 Shanshan Deng1,2
Shanshan Deng1,2 Michael J. Clowers1,3
Michael J. Clowers1,3 Walter V. Velasco1
Walter V. Velasco1 Marco Ramos-Castaneda1
Marco Ramos-Castaneda1 Seyed Javad Moghaddam1,3*
Seyed Javad Moghaddam1,3*- 1Department of Pulmonary Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Department of Respiratory and Critical Care Medicine, Second Affiliated Hospital of Xi'an Jiaotong University, Xi'an, China
- 3MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, United States
Kirsten rat sarcoma viral oncogene (K-ras) is a well-documented, frequently mutated gene in lung cancer. Since K-ras regulates numerous signaling pathways related to cell survival and proliferation, mutations in this gene are powerful drivers of tumorigenesis and confer prodigious survival advantages to developing tumors. These malignant cells dramatically alter their local tissue environment and in the process recruit a powerful ally: inflammation. Inflammation in the context of the tumor microenvironment can be described as either antitumor or protumor (i.e., aiding or restricting tumor progression, respectively). Many current treatments, like immune checkpoint blockade, seek to augment antitumor inflammation by alleviating inhibitory signaling in cytotoxic T cells; however, a burgeoning area of research is now focusing on ways to modulate and mitigate protumor inflammation. Here, we summarize the interplay of tumor-promoting inflammation and K-ras mutant lung cancer pathogenesis by exploring the cytokines, signaling pathways, and immune cells that mediate this process.
Introduction
A staggering 23% of cancer-related deaths in the Unites States can be imputed to lung cancer, which translates to upwards of 140,000 deaths per year (1). In 90% of cases, lung cancer is caused by cigarette smoke and subsequent DNA mutations (2). One commonly mutated gene, particularly in lung adenocarcinoma (LUAD), is Kirsten rat sarcoma viral oncogene (K-ras) (3). In the wild type state, K-ras hydrolyzes GTP and activates the Raf-MEK-ERK signaling cascade leading to cell survival, cell cycle progression, and cell polarity (4, 5). However, once mutated, overactive K-ras drives these processes independently of upstream signals, thereby heavily contributing to tumorigenesis (4). Logically, efforts have been made to pharmacologically target K-ras, yet all treatments tried so far have proven fruitless (6). Although downstream effectors of K-ras signaling (e.g., MEK) are being explored as alternate targets (7), another potential therapeutic modality is emerging: targeting tumor-promoting inflammation.
Unlike other oncogenic changes that have already become attractive drug targets, such as EGFR mutations and ALK, ROS and RET rearrangements, K-ras mutations are still viewed as “undruggable.” However, studies have shown that K-ras mutant lung cancer is strongly associated with inflammation which renders a new target for tackling this disease. Inflammation has been recognized as a key promoter of cancer initiation and progression (8). The immune system can often deleteriously impact antitumor responses by producing factors that aid mutagenesis, tumorigenesis, and immune suppression (8). Through the secretion of cytokines, tumor cells can reprogram the tumor microenvironment (TME), recruit immune cells, and then sway these cells toward non-productive immune responses while simultaneously dampening cytotoxic responses (9). The end result, as summarized in Figure 1, yields a protumor TME littered with molecules and cells helping the cancer survive and resist treatment.
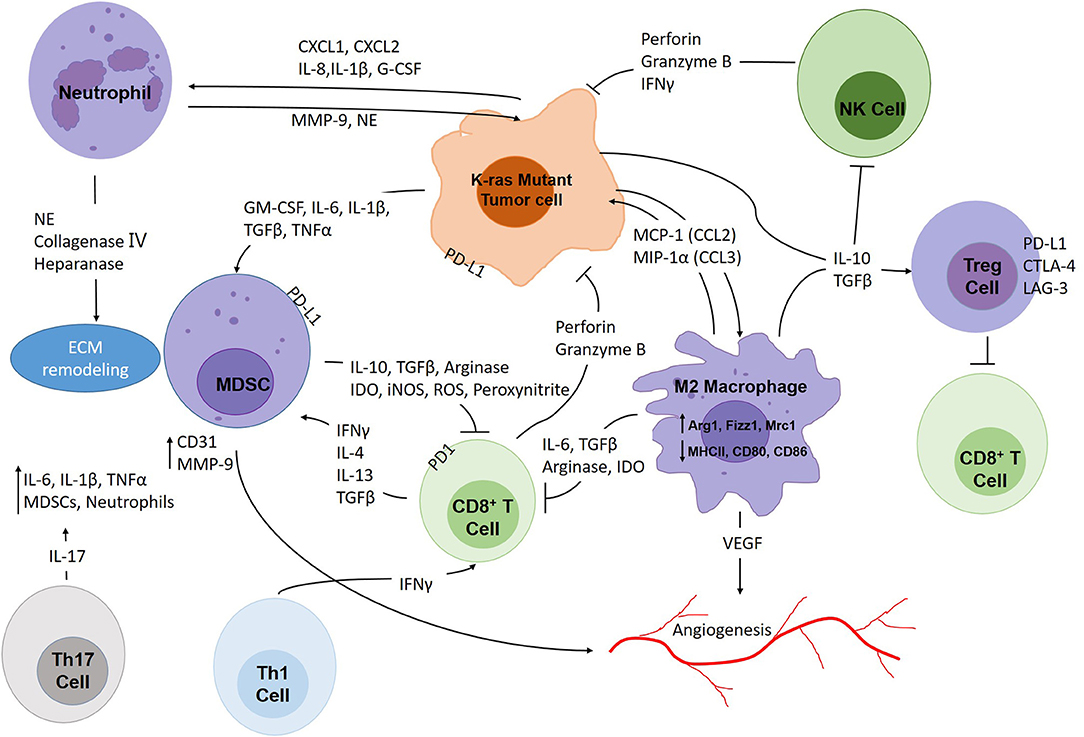
Figure 1. The intricate interplay within the K-ras mutant lung cancer tumor microenvironment. Tumors are constantly infiltrated with immune cells which make up important components of the TME. Tumor cells attract immunosuppressive immune cells such as M2 macrophages, neutrophils, MDSCs, Th17 and Treg cells through secretion of soluble factors (CCL2, CCL3, CXCL1, CXCL2, IL-1β, IL-6, IL-8, IL-10, TGFβ, G-CSF, etc.). These cells promote tumor growth, angiogenesis, and at the same time, protect tumor cells from cytotoxic effects. CD8+ T cells and NK cells attack tumor cells through secretion of perforin, granzyme B and IFNγ, and Th1 cells act as important assistants for CD8+ T cells. However, tumor cells, MDSCs and Treg cells could render cytotoxic immune cells incompetent through expressing immune checkpoint molecules. Moreover, immunosuppressive cells also produce soluble factors that exhaust CD8+ T cells and NK cells, such as arginase, IDO, iNOS, TGFβ, etc.
Using genetically modified mouse models, researchers have found that lung cancers with both K-ras and EGFR mutations display immune responses with 3–5 fold increase in total infiltrating CD45+ cells compared with normal lung tissue (10). However, EGFR mutant lung cancers exhibit myeloid cell recruitment, but no CD8+ immune response was observed, suggesting that EGFR mutations may not be able to generate a sufficient antigen-driven immune response (10). In contrast, both pre- and clinical data have shown that K-ras mutant lung cancers demonstrate significant infiltration of multiple inflammatory cells, such as myeloid cells, CD8+ T cells, regulatory T cells (Tregs), IL-17-producing lymphocytes, and inflammatory cytokines (e.g., IL-6, IL-8, CXCL1) (10, 11). The reason why K-ras mutant lung cancer could generate a stronger CD8+ T cell response might be the fact that it is strongly associated with cigarette smoke and a high mutation burden that could generate an antigen-specific response. Moreover, K-ras mutations have been reported to be associated with increased PD-L1 expression (12). Patients with K-ras/Tp53 co-mutations exhibit higher PD-L1 and a higher PD-L1+/CD8A+ ratio, reaping remarkable benefits from PD-1 inhibitors (13).
