 Jill Nehrbas1,2
Jill Nehrbas1,2 Ding-Wen Chen
Ding-Wen Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 14 February 2020
Sec. Pediatric Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00090
This article is part of the Research TopicMicroenvironment and Therapy-Resistance in LeukemiasView all 8 articles
Extracellular vesicle (EV) trafficking provides for a constitutive mode of cell-cell communication within tissues and between organ systems. Different EV subtypes have been identified that transfer regulatory molecules between cells, influencing gene expression, and altering cellular phenotypes. Evidence from a range of studies suggests that EV trafficking enhances cell survival and resistance to chemotherapy in solid tumors. In acute myeloid leukemia (AML), EVs contribute to the dynamic crosstalk between AML cells, hematopoietic elements and stromal cells and promote adaptation of compartmental bone marrow (BM) function through transport of protein, RNA, and DNA. Careful analysis of leukemia cell EV content and phenotypic outcomes provide evidence that vesicles are implicated in transferring several known key mediators of chemoresistance, including miR-155, IL-8, and BMP-2. Here, we review the current understanding of how EVs exert their influence in the AML niche, and identify research opportunities to improve outcomes for relapsed or refractory AML patients.
Acute myeloid leukemia (AML) is a genetically heterogenous disease that arises from abnormal proliferation of hematopoietic stem cells (HSCs) (1). Although most patients respond to current treatment strategies, a majority of patients will ultimately experience relapse (2). Patient survival rates remain low due largely to high rates of relapse as a consequence of intrinsic and extrinsic resistance (3). Survival of residual leukemic cells that give rise to relapse is typically attributed to clonal genetic adaptations that may precede treatment or emerge during chemotherapy (4). Extrinsic mechanisms that actively confer protection of leukemic cells from complete elimination in the bone marrow (BM) following therapy are increasingly recognized for their role in chemo-resistance and clonal persistence; examples include nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-kB) mutation, drug efflux pump activation, and adaptive action of microRNA (miRNA) mediated cell-cell crosstalk mechanisms (5, 6).
Extracellular vesicles (EVs) are membrane-bound particles secreted from cells, carrying a variety of nucleic acid and protein cargo active in cell-cell communication (7). As a constitutive cellular mechanism for the transport of bioactive cargo, EVs promote therapy resistance in the bone marrow niche (8, 9). Now EV content and functional analyses are beginning to shed light on the some key mediators involved in chemoresistance such as miRNA-155 (miR-155), interleukin 8 (IL-8), and bone morphogenic protein 2 (BMP-2) (10–12). In this review, we critically examine the current evidence connecting EV trafficking and resistance to both chemo- and immunotherapies, while highlighting key areas of research.
AML is a genetically heterogeneous disease characterized by the successive acquisition of mutations in HSCs that cause unchecked proliferation and a coincident differentiation arrest (1). At diagnosis, patients typically present with symptoms arising from leukocytosis, anemia, and thrombocytopenia. At an annual incidence of 4.3 per 100,000 persons, AML is the most common acute leukemia in adults, and second to acute lymphoid leukemia (ALL) in children (13). The overall 5-year survival rate is <50% in young adults, which drastically decreases in elderly patients with a 2-year survival rate of <20% (3). With well over 200 identified molecular lesions illustrating overall disease heterogeneity, some common recurring cytogenetic abnormalities are seen, including translocations between chromosomes 8 and 21, deletions in chromosomes 5 and 7, and inversions in chromosome 16 (3). Likewise, activating mutations in several oncogenes have been shown to alter key components in cell cycle regulation such as tumor protein p53, fms-like tyrosine kinase 3 (FLT-3), WNT, and MYC (3). Individually and in combination, these genetic abnormalities carry important information for both diagnostic and treatment stratification of AML. For example, depending on genetic context, mutations in nucleophosmin (NPM1) found in about 30% of adult AML cases can denote more favorable outcomes for patients (14). Meanwhile, patients with FLT-3 internal tandem duplication (FLT-3 ITD) account for ~25% of AML cases and generally denote a poor prognosis, except in combination with NPM1 (15). Other oncogene mutations such as mutations in the RAS family of proteins, accounting for around 15% of AML cases, are associated with a mixed prognostic impact (16). In spite of the genetic heterogeneity, current AML treatment typically relies on a backbone of cytarabine and anthracyclines in use since the 1970s, with the more recent addition of hypomethylating agents for patients over the age of 65 (17). Critically, whereas most patients initially respond to these treatments, many experience relapse or develop refractory disease (18). While HSC transplant can be a therapeutic option, the overall survival remains between 20 and 60%, due to ineffective salvage treatment regimens (17, 19). Clearly, a better understanding of the barriers to elimination of residual AML cells from their sanctuary in the BM holds untapped therapeutic opportunities for patients.
Development of drug resistance during chemotherapy is a primary challenge to sustaining AML remission in patients. This can occur as a direct consequence of mutations, or indirectly through signaling pathways or enzyme activities that lead to cancer cell protection (6). Conceptually, clonal evolution under therapy can lead to the emergence of more chemo-resistant clones, that further fuel cell growth and boost the survival advantage (4, 20). For example, some patients acquire FLT-3 ITD mutations during the course of treatment (21, 22). Interestingly, the observation that treatment through tyrosine kinase inhibition efficiently clears peripherally circulating blasts, but not those in the in the BM does not reflect intrinsic genetic events (23).
Non-genetic chemoresistance mechanisms such as p-glycoprotein (P-gp) overexpression, an ATPase efflux pump that export drugs or their active metabolites, are also correlated with poor disease outcomes (24). These direct effects are compounded by the downstream activation of NF-κB signaling that controls cell proliferation and counters apoptosis (6). Mechanistically, NF-κB overactivation protects AML blasts from apoptosis as a result of increased expression of pro-survival BCL-2 proteins (25). Another common mechanism of chemoresistance involves glutathione s-transferase (GST) overexpression, an enzyme that typically protects against reactive electrophiles and DNA damage (26). When overexpressed in cancer, GST has been shown to increase chemoresistance, possibly by catalyzing the binding of glutathione to chemotherapy drugs to minimize their effects, preventing drugs from attacking DNA, or deactivating cisplatin, a common platinum-based component of many chemotherapies that targets DNA in cancer cells (6, 27, 28).
