 Harriet E. D. Southgate
Harriet E. D. Southgate Lindi Chen1
Lindi Chen1 Deborah A. Tweddle
Deborah A. Tweddle
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 03 April 2020
Sec. Pediatric Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00371
This article is part of the Research Topic Novel Molecular Targets and Therapies for Pediatric Solid Tumors View all 14 articles
Despite intensive multimodal therapy, the survival rate for high risk neuroblastoma (HR-NB) remains <50%. Most cases initially respond to treatment but almost half will subsequently relapse with aggressive treatment resistant disease. Novel treatments exploiting the molecular pathology of NB and/or overcoming resistance to current genotoxic therapies are needed before survival rates can significantly improve. DNA damage response (DDR) defects are frequently observed in HR-NB including allelic deletion and loss of function mutations in key DDR genes, oncogene induced replication stress and cell cycle checkpoint dysfunction. Exploiting defects in the DDR has been a successful treatment strategy in some adult cancers. Here we review the genetic features of HR-NB which lead to DDR defects and the emerging molecular targeting agents to exploit them.
Neuroblastoma (NB) is a rare childhood cancer derived from cells of the embryonal neural crest. Many cancers, including NB, show a high number of chromosomal genetic abnormities, including rearrangement of chromosomes or gains and losses of whole or parts of chromosomes and less frequently changes to the nucleic acid sequence. This dynamic process is known as genome instability and is described as an enabling characteristic allowing cancer cells to acquire six major hallmarks required for survival and proliferation, first outlined by Hanahan and Weinburg in 2000 and updated in 2011. These hallmarks include self-sufficiency in growth signals, insensitivity to anti-growth signals, infinite replication, evasion of apoptosis, angiogenesis, and tissue invasion and metastasis (1, 2).
Genomic instability can arise due to defects in the DNA damage response (DDR) (3). The DDR is a highly orchestrated network which signals DNA damage to cell cycle checkpoints (G1/S, intra-S and G2/M) resulting in cell cycle arrest (4). Dysregulation of cell cycle control allows for mutations to accumulate. The combination of genetic instability and loss of cell cycle control results in a “mutator phenotype” in which mutations are frequently established and maintained (5). Loss of G1 checkpoint control, through mutations in the TP53 or RB tumor suppressor genes, activation of oncogenes such as Ras or MYC, or imbalance in G1/S cyclins, cyclin-dependent kinases (CDKs) and their inhibitors, is a common feature of cancer cells (6), making these cells dependent on the G2 checkpoint for survival after DNA-damaging treatments.
Another cause of chromosome instability is DNA replication stress (7), a state in which the DNA replication machinery cannot maintain the rate of DNA synthesis resulting in increased replication fork stalling and collapse (8, 9). Replication stress is common in NB, and many other cancers, due to the overexpression of oncogenes driving rapid proliferation and loss of G1 checkpoint control, and provides an exploitable cancer-specific defect. Targeting the DDR can not only exploit cancer-specific defects, but could also overcome resistance to cytotoxic chemo- and radiotherapy resulting from upregulation of DNA repair pathways in cancer (10).
NB is the commonest extra-cranial malignant solid tumor of infancy and accounts for 8% of all childhood (0–14 years) cancers in the UK (11). Around 100 new cases are diagnosed each year in the UK. Tumors usually appear in very young children, the median age of diagnosis being 17 months (12). NB is a neuroendocrine tumor derived from precursor cells of the sympathetic nervous system resulting in tumors in the adrenal glands or sympathetic ganglia (13). Most NB tumors present in the abdomen but can also appear in the neck, chest or pelvis in paraspinal regions. Tumors are highly heterogeneous both phenotypically and clinically, with outcome varying from maturation or spontaneous regression to aggressive progression (14). In addition to a variety of molecular markers associated with outcome (discussed in section Genetics of Neuroblastoma), studies have shown that the degree of tumor cell differentiation is related to patient survival (15). Pathologically, tumors show varying degrees of differentiation from NB which is predominantly composed of undifferentiated or poorly differentiated small round tumor cells to ganglioneuroblastoma intermixed, which consists of both immature cells and terminally differentiated ganglion cells to a mature ganglioneuroma (13). Tumors showing a higher degree of cell differentiation usually have a better prognosis than undifferentiated tumors. Tumor differentiation, age at diagnosis, tumor stage and molecular abnormalities are variables used to classify NB into risk groups which define treatment strategies (discussed in section Neuroblastoma Risk Stratification).
Amplification of the MYCN oncogene, either as intra-chromosomal homogenously staining regions (HSRs) or as extrachromosomal double minutes (16), is seen in around 20% of all NB cases and is one of the strongest unfavorable prognostic markers (17). The frequency of MYCN amplification increases to around 50% in the high-risk group (18). MYCN is a member of the MYC family of proto-oncogenes which also includes c-MYC and MYCL (19–21). The MYC family of proteins are basic-helix–loop–helix-leucine zipper (bHLH-LZ) transcription factors which mediate mitogen signaling by regulating transcription of target genes involved in metabolism, protein biosynthesis, cell cycle regulation, DNA repair, cell adhesion, and the cytoskeleton (22, 23). They therefore have a critical role in cellular proliferation, differentiation, apoptosis, and oncogenesis. In contrast to c-MYC, which is expressed in a variety of embryonal and adult tissues, expression of MYCN is restricted to the developing nervous system and only a few other sites (24–26). Ectopic expression of MYCN drives cell proliferation but also leads to sensitization to apoptosis through activation of the tumor suppressor protein p53 (27), therefore mechanisms to evade MYCN induced apoptosis are essential for NB development [reviewed by (28)]. This may be achieved by loss of expression of the initiator caspase, caspase 8 (29–31), which mediates the extrinsic death receptor apoptosis pathway (32, 33). A functional MYCN/c-MYC signature also characterizes a fraction of aggressive NB without MYCN amplification (34, 35), which suggests that increased MYC activity is a main driver of aggressiveness in neuroblastoma.
Increased expression of MYC oncogenes drives rapid, erroneous replication leading to replication stress (36).
Many diploid and tetraploid NB tumors show numerous non-random structural chromosome alterations, such as deletion of chromosomes 1p, 3p, 4p, 11q, and gain of 1q, 2p, and 17q, which are associated with poor prognosis (37–39). Gain of chromosome 17q and loss of chromosome 1p are observed in half and a third of NB cases, respectively, and correlate with MYCN amplification and poor prognosis (40, 41). 11q loss is also observed in about third of NB tumors and is a marker of poor prognosis independent of MYCN status (discussed in section 11q Loss) (40). Chromosome 2p is the location of both the MYCN and ALK genes (discussed in section ALK and MAP Kinase Pathways) (42), therefore gain of 2p could contribute to overexpression of both of these genes. In general, the presence of structural chromosome alterations, in contrast to whole chromosome gains or losses (numerical chromosome alterations), is associated with advanced stage of disease and inferior outcome due to the former being associated with genomic instability whereas the latter is associated with mitotic defects (38).
A common structural chromosome aberration is 11q loss, which is seen in around 30–40% of NBs. Many high risk, non-MYCN amplified NB tumors show 11q deletion. MYCN amplification and 11q loss rarely occur together, suggesting a degree of mutual exclusivity. The smallest region of overlap in 11q deletions has been reported between 11q14 and 11q23 (43) including genes such as CADM1 (11q23.3), and 4 genes involved in the DDR: ATM (11q22.3), CHK1 (11q24.2), MRE11 (11q21), and H2AFX (11q23.3), which have been functionally tested as candidate genes responsible for driving NB tumorigenesis [Figure 1; (44)]. No mutation or hyper-methylation was found in the other allele of these genes in most cases (44), however loss of one copy via 11q deletion could contribute to tumorigenesis due to haploinsufficiency.
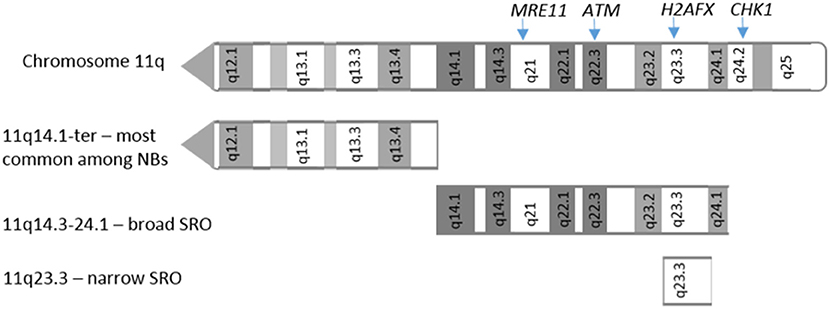
Figure 1. 11q deletions in neuroblastoma. Adapted from Mlakar et al. (44). Location of 11q arm deletions observed in neuroblastoma tumors. SRO: shortest region of overlap.
Homozygous germline mutations in ATM (ataxia-telangiectasia mutated) cause ataxia telangiectasia (A-T), a recessive genetic disease characterized by cerebellar degeneration, chromosomal instability and cancer predisposition. ATM is a key DDR protein which signals to DNA repair machinery and leads to cell cycle arrest in response to double strand DNA breaks (DSBs) [reviewed in (45)]. Furthermore, somatic mutations of ATM have been identified in many cancer types, most commonly lymphoid malignancies (46), suggesting that ATM loss contributes to tumorigenesis. ATM deficiency results in impaired cell cycle arrest through disrupted signaling to p53 (discussed section p53 Pathway) (47, 48), enabling mutations to accumulate in the genome.