In contrast with other subgroups of NSCLC which are deemed “immune-cold,” the future direction for tackling K-ras mutant lung cancer might rest on controlling tumor-promoting inflammation. In order to combat this inflammatory environment, it is necessary to take into account the main signaling pathways involved, the cytokines and other soluble factors present in the TME, and the immune cells that are recruited. In this review, we describe the interplay between these components in the TME and highlight ways in which future cancer therapeutics may target tumor-promoting inflammation in K-ras mutant lung cancer.
Inflammatory Signaling Pathways
Signaling downstream of K-ras plays a pivotal role in K-ras-driven non-small cell lung cancer (NSCLC), especially LUAD. In fact, studies revolving around this oncogene have identified STAT3 and NF-κB as key pathways. In this section we will cover the importance of each of these pathways and their contributions to understanding the mechanisms behind this increasingly important disease.
STAT3
An important signaling mediator downstream and associated with K-ras mutations is signal transducer and activator of transcription (STAT), particularly STAT3. STAT3 is triggered via several cytokines and growth factors; one, which we discuss later, is IL-6 (14). When IL-6 binds to its receptor, it can activate Janus kinase non-receptor tyrosine kinases (JAKs) 1 and 2 (14). These JAKs will cause receptor phosphorylation, which will produce docking sites for signaling molecules, mainly STAT3 (15). Ultimately, STAT3 will dimerize and translocate to the nucleus in order to transcribe several target genes involved in cellular processes like apoptosis, angiogenesis, and metastasis (14).
The role that STAT3 plays in lung cancer is controversial due to opposite findings by various researches. Mohrherr et al. demonstrate the presence of JAK1 and JAK2 in human LUAD that positively correlates with disease progression and K-ras activity. In their mouse models, they administered a JAK1/2 selective tyrosine kinase inhibitor called ruxolitinib. With the administration of this drug they found a reduction in tumor cell proliferation and effectively ameliorated the tumorigenic phenotype in both immunocompetent and immunodeficient mouse models of K-ras-driven LUAD (16). Another research group, Grabner et al. has also interrogated the role of STAT3 during K-ras-driven lung tumorigenesis using the Cre-inducible K-rasG12D knock-in lung cancer mouse model as well as a human xenograft model. Their findings contrastingly indicate that STAT3 functions as a tumor suppressor in both models. When they genetically eliminated STAT3 in mouse lung tumors and human LUAD cell line A549, they found increased tumor growth, higher tumor grade, increased vascularization, and significantly reduced survival. These results are further supported by clinical findings where activation and expression of STAT3 was decreased in mutant K-ras patient samples compared to mutant EGFR and wild type K-ras tumors. Essentially, higher-grade tumors showed a significant reduction in STAT3 expression when compared to their low-grade tumor counterparts. In addition, low STAT3 expression had worse overall survival when compared to patients with higher STAT3 expression levels (17, 18).
Timing also contributes to the controversy of this disease. One particular study demonstrates how lung epithelial deletion of STAT3 in mice before the induction of cancer by urethane, a smoke carcinogen, resulted in increased lung tissue damage, inflammation, and tumorigenesis, while ablation of lung epithelial STAT3 after establishing lung cancer inhibited this tumorigenic process (19). These results seem to indicate that tumor heterogeneity is an important aspect in K-ras-driven LUAD. Even though K-ras mutations may share some common signaling, some K-ras mutations such as K-rasG12C tumors show greater dependence on the Raf-MEK-ERK pathway compared with K-rasG12D suggesting they may be more sensitive to MEK inhibitors as assessed by Li et al. (20).
While acquisition of these tumorigenic processes are often found in K-ras mutant mice, a previously unknown function of STAT3 in regards to epithelial identity and differentiation was recently discovered. This study associates aggressive tumor behavior and acquirement of mesenchymal-like phenotypes with the loss of STAT3 function, while persistent STAT3 activation bestows a differentiated epithelial morphology to cells that can affect their potential for tumorigenesis (21). Receptor-interacting serine/threonine protein kinase 4 (RIP4) is an ankyrin repeat-containing kinase that is in charge of keratinocyte differentiation and delays cell migration during wound healing. It has recently been found to be a likely regulator of tumor differentiation in LUAD and contributes to epithelial identity and differentiation (22). This study demonstrated that poorly differentiated tumors correlate with low expression of RIP4, whereas high expression correlates with better overall survival. Cells from their in vitro studies associate reduced RIP4 expression with elevated activation of STAT3 signaling and had an overall increased capacity for tissue invasion. In comparison, overexpression of RIP4 inhibited STAT3: after tail vein injections of RIP4-overexpressing cells, tissue invasion and tumor formation were reduced, which was restored by co-expression of STAT3 (22).
Our own group has interestingly shown a gender-specific role for lung epithelial STAT3 signaling in the pathogenesis of K-ras-driven LUAD. Decreased tumorigenesis was found in female mice lacking epithelial STAT3, yet loss of epithelial STAT3 in male littermates led to an opposite effect of enhanced malignancy, an effect driven by induction of an NF-κB-mediated IL-6/CXCL2 associated neutrophilic response and reduction of immune-mediated cytotoxicity (23). Zhou et al. used mouse models of myeloid-specific STAT3 deletion to highlight the importance of STAT3 as a major driver of myeloid-derived suppressor cell (MDSC) and macrophage pro-tumorigenic states. They found that the antitumor T helper 1 (Th1) and CD8+ T cells shared an inverse relationship in the development of lung cancer. Promotion of tumorigenesis was caused by induction of Tregs, inhibition of dendritic cells (DCs), and polarization of macrophages toward a pro-tumorigenic M2 phenotype due to activation of STAT3 in MDSCs and macrophages. Conversely, deletion of myeloid STAT3 boosted antitumor immunity and suppressed lung tumorigenesis (24).
A great amount of effort has gone into the development and identification of STAT3 inhibitors that can be applied in a clinical setting. The first ones developed were direct inhibitors of STAT3, which bind to the SH2 domain of STAT3, disrupting STAT3 dimerization and DNA-binding activity (25). However, their use has been limited in patients with NSCLC since studies showed issues with tolerability (26). The use of antisense oligonucleotides, most notably AZD9150, has emerged to provide an alternate approach to inhibition of STAT3 and has shown promising results when compared to direct STAT3 inhibitors as they mitigate end-organ damage and other adverse effects (27). Indeed, with the favorable safety profile and preliminary data, further evaluation of this therapy should be investigated in order to proceed to its use in a clinical setting.
NF-κB
Another frequently activated pathway in NSCLC is the nuclear factor-κB (NF-κB) transcription factor pathway. Five members compose this dimeric transcription factor including: RelA (p65), RelB, c-Rel, p50/p105, and p52/p100 (28). These five members are capable of forming diverse homo- and heterodimers in order to variably control gene expression which is directed by signaling from cytokines, bacterial and viral byproducts, stressful stimuli, and growth factors (29). In naïve cells, the NF-κB complex is kept in a dormant state through its interaction with inhibitor of κB (IκB) proteins. IκB is phosphorylated by the IκB kinase (IKK) complex due to cytokine signaling or other relevant stimuli and afterwards undergoes rapid degradation. NF-κB subunits are freed and then released into the nucleus where they control various gene transcription targets that are crucial in cell proliferation, cell survival, inflammation, and immune responses (30, 31).
When looking at data obtained from lung cancer patients, high levels of NF-κB activation in NSCLC was significantly associated with TNM stages: In particular, NF-κB p65 expression level was significantly increased in TNM stages III and IV when compared to stages I and II (32). Additionally, the presence of nuclear RelA and cytoplasmic phosphorylated IκB (pIκB) significantly correlated with poor patient prognosis and survival (33). Song et al. have interrogated the mechanisms behind the IκB complex specifically IKKα which is essential for NF-κB activation. They found that its inhibition upregulates NOX2 and downregulates NRF2, leading to reactive oxygen species (ROS) accumulation and blockade of cell senescence which ultimately accelerates LUAD development (34). Their work demonstrates a unique pathogenesis mechanism mediated through ROS. Our own studies have likewise shown that NF-κB is activated in tumor and surrounding inflammatory cells in our K-ras-driven mouse model of LUAD (35). Bassères et al. also demonstrate that NF-κB is important in K-ras-driven tumorigenesis because the absence of p65/RelA significantly impairs K-ras-driven lung tumorigenesis. Also, inhibition of IKKβ expression stops NF-κB activation in K-ras-driven lung cells (31). The researchers further support the importance of the IκB complex by administering an IKKβ inhibitor in primary human lung epithelial cells transformed by K-ras and K-ras-mutant lung cancer cell lines. Afterwards, they tested this drug in mouse models of K-ras-driven LUAD which resulted in smaller and lower grade tumors than mice treated with placebo in conjunction with reduced angiogenesis and inflammation (31). These studies point toward targeting IKKα and IKKβ as potential therapeutic approaches for K-ras-driven LUAD.