Evidence has also shown that cellular microRNAs are highly involved in the development of drug resistance in AML (29). For example, CXCR4-mediated signaling has been shown to cause chemoresistance in AML cells by downregulating miR-let-7a, which increases transcriptional activation of MYC oncogene and BCL-XL in AML cells (30). Several miRNAs that bind to DNA damage regulatory proteins are overexpressed in AML. For example, overexpression of miR-181a in AML cells downregulates ATM, a critical checkpoint kinase required for cell cycle arrest, and in turn promotes cancer cell growth (31). miR-128 is also overexpressed in AML, which downregulates RAD51 and reduces DNA damage response (32). Downregulation in a number of different miRNAs such as miRs−15a (33),−15b (34),−125b-5p (35),−139-5p (36),−145 (37), and−181a (38) have been reported in other forms of cancer, which collectively suggests that elevated BCL2 translation plays a major role in resistance development. Intriguingly, EV secretion may alter leukemogenic properties in a cell autonomous fashion and promotes expansion and persistence of leukemia initiating cells (39).
The bone marrow compartment comprises a range of cell types that form HSC supportive microenvironments: endosteal (comprised of osteoblasts, osteocytes, and osteoclasts) and vascular (comprised of endothelial cells and megakaryocytes) niches (Figure 1A). The endosteal niche in the internal surface of the bone helps maintain stem cell quiescence through the binding of osteoblasts to HSCs (40), while the vascular niche promotes the proliferation and differentiation of HSCs through crosstalk between endothelial cells and HSCs (40). Through successive adaptation of these niches, the BM microenvironment contributes to leukemia cell proliferation and survival (Figure 1B) (41–43). In the endosteal niche, AML cells may induce mesenchymal stromal cells (MSCs) toward an accelerated osteoblastic differentiation where accumulation of these immature osteoblasts can lead to an overall decrease in the number of functional osteoblasts (5). One study showed that AML and myelodysplastic syndrome patients had an osteoblast count 55% lower than healthy controls, consistent with observations in mice bearing acute leukemia, that exhibited osteoblast deficits inversely proportional to circulating blast burden and survival (44).
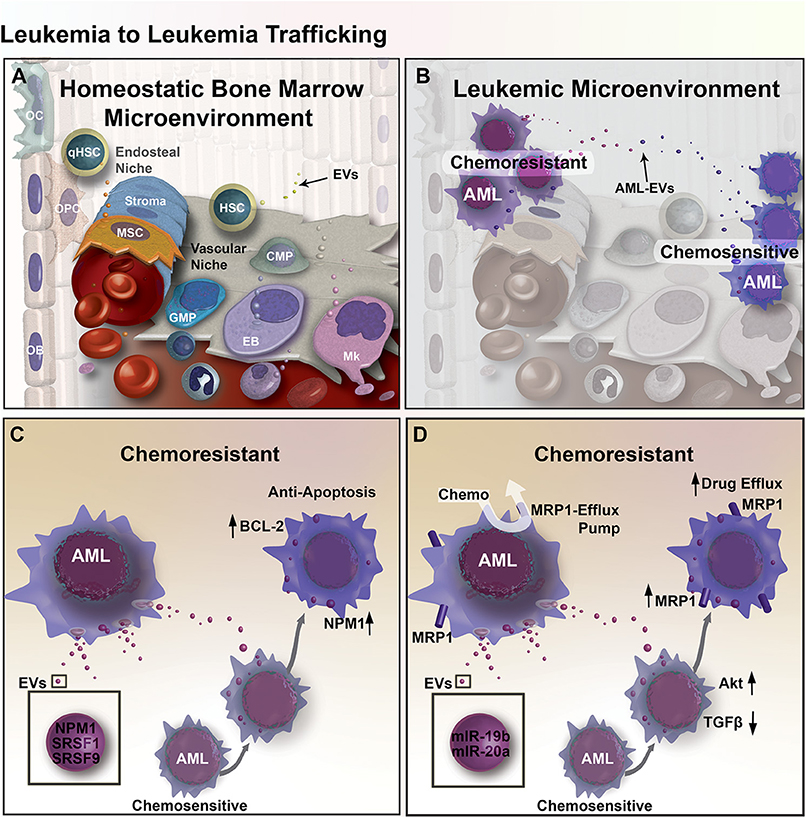
Figure 1. EV mediated transfer of chemoresistance between leukemia cells in the BM microenvironment. (A) Diagram of the BM microenvironment, composed of the hematopoietic niche (right) stromal compartment (left). Hematopoietic Stem and Progenitor Cell (HSPC) give rise to Common Myeloid Progenitors (CMP), Granulo-Monocytic Progenitor Cells (GMP), Erythroblasts (EB), Megakaryocytes (Mk), and many other cell types that populate the cells of the blood. In the stromal compartment, Mesenchymal Stromal Cells (MSC) give rise to Osteoprogenitor Cells (OPC) and Osteoblasts (OB), together these cells function to form bone and regulate hematopoiesis in part through EV-mediated signaling. (B) Expansion of leukemic cells results in microenvironmental dysregulation. EV trafficking between AML cells transfers regulatory factors that induce resistance to chemotherapy. (C) Chemo-experienced AML cells shed EVs containing NMP1, SRSF1, and SRSF9, which increase apoptosis resistance through upregulation of BCL-2 and NPM1 in unexperienced recipient AML cells. (D) EVs from chemo-experienced AML cells also contain miR-19b and−20a, which reduce TGF-β signaling and increase Akt signaling and the expression of MRP1 chemo-efflux pump in recipient AML cells.
Emerging evidence also suggests that AML blasts localize and integrate with the BM vascular niche. A study by Cogle et al. showed that some AML cells have the capacity to fuse with vascular endothelium (vascular-AML), acquiring several endothelial characteristics before assuming a state of quiescence (45). Moreover, these vascular-AML cells remain leukemogenic and were able to give rise to leukemia following transplantation into healthy mice, suggesting that AML cells that have integrated into the vascular niche may be implicated in relapse (45). Within the BM microenvironment, crosstalk between AML cells and BM stromal cells (BMSCs) also plays a role in chemoresistance development. For example, AML cells can induce secretion of growth arrest-specific gene 6 (GAS6) in BMSCs, which protects AML cells from cell death (46). More recently, findings further illustrate the significance of BMSC-AML interactions, whereby AML cells remodel the endosteal vascular niche by producing pro-inflammatory and anti-angiogenic cytokines that results in the loss of endosteal blood vessels and BMSCs surrounding blood vessels and bones (47). These endosteal AML cells were found to have elevated expression of tumor necrosis factor (TNF) and CXCL2, which are involved in vascular destruction and angiogenesis inhibition, respectively. In addition, AML-induced remodeling of the niche disrupts HSC homing, and reduces the efficiency of drug transport, increasing the risk of chemoresistance (48).