CHK1, MRE11, and H2FAX also encode proteins integral to the DDR. Chk1 is the primary target of ataxia-telangiectasia and rad3-related (ATR) kinase which, similar to ATM, signals to DNA repair and cell cycle checkpoints in response to DNA damage. Mre11 is part of the Mre11-Rad50-Nbls1 (MRN) complex, a multiprotein complex which regulates repair of DSBs. In contrast to ATM, Mre11 is essential for cell survival and homozygous hypomorphic Mre11 mutations lead to a genetic disease which is phenotypically similar to A-T called ataxia telangiectasia-like disorder (49). H2AFX encodes a variant of histone 2A which can be phosphorylated. H2AX is phosphorylated in response to DSB formation by DDR kinases, such as ATM, which results in the recruitment of DNA repair machinery and chromatin remodeling complexes and the amplification of the signal by spreading along chromatin (50). H2AX deficiency leads to chromosome instability.
Overall, heterozygous loss of 11q results in loss of DDR proteins which leads to chromosome instability allowing cancer to develop.
The tumor suppressor protein p53 is one of the main downstream targets of the ATM signaling pathway. p53 is a transcription factor that has a crucial role in maintaining genome integrity and tumor suppression and is activated in response to a variety of intra- and extra-cellular stresses, including DNA damage, oncogene activation, oxidative stress, deficient growth factors/signals, ribonucleotide depletion, and hypoxia (51). Activated p53 regulates transcription of genes involved in cell cycle arrest, apoptosis, senescence, DNA repair, differentiation, angiogenesis, metastasis and metabolism (52–54). TP53, is the most frequently mutated gene in human cancer (55).
Loss of p53 function is also seen in tumors with wild-type p53 (52). Upregulation of MDM2, an E3 ubiquitin ligase which targets p53 for degradation (56), or loss of p14ARF function, which inhibits MDM2 (57, 58), causes decreased p53 stability and reduced p53 function (Figure 2).
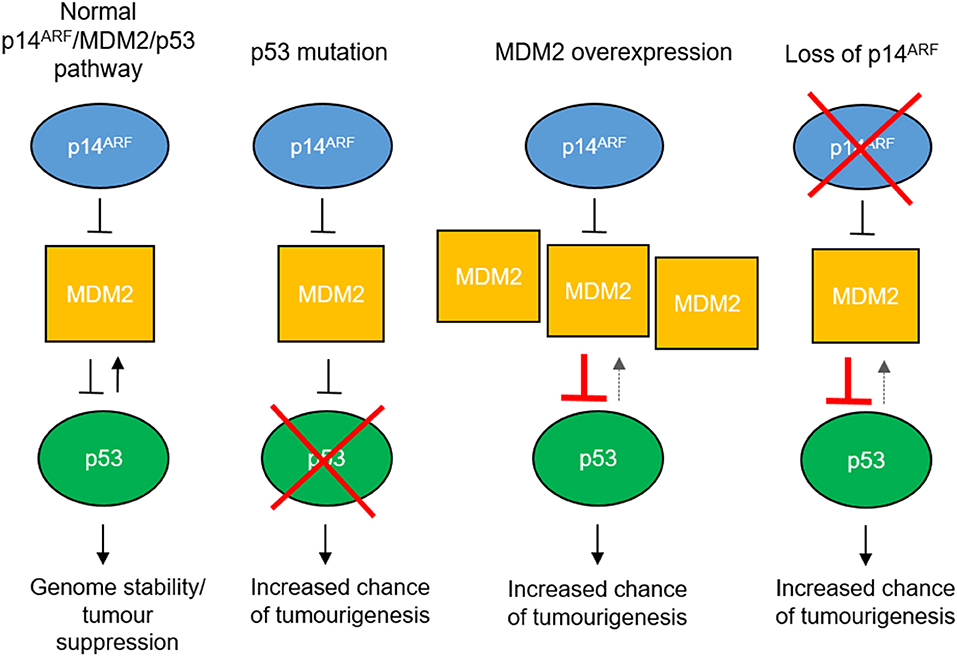
Figure 2. Mechanisms of p53 pathway dysfunction. The p53 pathway can be disrupted in cancer by mutation of the TP53 gene, overexpression of MDM2 e.g., by gene amplification or loss of p14ARF expression by gene methylation or deletion.
In neuroblastoma, TP53 mutations are rare at diagnosis, however aberrations in the p53 pathway are observed more frequently at relapse (59–62), where around 50% of relapsed cases analyzed show TP53 mutation, MDM2 amplification or p14ARF inactivation, suggesting that p53 inactivation could be a contributor to acquired drug resistance. In addition, TP53 is located on chromosome 17p13.1 and allelic loss of 17p has been observed in both NB cell lines and tumors (63, 64), more frequently in cell lines derived at relapse (61), indicating that 17p loss could be a mechanism by which p53 function is reduced.
Telomere maintenance is essential for establishment of high risk NB (65). High telomerase expression indicates increased invasiveness and poor prognosis (66), comparable to MYCN amplified tumors (67). Rearrangements at the TERT gene locus (5p15.33) are frequent in NB resulting in overexpression of the TERT gene and subsequent increased telomerase expression (67, 68). In contrast to other cancers, mutations in the TERT promoter region are rare in NB primary tumors and cell lines (69) and TERT activation is most likely achieved by amplification or juxtapositions of TERT to strong enhancer elements (70, 71). TERT is known to be a transcriptional target of MYCN (72), and MYCN amplified NB cells show increased TERT expression in comparison to non-MYCN amplified in the absence of TERT rearrangements (70).
In high risk NB tumors which do not express telomerase, a recombination mediated mechanism known as alternate lengthening of telomeres (ALT) is activated. ALT activity in NB is associated with mutations in the α-thalassaemia/mental retardation syndrome X-linked (ATRX) gene (73–75). Loss of function mutations in ATRX are among the most common genetic lesions in NB (73, 76). ATRX encodes an RNA-helicase which plays a role in chromatin remodeling, nucleosome assembly and telomere maintenance (77). ATRX mutation has been shown to be mutually exclusive with MYCN amplification (78) and is often seen in tumors from older patients, such as adolescent or young adult but have also been observed in children over 5 years, and is associated with a chronic or indolent disease course (73). Identifying ATRX mutations could define a subset of NB cases in which the ALT pathway could be targeted to improve treatment. Recently, loss of ATRX function has not only been shown to be mutually exclusive with MYCN amplification, but also incompatible with overexpression of the MYCN protein due to intolerable levels of replication stress (79). This suggests potential synthetically lethal approaches that could be explored by targeting ATRX function in MYCN-driven tumors, or inducing MYCN-related metabolic changes in ATRX mutant NB.
Activating mutations in the anaplastic lymphoma kinase (ALK) gene have been reported in 50% of familial NB (familial NB is rare accounting for around 2% of NB cases) and between 8 and 10% of sporadic neuroblastoma, across all risk groups and occurring more frequently at relapse (42, 78, 80–82). In neuroblastoma, the constitutive activation of ALK, and subsequent downstream pathways, have been shown to be involved in cell proliferation, inducing replication stress, migration, and invasion (83). As well-mutations, aberrant ALK activity has also been reported through ALK amplification (84, 85). ALK amplification has been shown to be accompanied by MYCN amplification and there is evidence that ALK activation accelerates MYCN driven tumorigenesis in animal models (84, 85). ALK is a receptor tyrosine kinase (RTK) specifically expressed in the developing nervous system (86, 87). Like other RTKs, ligand binding leads to receptor activation by dimerization and auto-phosphorylation, recruitment of adaptor proteins and downstream signal transduction through pathways such as PI3K/AKT, RAS/MAPK, and JAK/STAT (88–90). In addition to ALK aberrations, mutations in components of the RAS-ERK/MAPK pathway are frequently observed at relapse and are likely contributors to therapy resistance (91). These mutations include activating mutations in BRAF, RAS (KRAS and HRAS), and PTPN11 (78), which encodes the tyrosine phosphatase SHP-2, and inactivating mutations in the NF1 tumor suppressor gene (91), a negative regulator of RAS (92). Activation of the RAS signaling pathway leads to replication stress by driving DNA replication, by a similar mechanism to MYCN overexpression (93, 94).
At diagnosis, NB cases are categorized into three risk groups, low, intermediate and high risk according to the International Neuroblastoma risk group (INRG) classification system on the basis of age at diagnosis, tumor stage, histopathology and molecular abnormalities including MYCN status and DNA copy number abnormalities (17). The probability of disease free survival for each group is 95–100%, 85–90%, and <50%, respectively (95). High risk NB (HR-NB) accounts for around 50% of all NB cases (17) and, despite intensive multi-modal therapy, only 50% of patients with HR-NB are cured (96). New treatments and a better understanding of drug resistance are needed before these survival rates can significantly improve. Risk category defines which treatment strategy to follow and correlates with outcome, with low risk showing the best outcome and high risk showing the poorest (97).
Treatment strategies in NB are defined by risk classification. Low risk disease will often spontaneously regress and generally results in a good outcome with clinical observation or surgical resection alone. For intermediate risk, treatment regimens are response dependent and vary from 4 to 8 cycles of conventional chemotherapy, which is often at lower doses than high risk regimens, and the primary tumor is surgically resected where possible.