NF-κB may originate from myeloid cells, specifically macrophages where it plays a crucial role in mediating cytokine synthesis and secretion (36). Therefore, it is reasonable to think that myeloid cell-derived NF-κB may promote lung cancer through a mechanism of inflammatory cytokine secretion, which ultimately would lead to an inflammatory microenvironment predisposed to cancer. Inhibiting NF-κB in myeloid cells significantly decreased inflammatory chemokines and cytokines that were induced by cigarette smoke including tumor necrosis factor (TNF), CCL2, CCL3, and IL-6 as well as inflammatory cell infiltrate, which is associated with reduced size and multiplicity of the lung tumors (37, 38).
NF-κB is an important signal for humans to defend themselves from environmental insults, and it has important roles in both innate and adaptive immunity. Therefore, systemic administration of NF-κB inhibitors may negatively affect the host immune response and actually be detrimental to patient health. This is why currently there is no standalone therapy and in fact combination treatments are a better alternative for lung cancer chemoprevention (30, 39).
Cytokines and Their Receptors
Lung cancer has been associated with many cytokine signatures that aid in the survival of tumor cells. As a rule of thumb, cytokines that promote type 1 immunity are anti-tumorigenic, whereas those that lead to type 2/3 immunity or immunosuppression are typically pro-tumorigenic. The complexity of the TME response has shown us that many cytokines play different roles in tumor cell death or nurturing, and our understanding of how this complex network behaves could help us devise different therapeutic strategies. In this section, we briefly review the role of six important cytokines in the pathogenesis of lung cancer.
Interleukin 1β
Interleukin 1β (IL-1β) is a member of the IL-1 family of proteins. Two receptors have been reported to interact with IL-1β: type I and type II IL-1 receptors (IL-1RI/II). It is believed that both receptors mediate different actions with IL-1RI as the main signal transducer and IL-1RII as a decoy factor. Upon complex formation, the IL-1RI cytosolic domain signals through different adaptor proteins such as myeloid differentiation primary response gene 88 (MYD88) and interleukin 1 receptor-activated protein kinases 1, 2, and 4 (IRAK1/2/4). These proteins help in signal transduction of several pathways including NF-κB, PI3K/Akt/PKB, MAPK, and mTOR (40, 41).
IL-1β is expressed by many cells including natural killer (NK) cells (42), macrophages (43), endothelial cells (44), neutrophils (45), T cells, and fibroblasts (46). It has been traditionally associated with the promotion of inflammation during acute and chronic tissue injuries. Many of its immunological functions include promotion of monocyte to conventional DC differentiation, macrophage polarization toward an M1-like antitumor phenotype, and activated B cell clonal expansion and differentiation into plasma cells. IL-1β secreted by activated antigen presenting cells (APCs) induces type 1 responses by increasing interferon gamma (IFNγ) producing cytotoxic T lymphocytes (CTLs) and increasing the polarization of T cells toward Th1 (47).
Despite benefiting the type 1 immune response, IL-1β often exerts adverse effects in the context of cancer. Cytokine profiling of patients with NSCLC has shown increased levels of IL-1β in tumor specimens (48). In a NSCLC cancer study of patients treated with radiotherapy, increased levels of IL-1β showed a direct correlation with worse overall survival compared to patients with lower levels (49). Mechanistically, it has been shown in different types of cancer models that IL-1β augments intratumoral immunosuppressive macrophages and increases levels of VEGF and fibroblast growth factor (FGF), supporting angiogenesis and metastasis (50, 51). Moreover, IL-1β-deficient mice show increased DC infiltration and increased CD8+ lymphocytes supporting antitumor cytotoxic responses (52). GATA2, a transcription factor directly known to regulate the IL-1β signaling pathway, has been shown to function as a link between IL-1β signaling and tumor cell proliferation, since GATA2 genetic depletion diminishes tumor burden and progression in K-ras-induced lung cancer models (53). Additionally, urethane-induced lung cancer mouse models, in which myeloid NF-κB was inhibited, displayed increased levels of IL-1β and cell proliferation (54).
Recently, clinical trials targeting IL-1β in patients with atherosclerosis using the monoclonal antibody canakinumab (anti-IL-1β) have shown a direct correlation with decreased incidence of lung cancer, decreased mortality, and better prognosis when compared to patients treated with placebo (55). In addition, new ongoing clinical trials for patients with NSCLC are targeting IL-1β. A phase II clinical trial (NCT03968419) is combining canakinumab with pembrolizumab (anti-PD-1) (56), and a phase III clinical trial (NCT03631199) is exploring the safety and efficacy of pembrolizumab and conventional chemotherapy with or without canakinumab.
Interleukin 6
The glycosylated polypeptide interleukin 6 (IL-6) is the main member of the IL-6 family cytokines. Its receptor (IL-6R) dimerizes with another IL-6/IL6R complex and forms a hexameric structure that requires glycoprotein-130 (gp130, also called IL6-Rβ) membrane protein dimers (57). Trans-signaling occurs when IL-6 binds to a soluble form of IL-6R (sIL-6R) making a complex that later can bind to intramembranous gp130. Signal transduction can proceed through different pathways, with JAK/STAT (mainly JAK2/STAT3) predominating, though other pathways such as Raf-MEK-ERK, mTOR, Akt, and PI3K also play roles in cell survival, cell proliferation, and protein synthesis (58).
IL-6 expression is triggered by different inflammatory stimuli that are associated with tissue stress or damage (e.g., ROS, ultraviolet radiation, or ionizing radiation) and is mainly regulated by activation of the NF-κB pathway. IL-6 has been shown to induce immune cell recruitment, modify the TME (59), and help tumor cells with functions such as survival, apoptosis, angiogenesis, invasiveness, and metabolism. Immunological changes are also a hallmark of IL-6 expression, mainly characterized by protumoral changes with a decrease in CD8+ T cell responses and cytotoxicity (60), M1 to M2 macrophage polarization, induction of T helper 17 (Th17) and Treg cell responses, as well as increased migration of MDSCs. There is also evidence supporting a role for IL-6 in promoting epithelial-mesenchymal transition (EMT) (61).
Overexpression of IL-6 has been found in most cancer types, particularly NSCLC (62, 63), and has been shown to have an inverse correlation with patient prognosis (64, 65) and higher resistance to chemotherapy (66). NSCLC patients with higher TMN staging and worse prognosis exhibited higher serum levels of IL-6 compared to patients with lower disease stages (67–69). In lung cancer mouse models, blocking or deleting IL-6 reveals a delay in tumor progression and metastasis through deactivation of the IL-6/STAT3 pathway and cell proliferation regulator cyclin D1, as well as through enhancement of tumor cell apoptosis (70). Mice with K-ras mutant lung tumors treated with anti-IL-6 antibodies have shown an overall reduction in tumor burden and a shift to a TME with a less proliferative and a less protumor inflammatory context: fewer Th17 cells, fewer Tregs, and more M1-type polarization (62). Paradoxically, some models show evidence of accelerated tumorigenesis in early stages of IL-6 depletion (70, 71). However, therapeutic antibodies against IL-6R have decreased tumorigenesis and increased apoptosis in numerous human cancer cell lines synergistically with other targeted therapies such as EGFR inhibitors (71, 72). A clinical trial (NCT03337698) using tocilizumab (anti-IL-6R) is underway in patients with metastatic disease who have or have not received previous therapy with conventional chemotherapy plus anti-PD-1/PD-L1 therapy (73).