Several mechanisms of intercellular communication contribute to the conversion of BM from a homeostatic-supportive compartment to an oncogenic compartment (Table 1). Traditionally, cells communicate via soluble factors such as cytokines, neurotransmitters, hormones, and growth factors, where they act in a juxtacrine, paracrine, or endocrine manner. In recent years, EVs have emerged as a novel mechanism of intercellular communication. Based on their method of biogenesis, content, and physical properties, EVs can be broadly categorized into exosomes, exomeres, microvesicles, and apoptotic bodies (52, 53). Like other types of EVs, exosomes sized at up to 120 nm, are equipped to traffic proteins, lipids, mRNAs, multiple types of non-coding RNAs, DNAs, and chemokines/cytokines between cells (52). Observations in multiple types of cancer (Table 2) indicate that cells with malignant potential release exosomes at an increased rate, selectively capturing certain molecules that promote the proliferation of cancer cells, metastasis, and drug resistance (59). Exosomes have been implicated in the transformation of BM microenvironment into a leukemia-permissive space. For example, one recent xenograft study revealed that AML-derived exosomes upregulate DKK1 in BMSCs, suppressing hematopoiesis and inhibiting osteoblast differentiation via Wnt pathway signals (43). Specifically, these AML derived exosomes downregulate gene expression that otherwise sustains normal hematopoiesis (Igf1, Cxcl12, Kitl, and Il-7), and genes involved in normal bone development (Ocn and Col1A1). In addition to playing a key role in transforming the BM niche, exosomes may in part contribute toward the protection of leukemia blasts during chemotherapy treatment (43).
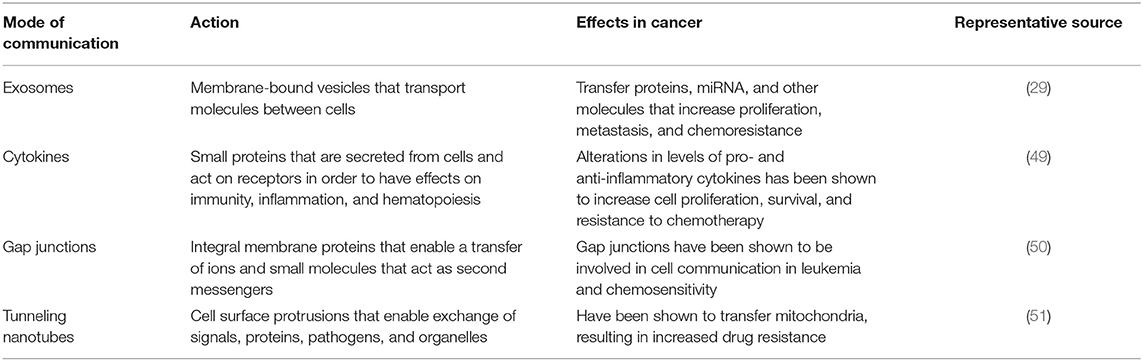
Table 1. Modes of cell-cell communication and their potential role in drug resistance.

Table 2. Transfer of molecular cargo via exosomes in cancer.
Another well-characterized mechanism of cell-cell crosstalk is the transmission of signals involving direct cell-cell contacts such as gap junctions composed of membrane protein called connexins, that transfer ions and small molecules serving as second messengers in a neighboring cell. Connexins have been shown to act as either tumor promoters or suppressors in breast cancer, prostate cancer, and brain cancer (60–63). A more recently described method of direct cell-cell communication is through tunneling nanotubes (TNTs), cell membrane protrusions composed of F-actin that transiently connect cells to exchange proteins, pathogens, or even organelles. Formation of TNTs appears to increase in cells under stress, potentially increasing survival through the exchange of mitochondria (64, 65). TNT trafficking of bioactive cargo influences differentiation, metabolism and immune response (65).
An elegant study in glioma reported by Al-Nedawi provided the first compelling evidence that shows EV-mediated intratumoral transfer of functional resistance factors between differentially chemo-sensitive cancer cells (66). In recent years, emerging evidence has shown that EV-mediated resistance transfer also exists in AML (Figure 1B), where proteins and miRNAs can be transported by EVs between AML cells and elicit differential gene expression profiles and cell activity in recipient AML cells (67, 68). More specifically, a co-culture experiment showed that chemo-resistant AML cells can induce chemoresistance in chemo-sensitive AML cells by triggering anti-apoptotic protein BCL-2 upregulation in chemo-sensitive AML cells, while chemo-sensitive AML cells could not induce BCL-2 upregulation in other chemo-sensitive AML cells (67). Aberrant expression of apoptosis-regulating proteins may allow AML cells to escape apoptosis, which can also be used to predict minimal residual disease (69). In addition to anti-apoptotic effects, upregulation of BCL-2 family of proteins also have implications in reducing unfolded protein response (UPR) stress in cancer cells through IRE1α pro-survival pathway activation (67). Activation of the IRE1α pathway involves the splicing of Xbp1, leading to endoplasmic reticulum (ER) chaperone synthesis and ER-associated protein degradation complex formation that ultimately supports cancer cells in adapting to ER-stress (70). Proteomic analysis of secretomes derived from both apoptotic-resistant and apoptosis-sensitive AML cells reveal an increase of apoptosis-regulating proteins are present in the secretome of apoptosis-sensitive AML cells (67). Furthermore, functional clusters of proteins within apoptosis-resistant cell-derived exosomes were associated with gene ontology (GO) terms such as non-coding RNA (ncRNA) metabolism, DNA replication and repair, translation elongation, and mRNA splicing. In addition, these exosomes contained NPM1, a protein associated with increased apoptosis-resistance. Meanwhile, exosomes from apoptosis-sensitive cells were more strongly associated with inflammatory and stress response GO terms. These findings suggest that apoptosis resistance can be transferred from apoptosis-resistant to apoptosis-sensitive AML cells via exosomes (Figure 1C) (67).
EV-mediated transfer of miRNAs is known to increase chemoresistance in several types of cancer (68, 71, 72). Exosomes derived from BMSCs of AML patients express different miRNA profile compared to healthy controls (10). More specifically, analysis of BMSC-derived exosomes from eight AML patients revealed that miR-155 and miR-375 are consistently enriched in exosomes, unlike exosomes derived from healthy donor BMSCs. Furthermore, the effect of these miRNAs in chemoresistance was demonstrated in a tyrosine kinase inhibitor AC220 challenge, where Molm14 AML cells pretreated with AML BMSC-derived exosomes gained resistance to tyrosine kinase inhibitor and conferred AML cell protection (10).
EV-mediated resistance transfer was also demonstrated in a separate study, where multidrug-resistant AML cells were able to transfer chemoresistance properties to chemo-sensitive AML cells via EV and induce the expression of drug efflux pump multidrug resistance protein 1 (MRP-1) (68). Tracking of fluorescently-labeled EVs derived from chemo-resistant cells showed that those EVs were uptaken by 85% of chemo-sensitive AML cells, which provided increased resistance to daunorubicin. Exposing EV-treated chemo-sensitive AML cells with MRP-1 inhibitor led to increased intracellular retention of daunorubicin following daunorubicin treatment. However, it is unclear whether the exosomes directly transferred MRP-1 or MRP-1 regulatory factors. To better understand the role of exosomes in this gain of chemoresistance, a miRNA analysis of exosomes was performed and showed that exosomes from chemo-resistant cells, in comparison to exosomes from chemo-sensitive cells, contained four-fold more miR-19b and miR-20a, which are often overexpressed in cancer. Both miRNAs play a role in the inhibition of TGF-β signaling (73). Additionally, research suggests that elevated levels of both miRNAs may activate PI3 kinase/Akt signaling, which could lead to the overexpression of MRP-1, supporting the possibility that exosomes mediate the transfer of chemoresistance via miRNAs transport (Figure 1D) (68, 74).
