrexed. J Control Release. (2015) 220:29–36. doi: 10.1016/j.jconrel.2015.10.019
57. Meerang M, Boss A, Kenkel D, Broggini-Tenzer A, Berard K, Lauk O, et al. Evaluation of imaging techniques for the assessment of tumour progression in an orthotopic rat model of malignant pleural mesotheliomadagger. Eur J Cardiothorac Surg. (2015) 47:e34–41. doi: 10.1093/ejcts/ezu393
58. Iochmann S, Lerondel S, Blechet C, Lavergne M, Pesnel S, Sobilo J, et al. Monitoring of tumour progression using bioluminescence imaging and computed tomography scanning in a nude mouse orthotopic model of human small cell lung cancer. Lung Cancer. (2012) 77:70–6. doi: 10.1016/j.lungcan.2012.01.009
59. Nayak TK, Bernardo M, Milenic DE, Choyke PL, Brechbiel MW. Orthotopic pleural mesothelioma in mice: SPECT/CT and MR imaging with HER1- and HER2-targeted radiolabeled antibodies. Radiology. (2013) 267:173–82. doi: 10.1148/radiol.12121021
60. Behbehani AM, Hunter WJ, Chapman AL, Lin F. Studies of a human mesothelioma. Hum Pathol. (1982) 13:862–6. doi: 10.1016/S0046-8177(82)80083-X
61. Reale FR, Griffin TW, Compton JM, Graham S, Townes PL, Bogden A. Characterization of a human malignant mesothelioma cell line (H-MESO-1): a biphasic solid and ascitic tumor model. Cancer Res. (1987) 47:3199–205.
62. Gueugnon F, Leclercq S, Blanquart C, Sagan C, Cellerin L, Padieu M, et al. Identification of novel markers for the diagnosis of malignant pleural mesothelioma. Am J Pathol. (2011) 178:1033–42. doi: 10.1016/j.ajpath.2010.12.014
63. Jean D, Thomas E, Manie E, Renier A, de Reynies A, Lecomte C, et al. Syntenic relationships between genomic profiles of fiber-induced murine and human malignant mesothelioma. Am J Pathol. (2011) 178:881–94. doi: 10.1016/j.ajpath.2010.10.039
64. Tranchant R, Quetel L, Montagne F, De Wolf J, Meiller C, De Koning L, et al. Assessment of signaling pathway inhibitors and identification of predictive biomarkers in malignant pleural mesothelioma. Lung Cancer. (2018) 126:15–24. doi: 10.1016/j.lungcan.2018.10.015
65. Zeng L, Buard A, Monnet I, Boutin C, Fleury J, Saint-Etienne L, et al. In vitro effects of recombinant human interferon gamma on human mesothelioma cell lines. Int J Cancer. (1993) 55:515–20. doi: 10.1002/ijc.2910550331
66. Zeng L, Fleury-Feith J, Monnet I, Boutin C, Bignon J, Jaurand MC. Immunocytochemical characterization of cell lines from human malignant mesothelioma: characterization of human mesothelioma cell lines by immunocytochemistry with a panel of monoclonal antibodies. Hum Pathol. (1994) 25:227–34. doi: 10.1016/0046-8177(94)90192-9
67. Chernova T, Sun XM, Powley IR, Galavotti S, Grosso S, Murphy FA, et al. Molecular profiling reveals primary mesothelioma cell lines recapitulate human disease. Cell Death Differ. (2016) 23:1152–64. doi: 10.1038/cdd.2015.165
68. Smeele P, d'Almeida SM, Meiller C, Chene AL, Liddell C, Cellerin L, et al. Brain-derived neurotrophic factor, a new soluble biomarker for malignant pleural mesothelioma involved in angiogenesis. Mol Cancer. (2018) 17:148. doi: 10.1186/s12943-018-0891-0
69. Jongsma J, van Montfort E, Vooijs M, Zevenhoven J, Krimpenfort P, van der Valk M, et al. A conditional mouse model for malignant mesothelioma. Cancer Cell. (2008) 13:261–71. doi: 10.1016/j.ccr.2008.01.030
70. Jean D, Jaurand MC. Mesotheliomas in Genetically engineered mice unravel mechanism of mesothelial carcinogenesis. Int J Mol Sci. (2018) 19:2191. doi: 10.3390/ijms19082191
71. Kukuyan AM, Sementino E, Kadariya Y, Menges CW, Cheung M, Tan Y, et al. Inactivation of Bap1 cooperates with losses of Nf2 and Cdkn2a to drive the development of pleural malignant mesothelioma in conditional mouse models. Cancer Res. (2019) 79:4113–23. doi: 10.1158/0008-5472.CAN-18-4093
72. Ruggeri BA, Camp F, Miknyoczki S. Animal models of disease: pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem Pharmacol. (2014) 87:150–61. doi: 10.1016/j.bcp.2013.06.020