Interleukin 8
Interleukin 8 (IL-8) is also called neutrophil chemotactic factor or CXCL8. Its receptors, CXCR1 and CXCR2, are G-protein family members expressed mainly on neutrophils but also cells like fibroblasts, neurons, and epithelial cells (74). IL-8 expression is initiated and regulated by the NF-κB pathway, with its strongest activators being IL-1β and TNF (75). It is mainly secreted by immune cells such as macrophages, neutrophils, T cells, and non-immune cells like epithelial and endothelial cells. IL-8 secretion has several physiologic functions including chemotaxis, induction of phagocytosis, degranulation of neutrophils, DC migration, and potentiation of acute inflammatory reactions (76).
Overexpression of IL-8 and its receptors have been shown to play a pivotal role in promoting tumorigenesis in several neoplasia such as gliomas (77), cervical cancer (78), colorectal cancer (79), and lung cancer (17). Its main roles comprise autocrine and paracrine alterations of the TME, with one main mechanism being the recruitment of MDSCs (80). Other roles include angiogenesis through promotion of VEGF expression (81), induction of tumor growth, and facilitation of metastasis (82). Furthermore, IL-8 plays a central role in the promotion and induction of lung cancer cell proliferation (83), which is achieved through EGFR transactivation and increased MAPK pathway signaling (84).
High levels of IL-8 have been described within tumors of lung cancer patients with K-ras mutations, correlating with decreased disease-free survival and overall survival when compared to patients with low levels of IL-8 (85). K-ras mutant IL-8-overexpressing cell lines showed a sharp decrease in IL-8 expression when treated with K-ras-shRNA and MEK inhibitors, suggesting a direct relationship between activating K-ras mutations and upregulation of IL-8 expression. In these cell lines, inhibiting IL-8 led to decreased cell proliferation and migration, further indicating a role for IL-8 in promoting K-ras mutant lung cancer (86). Similarly, studies in which the CXCR2 receptor was inhibited through the use of neutralizing antibodies or selective inhibitors have shown a decrease in tumorigenesis, suppression of neutrophil migration, and induction of apoptosis in vascular endothelial cells in K-ras mutant mice (87, 88). Additionally, several NSCLC non-K-ras mutant cell lines where shown to have an increased proliferative rate and increased growth through the action of IL-8 in an endocrine fashion (83). Furthermore, high levels of IL-8 have been associated with resistance to lung cancer therapies such as EGFR inhibitors (89), conventional chemotherapy (90), and anti-PD-1 immunotherapy (91).
Interleukin 17A
Interleukin 17A (IL-17) is a 155 amino acid glycoprotein, which is secreted mainly by Th17 cells as a homodimer (92, 93). It can also be produced by other cells, such as CD8+ T cells, NK cells, type 3 innate lymphoid cells (ILC3s), γδ T cells and in some cases epithelial cells (94–96). Its receptor, IL-17R, is ubiquitously expressed and usually forms multimeric complexes (97).
IL-17-mediated responses have been shown to produce tumorigenic effects in the early stages of multiple cancers, including lung (98), gastric (99), and prostate cancer (100). Specifically, IL-17 acts to recruit MDSCs (101) through increased levels of IL-8 and encourages angiogenesis in tumor-surrounding endothelial cells and fibroblasts via production of VEGF, TGF-β, CXCL1, and CXCL5 (102). IL-17 also elevates levels of IL-1β, IL-6, and TNF, and its signaling activates the NF-κB and MAPK pathways (103, 104). Paradoxically IL-17 has also shown antitumor activity, in which IL-17-mediated responses had higher effects than Th1-driven responses in ovarian carcinomas. This antitumor response was highly dependent on high levels of IFNγ, IL-21, IL-22, and chemokines such as CCL20 (103, 105).
Several studies have shown that increased IL-17 levels in patients with lung cancer are associated with poor prognosis and higher TNM staging (106). The use of lung cancer murine models confirmed an increased infiltration of Th17 cells in tumors. These cells promoted tumorigenesis through production of IL-17 and subsequent recruitment of myeloid cells. IL-17 deletion reduced K-ras mutant lung tumorigenesis, tumor cell proliferation, and angiogenesis (98). This was associated with decreased levels of inflammatory mediators such as IL-6 and different chemokines related to neutrophil and monocyte migration such as CXCL1, CXCL2, CCL2, and GM-CSF, resulting in fewer myeloid cells (98). IL-17 has also been shown to promote metastasis of NSCLC xenograft models due to its direct association with the IL-6/STAT3 pathway (106). Recently IL-17 has been found to promote resistance to immune checkpoint blockade in lung cancer through a neutrophil-dependent modification of the lung TME (107).
Interleukin 22
Interleukin 22 (IL-22) is classified inside the IL-10 related cytokines. It is secreted primarily by Th17 cells (108); however, other immune cells such as γδ T cells, natural killer T cells, neutrophils, macrophages, and ILC3s secrete IL-22 (109). IL-22 expression is highly dependent on the activation of aryl hydrocarbon receptor (AHR) (110) and transcription of STAT3. Signaling pathways activated by IL-22 include the JAK-STAT pathway (particularly JAK1/2) resulting in the activation of STAT1, STAT3, and STAT5 (111, 112). Other pathways activated include MAPK, mTOR, Akt, and PI3K.
Recently it has been described that IL-22 is secreted in high amounts in different cancers of the lung, liver, skin, and colon (56, 113, 114). It has been shown to be upregulated in tumor tissues and bronchoalveolar lavage fluid of patients with recurrent NSCLC, and its abrogation decreases the proliferative and migratory capabilities of tumor cells (115, 116). IL-22R overexpression in NSCLC has also been associated with poor prognosis in patients with LUAD (117). In mice, it has been shown that IL-22 also increases levels of other cytokines such as IL-6, IL-10, TGF-β, and TNF, inducing an immunosuppressive environment. Studies in a murine K-ras mutant lung cancer model have confirmed the promoting role of IL-22 in tumorigenesis and its pro-inflammatory function that is mediated through activation of different cytokines including IL-6 and IL-17 (113). It has also been shown to trigger upregulation of stemness markers (113). Increased expression of IL-22 and its receptor IL-22R1 and increased activation of STAT3 in human lung cancer cell lines mediate resistance to standard chemotherapy (118), which could be linked to the role of IL-22 in induction of stemness properties.
Tumor Necrosis Factor
TNF is comprised of a 233 amino acid homodimeric transmembrane protein (119). It is mostly produced by immune cells such as CD4+ T cells, mast cells, eosinophils, neutrophils, NK cells, and macrophages (120). TNF is a cytotoxic molecule that serves as an acute phase reaction protein that chemotactically attracts neutrophils and increases levels of endothelial adhesion proteins (120, 121). Several pathways are activated by TNF including NF-κB, Akt/PKB, MAPK, and the apoptosis pathways (73, 119, 122, 123). TNF expression is ubiquitous in different organs including lung epithelial cells, fibroblasts, and stromal tissue (121).
Chronic increase of TNF has been historically related to cancer-associated cachexia due to its effects on hypothalamic structures (124), and high serum TNF levels have been associated with worse prognosis in lung cancer (125, 126). TNF increases the number of MDSCs and neutrophils in the TME, leading to increased tumor growth, angiogenesis, and amplification of inflammation (127, 128). To model these effects, murine studies have used epithelial-specific TNF overexpression and deletion using a TNF knockout mouse in a K-ras mutant model of lung cancer to show the importance of TNF in recruiting MDSCs and promoting lung cancer (129, 130). It has also been shown that TNF neutralization impairs inflammatory cell migration and angiogenesis with an overall decrease in tumor size but not number in a urethane-induced lung cancer mouse model (131). Efforts to combine TNF agonists with other cancer therapies such as 17-allylamino-17-demethoxygeldanamycin (17AAG) have been tested in several human cancer cell lines in vitro (132).
Currently anti-TNF therapy is being used in different types of cancers, including lung cancer, and has been shown to enhance chemotherapeutic effect by decreasing inflammation, proliferation, and metastasis (133). Clinical trials using anti-TNF with or without chemotherapy have shown prolonged disease eradication in patients with ovarian cancer (134) and breast cancer (135).