In addition to exosome-mediated transfer of chemoresistance phenotypes between chemo-resistant and chemo-sensitive leukemic cells, exosomes can also transport cargo from AML cells to BMSCs that may contribute to the formation of a leukemia-supportive BM microenvironment (Figure 2A). A recent study reported that treatment of AML cells with BMSC-derived exosomes modestly decreased etoposide-induced apoptosis (11). However, in AML cells and BMSCs co-culture, levels of pro-inflammatory cytokine IL-8 significantly increased in BMSCs and protected AML cells from etoposide-induced apoptosis (Figure 2B). Furthermore, inhibition of exosome secretion using an inhibitor of the EV budding regulator neutral sphingomyelinase (GW4869) resulted in a reduction of IL-8 secretion from BMSCs in a BMSC-AML co-culture setting, which had no effect on IL-8 secretion levels in BMSCs cultured alone (75). This finding provides evidence that AML cells secrete exosomes that induce BMSCs to release IL-8, which in turn makes AML cells more resistant to etoposide (11). Similarly, in a study of chronic myelogenous leukemia (CML), IL-8 secreted from BMSCs treated with CML exosomes binds to two transmembrane domain receptors, CXCR-1 and CXCR-2, and increased cell adhesion, motility, and survival of CML cells (76).
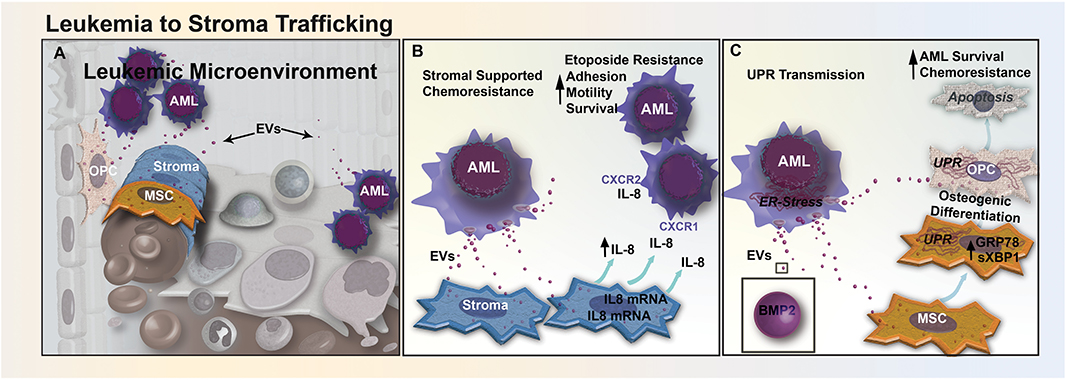
Figure 2. EV trafficking between leukemia and stromal cells reduces sensitivity to chemotherapeutics. (A) Bidirectional trafficking between leukemia and stromal cells leads to alterations in both the cellular composition and secretome of the stromal compartment. (B) In CML, EV-mediated signaling upregulates the secretion of IL-8 from BM stromal cells. Increased IL-8 in the leukemic microenvironment increases CML cell adhesion, motility, and survival through CXCR1 and CXCR2 engagement, and promotes resistance to etoposide. (C) AML cells from the nutrient-depleted leukemic microenvironment exhibit marked endoplasmic reticulum (ER) stress and upregulation of BMP-2. AML-EVs transfer BMP-2 and ER-stress to stromal MSCs and OPCs, activating the unfolded protein response pathway (UPR). UPR activation induces osteogenic differentiation in MSCs and causes increased apoptosis in osteoprogenitor cells, altering the cellular composition of the BM, AML survival, and response to chemotherapy.
Our own recent study suggests that AML exosomes may also modulate the microenvironment by transfer of ER stress responses via exosomes in BMSCs (12). During chemotherapy, leukemic cells can become deprived of oxygen, nutrients, and intracellular calcium, leading to the accumulation of unfolded proteins within the ER (77). This leads to activation of the UPR, decreasing the synthesis of new proteins, increasing unfolded protein degradation and protein folding chaperone levels, while helping cancer cells evade chemo-induced apoptosis (77). We have shown that the transfer of EVs from AML cells to BMSCs can enhance extrinsic chemoresistance and propose that AML exosomes trigger UPR by transferring BMP-2, a protein known to be upregulated in AML (Figure 2C). The trafficking of BMP-2 may cause increased osteogenic differentiation in MSCs, and an increase AML growth by inducing connective tissue growth factor (CTGF) expression in MSCs (42). In our study, ELISA analysis of EVs released from Molm-14 cells treated with Thapsigargin, a drug that induces UPR by inhibiting sarcoplasmic/endoplasmic reticulum Ca2+-ATPase, showed higher levels of BMP-2 than EVs released from untreated Molm-14 cells (12). Additional evidence supports that AML EVs traffic to the ER of MSCs and OPCs in vitro and induce upregulation of both GRP78, a key chaperone protein involved in UPR, and spliced Xbp1, a transcription factor for chaperones and ER stress sensors (12). Finally, high levels of BMP signaling have been linked to elevated expression of anti-apoptotic genes (42). Mechanistically, BMP action may involve additional cellular targets, as have been identified in CML where BMP-2 and BMP-4 were found to promote overexpression of the BMPR1a and altered downstream signaling in leukemic stem cells (78). Therapeutically, BMP-mediated leukemic myeloid progenitor expansion can be rescued through neutralization of circulating BMP-2 and BMP-4 proteins using soluble BMP receptor acting as a decoy. Taken together, these observations suggest that BMP-2 trafficked by exosomes influences recipient cell ER stress responses, increasing AML cell survival by altering gene expression and driving osteogenic MSC differentiation.