HR-NB is currently treated with a number of different DNA damaging agents during induction and consolidation according to the previous European High risk NB trial (HR-NBL1, NCT01704716) including cisplatin, carboplatin, etoposide, vincristine, cyclophosphamide, topotecan and doxorubicin during induction, and high dose busulfan and melphalan myeloablative therapy with autologous stem cell rescue followed by local radiotherapy to the site of the primary tumor during consolidation.
Immunotherapy with the anti-GD2 chimeric mono-clonal antibody Dinutuximab was approved in 2015 by the Food and Drug Administration (FDA) for maintenance treatment in combination with GM-CSF and 13-cis retinoic acid for pediatric HR-NB patients (98). Dinutuximab beta was also recommended by the National Institute for Health and Care Excellence (NICE) in 2018 for maintenance treatment of HR-NB.
With these regimens the majority of patients will respond to treatment but over 50% of cases will relapse and very few relapsed patients can then be cured (96, 99). Presently at relapse patients are given a backbone chemotherapy of temozolomide and irinotecan to which new agents are added.
Due to the intensive treatment of high risk disease, surviving patients often suffer from multiple sequelae (100). Selective inhibitors of cancer specific aberrant pathways have the potential to replace these conventional chemotherapeutics or decrease the dose required for therapeutic effect, thus reducing the toxic side effects of HR-NB treatment.
The differential response of cancers to current anti-cancer therapies are likely to be dependent on the DNA damage response (DDR). Although some DDR dysfunction enables cancer development and increased therapeutic resistance (101), defects in particular pathways are exploitable with the appropriate conventional therapy or novel agents targeting components of the DDR [reviewed by (102)], selectively killing the cancer cells. Cells from HR-NB tumors show a high degree of chromosome instability in the form of segmental chromosome aberrations, including allelic gains, and losses of chromosomes and regional amplifications (38).
The DDR is a highly orchestrated signaling system which detects DNA damage and signals to cellular responses including cell cycle checkpoint arrest, DNA repair, and apoptosis (103). It has evolved to allow cells to survive high levels of endogenous and environmental DNA damage and prevent damaged DNA being copied and passed on to daughter cells.
DNA damage sensors initiate cell cycle arrest by the activation of downstream signaling pathways. Two of these sensors are ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR). The primary targets of ATM and ATR are the checkpoint kinases CHK2 and CHK1, respectively, which signal to checkpoint arrest by regulation of the proteins involved in cell cycle progression, demonstrated in Figure 3.
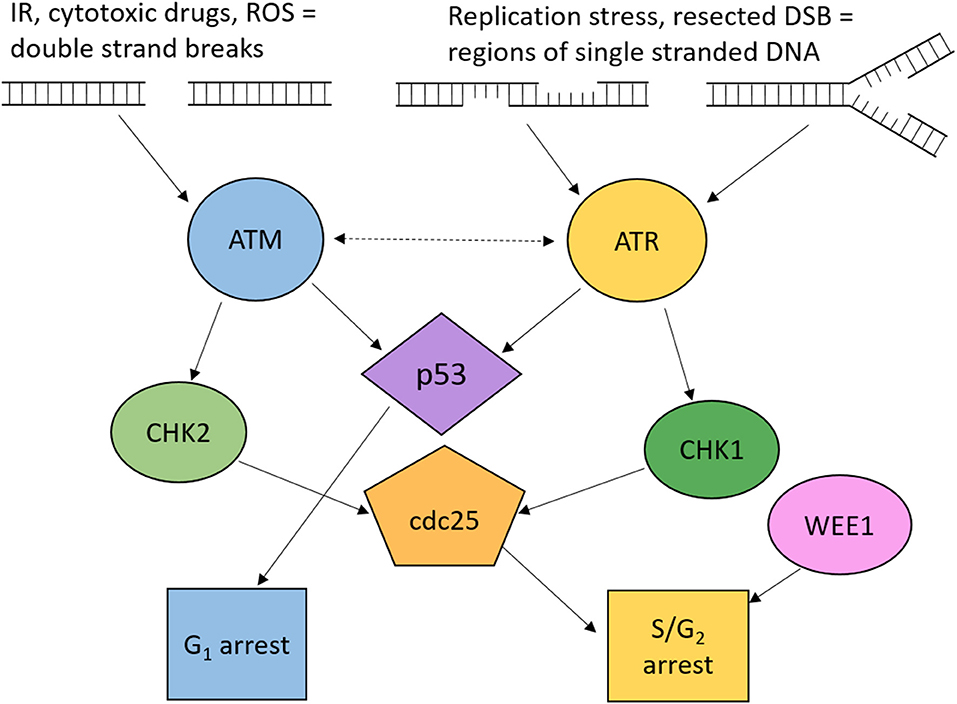
Figure 3. Overview of ATM and ATR signaling to cell cycle checkpoint arrest. Cell cycle arrest is induced through ATM and ATR dependent phosphorylation of p53, CHK1, and CHK2. Active p53 induces G1 arrest. Active CHK1 and CHK2 phosphorylate Cdc25 phosphatases resulting in S and G2 arrest. Wee1 kinase is also key to maintaining G2 cell cycle arrest. ROS, reactive oxygen species; DSB, double strand breaks; IR, ionizing radiation.
ATM is activated in response to DSBs and plays a crucial role in the activation of the G1/S cell cycle checkpoint which is primarily mediated through p53 activity. ATM can also signal to S and G2/M checkpoints via CHK2. CHK2 phosphorylates cdc25A, preventing S phase progression, and cdc25C, preventing the transition into mitosis [Figure 3; (104)].
ATR is activated by regions of single-stranded DNA (ssDNA), such as regions at stalled replication forks or formed by resection during DNA repair by homologous recombination repair (HRR) (45). It signals to CHK1 which phosphorylates cdc25A and cdc25C, leading to their inhibition (105). Although many ATR substrates overlap with ATM, loss of ATR is embryonically lethal, whereas loss of ATM is viable, therefore some roles of ATR are essential. One of the essential functions is the role of ATR in survival through replication stress. In addition to inducing cell cycle arrest, ATR prevents replication origin firing, thus reducing the number of active forks, maintains stability of stalled replication forks and promotes replication restart (106).
Endogenous DNA damage is repaired by a number of pathways specific to the type of damage sustained. These pathways work together to ensure any damage to DNA is repaired with high fidelity to maintain genome integrity. DNA lesions caused by DNA damaging chemotherapeutic agents are also repaired by these pathways. Table 1 outlines the mechanism by which cytotoxic agents used in the treatment of HR-NB inflict DNA damage and the pathways involved in subsequent DNA repair.
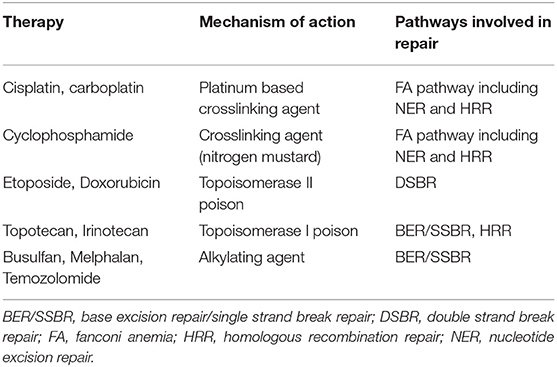
Table 1. DNA damaging mechanism of chemotherapeutic agents used in the treatment of high risk NB (107).
DNA lesions affecting one strand, such as single strand breaks (SSBs), base deamination, oxidation, methylation or loss, bulky DNA adducts or intra-strand cross links are repaired by base excision repair (BER) (108), SSB repair (SSBR) (109), and nucleotide excision repair (NER) (110). Poly (ADP-ribose) polymerases 1 and 2 (PARP1 and PARP2) signaling is required for efficient SSBR (111) for which many inhibitors have been developed (section Cell Cycle Checkpoint Signaling and Replication Stress).
Errors made during replication, known as mismatches, are repaired by the mismatch repair (MMR) pathway. In this pathway, errors in the newly synthesized strand are removed and the DNA is resynthesized by the replication machinery (112). Bulky abducts which remain unrepaired at DNA replication are bypassed by a process known as translesion synthesis (TLS), which allows restoration of the double stranded DNA prior to NER (113).
DNA damage affecting both DNA strands, such as double strand breaks (DSBs), replication stress and interstrand cross-links are repaired by non-homologous end joining (NHEJ) (114), homologous recombination repair (HRR) (115) and the Fanconi anemia (FA) pathway. The FA pathway is required for the repair of interstrand cross-links and is mediated by the large multimeric FA complex. The TLS, HRR, and NER pathways are also required to repair the DNA after excision of the cross-link from one DNA strand (116).
Figure 4 shows an overview of DDR defects in HR-NB. Among the most common lesion is allelic loss of chromosome 11q. Many DDR proteins are encoded on 11q and are included within the smallest region of overlap, including ATM, CHK1, MRE11, and H2AFX. Although no mutation or hyper-methylation (silencing) was found in the other allele of these genes in most cases (44), loss of one copy via 11q deletion could result in reduced expression of these proteins, compromising DNA damage signaling and DSB repair, and contributing to replication stress.