Immune Cells
As we have previously mentioned, inflammation significantly augments tumorigenesis (8) and in the process recruits various immune cells. Tumors are constantly infiltrated with immune cells with diverse functions which make up important components of the TME. Cells with cytotoxic activities, such as CD8+ T lymphocytes and NK cells, generally exert antitumor functions, whereas Tregs, Th17 cells, MDSCs, neutrophils, and macrophages are usually pro-tumorigenic. Lung cancer driven by K-ras activation in the airway epithelium elicits a formidable inflammatory response distinguished by macrophagic and neutrophilic infiltration accompanied by increased chemokines like CCL2, CXCL1, and CXCL2 (11). In this section, we will discuss the function and importance of each immune cell in the pathogenesis of K-ras mutant lung cancer.
Macrophages
Macrophages are innate immune cells resident in tissues originating from peripheral, circulating monocytes. They are involved in the detection, phagocytosis, and destruction of foreign organisms. They also present antigens to T cells and initiate inflammation through secreting cytokines that activate other immune cells. As crucial drivers of chronic inflammation, they have been reported to be involved in almost every step of cancer progression including early carcinogenesis, metastatic progression, and therapeutic resistance. Tumor-associated macrophages (TAMs) constitute the majority of infiltrating immune cells in tumors, and their presence usually negatively correlates with clinical outcome (136). In lung cancer, higher TAM density is related to worse patient survival, and patients with recurrent disease display elevated macrophage infiltrates in their primary tumors (137, 138).
Activated macrophages are classified into two phenotypes by their roles: pro-inflammatory M1 type or anti-inflammatory M2 type (139). During early tumorigenesis, M1 macrophages play a role in the elimination of more immunogenic tumor cells, whereas less antigenic tumor cells survive and skew macrophages toward the M2 phenotype to promote tumor survival and metastasis through angiogenesis, EMT, and immune evasion (136, 140–142).
In an analysis of infiltrated immune cells in subtypes of lung cancer, researchers found that LUADs with K-ras or EGFR mutations were more densely infiltrated by cells of myeloid lineage, whereas small cell lung cancers (SCLCs) were mostly infiltrated by T cells. Moreover, in K-ras mutant LUAD macrophage content increases with time (10). This finding suggests that K-ras mutated cancer cells can modulate the TME to facilitate their growth through recruiting and remodeling macrophages. Actually, lung epithelial activation of K-ras signaling led to LUAD development and pronounced pulmonary inflammation with a 14-fold increase in macrophage content and a 100-fold increase in neutrophil count (11). Further analysis showed that macrophage chemoattractants MCP-1 (CCL2) and MIP-1α (CCL3) and neutrophil chemoattractants MIP-2 (CXCL2) and KC (CXCL1) were significantly elevated, and these chemokines were secreted by K-ras activated tumor cells (11). After being recruited to tumor sites, these macrophages were reshaped into an M2 protumor phenotype (i.e., increased Arg1, Fizz1, and Mrc1 expression) to promote tumor progression (62).
Macrophages promote tumor progression through multiple mechanisms such as boosting angiogenesis, stimulating proliferation and EMT, remodeling the extracellular matrix (ECM), enhancing immunosuppression, and inhibiting antitumor cytotoxic activities (136, 143). In NSCLC patients, tumor-infiltrating macrophage density correlated significantly and positively with intratumor microvessel counts and negatively with patient survival. Recent studies using K-ras mutant LUAD models found that skewing from M2 to M1 in the TME resulted in reduced angiogenesis (143), and knocking out TLR9 in mononuclear cells led to reduced microvessel density and lower VEGF expression (144). Macrophages also undergo bidirectional crosstalk with lung cancer cells through CCR2-CCL2 and CX3CR1-CX3CL1 signaling axes to facilitate tumor growth as well as establish a nurturing tumor microvasculature and metastasis (145).
On the other hand, M2 macrophages promote an immunosuppressive TME, as remodeling from M2 to M1 results in an attenuated immunosuppressive environment and boosts the efficiency of antitumor T cell function characterized by lower PD-L1 expression and decreased levels of IL-6 and TGF-β (143). Sustained activation of NF-κB signaling led to chronic inflammation and adenoma development accompanied by increased M2 macrophage infiltration, and these macrophages induced Treg differentiation through expression of IL-10 and TGF-β (146). In a cigarette smoke-induced K-ras mutant LUAD mouse model, myeloid cells were able to promote LUAD cell proliferation, angiogenesis, IL-6/STAT3 signaling, and infiltration of neutrophils (37). Furthermore, M2 macrophages maintain an immunosuppressive TME through downregulation of MHC II and costimulatory proteins (CD80 and CD86) and increased arginase (Arg1) and indoleamine 2,3-dioxygenase (IDO) activity to prevent the activation of antitumor immunity (147).
Neutrophils
Chemotactic factors like IL-8, which we discussed earlier, that are produced in inflamed tissues are instrumental in recruiting neutrophils. These cells are the backbone of pathogen clearance, phagocytosing their targets while releasing antimicrobial proteins, proteases, and even their own DNA into the microenvironment in an effort to mitigate infection (136, 148).
It has been widely recognized that tumor-associated neutrophils (TANs) play an important role in cancer promotion (149). High levels of neutrophil infiltration and high neutrophil/lymphocyte ratio are negatively associated with prognosis in different malignancies including lung cancer (150–152). Similar to M1 and M2 macrophages, neutrophils are also thought to exist in two distinct phenotypes, “N1” and “N2” (153); however, there are no biomarkers yet to specifically identify these two phenotypes (136). N2 neutrophils are pro-tumorigenic by influencing angiogenesis and immune surveillance, secreting various cytokines, and generating ROS (154). Under certain conditions such as TGF-β blockade, TANs can take on an antitumor N1 phenotype (149).
TANs are recruited to the TME through cytokines and chemokines secreted by cancer cells and stromal cells, such as TGF-β, TNF, and CXCL1/2/5 (155–157). TANs are a crucial component of the lung cancer TME. Actually, neutrophils comprised 20% of all CD45+ cells in NSCLC samples, and they were predominantly located in tumor stroma (158). Our previous studies and others using K-ras mutant LUAD models have found that neutrophils make up a significant portion of immune cells in the TME, and lung epithelial K-ras activation significantly increases neutrophil infiltration through CXCR2 ligands CXCL1/2 (11, 35, 87, 88), with neutrophil infiltration increasing with time (10).
TANs are involved in tumor initiation and progression through promoting angiogenesis, remodeling the ECM, producing ROS, and modulating immunity (149). Neutrophils can promote lung carcinogenesis through secreting MMP-9 to prevent apoptosis (159). They also secrete neutrophil elastase (NE) that activates Akt signaling to potentiate lung cancer growth (160). Neutrophils likewise promote angiogenesis through secretion of chemokines and MMPs, which can switch on angiogenesis through counteracting anti-angiogenic molecules and promoting the release of VEGF (161). NE, collagenase IV, and heparanase produced by neutrophils degrade the ECM and assist tumor cell extravasation during the metastatic process (162, 163). Neutrophils also play a major role in reshaping the leukocyte component in the TME. Using NSCLC specimens, Kargl and colleagues (158) found a strong negative correlation between neutrophils and CD8+/CD4+ lymphocytes, suggesting lymphocytes are able to suppress neutrophils. Since neutrophil-derived NE promotes cancer development, a genetic knockout of NE resulted in reduced tumor burden, delayed cancer progression, lower proliferation rate, lower angiogenesis, and decreased levels of IL-6 and TGF-β (88). On the other hand, NE is also reported to accelerate tumor growth through skewing the PI3K axis toward tumor cell proliferation (160).
IL-17 in the K-ras mutant LUAD microenvironment induces epithelial secretion of CXCL2 and G-CSF to recruit neutrophils to the TME and exert their tumor-promoting functions (98). Neutrophils also reduce T cell homing, impair anti-PD-1 efficacy, and alter angiogenesis, leading to hypoxia, and sustained Snail expression in lung cancer cells (164). Besides the intrinsic inflammation caused by K-ras mutation, extrinsic inflammation, like airway inflammation in chronic obstructive pulmonary disease (COPD), can shift inflammation from macrophage-predominant to neutrophil-predominant and significantly promote lung tumor growth, which further emphasizes the cancer-promoting role of neutrophils (35).