While several chemoresistance mechanisms in leukemia involve the direct delivery of critical molecules via exosomes, resistance can also arise through immune dysregulation. For example, exosomes can reduce the efficacy of adoptive natural killer (NK) cell therapy in AML patients through interaction with activated NK-92 cells (79). More specifically, exosomes appeared to reduce the efficacy of activated NK-92 by transporting inhibitory ligands to NK-92 surface receptors, as demonstrated through a co-incubation study that exosomes derived from AML patients with NK-92 cells resulted in a 40% reduction of NKG2D receptor expression on NK-92 cell surface. As NKG2D receptor is involved in initiating a cytotoxic and cytokine response against threats, and inhibition of this receptor results in a reduction in cytotoxicity of NK-92 cells against AML blasts (Figure 3A). Exosome delivery of TGF-β to NK-92 cells is believed to be in part responsible for the decrease in NKG2D through TGFβRI/II pathway activation (79). Conceptually, exosomes may also contribute toward immunotherapy resistance through binding of antibodies to their surface. One study suggested that in CLL, exosomes may lower the bioavailability of rituximab, a common immunomodulatory antibody that targets the CD20 epitope on B-cells. Exosomal binding of anti-CD20 reduces circulating levels of rituximab, which in turn protects lymphocytic leukemia cells from anti-CD20 mediated opsonization (Figure 3B) and may explain why a number of CLL patients develop resistance to rituximab treatment (80).
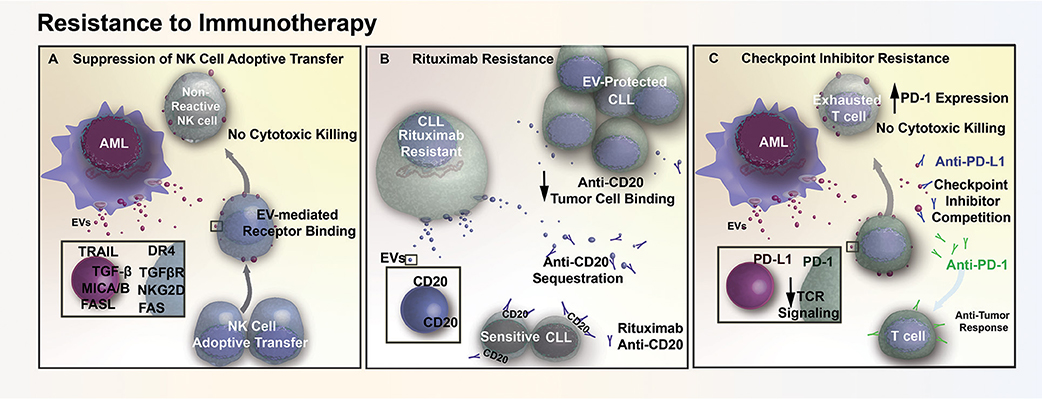
Figure 3. EV mediated resistance to immunotherapy. (A) AML EVs contain numerous immunosuppressive ligands (TRAIL, FASL, MICA/B) that reduce natural killer (NK) cell reactivity through receptor mediated binding. This EV-mediated signaling interferes with cell-based therapy, diminishing cytotoxic killing of tumor cells following adoptive transfer of NK cells. (B) EVs in CLL contain surface CD20, which acts as a decoy by sequestering Rituximab (anti-CD20) and preventing therapeutic antibodies from binding and opsonizing the tumor cells. (C) AML cells release EVs that contain the immunosuppressive ligand PD-L1. The transfer of PD-L1 via EVs reduces T cell activation in response to TCR stimulus, while also acting as decoys that compete with checkpoint inhibitor binding and prevent therapeutic antibodies from reaching their intended target.
AML cells also release exosomes that contain a potent immunosuppressive protein, programmed death-receptor ligand 1 (PD-L1) (79). PD-L1 binding to its cognate receptor, programed death-receptor 1 (PD-1), in both leukemia and solid tumors are able to suppress T cell activation in response to T cell receptor stimulation (81, 82). Expression of PD-L1 by tumor cells prevents T cell- and NK cell-mediated immune recognition and clearance, which increases the number of T cells with an “exhausted” and unreactive phenotype. It has been shown in both prostate cancer and melanoma that exosome-bound PD-L1 contributes to T cell suppression in vitro and in vivo. Additionally, exosomal PD-L1 has been shown to act as a decoy, sequestering anti-PD-L1 checkpoint inhibitors and outcompeting the anti-PD-1 checkpoint blockade for binding sites on cytotoxic CD8+ T cells (83, 84). Similar to prostate and melanoma models, we have found that AML EVs suppress T cell activation and adsorb anti-PD-L1 antibodies, suggesting that EVs may also contribute to immune checkpoint inhibitor resistance in AML (Figure 3C) (Butler, unpublished).
Recent research has uncovered numerous mechanisms through which EVs modulate proliferation, migration, and survival of malignant cells. While studies have implicated several specific proteins, cytokines, mRNAs, and miRNAs in the EV-mediated transfer of chemoresistance, the full spectrum of EV cargo involved in extrinsic chemoresistance remains to be fully defined. Additionally, the mechanisms through which many of these transferred miRNAs and proteins influence cell survival during chemotherapy are not fully understood. Investigation into leukemic stem cells and their role in transferring functional resistance factors similarly remains a potential focus for future AML EV research. In vivo trafficking of EVs could also provide additional perspectives on how the chemoresistance phenotype is being transferred (85). While phenotypic changes have been observed due to the trafficking of miRNAs and other molecules via EVs, these EVs may additionally influence cells through novel ligand-receptor mediated mechanisms with EV surface molecules. Here, a recent study provides an elegant approach to EV surface protein profiling, and it would be valuable to investigate whether EVs exhibit preferential targeting based on surface epitopes, and how this may alter chemoresistance (86). Recent work suggesting EV involvement in metastatic dissemination and functional conversion of other tissues is instructive, and may hold insight into the spread of AML to extramedullary sites (87). It will be critical to improve our understanding of dose and potency of EVs needed to confer drug resistance and whether a certain tumor burden must first exist, before AML-derived EVs have a significant influence on the leukemic niche. With numerous pathways implicated in the development of extrinsic chemoresistance, selectively targeting EV biogenesis or uptake represents a potential way to prevent intracellular leukemia cell survival signaling. As a proof-of-concept toward EV-uptake inhibition as a therapeutic strategy, Ortiz et al. recently showed that tumor EV-mediated pre-metastatic niche conversion can be prevented pharmacologically via administration of an EV-uptake inhibitor (87). On the other hand, Rab27 alpha and -beta (Rab27a/b) have both been identified as a critical modulators of EV biogenesis and secretion (39, 88). Thus, blocking EV biogenesis via loss of Rab27a/b function (89) represents a potential therapeutic approach. The ultimate goal, of course, is not so much to suppress EV trafficking per se, but rather to exploit our understanding of EV biology to identify cellular targets to overcome chemoresistance and achieve sustained long-term remissions. As an example, we previously have identified the role of miR-155, highly abundant in AML-EV, in the suppression of residual hematopoiesis in the AML BM (90). That study also introduced a discovery approach for novel EV-miRNA targets utilizing of molecular (RNA-induced silencing complex trap) and STING database (bioinformatic methodologies). Such an approach enables a broader understanding of complex AML-EV-mediated signaling and novel candidate targets (90). From a disease standpoint, minimizing and reversing the functional adaptation of the BM into a leukemia reinforcing compartment provides untapped opportunities to improve treatment outcomes in AML patients.