73. Singh M, Murriel CL, Johnson L. Genetically engineered mouse models: closing the gap between preclinical data and trial outcomes. Cancer Res. (2012) 72:2695–700. doi: 10.1158/0008-5472.CAN-11-2786
74. Disselhorst MJ, Quispel-Janssen J, Lalezari F, Monkhorst K, de Vries JF, van der Noort V, et al. Ipilimumab and nivolumab in the treatment of recurrent malignant pleural mesothelioma (INITIATE): results of a prospective, single-arm, phase 2 trial. Lancet Respir Med. (2019) 7:260–70. doi: 10.1016/S2213-2600(18)30420-X
75. Fennell DA, Kirkpatrick E, Cozens K, Nye M, Lester J, Hanna G, et al. CONFIRM: a double-blind, placebo-controlled phase III clinical trial investigating the effect of nivolumab in patients with relapsed mesothelioma: study protocol for a randomised controlled trial. Trials. (2018) 19:233. doi: 10.1186/s13063-018-2602-y
76. Mansfield AS, Zauderer MG. Nivo-lution in mesothelioma. Clin Cancer Res. (2019) 25:5438–40. doi: 10.1158/1078-0432.CCR-19-1836
77. Moalli PA, MacDonald JL, Goodglick LA, Kane AB. Acute injury and regeneration of the mesothelium in response to asbestos fibers. Am J Pathol. (1987) 128:426–45.
78. Jackaman C, Bundell CS, Kinnear BF, Smith AM, Filion P, van Hagen D, et al. IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol. (2003) 171:5051–63. doi: 10.4049/jimmunol.171.10.5051
79. Davis MR, Manning LS, Whitaker D, Garlepp MJ, Robinson BW. Establishment of a murine model of malignant mesothelioma. Int J Cancer. (1992) 52:881–6. doi: 10.1002/ijc.2910520609
80. Craighead JE, Akley NJ, Gould LB, Libbus BL. Characteristics of tumors and tumor cells cultured from experimental asbestos-induced mesotheliomas in rats. Am J Pathol. (1987) 129:448–62.
81. Roulois D, Deshayes S, Guilly MN, Nader JS, Liddell C, Robard M, et al. Characterization of preneoplastic and neoplastic rat mesothelial cell lines: the involvement of TETs, DNMTs, and 5-hydroxymethylcytosine. Oncotarget. (2016) 7:34664–87. doi: 10.18632/oncotarget.8970
82. Nader JS, Abadie J, Deshayes S, Boissard A, Blandin S, Blanquart C, et al. Characterization of increasing stages of invasiveness identifies stromal/cancer cell crosstalk in rat models of mesothelioma. Oncotarget. (2018) 9:16311–29. doi: 10.18632/oncotarget.24632
83. Shi Y, Hollenstein A, Felley-Bosco E, Fraefel C, Ackermann M, Soltermann A, et al. Bioluminescence imaging for in vivo monitoring of local recurrence mesothelioma model. Lung Cancer. (2011) 71:370–1. doi: 10.1016/j.lungcan.2010.12.020
84. Crisanti MC, Wallace AF, Kapoor V, Vandermeers F, Dowling ML, Pereira LP, et al. The HDAC inhibitor panobinostat (LBH589) inhibits mesothelioma and lung cancer cells in vitro and in vivo with particular efficacy for small cell lung cancer. Mol Cancer Ther. (2009) 8:2221–31. doi: 10.1158/1535-7163.MCT-09-0138
85. Laszlo V, Valko Z, Ozsvar J, Kovacs I, Garay T, Hoda MA, et al. The FAK inhibitor BI 853520 inhibits spheroid formation and orthotopic tumor growth in malignant pleural mesothelioma. J Mol Med. (2019) 97:231–42. doi: 10.1007/s00109-018-1725-7
86. Pulito C, Korita E, Sacconi A, Valerio M, Casadei L, Lo Sardo F, et al. Dropwort-induced metabolic reprogramming restrains YAP/TAZ/TEAD oncogenic axis in mesothelioma. J Exp Clin Cancer Res. (2019) 38:349. doi: 10.1186/s13046-019-1352-3
87. Abe S, Kaneko MK, Tsuchihashi Y, Izumi T, Ogasawara S, Okada N, et al. Antitumor effect of novel anti-podoplanin antibody NZ-12 against malignant pleural mesothelioma in an orthotopic xenograft model. Cancer Sci. (2016) 107:1198–205. doi: 10.1111/cas.12985
88. Golfier S, Kopitz C, Kahnert A, Heisler I, Schatz CA, Stelte-Ludwig B, et al. Anetumab ravtansine: a novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. (2014) 13:1537–48. doi: 10.1158/1535-7163.MCT-13-0926