Myeloid-Derived Suppressor Cells
MDCSs represent a diverse collection of immature myeloid lineage cells that together contribute to negative regulation of the immune response during cancer and chronic inflammation (165). In mice, MDSCs are recognized as CD11b+GR1+, whereas the human phenotype is CD11b+CD14−CD33+ (165). MDSCs can be further classified as polymorphonuclear (PMN-MDSCs) and mononuclear (M-MDSCs) (166). The expansion and activation of MDSCs are influenced by tumor cells, activated T cells, stromal cells, and active molecules released by these cell populations (165). Tumor-secreted factors such as GM-CSF, IL-1β, IL-6, TGF-β, and TNF mediate expansion of MDSCs (167–171). T cells and tumor stromal cells activate MDSCs through TLRs, IFNγ, IL-4, IL-13, and TGF-β (165).
MDSCs are present in activated states and are potent suppressors of T cell functions (165). The main mediators of MDSC immunosuppressive functions are Arg1, iNOS, TGF-β, IL-10, COX2, and IDO (166). MDSCs express high levels of Arg1 and iNOS, both of which directly inhibit T cell function (172, 173). MDSCs generate oxidative stress by producing ROS, and inhibition of ROS in MDCSs completely abrogated the inhibition on T cells (174). Peroxynitrite is a potent oxidant produced by the body, and peroxynitrite levels are elevated at sites where MDSCs and inflammatory cell correspondingly amass. Direct contact of peroxynitrite with T cells results in T cell receptor and CD8 nitration, which renders T cells unresponsive (175). MDSCs release IDO, which polarizes APCs toward a tolerant phenotype. MDSCs also express the immune checkpoint molecule PD-L1, which binds to PD-1 on T cells resulting in T cell exhaustion (176). MDSCs also promote tumor progression through interaction with other immune cells. They produce IL-10 to inhibit DC function (177), polarize macrophages toward M2 (178), and recruit Tregs via IL-10 and TGF-β (179).
MDSCs play an important part in the TME of K-ras mutant LUAD. Oncogenic K-ras induced GM-CSF is capable of activating MDSCs, and MDSCs can directly promote angiogenesis or accumulate Tregs to enhance K-ras mutant tumor growth (82, 88). COPD-like airway inflammation in K-ras mutant LUAD can increase TNF levels, which induces a robust accumulation of MDSCs. Recruitment of MDSCs results in increased angiogenic markers CD31 and MMP-9 and increased TGF-β and IL-6 levels that induce an immune-suppressive Treg response (129). It has also been shown that IL-6 inhibition in K-ras mutant LUAD significantly reduced the MDSC population, as well as levels of Arg1, IDO, CXCL1, and IL-17 in these cells, and at the same time switched the T cell phenotype from a protumor Treg/Th17 to an antitumor Th1/CD8 T cell response (62).
T Helper 17 Cells
Th17 cells, a T helper cell subgroup that functionally differs from conventional Th1 and Th2 cells (180), have been associated with a wide range of inflammatory diseases including COPD. The role of Th17 cells varies in different cancer types and is currently uncertain due to evidences on both anti- and protumor functions. Th17 cells have been reported to play antitumor roles as their existence in patients correlates with lower clinical stages in prostate cancer (181) and longer survival in SCLC (182). In hepatocarcinoma (183) and pancreatic carcinoma (184), Th17 cells from patients could produce IFNγ and have cytotoxic activities, which also suggests an antitumor role of Th17 cells. The effects of Th17s might vary temporally, as Th17 cells have been found to increase in early stage NSCLC tumors compared with tumor-free parenchyma. However, in advanced stage NSCLC, Th17 infiltration in positive lymph nodes is inversely related to PD-1+CD4+ T cells (185). In NSCLC, Th17 cells have been widely reported to promote tumor progression: levels of IL-17, a major cytokine from Th17 cells, are significantly higher in NSCLC patients than non-cancer patients (186), and high levels of IL-17 are positively related to lymph node metastasis and advanced staging (106). On the other hand, Th17 cells might paradoxically have antitumor functions in NSCLC: high Th17 cell counts in pleural effusion are related to better survival of NSCLC patients (187), and IL-21 secreted by Th17 cells could induce the expansion of cytotoxic CD8+ T cells (188).
As we have previously discussed in the cytokine section, Th17 cells produce IL-17 to promote tissue inflammation and modulate the lung TME through secreting pro-inflammatory cytokines and chemokines (98). Th17 differentiation is usually triggered by IL-23 (189), and after activation, Th17 cells produce IL-17 and IL-22 to induce the production of pro-inflammatory factors including IL-1β, IL-6, and TNF (190). K-ras mutations lead to elevated IL-17-producing T cells (10). In our previous study of K-ras mutant LUAD concurrent with COPD-related airway inflammation (98), IL-17 deficient (il17a−/−) mice have lower tumor proliferation and vascular density. Reduced tumorigenesis was associated with lower infiltration of Gr-1+CD11b+ myeloid cells (MDSCs) in il17a−/− mice suggesting Th17 cells are involved in recruiting immunosuppressive cells. Different lung cancer subtypes and oncogenic mutations might generate distinct inflammatory responses. Currently, evidence on antitumor roles of Th17 cells in K-ras mutant lung cancer still remains scarce, and more studies are needed.
Regulatory T Cells
As the name suggests, regulatory T cells (Tregs) are T cells that play a role in regulating or suppressing the immune system and preventing autoimmune disease. Tregs are typically characterized as CD4+CD25+ and express the nuclear transcription factor FoxP3. Treg infiltration is related to the progression of lung cancers. NSCLC specimens have been found to have a significant increase in Tregs compared with non-tumor lung tissue (158). A clinical study revealed that circulating Treg numbers rose alongside disease stage and metastasis (191). Treg levels in NSCLC tumors and peripheral blood are linked to increased risk of recurrence and poor survival (192, 193).
Tregs induce immunosuppression through contact-dependent mechanisms such as expressing co-inhibitory molecules like cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), PD-1, PD-L1, lymphocyte-activation protein 3 (LAG-3), CD39/73, or through contact-independent mechanisms by producing immunosuppressive molecules like IL-10, TGF-β, adenosine, prostaglandin E2 (PGE2), and IL-35 (194, 195). In mouse LUADs, Treg depletion resulted in tumor cell death and increased levels of granzyme A, granzyme B, perforin, and IFNγ in infiltrating CD8+ T cells, suggesting Tregs inhibit the antitumor function of CD8+ T cells (196). In lung tumors, Tregs are also associated with the expression of angiogenic and metastatic potentiator cyclooxygenase-2 (COX-2), suggesting Tregs are involved in tumor cell dissemination (192). It has been shown that Tregs inhibit NK cell-mediated cytotoxicity in a TGF-β dependent manner in a mouse Lewis lung carcinoma model, and depletion of Tregs restored NK cell anti-metastatic activities (197).
Tregs play an important immunosuppressive role in the K-ras mutant LUAD microenvironment. In a study of immune cells in different subtypes of lung cancer, researchers found that K-ras mutations give rise to elevated Treg populations, and they are the only non-myeloid lineage population to expand over the course of tumor development (10). In contrast to their wild-type K-ras cousins, tumor cells bearing K-ras mutations induce suppressive Tregs by enhancing the secretion of IL-10 and TGF-β. Conversely, inhibition of K-ras reduces the Treg population in K-ras driven lung tumorigenesis even before tumor formation (198). Using antibodies against CD25 and FR4 depletes Tregs and reduces K-ras induced oncogenesis (199). In a mouse model with K-ras and p53 co-mutations, an epigenetic modifier (BET bromodomain inhibitor) significantly reduced the Treg population while increasing Th1 infiltration and antitumor effect (200). Another group using a K-ras-driven LUAD model found that a DNA damage signaling kinase inhibitor, AZD6738, successfully reduced radiation-induced Treg proliferation and infiltration while enhancing CD8+ T cell activity (201).
CD8+ T Cells and T Helper 1 Cells
CD8+ T cells are the most prominent antitumor cells. Activated CD8+ T cells, referred to as CTLs, exert antitumor effects by producing perforin and granzyme containing granules (202, 203). CD4+ Th1 cells mediate antitumor function through secretion of pro-inflammatory cytokines such as IL-2 and IFNγ, which promote T cell priming, activation, and CTL cytotoxicity (204). Optimal T cell-based antitumor immunity necessitates both cytotoxic CD8+ T cells and Th1 cells to improve the efficacy of the antitumor response (205). CD8+ T and Th1 cell infiltrations in tumors are positively correlated with better prognosis (206).