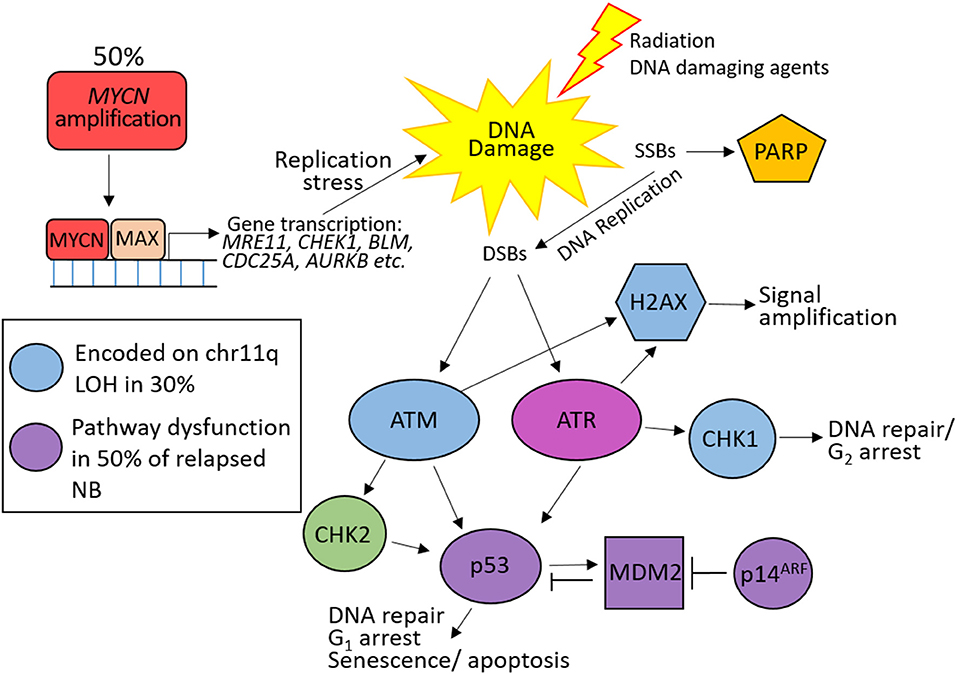
Figure 4. DNA damage response defects in high risk neuroblastoma (HR-NB). 50% of HR-NB are MYCN amplified resulting in increased expression of DNA repair genes (MRE11, CHEK1, BLM etc.) and genes driving proliferation (CDC25A, AURKB etc.), leading to replication stress and DNA damage through replication errors. 30% harbor 11q loss of heterozygosity (LOH) resulting in reduced expression of many proteins involved in the response to double strand breaks (blue). p53 pathway (purple) dysfunction is common at NB relapse leading to defective G1 checkpoint arrest.
Around 50% of HR-NB have an amplification of the MYCN oncogene, which drives proliferation and causes replication stress (117). MYCN also transcriptionally upregulates many proteins involved in DNA DSB repair, including components of the MRE11-RAD50-NBLS1 (MRN) complex (118, 119), alternative NHEJ (alt-NHEJ) (120), and Bloom syndrome (BLM) helicase (121), and the cell cycle checkpoint protein CHK1 (117, 122). Upregulation of these genes likely provide MYCN-driven tumors the ability to tolerate higher levels of DNA damage and replication stress. 11q loss is rarely observed in MYCN–amplified NB therefore this subset of HR-NB could show benefit from treatments targeting ATM (which is activated by MRN) and/or CHK1.
Loss of G1 checkpoint control in NB, through reduced ATM expression, loss of p53 function, and overexpression of MYCN (promotes premature S phase entry and increases replication stress) result in intra-S and G2/M checkpoint dependency in these cells, in order to prevent mitosis with damaged DNA, and are therefore especially vulnerable to its inhibition. In addition to ATM loss, MYCN induces ATM downregulation by miR-421 (123). Targeting tumor specific DDR defects with PARP and ATR inhibitors in the treatment of HR-NB could potentially increase survival in this risk group.
Inhibition of PARP, the enzyme which promotes the repair of DNA single strand breaks, selectively kills cells defective in homologous recombination repair (HRR), e.g., due to BRCA mutation. This is due to synthetic lethality when the function of two complementary pathways are inactivated. In normal cells, blocking the repair of SSBs by PARP inhibition will result in a single ended DSB when the DNA replication machinery reaches this lesion, which is then repaired by HRR. Cancer cells defective in HRR cannot repair this break resulting in cell death. Four PARP inhibitors have been approved to date, Lynparza (olaparib, Astra Zeneca), Rubraca (rucaparib, Clovis oncology), and Zejula (niraparib, Tesaro), for the treatment of platinum sensitive ovarian cancer (124) and Talzenna (talazoparib, Pfizer) for the treatment of germline BRCA mutated, HER2 negative breast cancer (125). Other PARP inhibitors, veliparib (Abbvie), and pamiparib (BeiGene) are being investigated in clinical trials, with veliparib having advanced to phase 3.
There is accumulating evidence in favor of introducing PARP inhibitors to HR-NB treatment regimens. In 2009, we showed that the PARP inhibitor rucaparib potentiated the cytotoxic effect of temozolomide and topotecan in both in vitro and in vivo models of NB (126). Subsequent studies have also shown that PARP inhibition increases sensitivity to a variety chemotherapeutic agents and to ionizing radiation in preclinical models of NB (127, 128).
PARP inhibitors have been reported to be synthetically lethal in cells with 11q deletions and ATM mutations in lymphoid tumors (129). Recent studies in preclinical models of NB have also shown that 11q loss confers sensitivity to PARP inhibitors (130, 131), further supporting the hypothesis that heterozygous loss of ATM and other DDR genes determines sensitivity to PARP inhibition.
In addition to 11q, Colicchia et al. showed that PARP inhibition enhances replication stress in MYCN amplified cells and leads to increased cell death through mitotic catastrophe as these cells enter S-phase with damaged DNA (132). The mechanism suggested in this paper defines another subgroup of HR-NB tumor whereby PARP inhibitors might be beneficial therapeutically.
Early phase international clinical trials are currently testing the efficacy of PARP inhibitors (PARPi) for the treatment of childhood solid tumors with defects in DDR genes, including NB. These are summarized in Table 2.

Table 2. PARP inhibitors currently in pediatric clinical trials.
However, it is worth noting that the combination of PARPi with conventional chemotherapy in adults leads to increased hematological toxicity (133), with doses of the PARP inhibitor and cytotoxic chemotherapy combination subsequently being reduced. This observation is reflected in the results of a pediatric trial combining veliparib (ABT-888) with temozolomide in brain tumors (134), where the main dose limiting toxicity was myelosuppression. In the case of NB (and other pediatric tumors), a reduction in chemotherapy doses when combined with a PARP inhibitor might be advantageous in reducing the long term toxicity of these drugs, if efficacy is maintained.
Replication stress and defects in G1 cell cycle control render cells highly dependent on ATR and hence should be sensitive to its inhibition (135). Four inhibitors of ATR are now in clinical trials: M6620 (berzosertib, formally VX-970, Merck), M4344 (formally VX-803, Merck), AZD6738 (ceralasertib, Astra Zeneca), and BAY1895344 (Bayer) (Table 3).
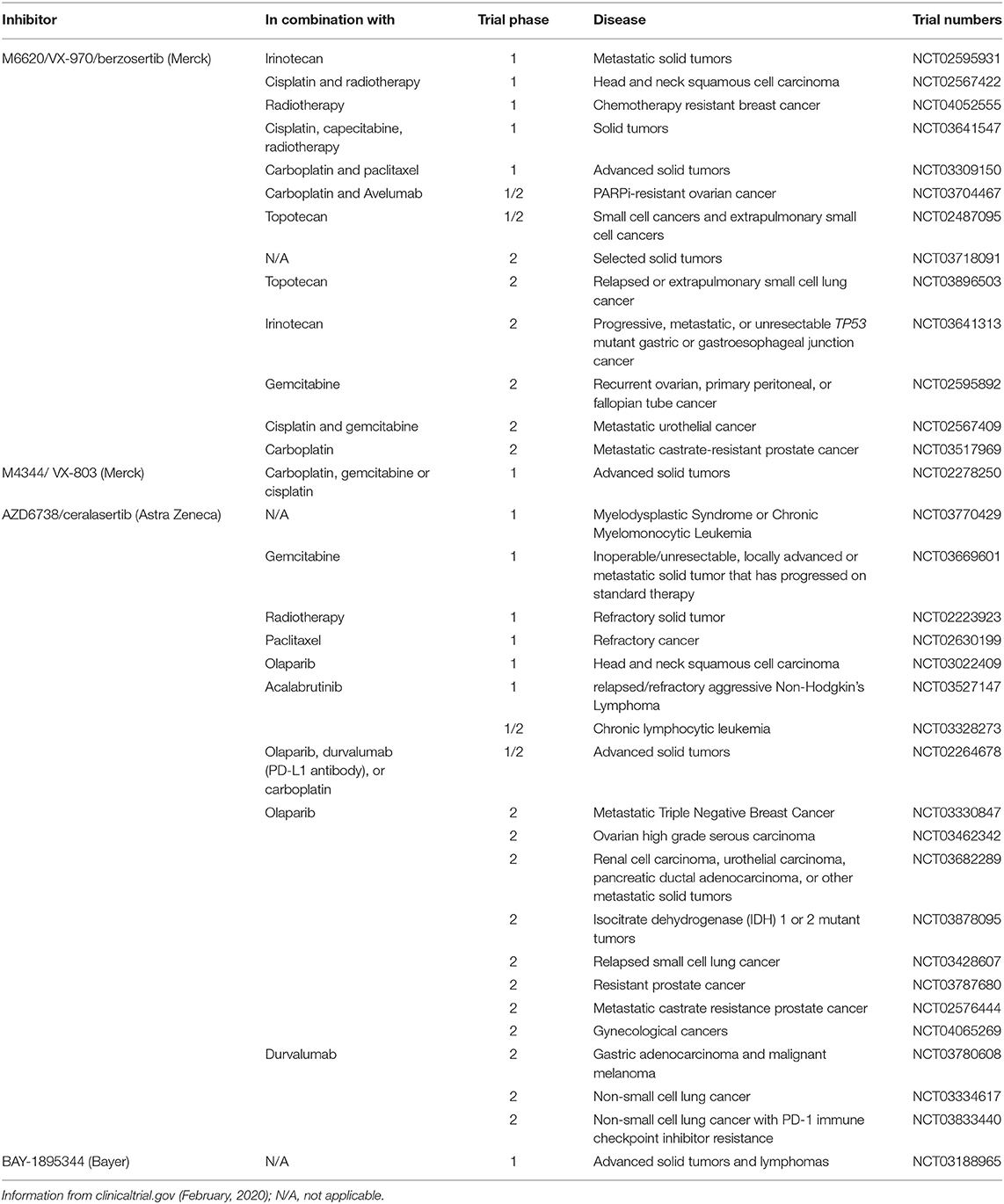
Table 3. ATR inhibitors currently in adult clinical trials.