89. Krishnan H, Rayes J, Miyashita T, Ishii G, Retzbach EP, Sheehan SA, et al. Podoplanin: an emerging cancer biomarker and therapeutic target. Cancer Sci. (2018) 109:1292–9. doi: 10.1111/cas.13580
90. Scales SJ, Gupta N, Pacheco G, Firestein R, French DM, Koeppen H, et al. An antimesothelin-monomethyl auristatin e conjugate with potent antitumor activity in ovarian, pancreatic, and mesothelioma models. Mol Cancer Ther. (2014) 13:2630–40. doi: 10.1158/1535-7163.MCT-14-0487-T
91. Zhang YF, Phung Y, Gao W, Kawa S, Hassan R, Pastan I, et al. New high affinity monoclonal antibodies recognize non-overlapping epitopes on mesothelin for monitoring and treating mesothelioma. Sci Rep. (2015) 5:9928. doi: 10.1038/srep09928
92. Zhang J, Khanna S, Jiang Q, Alewine C, Miettinen M, Pastan I, et al. Efficacy of anti-mesothelin immunotoxin RG7787 plus nab-paclitaxel against mesothelioma patient-derived xenografts and mesothelin as a biomarker of tumor response. Clin Cancer Res. (2017) 23:1564–74. doi: 10.1158/1078-0432.CCR-16-1667
93. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
94. Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. (2010) 70:9053–61. doi: 10.1158/0008-5472.CAN-10-2880
95. Jung J, Seol HS, Chang S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res Treat. (2018) 50:1–10. doi: 10.4143/crt.2017.307
96. Chahinian AP, Beranek JT, Suzuki Y, Bekesi JG, Wisniewski L, Selikoff IJ, et al. Transplantation of human malignant mesothelioma into nude mice. Cancer Res. (1980) 40:181–5.
97. Wu L, Allo G, John T, Li M, Tagawa T, Opitz I, et al. Patient-derived xenograft establishment from human malignant pleural mesothelioma. Clin Cancer Res. (2017) 23:1060–7. doi: 10.1158/1078-0432.CCR-16-0844
98. Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity. (1995) 2:223–38. doi: 10.1016/1074-7613(95)90047-0
99. Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia. (2015) 17:1–15. doi: 10.1016/j.neo.2014.12.004
100. Kim H, Phung Y, Ho M. Changes in global gene expression associated with 3D structure of tumors: an ex vivo matrix-free mesothelioma spheroid model. PLoS ONE. (2012) 7:e39556. doi: 10.1371/journal.pone.0039556
101. Barbone D, Van Dam L, Follo C, Jithesh PV, Zhang SD, Richards WG, et al. Analysis of gene expression in 3D spheroids highlights a survival role for ASS1 in mesothelioma. PLoS ONE. (2016) 11:e0150044. doi: 10.1371/journal.pone.0150044
102. Barbone D, Yang TM, Morgan JR, Gaudino G, Broaddus VC. Mammalian target of rapamycin contributes to the acquired apoptotic resistance of human mesothelioma multicellular spheroids. J Biol Chem. (2008) 283:13021–30. doi: 10.1074/jbc.M709698200
103. Barbone D, Cheung P, Battula S, Busacca S, Gray SG, Longley DB, et al. Vorinostat eliminates multicellular resistance of mesothelioma 3D spheroids via restoration of noxa expression. PLoS ONE. (2012) 7:e52753. doi: 10.1371/journal.pone.0052753
104. Daubriac J, Fleury-Feith J, Kheuang L, Galipon J, Saint-Albin A, Renier A, et al. Malignant pleural mesothelioma cells resist anoikis as quiescent pluricellular aggregates. Cell Death Differ. (2009) 16:1146–55. doi: 10.1038/cdd.2009.32
105. Gerogianni I, Pitaraki E, Jagirdar RM, Kouliou O, Giannakou L, Giannopoulos S, et al. 2-Deoxy-glucose enhances the effect of cisplatin and pemetrexed in reducing malignant pleural mesothelioma cell proliferation but not spheroid growth. Anticancer Res. (2019) 39:3809–14. doi: 10.21873/anticanres.13530
106. Lei H, Hofferberth SC, Liu R, Colby A, Tevis KM, Catalano P, et al. Paclitaxel-loaded expansile nanoparticles enhance chemotherapeutic drug delivery in mesothelioma 3-dimensional multicellular spheroids. J Thorac Cardiovasc Surg. (2015) 149:1417–24. doi: 10.1016/j.jtcvs.2015.02.020
107. Linot C, Poly J, Boucard J, Pouliquen D, Nedellec S, Hulin P, et al. PEGylated anionic magnetofluorescent nanoassemblies: impact of their interface structure on magnetic resonance imaging contrast and cellular uptake. ACS Appl Mater Interfaces. (2017) 9:14242–57. doi: 10.1021/acsami.7b01737