Despite being monitored by these antitumor T cells, cancer cells still manage to prosper through exploiting an immunosuppressive TME. CD8+ T cells have been found to have an exhausted phenotype in the TME. Exhausted CD8+ T cells are characterized by high levels of inhibitory receptors (PD-1/CTLA-4/TIM-3/LAG-3//BTLA/TIGIT) and production of fewer effector cytokines (IL-2/IFNγ/TNF/GzmB) that lead a diminished ability to eliminate cancer cells and their final deletion in the TME (207, 208). In the TME, immune cells such as MDSCs and tumor-associated DCs express high levels of co-inhibitory molecules that directly suppress T cell function. Tregs and M2 macrophages produce adenosine, IL-10, and TGF-β to induce T cell exhaustion (208). Th1 cell maturation involves the consecutive activation of the transcription factors STAT1, T-bet, and STAT4. Compared with peripheral blood lymphocytes, tumor-infiltrating lymphocytes (TILs) in human head and neck squamous cell carcinoma had lower Th1 differentiation and activation, which was mechanistically regulated through suppressed activation of STAT1, T-bet, and Th1 cytokine secretion by PD-1 signaling (205).
In lung cancer patients, TILs were found to have reduced levels of perforin and granzyme, suggesting the TILs were dysfunctional (209). Fewer exhausted T cells as indicated by a low PD-1 to CD8 ratio provides a favorable immune microenvironment that aids patient survival post-resection and response to immunotherapy in advanced NSCLC (210). K-ras mutant LUAD displays greater CD8+ T cell content than EGFR mutant LUAD; however, CD8+ T cell proliferation in K-ras-driven LUAD is lower, suggesting an exhausted phenotype of T cells in this context (10). K-ras activated lung cancer cells orchestrate the TME through secreting cytokines IL-6, IL-10, IL-17, TGF-β, and TNF, which can either suppress antitumor T cells directly or stimulate immune cells such as MDSCs, Th17s, M2 macrophages, and Tregs to inhibit Th1/CD8+ T cell function (62, 88, 98, 129), as we have described above.
Natural Killer Cells
NK cells are the innate relatives of CTLs that likewise eliminate infected or transformed cells through cytolytic mechanisms. These cells were first noticed for their ability to kill tumor cells without any priming or prior activation, in contrast to CTLs, which need priming by APCs (211). NK cells are required for effective tumor surveillance. NK cell infiltration in tumor biopsies is a favorable prognostic marker in cancer patients (212, 213). Upon activation, NK cells attack tumor cells through releasing cytotoxic perforin and granzyme (214) and activating apoptosis in tumor cells through the production of IFNγ and TNF or via direct cell contact through death receptor-mediated pathways such as the TRAIL and FASL pathways (136, 215). NK cells also modulate the activity of other leukocytes. They produce IFNγ for Th1 priming (216) and polarize macrophages toward an M1 phenotype (217).
In normal lung tissue, NK cell counts are comparable to those of macrophages and granulocytes. However, as myeloid cells expand with LUAD development, NK cell populations remain unaltered, and in K-ras mutant LUAD, NK cell counts even decrease with time (10). NKG2D is a stimulatory receptor expressed on NK cells, which upon activation can stimulate NK cells; however, surface expression of NKG2D on NK cells decreases significantly in K-ras and p53 tumor-bearing mice but remains unchanged in EGFR mutant LUAD (164). Soluble factors that are abundant in K-ras mutant LUAD, such as MDSC-, macrophage- and tumor cell-derived TGF-β, PGE2, IDO, and IL-10, also inhibit NK cell function (218). Cong and colleagues found that NK cells inhibited K-ras LUAD initiation but gradually lost their antitumor effect, that this was associated with impaired metabolism caused by aberrant fructose-1,6-bisphosphatase (FBP1) expression in NK cells, and that FBP1 inhibition could partially restore NK cell function (219).
Innate Lymphoid Cells
Innate lymphoid cells (ILCs) are newly defined members of the lymphocyte population that derive from common lymphoid progenitors but lack specific antigen receptors (220). They play important roles in tissue homeostasis and regulate the host immune response under infection and inflammation (221). ILCs can be defined into four subgroups: NK cells, which we have already reviewed, and ILC1, ILC2, and ILC3 which function like T helper cells (Th1, Th2, and Th17, respectively) (220). NK cells have been extensively studied in various cancers. Due to their relatively recent discovery, roles of others ILCs in cancer cell biology is still an emerging research field, and there has been several reviews about ILC functions in human cancers (220, 222, 223).
Reports on ILCs in lung cancer are still limited. Researchers have found that ILC1 cells remained largely unchanged in patients' NSCLC tissues compared with patients' own normal lung tissues; however, ILC2 cells decreased in NSCLC samples while ILC3 cells increased dramatically (224), suggesting that ILC2 and ILC3 cells might affect lung cancer growth. Further analysis showed that the amount of infiltrated ILC3 cells correlated with the density of intratumoral tertiary lymphoid structures, and these ILC3 cells can be directly activated by cancer cells via the NKp44 receptor. Once activated, the ILC3s secret IL-2, which stimulates expansion of tumor-specific lymphocytes; however, since ILC3s also provide an innate source of IL-22 which might favor tumor growth, their role in NSCLC still remains to be identified (224). On the other hand, ILC3s have also been found to promote tumor growth: in squamous cell lung cancer, IL-23 can convert ILC1s to ILC3s and promote IL-17-mediated tumor growth. Clinically, numbers of ILC3s and levels of IL-17 are correlated with poor prognosis. Notably, these effects did not occur in adenocarcinoma, suggesting that the roles of these cells are quite heterogeneous among cancer types (225). ILC2s have been recently reported to secret amphiregulin, an EGFR ligand, which can promote lung tumor cell growth, facilitate resistance to apoptosis (226), and even interfere with antitumor immunity through stimulating Tregs and establishing an immunosuppressive TME (227). However, a study on ILC2s found that these cells reside in the lung and produce IL-5 to recruit eosinophils that could prevent tumor metastasis to the lung (228). In all, study on ILC functions in lung cancer and K-ras mutant LUAD is still limited, and more evidence is needed to illustrate their roles in lung cancer.
Clinical Insights
The TME is a complex system, with tumor cells and various immune cells interacting with each other to orchestrate either an antitumor or protumor immune response. Understanding and manipulating the crosstalk between different immune cells is of great importance as the subsequent immune response could strongly impact patient outcome. Clinical trials on improving therapeutic efficacy have focused on enhancing antitumor cytotoxic activity or dampening protumor immunosuppressive responses. Since there has not been any study exclusively focused on K-ras mutant LUAD, here we are reviewing studies containing NSCLC cohorts.
In regards to enhancing antitumor immunity, the most promising field of research is immune checkpoint blockade (e.g., anti-PD1/PD-L1, anti-CTLA4), which blocks inhibitory signaling and results in activation of T cell effector function; since this topic has already been widely reviewed (229–232), we will not discuss it extensively here. DCs are powerful APCs for the induction of antigen-specific T cell responses, and DC vaccines have been introduced as a new therapeutic strategy in NSCLC as they prime and activate CD8+ T cells (233). Takahashi et al. reported that DC vaccines pulsed with Wilms' tumor protein-1 (WT1) peptide could improve survival in patients with advanced NSCLCs (234). Ge et al. found that DC vaccines pulsed with survivin and mucin1 (MUC1) induced modest antitumor activity and improved quality of life for patients with stage I-III NSCLC (235). Lymphocyte (particularly helper T cell) abundance and activity depends on the availability of endogenous IL-2, which is severely impaired in lung cancer (236). In a Phase II randomized study, Masotti et al. found that preoperative administration of recombinant human IL-2 (rhIL-2) increased CD8+ T cell, T helper cell, and NK cell numbers in NSCLC patients (237).