Both amplification of MYCN and impaired ATM function, which result in replication stress and defects in G1 cell cycle control, are known determinants of sensitivity to ATR inhibitors (105, 136). There is some evidence that chemosensitization by ATR inhibitors relies on a dysfunctional p53 pathway, and therefore a defective G1/S checkpoint (105, 137). p53 pathway dysfunction is rare in NB at diagnosis but frequent abnormalities are observed at relapse (59). Collectively, MYCN amplification and allelic 11q deletion are observed in 70–80% of HR-NB tumors (44), suggesting a large group of HR-NB patients may benefit from treatment with ATR inhibitors.
Inhibition of ATR has been shown to mediate sensitivity to PARP inhibition (138, 139). PARP inhibition results in DNA DSBs in S-phase, which require activity of ATR signaling to S phase cell cycle arrest and HRR for repair. ATR inhibition has also been shown to overcome acquired resistance to PARP inhibitors (140, 141). In theory, the combination with ATR inhibitors should potentiate the cytotoxic effects of PARP inhibitors in the treatment of NB.
It has been suggested that cancer cells which maintain their telomeres by alternative lengthening have increased sensitivity to ATR inhibition (142). ALT is found in 50% of NB cells which harbor loss of function mutations or intragenic deletions in ATRX (74, 75). ATRX mutation has been observed in around 25% of HR-NB (73). However, it has subsequently been reported that ALT is not an independent determinant of ATR inhibitor sensitivity (143). At present, it is unclear whether ATRX loss of function will define a subset of NB cells sensitive to ATR inhibition due to telomere maintenance by ALT.
The efficacy of the ATR inhibitor clinical candidate from Merck, M6620 (formerly VX-970, Vertex), has recently been tested alone and in combination with cisplatin and melphalan in a range of pediatric solid tumor cell lines and xenograft models including NB (144). This study showed that M6620 had limited single agent cytotoxicity but potentiated the cytotoxic effects of cisplatin and melphalan in the majority of cell lines tested. Although limited, this study indicated that ATR inhibitors could potentially be beneficial when used in combination with existing chemotherapeutic regimens.
Further studies are required to determine which molecular abnormalities confer sensitivity to ATR inhibition in NB.
CHK1 kinase is the direct downstream effector of ATR. MYCN amplified NB cell lines show sensitivity to CHK1 inhibitors as single agents (145, 146) and as a chemosensitizer to cytotoxic agents (145, 147). Although CHK1 inhibitors have been in clinical development for many years, many compounds have been discontinued before Phase 3 trials due to toxicities. Two CHK1 inhibitors, Prexasertib (LY2606368; Eli Lilley) and SRA737 (Sierra Oncology) are currently being tested in clinical trials, with Prexasertib having entered a Phase 1 clinical trial in pediatric solid tumors (NCT0280865, NCT04023669).
WEE1 is a key kinase in the activation of the S and G2/M cell cycle checkpoints in response to DNA damage. Phosphorylation of CDK1 by WEE1 keeps CDK1 in an inactive state, thus preventing entry into mitosis (148). Single agent treatment with the WEE1 inhibitor Adavosertib was shown to be effective in both in vitro and in vivo preclinical NB models (145). In the same study, Adavosertib was shown to be synergistic with the CHK1 inhibitor MK-8776, the topoisomerase I poison SN-38 (active metabolite of irinotecan) and gemcitabine.
Adavosertib is currently the only WEE1 inhibitor in clinical development and has advanced into Phase II clinical trials for the treatment of pediatric solid tumors including NB (Table 4).
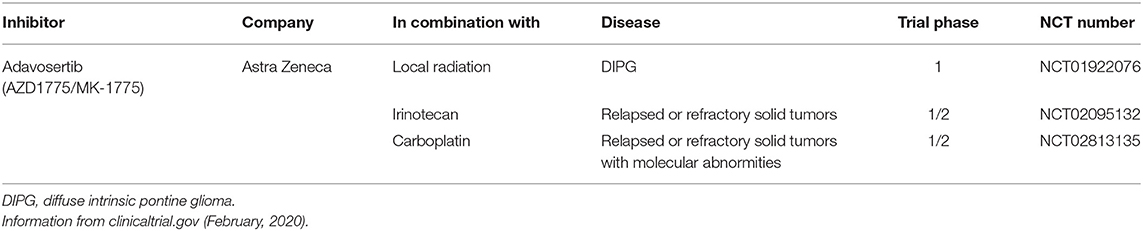
Table 4. Current pediatric clinical trials of the WEE1 inhibitor Adavosertib.
Since ATR inhibition has been shown to overcome PARPi resistance by abrogating the G2 checkpoint, there are many clinical trials testing this combination. PARP inhibition increases replication stress (132), which would also render cells dependent on ATR inhibition. A summary of currently listed clinical trials involving a combination of PARP and ATR inhibitors is listed in Table 5.
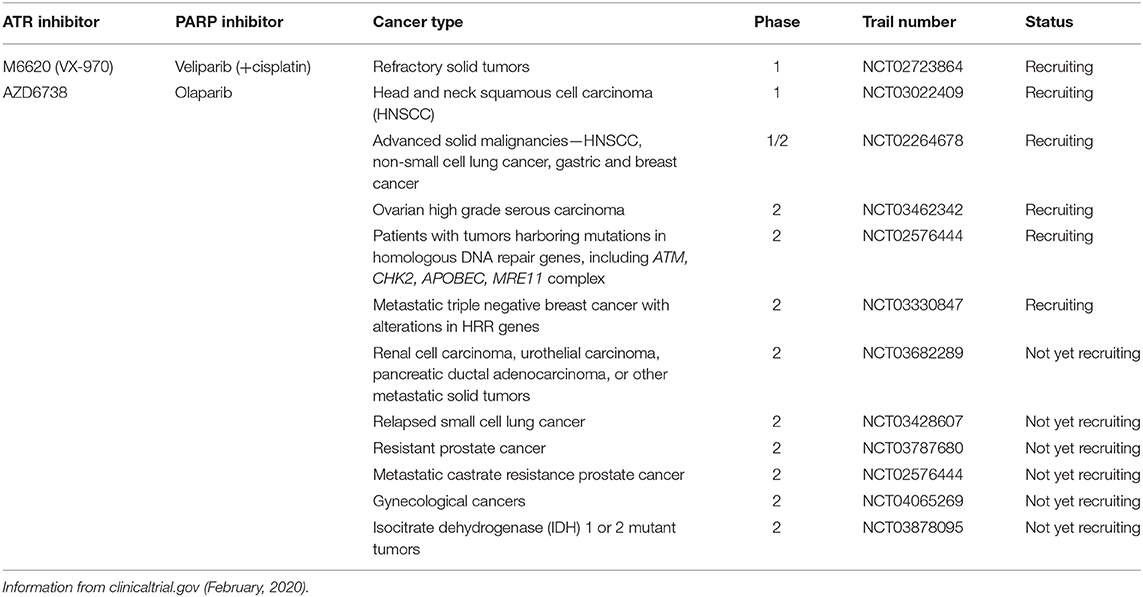
Table 5. ATR and PARP inhibitor combinations in adult clinical trials.
Improving survival rates for HR-NB remains a challenge in pediatric oncology. If long term survival is achieved, high risk patients are often left with severe sequelae as a result of high dose chemotherapy, and relapse is common. Many features of HR-NB suggest that subsets of these tumors will be sensitive to DDR inhibitors. Mutation or loss of genes such as ATM or others involved in HRR suggest sensitivity to PARP inhibition. Around half of HR-NB tumors are MYCN-amplified, which would lead to sensitivity to ATR inhibition due to oncogene induced replication stress. In addition, the frequent loss of G1 checkpoint control in HR-NB, by MYCN amplification and p53 pathway loss at relapse, provide a rationale for treatment with G2 checkpoint targeting agents (ATR, CHK1, and/or WEE1 inhibitors). Further work to identify predictive biomarkers of sensitivity to DDR inhibitors in NB will better stratify patients who might benefit from these agents.