JN and PK conceived, wrote, and edited the manuscript. JB designed figures and wrote and edited the manuscript. D-WC wrote and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge the many investigators working in the field, not all of whom we were able to cite due to space limitations.
1. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. (2016) 6:e441. doi: 10.1038/bcj.2016.50
2. Szer J. The prevalent predicament of relapsed acute myeloid leukemia. ASH Educ Prog Book. (2012) 2012:43–8. doi: 10.1182/asheducation.V2012.1.43.3798183
3. Prada-Arismendy J, Arroyave JC, Rothlisberger S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. (2017) 31:63–76. doi: 10.1016/j.blre.2016.08.005
4. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. (2012) 481:506–10. doi: 10.1038/nature10738
5. Ghiaur G, Wroblewski M, Loges S. Acute myelogenous leukemia and its microenvironment: a molecular conversation. Semin Hematol. (2015) 52:200–6. doi: 10.1053/j.seminhematol.2015.03.003
6. Zhang J, Gu Y, Chen B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. (2019) 12:1937–45. doi: 10.2147/OTT.S191621
7. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
8. Huan J, Hornick NI, Shurtleff MJ, Skinner AM, Goloviznina NA, Roberts CT Jr, et al. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. (2013) 73:918–29. doi: 10.1158/0008-5472.CAN-12-2184
9. Butler JT, Abdelhamed S, Kurre P. Extracellular vesicles in the hematopoietic microenvironment. Haematologica. (2018) 103:382–94. doi: 10.3324/haematol.2017.183335
10. Viola S, Traer E, Huan J, Hornick NI, Tyner JW, Agarwal A, et al. Alterations in acute myeloid leukaemia bone marrow stromal cell exosome content coincide with gains in tyrosine kinase inhibitor resistance. Br J Haematol. (2016) 172:983–6. doi: 10.1111/bjh.13551
11. Chen T, Zhang G, Kong L, Xu S, Wang Y, Dong M. Leukemia-derived exosomes induced IL-8 production in bone marrow stromal cells to protect the leukemia cells against chemotherapy. Life Sci. (2019) 221:187–95. doi: 10.1016/j.lfs.2019.02.003
12. Doron B, Abdelhamed S, Butler JT, Hashmi SK, Horton TM, Kurre P. Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia. (2019) 33:918–30. doi: 10.1038/s41375-018-0254-2
13. Bloomfield CD, Marcucci G, Dohner K, Dohner H. Acute myeloid leukemia. Introduction. Semin Oncol. (2008) 35:324–5. doi: 10.1053/j.seminoncol.2008.04.016
14. Moarii M, Papaemmanuil E. Classification and risk assessment in AML: integrating cytogenetics and molecular profiling. Hematology Am Soc Hematol Educ Program. (2017) 2017:37–44. doi: 10.1182/asheducation-2017.1.37
15. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. (2019) 33:299–312. doi: 10.1038/s41375-018-0357-9
16. DiNardo CD, Cortes JE. Mutations in AML: prognostic and therapeutic implications. Hematology Am Soc Hematol Educ Program. (2016) 2016:348–55. doi: 10.1182/asheducation-2016.1.348
17. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol. (2017) 18:17. doi: 10.1007/s11864-017-0456-2
18. Ramos NR, Mo CC, Karp JE, Hourigan CS. Current approaches in the treatment of relapsed and refractory acute myeloid leukemia. J Clin Med. (2015) 4:665–95. doi: 10.3390/jcm4040665
19. Tsirigotis P, Byrne M, Schmid C, Baron F, Ciceri F, Esteve J, et al. Relapse of AML after hematopoietic stem cell transplantation: methods of monitoring and preventive strategies. A review from the ALWP of the EBMT. Bone Marrow Transplant. (2016) 51:1431–8. doi: 10.1038/bmt.2016.167
20. Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia. (2005) 19:1345. doi: 10.1038/sj.leu.2403838
21. Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. (2002) 100:2393–8. doi: 10.1182/blood-2002-02-0420
22. Moore AS, Faisal A, De Castro DG, Bavetsias V, Sun C, Atrash B, et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: a model for emerging clinical resistance patterns. Leukemia. (2012) 26:1462. doi: 10.1038/leu.2012.52
23. Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. (2005) 105:54–60. doi: 10.1182/blood-2004-03-0891
24. Yeung CCS, Radich J. Predicting chemotherapy resistance in AML. Curr Hematol Malig Rep. (2017) 12:530–6. doi: 10.1007/s11899-017-0378-x
25. Zhou J, Ching YQ, Chng W-J. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: from molecular pathogenesis to therapeutic target. Oncotarget. (2015) 6:5490. doi: 10.18632/oncotarget.3545
26. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. (2013) 2013:972913. doi: 10.1155/2013/972913
27. Hatem E, El Banna N, Huang M-E. Multifaceted roles of glutathione and glutathione-based systems in carcinogenesis and anticancer drug resistance. Antioxid Redox Signal. (2017) 27:1217–34. doi: 10.1089/ars.2017.7134
28. Li S, Li C, Jin S, Liu J, Xue X, Eltahan AS, et al. Overcoming resistance to cisplatin by inhibition of glutathione S-transferases (GSTs) with ethacraplatin micelles in vitro and in vivo. Biomaterials. (2017) 144:119–29. doi: 10.1016/j.biomaterials.2017.08.021
29. Gabra MM, Salmena L. microRNAs and acute myeloid leukemia chemoresistance: a mechanistic overview. Front Oncol. (2017) 7:255. doi: 10.3389/fonc.2017.00255
30. Chen Y, Jacamo R, Konopleva M, Garzon R, Croce C, Andreeff M. CXCR4 downregulation of let-7a drives chemoresistance in acute myeloid leukemia. J Clin Invest. (2013) 123:2395–407. doi: 10.1172/JCI66553
31. Liu X, Liao W, Peng H, Luo X, Luo Z, Jiang H, et al. miR-181a promotes G1/S transition and cell proliferation in pediatric acute myeloid leukemia by targeting ATM. J Cancer Res Clin Oncol. (2016) 142:77–87. doi: 10.1007/s00432-015-1995-1
32. Lai T-H, Ewald B, Zecevic A, Liu C, Sulda M, Papaioannou D, et al. HDAC inhibition induces microRNA-182, which targets RAD51 and impairs HR repair to sensitize cells to sapacitabine in acute myelogenous leukemia. Clin Cancer Res. (2016) 22:3537–49. doi: 10.1158/1078-0432.CCR-15-1063