108. Phung YT, Barbone D, Broaddus VC, Ho M. Rapid generation of in vitro multicellular spheroids for the study of monoclonal antibody therapy. J Cancer. (2011) 2:507–14. doi: 10.7150/jca.2.507
109. Xiang X, Phung Y, Feng M, Nagashima K, Zhang J, Broaddus VC, et al. The development and characterization of a human mesothelioma in vitro 3D model to investigate immunotoxin therapy. PLoS ONE. (2011) 6:e14640. doi: 10.1371/journal.pone.0014640
110. Barbone D, Ryan JA, Kolhatkar N, Chacko AD, Jablons DM, Sugarbaker DJ, et al. The Bcl-2 repertoire of mesothelioma spheroids underlies acquired apoptotic multicellular resistance. Cell Death Dis. (2011) 2:e174. doi: 10.1038/cddis.2011.58
111. Echeverry N, Ziltener G, Barbone D, Weder W, Stahel RA, Broaddus VC, et al. Inhibition of autophagy sensitizes malignant pleural mesothelioma cells to dual PI3K/mTOR inhibitors. Cell Death Dis. (2015) 6:e1757. doi: 10.1038/cddis.2015.124
112. Follo C, Barbone D, Richards WG, Bueno R, Broaddus VC. Autophagy initiation correlates with the autophagic flux in 3D models of mesothelioma and with patient outcome. Autophagy. (2016) 12:1180–94. doi: 10.1080/15548627.2016.1173799
113. Kim KU, Wilson SM, Abayasiriwardana KS, Collins R, Fjellbirkeland L, Xu Z, et al. A novel in vitro model of human mesothelioma for studying tumor biology and apoptotic resistance. Am J Respir Cell Mol Biol. (2005) 33:541–8. doi: 10.1165/rcmb.2004-0355OC
114. Kuen J, Darowski D, Kluge T, Majety M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS ONE. (2017) 12:e0182039. doi: 10.1371/journal.pone.0182039
115. Tevis KM, Cecchi RJ, Colson YL, Grinstaff MW. Mimicking the tumor microenvironment to regulate macrophage phenotype and assessing chemotherapeutic efficacy in embedded cancer cell/macrophage spheroid models. Acta Biomater. (2017) 50:271–9. doi: 10.1016/j.actbio.2016.12.037
116. Rebelo SP, Pinto C, Martins TR, Harrer N, Estrada MF, Loza-Alvarez P, et al. 3D-3-culture: a tool to unveil macrophage plasticity in the tumour microenvironment. Biomaterials. (2018) 163:185–97. doi: 10.1016/j.biomaterials.2018.02.030
117. de Bono JS, Ashworth A. Translating cancer research into targeted therapeutics. Nature. (2010) 467:543–9. doi: 10.1038/nature09339
118. Moreno L, Pearson AD. How can attrition rates be reduced in cancer drug discovery? Exp Opin Drug Discov. (2013) 8:363–8. doi: 10.1517/17460441.2013.768984
119. Graham ML, Prescott MJ. The multifactorial role of the 3Rs in shifting the harm-benefit analysis in animal models of disease. Eur J Pharmacol. (2015) 759:19–29. doi: 10.1016/j.ejphar.2015.03.040
120. Jean-Quartier C, Jeanquartier F, Jurisica I, Holzinger A. In silico cancer research towards 3R. BMC Cancer. (2018) 18:408. doi: 10.1186/s12885-018-4302-0
121. Sun W, Luo Z, Lee J, Kim HJ, Lee K, Tebon P, et al. Organ-on-a-chip for cancer and immune organs modeling. Adv Healthc Mater. (2019) 8:e1801363. doi: 10.1002/adhm.201801363
122. Begley CG, Ellis LM. Drug development: raise standards for preclinical cancer research. Nature. (2012) 483:531–3. doi: 10.1038/483531a
Keywords: thoracic cancer, mesothelioma, molecular characteristics, tumor heterogeneity, preclinical models, cell models, animal models
Citation: Blanquart C, Jaurand M-C and Jean D (2020) The Biology of Malignant Mesothelioma and the Relevance of Preclinical Models. Front. Oncol. 10:388. doi: 10.3389/fonc.2020.00388
Received: 03 September 2019; Accepted: 04 March 2020;
Published: 25 March 2020.
Edited by:
Paul Baas, The Netherlands Cancer Institute (NKI), NetherlandsReviewed by:
Arnold Manfred Herskovic, Rush University, United StatesCopyright © 2020 Blanquart, Jaurand and Jean. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Didier Jean, ZGlkaWVyLmplYW5AaW5zZXJtLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.