As well as single agent efficacy of DDR inhibitors, there is mounting evidence to suggest that combining these agents with conventional chemotherapeutics or radiotherapy would permit lower doses to be given with the same effect due to chemo- and radio-sensitization.
Although inhibitors of these target proteins have been in adult trials for many years, the potential for their use in the treatment of pediatric tumors has only recently been explored. It is worth noting that even though a compound is effective in adult cancers, the same may not be true in the pediatric setting. It is also important to consider the potential long term toxicity of inhibiting the DDR, such as the development of secondary malignancies. Although unlikely to cause more off-target effects than current high dose chemotherapy regimens, the long-term toxicity of these agents in children is unknown and may take years to become apparent.
Nevertheless, exploiting defects in the DDR has the potential to lead to novel therapeutic options for a large subset of HR-NB patients for whom the prognosis is still unacceptably poor.
HS collected the data and wrote the manuscript. HS, LC, NC, and DT contributed to the design and content of the review, read, and approved the final manuscript for publication.
This work was supported by the Children's Cancer & Leukaemia Group and the Little Princess Trust (CCLGA 2017-12).
NC has received research funding from Aguoron Pharmaceuticals, Pfizer and Clovis Oncology for the development of rucaparib, BioMarin for studies on talazoparib and Tesaro for studies with niraparib and paid consultancy from Abbvie for veliparib. NC is inventor on the patent relating to the use of rucaparib in HRD cancer (WO2005012305A3) and Newcastle University receives royalty income from the sales of rucaparib that are shared with contributors to its development, including NC. NC does not take these royalties personally. NC has also received research funding from Vertex Pharmaceuticals and is currently in receipt of funding from Merck KGaA for work on ATR inhibitors and holds a patent for a pharmacodynamic biomarker of ATR inhibition (WO2014055756A1).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
3. Yao Y, Dai W. Genomic instability and cancer. J Carcinog Mutagen. (2014) 5:1000165. doi: 10.4172/2157-2518.1000165
4. Shaltiel IA, Krenning L, Bruinsma W, Medema RH. The same, only different – DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci. (2015) 128:607–20. doi: 10.1242/jcs.163766
5. Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. Human cancers express a mutator phenotype. Proc Natl Acad Sci USA. (2006) 103:18238–42. doi: 10.1073/pnas.0607057103
6. Massague J. G1 cell-cycle control and cancer. Nature. (2004) 432:298–306. doi: 10.1038/nature03094
7. Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. (2013) 494:492–6. doi: 10.1038/nature11935
8. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. (2014) 16:2–9. doi: 10.1038/ncb2897
9. Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. (2015) 15:276. doi: 10.1038/nrc3916
10. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, et al. DNA repair targeted therapy: the past or future of cancer treatment? Pharmacol Ther. (2016) 160:65–83. doi: 10.1016/j.pharmthera.2016.02.003
11. Stiller C. Childhood Cancer in Britain: Incidence, Survival, Mortality. Oxford: Oxford University Press (2007).
12. Maris JM. Recent advances in neuroblastoma. N Engl J Med. (2010) 362:2202–11. doi: 10.1056/NEJMra0804577
13. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers. (2016) 2:16078. doi: 10.1038/nrdp.2016.78
14. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, et al. The international neuroblastoma pathology classification (the shimada system). Cancer. (1999) 86:364–72. doi: 10.1002/(SICI)1097-0142(19990715)86:2<364::AID-CNCR21>3.0.CO;2-7
15. Fredlund E, Ringnér M, Maris JM, Påhlman S. High MYC pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc Natl Acad Sci USA. (2008) 105:14094–9. doi: 10.1073/pnas.0804455105
16. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. (2003) 3:203–16. doi: 10.1038/nrc1014
17. Cohn SL, Pearson ADJ, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The international neuroblastoma risk group (INRG) classification system: an INRG task force report. J Clin Oncol. (2009) 27:289–97. doi: 10.1200/JCO.2008.16.6785
18. Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-MYC in untreated human neuroblastomas correlates with advanced disease stage. Science. (1984) 224:1121–4. doi: 10.1126/science.6719137
19. Collins S, Groudine M. Amplification of endogenous MYC-related DNA sequences in a human myeloid leukaemia cell line. Nature. (1982) 298:679–81. doi: 10.1038/298679a0
20. Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, et al. L-myc, a new MYC-related gene amplified and expressed in human small cell lung cancer. Nature. (1985) 318:69–73. doi: 10.1038/318069a0
21. Schwab M, Alitalo K, Klempnauer K.-H., Varmus HE, Bishop JM, Gilbert F, et al. Amplified DNA with limited homology to MYC cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. (1983) 305:245–8. doi: 10.1038/305245a0
22. Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F, et al. The c-MYC target gene network. Semin Cancer Biol. (2006) 16:253–64. doi: 10.1016/j.semcancer.2006.07.014
23. Eilers M, Eisenman RN. MYC's broad reach. Genes Dev. (2008) 22:2755–66. doi: 10.1101/gad.1712408
24. Downs KM, Martin GR, Bishop JM. Contrasting patterns of MYC and N-MYC expression during gastrulation of the mouse embryo. Genes Dev. (1989) 3:860–9. doi: 10.1101/gad.3.6.860
25. Hirning U, Schmid P, Schulz WA, Rettenberger G, Hameister H. A comparative analysis of N-MYC and C-MYC expression and cellular proliferation in mouse organogenesis. Mech Dev. (1991) 33:119–25. doi: 10.1016/0925-4773(91)90078-K
26. Ma M, Zhao K, Wu W, Sun R, Fei J. Dynamic expression of N-MYC in mouse embryonic development using an enhanced green fluorescent protein reporter gene in the N-MYC locus. Dev Growth Differ. (2014) 56:152–60. doi: 10.1111/dgd.12115
27. Chen L, Iraci N, Gherardi S, Gamble LD, Wood KM, Perini G, et al. p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. (2010) 70:1377–88. doi: 10.1158/0008-5472.CAN-09-2598
28. Hogarty MD. The requirement for evasion of programmed cell death in neuroblastomas with MYCN amplification. Cancer Lett. (2003) 197:173–9. doi: 10.1016/S0304-3835(03)00103-4
29. Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: an important component of programmed cell death. J Mol Med. (2001) 79:428–36. doi: 10.1007/s001090100233
30. Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. (2006) 439:95–9. doi: 10.1038/nature04323
31. McKee AE, Thiele CJ. Targeting caspase 8 to reduce the formation of metastases in neuroblastoma. Expert Opin Ther Targets. (2006) 10:703–8. doi: 10.1517/14728222.10.5.703
32. Crowder RN, El-Deiry WS. Caspase-8 regulation of trail-mediated cell death. Exp Oncol. (2012) 34:160–4.
33. Kaufmann T, Strasser A, Jost PJ. Fas death receptor signalling: roles of Bid and XIAP. Cell Death Differ. (2012) 19:42–50. doi: 10.1038/cdd.2011.121
34. Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans MEC, et al. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci USA. (2012) 109:19190–5. doi: 10.1073/pnas.1208215109
35. Westermann F, Muth D, Benner A, Bauer T, Henrich KO, Oberthuer A, et al. Distinct transcriptional MYCn/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. (2008) 9:R150. doi: 10.1186/gb-2008-9-10-r150
36. Rohban S, Campaner S. MYC induced replicative stress response: how to cope with it and exploit it. Biochim Biophys Acta. (2015) 1849:517–24. doi: 10.1016/j.bbagrm.2014.04.008
37. Schleiermacher G, Michon J, Ribeiro A, Pierron G, Mosseri V, Rubie H, et al. Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCn-non-amplified localised unresectable/disseminated neuroblastoma (a siopen collaborative study). Br. J. Cancer. (2011) 105:1940–8. doi: 10.1038/bjc.2011.472
38. Schleiermacher G, Mosseri V, London WB, Maris JM, Brodeur GM, Attiyeh E, et al. Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the inrg project. Br J Cancer. (2012) 107:1418–22. doi: 10.1038/bjc.2012.375
39. Defferrari R, Mazzocco K, Ambros IM, Ambros PF, Bedwell C, Beiske K, et al. Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer. (2015) 112:290–5. doi: 10.1038/bjc.2014.557
40. Attiyeh EF, London WB, Mossé YP, Wang Q, Winter C, Khazi D, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. (2005) 353:2243–53. doi: 10.1056/NEJMoa052399
41. Bown N, Cotterill S, Łastowska M, O'Neill S, Pearson AD, Plantaz D, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med. (1999) 340:1954–61. doi: 10.1056/NEJM199906243402504
42. Mossë YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as the major familial neuroblastoma predisposition gene. Nature. (2008) 455:930–5. doi: 10.1038/nature07261
43. Maris JM, Guo C, White PS, Hogarty MD, Thompson PM, Stram DO, et al. Allelic deletion at chromosome bands 11q14–23 is common in neuroblastoma. Med Pediatr Oncol. (2001) 36:24–7. doi: 10.1002/1096-911X(20010101)36:1<24::AID-MPO1007>3.0.CO;2-7
44. Mlakar V, Mlakar SJ, Lopez G, Maris JM, Ansari M, Gumy-Pause F. 11q deletion in neuroblastoma: a review of biological and clinical implications. Mol Cancer. (2017) 16:114. doi: 10.1186/s12943-017-0686-8
45. Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. (2013) 5:a012716. doi: 10.1101/cshperspect.a012716
46. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. (2016) 15:1781–91. doi: 10.1158/1535-7163.MCT-15-0945
47. Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. (1992) 71:587–97. doi: 10.1016/0092-8674(92)90593-2
48. Weber AM, Drobnitzky N, Devery AM, Bokobza SM, Adams RA, Maughan TS, et al. Phenotypic consequences of somatic mutations in the ataxia-telangiectasia mutated gene in non-small cell lung cancer. Oncotarget. (2016) 7:60807–22. doi: 10.18632/oncotarget.11845
49. Delia D, Piane M, Buscemi G, Savio C, Palmeri S, Lulli P, et al. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. HumMol Genet. (2004) 13:2155–63. doi: 10.1093/hmg/ddh221
50. Podhorecka M, Skladanowski A, Bozko P. H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucleic Acids. (2010) 2010:920161. doi: 10.4061/2010/920161
51. Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. (2007) 26:1306–16. doi: 10.1038/sj.onc.1210263
52. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. (2000) 408:307–10. doi: 10.1038/35042675
53. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. (2015) 356:197–203. doi: 10.1016/j.canlet.2013.12.025
54. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. (2014) 14:359–70. doi: 10.1038/nrc3711
55. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer. (2011) 2:466–74. doi: 10.1177/1947601911408889
56. Zhao Y, Yu H, Hu W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim Biophys Sin. (2014) 46:180–9. doi: 10.1093/abbs/gmt147
57. Agrawal A, Yang J, Murphy RF, Agrawal DK. Regulation of the p14ARF-MDM2-p53 pathway: an overview in breast cancer. Exp Mol Pathol. (2006) 81:115–22. doi: 10.1016/j.yexmp.2006.07.001
58. Ozenne P, Eymin B, Brambilla E, Gazzeri S. The ARF tumor suppressor: structure, functions and status in cancer. Int J Cancer. (2010) 127:2239–47. doi: 10.1002/ijc.25511
59. Carr-Wilkinson J, O'Toole K, Wood KM, Challen CC, Baker AG, Board JR, et al. High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin Cancer Res. (2010) 16:1108–18. doi: 10.1158/1078-0432.CCR-09-1865
60. Carr J, Bell E, Pearson ADJ, Kees UR, Beris H, Lunec J, et al. Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. (2006) 66:2138–45. doi: 10.1158/0008-5472.CAN-05-2623
61. Tweddle DA, Malcolm AJ, Bown N, Pearson ADJ, Lunec J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. (2001) 61:8–13.