33. Cittelly DM, Das PM, Salvo VA, Fonseca JP, Burow ME, Jones FE. Oncogenic HER2Δ16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis. (2010) 31:2049–57. doi: 10.1093/carcin/bgq192
34. Xia L, Zhang D, Du R, Pan Y, Zhao L, Sun S, et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer. (2008) 123:372–9. doi: 10.1002/ijc.23501
35. Yang D, Zhan M, Chen T, Chen W, Zhang Y, Xu S, et al. miR-125b-5p enhances chemotherapy sensitivity to cisplatin by down-regulating Bcl2 in gallbladder cancer. Sci Rep. (2017) 7:43109. doi: 10.1038/srep43109
36. Li Q, Liang X, Wang Y, Meng X, Xu Y, Cai S, et al. miR-139-5p inhibits the epithelial-mesenchymal transition and enhances the chemotherapeutic sensitivity of colorectal cancer cells by downregulating BCL2. Sci Rep. (2016) 6:27157. doi: 10.1038/srep27157
37. Ikemura K, Yamamoto M, Miyazaki S, Mizutani H, Iwamoto T, Okuda M. MicroRNA-145 post-transcriptionally regulates the expression and function of P-glycoprotein in intestinal epithelial cells. Mol Pharmacol. (2013) 83:399–405. doi: 10.1124/mol.112.081844
38. Li H, Hui L, Xu W. miR-181a sensitizes a multidrug-resistant leukemia cell line K562/A02 to daunorubicin by targeting BCL-2. Acta Biochim Biophys Sin. (2012) 44:269–77. doi: 10.1093/abbs/gmr128
39. Gu H, Chen C, Hao X, Wang C, Zhang X, Li Z, et al. Sorting protein VPS33B regulates exosomal autocrine signaling to mediate hematopoiesis and leukemogenesis. J Clin Invest. (2016) 126:4537–53. doi: 10.1172/JCI87105
40. Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. Br J Haematol. (2014) 164:767–78. doi: 10.1111/bjh.12725
41. Ryningen A, Wergeland L, Glenjen N, Gjertsen BT, Bruserud O. In vitro crosstalk between fibroblasts and native human acute myelogenous leukemia (AML) blasts via local cytokine networks results in increased proliferation and decreased apoptosis of AML cells as well as increased levels of proangiogenic Interleukin 8. Leuk Res. (2005) 29:185–96. doi: 10.1016/j.leukres.2004.06.008
42. Battula VL, Le PM, Sun JC, Nguyen K, Yuan B, Zhou X, et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. (2017) 2:90036. doi: 10.1172/jci.insight.90036
43. Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia. (2018) 32:575–87. doi: 10.1038/leu.2017.259
44. Krevvata M, Silva BC, Manavalan JS, Galan-Diez M, Kode A, Matthews BG, et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood. (2014) 124:2834–46. doi: 10.1182/blood-2013-07-517219
45. Cogle CR, Goldman DC, Madlambayan GJ, Leon RP, Masri AA, Clark HA, et al. Functional integration of acute myeloid leukemia into the vascular niche. Leukemia. (2014) 28:1978–87. doi: 10.1038/leu.2014.109
46. Ben-Batalla I, Schultze A, Wroblewski M, Erdmann R, Heuser M, Waizenegger JS, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. (2013) 122:2443–52. doi: 10.1182/blood-2013-03-491431
47. Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature. (2017) 545:98. doi: 10.1038/nature22311
48. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell. (2018) 22:64–77.e66. doi: 10.1016/j.stem.2017.11.006
49. Binder S, Luciano M, Horejs-Hoeck J. The cytokine network in acute myeloid leukemia (AML): a focus on pro- and anti-inflammatory mediators. Cytokine Growth Factor Rev. (2018) 43:8–15. doi: 10.1016/j.cytogfr.2018.08.004
50. Sinyuk M, Alvarado AG, Nesmiyanov P, Shaw J, Mulkearns-Hubert EE, Eurich JT, et al. Cx25 contributes to leukemia cell communication and chemosensitivity. Oncotarget. (2015) 6:31508–21. doi: 10.18632/oncotarget.5226
51. Wang J, Liu X, Qiu Y, Shi Y, Cai J, Wang B, et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J Hematol Oncol. (2018) 11:11. doi: 10.1186/s13045-018-0554-z
52. Samanta S, Rajasingh S, Drosos N, Zhou Z, Dawn B, Rajasingh J. Exosomes: new molecular targets of diseases. Acta Pharmacol Sin. (2018) 39:501–13. doi: 10.1038/aps.2017.162
53. Zhang H, Freitas D, Kim HS, Fabijanic K, Li Z, Chen H, et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat Cell Biol. (2018) 20:332–43. doi: 10.1038/s41556-018-0040-4
54. Santos JC, Lima NDS, Sarian LO, Matheu A, Ribeiro ML, Derchain SFM. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci Rep. (2018) 8:829. doi: 10.1038/s41598-018-19339-5
55. Zhang R, Xia Y, Wang Z, Zheng J, Chen Y, Li X, et al. Serum long non coding RNA MALAT-1 protected by exosomes is up-regulated and promotes cell proliferation and migration in non-small cell lung cancer. Biochem Biophys Res Commun. (2017) 490:406–14. doi: 10.1016/j.bbrc.2017.06.055
56. Zhang H, Wang Y, Bai M, Wang J, Zhu K, Liu R, et al. Exosomes serve as nanoparticles to suppress tumor growth and angiogenesis in gastric cancer by delivering hepatocyte growth factor siRNA. Cancer Sci. (2018) 109:629–41. doi: 10.1111/cas.13488
57. Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. (2014) 289:3869–75. doi: 10.1074/jbc.C113.532267
58. Hong CS, Muller L, Whiteside TL, Boyiadzis M. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front Immunol. (2014) 5:160. doi: 10.3389/fimmu.2014.00160
59. Zhang C, Ji Q, Yang Y, Li Q, Wang Z. Exosome: function and role in cancer metastasis and drug resistance. Technol Cancer Res Treat. (2018) 17:1533033818763450. doi: 10.1177/1533033818763450
60. Qin H, Shao Q, Curtis H, Galipeau J, Belliveau DJ, Wang T, et al. Retroviral delivery of connexin genes to human breast tumor cells inhibits in vivo tumor growth by a mechanism that is independent of significant gap junctional intercellular communication. J Biol Chem. (2002) 277:29132–8. doi: 10.1074/jbc.M200797200
61. Li X, Pan JH, Song B, Xiong EQ, Chen ZW, Zhou ZS, et al. Suppression of CX43 expression by miR-20a in the progression of human prostate cancer. Cancer Biol Ther. (2012) 13:890–8. doi: 10.4161/cbt.20841
62. Jin Z, Xu S, Yu H, Yang B, Zhao H, Zhao G. miR-125b inhibits Connexin43 and promotes glioma growth. Cell Mol Neurobiol. (2013) 33:1143–8. doi: 10.1007/s10571-013-9980-1
63. Aasen T, Leithe E, Graham SV, Kameritsch P, Mayan MD, Mesnil M, et al. Connexins in cancer: bridging the gap to the clinic. Oncogene. (2019) 38:4429–51. doi: 10.1038/s41388-019-0741-6
64. Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med. (2013) 11:94. doi: 10.1186/1479-5876-11-94
65. Ariazi J, Benowitz A, De Biasi V, Den Boer ML, Cherqui S, Cui H, et al. Tunneling nanotubes and gap junctions-their role in long-range intercellular communication during development, health, and disease conditions. Front Mol Neurosci. (2017) 10:333. doi: 10.3389/fnmol.2017.00333
66. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. (2008) 10:619–24. doi: 10.1038/ncb1725
67. Wojtuszkiewicz A, Schuurhuis GJ, Kessler FL, Piersma SR, Knol JC, Pham TV, et al. Exosomes secreted by apoptosis-resistant acute myeloid leukemia (AML) blasts harbor regulatory network proteins potentially involved in antagonism of apoptosis. Mol Cell Proteomics. (2016) 15:1281–98. doi: 10.1074/mcp.M115.052944
68. Bouvy C, Wannez A, Laloy J, Chatelain C, Dogne JM. Transfer of multidrug resistance among acute myeloid leukemia cells via extracellular vesicles and their microRNA cargo. Leuk Res. (2017) 62:70–6. doi: 10.1016/j.leukres.2017.09.014
69. van Stijn A, Feller N, Kok A, van der Pol MA, Ossenkoppele GJ, Schuurhuis GJ. Minimal residual disease in acute myeloid leukemia is predicted by an apoptosis-resistant protein profile at diagnosis. Clin Cancer Res. (2005) 11:2540–6. doi: 10.1158/1078-0432.CCR-04-1973
70. Sun H, Lin DC, Guo X, Kharabi Masouleh B, Gery S, Cao Q, et al. Inhibition of IRE1alpha-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget. (2016) 7:18736–49. doi: 10.18632/oncotarget.7702
71. Au Yeung CL, Co NN, Tsuruga T, Yeung TL, Kwan SY, Leung CS, et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. (2016) 7:11150. doi: 10.1038/ncomms11150
72. Patel GK, Khan MA, Bhardwaj A, Srivastava SK, Zubair H, Patton MC, et al. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br J Cancer. (2017) 116:609–19. doi: 10.1038/bjc.2017.18
73. Petrocca F, Vecchione A, Croce CM. Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor β signaling. Cancer Res. (2008) 68:8191–4. doi: 10.1158/0008-5472.CAN-08-1768
74. Tazzari PL, Cappellini A, Ricci F, Evangelisti C, Papa V, Grafone T, et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia. (2007) 21:427–38. doi: 10.1038/sj.leu.2404523
75. Menck K, Sönmezer C, Worst TS, Schulz M, Dihazi GH, Streit F, et al. Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J Extracell Vesicles. (2017) 6:1378056. doi: 10.1080/20013078.2017.1378056
76. Corrado C, Raimondo S, Saieva L, Flugy AM, De Leo G, Alessandro R. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett. (2014) 348:71–6. doi: 10.1016/j.canlet.2014.03.009
77. Rodvold JJ, Chiu KT, Hiramatsu N, Nussbacher JK, Galimberti V, Mahadevan NR, et al. Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci Signal. (2017) 10:eaah7177. doi: 10.1126/scisignal.aah7177
78. Laperrousaz B, Jeanpierre S, Sagorny K, Voeltzel T, Ramas S, Kaniewski B, et al. Primitive CML cell expansion relies on abnormal levels of BMPs provided by the niche and on BMPRIb overexpression. Blood. (2013) 122:3767–77. doi: 10.1182/blood-2013-05-501460
79. Hong CS, Sharma P, Yerneni SS, Simms P, Jackson EK, Whiteside TL, et al. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci Rep. (2017) 7:14684. doi: 10.1038/s41598-017-14661-w
80. Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood. (2015) 126:1106–17. doi: 10.1182/blood-2014-12-618025
81. Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. (2010) 116:2484–93. doi: 10.1182/blood-2010-03-275446
82. Boddu P, Kantarjian H, Garcia-Manero G, Allison J, Sharma P, Daver N. The emerging role of immune checkpoint based approaches in AML and MDS. Leuk Lymphoma. (2018) 59:790–802. doi: 10.1080/10428194.2017.1344905
83. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. (2018) 560:382–6. doi: 10.1038/s41586-018-0392-8
84. Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell. (2019) 177:414–27.e413. doi: 10.1016/j.cell.2019.02.016
85. Duarte D, Amarteifio S, Ang H, Kong IY, Ruivo N, Pruessner G, et al. Defining the in vivo characteristics of acute myeloid leukemia cells behavior by intravital imaging. Immunol. Cell Biol. (2019) 97:229–35. doi: 10.1111/imcb.12216
86. Wu D, Yan J, Shen X, Sun Y, Thulin M, Cai Y, et al. Profiling surface proteins on individual exosomes using a proximity barcoding assay. Nat Commun. (2019) 10:3854. doi: 10.1038/s41467-019-11486-1
87. Ortiz A, Gui J, Zahedi F, Yu P, Cho C, Bhattacharya S, et al. An interferon-driven oxysterol-based defense against tumor-derived extracellular vesicles. Cancer Cell. (2019) 35:33–45.e36. doi: 10.1016/j.ccell.2018.12.001
88. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. (2010) 12:19. doi: 10.1038/ncb2000
89. Guo D, Lui GY, Lai SL, Wilmott JS, Tikoo S, Jackett LA, et al. RAB27A promotes melanoma cell invasion and metastasis via regulation of pro-invasive exosomes. Int J Cancer. (2019) 144:3070–85. doi: 10.1002/ijc.32064
Keywords: acute myeloid leukemia, extracellular vesicles, chemoresistance, bone marrow microenvironment, stroma
Citation: Nehrbas J, Butler JT, Chen D-W and Kurre P (2020) Extracellular Vesicles and Chemotherapy Resistance in the AML Microenvironment. Front. Oncol. 10:90. doi: 10.3389/fonc.2020.00090
Received: 05 November 2019; Accepted: 17 January 2020;
Published: 14 February 2020.
Edited by:
Yong-mi Kim, Children's Hospital of Los Angeles, United StatesReviewed by:
Hinrich Peter Hansen, University of Cologne, GermanyCopyright © 2020 Nehrbas, Butler, Chen and Kurre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Kurre, a3VycmVwQGVtYWlsLmNob3AuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.