62. Chen L, Tweddle DA. p53, SKP2, and DKK3 as MYCN target genes and their potential therapeutic significance. Front Oncol. (2012) 2:173. doi: 10.3389/fonc.2012.00173
63. Vandesompele J, Michels E, De Preter K, Menten B, Schramm A, Eggert A, et al. Identification of 2 putative critical segments of 17q gain in neuroblastoma through integrative genomics. Int J Cancer. (2008) 122:1177–82. doi: 10.1002/ijc.23156
64. Schleiermacher G, Raynal V, Janoueix-Lerosey I, Combaret V, Aurias A, Delattre O. Variety and complexity of chromosome 17 translocations in neuroblastoma. Genes Chromosomes Cancer. (2004) 39:143–50. doi: 10.1002/gcc.10313
65. Hertwig F, Peifer M, Fischer M. Telomere maintenance is pivotal for high-risk neuroblastoma. Cell Cycle. (2016) 15:311–2. doi: 10.1080/15384101.2015.1125243
66. Onitake Y, Hiyama E, Kamei N, Yamaoka H, Sueda T, Hiyama K, et al. Telomere biology in neuroblastoma: telomere binding proteins and alternative strengthening of telomeres. J Pediatr Surg. (2009) 44:2258–66. doi: 10.1016/j.jpedsurg.2009.07.046
67. Kawashima M, Kojima M, Ueda Y, Kurihara S, Hiyama E. Telomere biology including TERT rearrangements in neuroblastoma: a useful indicator for surgical treatments. J Pediatr Surg. (2016) 51:2080–5. doi: 10.1016/j.jpedsurg.2016.09.042
68. Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. (2015) 47:1411–4. doi: 10.1038/ng.3438
69. Lindner S, Bachmann HS, Odersky A, Schaefers S, Klein-Hitpass L, Hero B, et al. Absence of telomerase reverse transcriptase promoter mutations in neuroblastoma. Biomed Rep. (2015) 3:443–6. doi: 10.3892/br.2015.463
70. Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. (2015) 526:700–4. doi: 10.1158/1538-7445.AM2015-LB-210
71. Kumps C, Fieuw A, Mestdagh P, Menten B, Lefever S, Pattyn F, et al. Focal DNA copy number changes in neuroblastoma target MYCN regulated genes. PLoS ONE. (2013) 8:e52321. doi: 10.1371/journal.pone.0052321
72. Mac SM, D'Cunha CA, Farnham PJ. Direct recruitment of N-MYC to target gene promoters. Mol Carcinog. (2000) 29:76–86. doi: 10.1002/1098-2744(200010)29:2<76::AID-MC4>3.0.CO;2-Y
73. Cheung N.-K.V., Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. (2012) 307:1062–71. doi: 10.1001/jama.2012.228
74. Bower K, Napier CE, Cole SL, Dagg RA, Lau LMS, Duncan EL, et al. Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of alternative lengthening of telomeres. PLoS ONE. (2012) 7:e50062. doi: 10.1371/journal.pone.0050062
75. Napier CE, Huschtscha LI, Harvey A, Bower K, Noble JR, Hendrickson EA, et al. ATRX represses alternative lengthening of telomeres. Oncotarget. (2015) 6:16543–58. doi: 10.18632/oncotarget.3846
76. Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. (2012) 483:589–93. doi: 10.1038/nature10910
77. Clynes D, Higgs DR, Gibbons RJ. The chromatin remodeller ATRX: a repeat offender in human disease. Trends Biochem Sci. (2013) 38:461–6. doi: 10.1016/j.tibs.2013.06.011
78. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. (2013) 45:279–84. doi: 10.1038/ng.2529
79. Zeineldin M, Federico S, Chen X, Fan Y, Xu B, Stewart E, et al. MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat Commun. (2020) 11:913. doi: 10.1038/s41467-020-14682-6
80. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. (2008) 455:967–70. doi: 10.1038/nature07398
81. Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol. (2014) 32:2727–34. doi: 10.1200/JCO.2013.54.0674
82. Carén H, Abel F, Kogner P, Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J. (2008) 416:153–9. doi: 10.1042/BJ20081834
83. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. (2014) 26:682–94. doi: 10.1016/j.ccell.2014.09.019
84. Zhu S, Lee J.-S., Guo F, Shin J, Perez-Atayde AR, Kutok JL, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. (2012) 21:362–73. doi: 10.1016/j.ccr.2012.02.010
85. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer cell. (2012) 22:117–30. doi: 10.1016/j.ccr.2012.06.001
86. Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. (1997) 14:439–49. doi: 10.1038/sj.onc.1200849
87. Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, et al. ALK, the chromosome 2 gene locus altered by the t (2;5) in non-hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene. (1997) 14:2175–88. doi: 10.1038/sj.onc.1201062
88. Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. (2013) 13:685–700. doi: 10.1038/nrc3580
89. Azarova AM, Gautam G, George RE. Emerging importance of ALK in neuroblastoma. Semin Cancer Biol. (2011) 21:267–75. doi: 10.1016/j.semcancer.2011.09.005
90. George RE, Sanda T, Hanna M, Fröhling S, Luther W, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. (2008) 455:975–8. doi: 10.1038/nature07397
91. Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. (2015) 47:864–71. doi: 10.1038/ng.3333
92. Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. (1996) 12:144–8. doi: 10.1038/ng0296-144
93. Sarni D, Kerem B. Oncogene-induced replication stress drives genome instability and tumorigenesis. Int J Mol Sci. (2017) 18:1339. doi: 10.3390/ijms18071339
94. Primo LMF, Teixeira LK. DNA replication stress: oncogenes in the spotlight. Genet Mol Biol. (2020) 43(1 Suppl. 1):e20190138. doi: 10.1590/1678-4685gmb-2019-0138
95. Davidoff AM. Neuroblastoma. Semin Pediatr Surg. (2012) 21:2–14. doi: 10.1053/j.sempedsurg.2011.10.009
96. Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev. (2013) 13:397–411. doi: 10.1038/nrc3526
97. Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KM, et al. Children's oncology group's 2013 blueprint for research: neuroblastoma. Pediatr Blood Cancer. (2013) 60:985–93. doi: 10.1002/pbc.24433
98. Gur H, Ozen F, Can Saylan C, Atasever-Arslan B. Dinutuximab in the treatment of high-risk neuroblastoma in children. Clin Med Insights Ther. (2017) 9:1179559X17719106. doi: 10.1177/1179559X17719106
99. Basta NO, Halliday GC, Makin G, Birch J, Feltbower R, Bown N, et al. Factors associated with recurrence and survival length following relapse in patients with neuroblastoma. Br J Cancer. (2016) 115:1048–57. doi: 10.1038/bjc.2016.302
100. Perwein T, Lackner H, Sovinz P, Benesch M, Schmidt S, Schwinger W, et al. Survival and late effects in children with stage 4 neuroblastoma. Pediatr Blood Cancer. (2011) 57:629–35. doi: 10.1002/pbc.23036
101. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. (2012) 12:587–98. doi: 10.1038/nrc3342
102. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev. (2012) 12:801–17. doi: 10.1038/nrc3399
103. Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. (2007) 28:739–45. doi: 10.1016/j.molcel.2007.11.015
104. Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. (2015) 149:124–38. doi: 10.1016/j.pharmthera.2014.12.001
105. Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res. (2015) 21:4780–5. doi: 10.1158/1078-0432.CCR-15-0479
106. Saldivar J, Cortez D, Cimprich K. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. (2017) 18:622–36. doi: 10.1038/nrm.2017.67
108. Fortini P, Dogliotti E. Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair(Amst). (2007) 6:398–409. doi: 10.1016/j.dnarep.2006.10.008
109. Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair. (2003) 2:955–69. doi: 10.1016/S1568-7864(03)00118-6
110. Spivak G. Nucleotide excision repair in humans. DNA Repair. (2015) 36:13–8. doi: 10.1016/j.dnarep.2015.09.003
111. Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. (2008).9:619–31. doi: 10.1038/nrg2380
112. Hsieh P, Zhang Y. The devil is in the details for DNA mismatch repair. Proc Natl Acad Sci USA. (2017) 114:3552–4. doi: 10.1073/pnas.1702747114
113. Tonzi P, Huang TT. Role of Y-family translesion DNA polymerases in replication stress: implications for new cancer therapeutic targets. DNA Repair(Amst). (2019) 78:20–6. doi: 10.1016/j.dnarep.2019.03.016
114. Mahaney BL, Meek K, Lees-Miller Susan P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. (2009) 417:639–50. doi: 10.1042/BJ20080413
115. Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem. (2018) 293:10524–35. doi: 10.1074/jbc.TM118.000372
116. Mouw KW, D'Andrea AD. Crosstalk between the nucleotide excision repair and fanconi anemia/BRCA pathways. DNA Repair. (2014) 19:130–4. doi: 10.1016/j.dnarep.2014.03.019
117. Gu L, Chu P, Lingeman R, McDaniel H, Kechichian S, Hickey RJ, et al. The mechanism by which MYCN amplification confers an enhanced sensitivity to a PCNA-derived cell permeable peptide in neuroblastoma cells. EBioMed. (2015) 2:1923–31. doi: 10.1016/j.ebiom.2015.11.016
118. Petroni M, Sardina F, Infante P, Bartolazzi A, Locatelli E, Fabretti F, et al. MRE11 inhibition highlights a replication stress-dependent vulnerability of MYCN-driven tumors. Cell Death Dis. (2018) 9:895. doi: 10.1038/s41419-018-0924-z
119. Petroni M, Sardina F, Heil C, Sahún-Roncero M, Colicchia V, Veschi V, et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ. (2016) 23:197–206. doi: 10.1038/cdd.2015.81
120. Newman EA, Chukkapalli S, Bashllari D, Thomas TT, Van Noord RA, Lawlor ER, et al. Alternative NHEJ pathway proteins as components of MYCN oncogenic activity in human neural crest stem cell differentiation: implications for neuroblastoma initiation. Cell Death Dis. (2017) 8:3208. doi: 10.1038/s41419-017-0004-9
121. Chayka O, D'Acunto CW, Middleton O, Arab M, Sala A. Identification and pharmacological inactivation of the MYCN gene network as a therapeutic strategy for neuroblastic tumor cells. J Biol Chem. (2015) 290:2198–212. doi: 10.1074/jbc.M114.624056
122. Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci USA. (2011) 108:3336–41. doi: 10.1073/pnas.1012351108
123. Hu H, Du L, Nagabayashi G, Seeger RC, Gatti RA. ATM is down-regulated by N-MYC-regulated microrna-421. Proc Natl Acad Sci USA. (2010) 107:1506–11. doi: 10.1073/pnas.0907763107
124. Ohmoto A, Yachida S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. OncoTargets Ther. (2017) 10:5195–208. doi: 10.2147/OTT.S139336
125. Hoy SM. Talazoparib: first global approval. Drugs. (2018) 78:1939–46. doi: 10.1007/s40265-018-1026-z
126. Daniel RA, Rozanska AL, Thomas HD, Mulligan EA, Drew Y, Castelbuono DJ, et al. Inhibition of poly(ADP-ribose) polymerase-1 enhances temozolomide and topotecan activity against childhood neuroblastoma. Clin Cancer Res. (2009) 15:1241–9. doi: 10.1158/1078-0432.CCR-08-1095
127. Nile DL, Rae C, Hyndman IJ, Gaze MN, Mairs RJ. An evaluation in vitro of PARP-1 inhibitors, rucaparib and olaparib, as radiosensitisers for the treatment of neuroblastoma. BMC cancer. (2016) 16:621. doi: 10.1186/s12885-016-2656-8
128. Norris RE, Adamson PC, Nguyen VT, Fox E. Preclinical evaluation of the PARP inhibitor, olaparib, in combination with cytotoxic chemotherapy in pediatric solid tumors. Pediatr Blood Cancer. (2014) 61:145–50. doi: 10.1002/pbc.24697
129. Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. (2010) 116:4578–87. doi: 10.1182/blood-2010-01-265769
130. Sanmartín E, Muñoz L, Piqueras M, Sirerol JA, Berlanga P, Cañete A, et al. Deletion of 11q in neuroblastomas drives sensitivity to PARP inhibition. Clin Cancer Res. (2017) 23:6875–87. doi: 10.1158/1078-0432.CCR-17-0593
131. Takagi M, Yoshida M, Nemoto Y, Tamaichi H, Tsuchida R, Seki M, et al. Loss of DNA damage response in neuroblastoma and utility of a PARP inhibitor. J Natl Cancer Inst. (2017) 109. doi: 10.1093/jnci/djx062
132. Colicchia V, Petroni M, Guarguaglini G, Sardina F, Sahún-Roncero M, Carbonari M, et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene. (2017) 36:4682–91. doi: 10.1038/onc.2017.40
133. Zhou JX, Feng LJ, Zhang X. Risk of severe hematologic toxicities in cancer patients treated with PARP inhibitors: a meta-analysis of randomized controlled trials. Drug Des Devel Ther. (2017) 11:3009–17. doi: 10.2147/DDDT.S147726
134. Su JM, Thompson P, Adesina A, Li X.-N., Kilburn L, Onar-Thomas A, et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a pediatric brain tumor consortium report. Neuro oncol. (2014) 16:1661–8. doi: 10.1093/neuonc/nou103
135. Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 axis in cancer therapy. Cancers. (2017) 9:41. doi: 10.3390/cancers9050041
136. Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM-Or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. (2011) 7:428–30. doi: 10.1038/nchembio.573
137. Schoppy DW, Brown EJ. Chk'ing p53-deficient breast cancers. J Clin Invest. (2012) 122:1202–5. doi: 10.1172/JCI63205
138. Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. (2008) 27:1368–77. doi: 10.1038/emboj.2008.61
139. Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. (2011) 105:372–81. doi: 10.1038/bjc.2011.243
140. Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. (2016) 7:76534–50. doi: 10.18632/oncotarget.12266
141. Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. (2017) 31:318–32. doi: 10.1101/gad.290957.116
142. Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science. (2015) 347:273–7. doi: 10.1126/science.1257216
143. Deeg KI, Chung I, Bauer C, Rippe K. Cancer cells with alternative lengthening of telomeres do not display a general hypersensitivity to ATR inhibition. Front Oncol. (2016) 6:186. doi: 10.3389/fonc.2016.00186
144. Kurmasheva RT, Kurmashev D, Reynolds CP, Kang M, Wu J, Houghton PJ, et al. Initial testing (stage 1) of M6620 (formerly VX-970), a novel ATR inhibitor, alone and combined with cisplatin and melphalan, by the pediatric preclinical testing program. Pediatr Blood Cancer. (2018) 65:e26825. doi: 10.1002/pbc.26825
145. Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, et al. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. (2013) 73:776–84. doi: 10.1158/0008-5472.CAN-12-2669
146. Lowery CD, Vanwye AB, Dowless M, Blosser W, Falcon BL, Stewart J, et al. The checkpoint kinase 1 inhibitor prexasertib induces regression of preclinical models of human neuroblastoma. Clin Cancer Res. (2017) 23:4354–63. doi: 10.1158/1078-0432.CCR-16-2876
147. Xu H, Cheung IY, Wei XX, Tran H, Gao X, Cheung NKV, et al. Checkpoint kinase inhibitor synergizes with DNA-damaging agents in G1 checkpoint-defective neuroblastoma. Int J Cancer. (2011) 129:1953–62. doi: 10.1002/ijc.25842
Keywords: neuroblastoma, DNA damage response, targeted therapy, PARP, ATR
Citation: Southgate HED, Chen L, Curtin NJ and Tweddle DA (2020) Targeting the DNA Damage Response for the Treatment of High Risk Neuroblastoma. Front. Oncol. 10:371. doi: 10.3389/fonc.2020.00371
Received: 24 September 2019; Accepted: 03 March 2020;
Published: 03 April 2020.
Edited by:
Karim Malik, University of Bristol, United KingdomReviewed by:
Madhu Kollareddy, University of Bristol, United KingdomCopyright © 2020 Southgate, Chen, Curtin and Tweddle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola J. Curtin, bmljb2xhLmN1cnRpbkBuZXdjYXN0bGUuYWMudWs=; Deborah A. Tweddle, ZGVib3JhaC50d2VkZGxlQG5ld2Nhc3RsZS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.