On the other hand, researchers have been working on mitigating protumor immunity in the TME. Albeituni et al. found that yeast-derived whole β-glucan particles (WGP) could reduce PMN-MDSCs in NSCLC patient blood, which may be a potent immune modulator of MDSC suppressive function (238). In a Phase Ia study including lung cancer patients, Kurose et al. found Treg depletion by KW-0761 (a humanized anti-human CCR4 monoclonal antibody) showed possible occurrence of an immune response and suggested that KW-0761 in conjunction with cancer vaccines or checkpoint inhibitors represents an enticing effort to augment the antitumor immune response (239). IDO inhibits T cell-mediated antitumor immunity in patients, and blocking this regulatory pathway might disinhibit T cells and improve tumor clearance. In a phase I clinical study with 15 stage III-IV NSCLC patients, Iversen et al. found IDO is frequently expressed in LUAD, and targeting IDO by a peptide vaccine reduced Tregs, enhanced CD8+ T cell function, and prolonged overall survival by 18.2 months (240).
To date, clinical trials besides checkpoint inhibitors (CPIs) are still limited. The interaction of immune cells in the TME is a multifaceted network, and more and more studies have been focused on combination strategies such as combining different CPIs, CPI with targeted therapy, or CPI with chemotherapy. Since each tumor is highly heterogeneous in each patient, individualized genomic and immune phenotypes are crucial to customize an optimum treatment strategy and evaluate therapeutic efficacy.
Conclusion
We wish to emphasize the intricate interplay within the K-ras mutant lung cancer TME between tumor cells and immune cells through numerous cytokines and inflammatory signaling cascades. Figure 1 provides a visual summary: in the center of this diverse network sit the tumor cells, secreting the soluble factors needed to recruit and reprogram immune cells by activation of target pathways. In this way, the tumor cells can inhibit antitumor responses and even persuade some immune cells to promote tumorigenesis. To forge future therapeutic strategies, we foresee the need for combinatorial treatments directed toward not only preventing tumor cell-intrinsic tumor initiating factors and immune suppression but protumor inflammation as well. Since the problem is multifaceted, it is only logical that the solution should be equally complex, and as personalized medicine evolves, the ability to design combined treatments tailored to the individual should become more readily attainable.
Author Contributions
SM, MR-C, WV, and SD participated in the conception and design of the review. MR-C, WV, SD, and MC wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
The authors were supported by National Cancer Institute (NCI) grant R01CA225977 awarded to SM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
2. Stellman SD, Takezaki T, Wang L, Chen Y, Citron ML, Djordjevic MV, et al. Smoking and lung cancer risk in American and Japanese men: an international case-control study. Cancer Epidemiol Biomarkers Prev. (2001) 10:1193–99.
3. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. (2008) 455:1069–75. doi: 10.1038/nature07423
4. Jančík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. BioMed Res Int. (2010) 2010:150960. doi: 10.1155/2010/150960
5. Barbacid M. Ras genes. Ann Rev Biochem. (1987) 56:779–827. doi: 10.1146/annurev.bi.56.070187.004023
6. Mayekar MK, BivonaTG. Current landscape of targeted therapy in lung cancer. Clin Pharmacol Therap. (2017) 102:757–64. doi: 10.1002/cpt.810
7. Jänne PA, Van Den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with kras-mutant advanced non–small cell lung cancer: the select-1 randomized clinical trial. JAMA. (2017) 317:1844–53. doi: 10.1001/jama.2017.3438
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
9. Srivastava MK, Andersson Å, Zhu L, Harris-White M, Lee JM, Dubinett S, et al. Myeloid suppressor cells and immune modulation in lung cancer. Immunotherapy. (2012) 4:291–304. doi: 10.2217/imt.11.178
10. Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung cancer subtypes generate unique immune responses. J Immunol. (2016) 197:4493–503. doi: 10.4049/jimmunol.1600576
11. Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene. (2006) 25:2105–112. doi: 10.1038/sj.onc.1209237
12. Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma: a systematic review and meta-analysis. JAMA Oncol. (2018) 4:210–6. doi: 10.1001/jamaoncol.2017.4427
13. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. (2017) 23:3012–24. doi: 10.1016/j.jtho.2016.11.504
14. O'Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Eng J Med. (2013) 368:161–70. doi: 10.1056/NEJMra1202117
15. Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. (2007) 117:3846–56. doi: 10.1172/JCI31871
16. Mohrherr J, Haber M, Breitenecker K, Aigner P, Moritsch S, Voronin V, et al. JAK-STAT inhibition impairs K-RAS-driven lung adenocarcinoma progression. Int J Cancer. (2019) 145:3376–88. doi: 10.1002/ijc.32624
17. Grabner B, Schramek D, Mueller KM, Moll HP, Svinka J, Hoffmann T, et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nature Commun. (2015) 6:6285. doi: 10.1038/ncomms7285
18. Grabner B, Moll HP, Casanova E. Unexpected oncosuppressive role for STAT3 in KRAS-induced lung tumorigenesis. Mol Cell Oncol. (2016) 3:e1036199. doi: 10.1080/23723556.2015.1036199
19. Zhou J, Qu Z, Yan S, Sun F, Whitsett JA, Shapiro SD, et al. Differential roles of STAT3 in the initiation and growth of lung cancer. Oncogene. (2015) 34:3804–14. doi: 10.1038/onc.2014.318
20. Li S, Liu S, Deng J, Akbay EA, Hai J, Ambrogio C, et al. Assessing therapeutic efficacy of MEK inhibition in a KRASG12C-driven mouse model of lung cancer. Clin Cancer Res. (2018) 24:4854–64. doi: 10.1158/1078-0432.CCR-17-3438
21. D'Amico S, Shi J, Martin BL, Crawford HC, Petrenko O, Reich NC. STAT3 is a master regulator of epithelial identity and KRAS-driven tumorigenesis. Genes Dev. (2018) 32:1175–87. doi: 10.1101/gad.311852.118
22. Kopparam J, Chiffelle J, Angelino P, Piersigilli A, Zangger N, Delorenzi M, et al. RIP4 inhibits STAT3 signaling to sustain lung adenocarcinoma differentiation. Cell Death Diff. (2017) 24:1761–71. doi: 10.1038/cdd.2017.81
23. Caetano MS, Hassane M, Van HT, Bugarin E, Cumpian AM, McDowell CL, et al. Sex specific function of epithelial STAT3 signaling in pathogenesis of K-ras mutant lung cancer. Nat Commun. (2018) 9:4589. doi: 10.1038/s41467-018-07042-y
24. Zhou J, Qu Z, Sun F, Han L, Li L, Yan S, et al. Myeloid STAT3 promotes lung tumorigenesis by transforming tumor immunosurveillance into tumor-promoting inflammation. Cancer Immunol Res. (2017) 5:257–68. doi: 10.1158/2326-6066.CIR-16-0073
25. Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. (2001) 276:45443–45455. doi: 10.1074/jbc.M107527200
26. Wong A, Soo RA, Tan D, Lee SC, Lim J, Marban P, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. (2015) 26:998–1005. doi: 10.1093/annonc/mdv026
27. Burel SA, Han SR, Lee HS, Norris DA, Lee BS, Machemer T, et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucl Acid Therapeut. (2013) 23:213–27. doi: 10.1089/nat.2013.0422
28. Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene. (2006) 25:6680–4. doi: 10.1038/sj.onc.1209954
29. Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
30. Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Ann Rev Immunol. (2009) 27:693–733. doi: 10.1146/annurev.immunol.021908.132641
31. Bassères DS, Ebbs A, Cogswell PC, Baldwin AS. IKK is a therapeutic target in KRAS-Induced lung cancer with disrupted p53 activity. Genes Cancer. (2014) 5:41. doi: 10.18632/genesandcancer.5
32. Tang X, Liu D, Shishodia S, Ozburn N, Behrens C, Lee JJ, et al. Nuclear factor-κB (nf-κB) is frequently expressed in lung cancer and preneoplastic lesions. Cancer Interdiscipl Int J Am Cancer Soc. (2006) 107:2637–46. doi: 10.1002/cncr.22315
33. Jin X, Wang Z, Qiu L, Zhang D, Guo Z, Gao Z, et al. Potential biomarkers involving IKK/RelA signal in early stage non-small cell lung cancer. Cancer Sci. (2008) 99:582–9. doi: 10.1111/j.1349-7006.2007.00713.x
34. Song NY, Zhu F, Wang Z, Willette-Brown J, Xi S, Sun Z, et al. IKKα inactivation promotes Kras-initiated lung adenocarcinoma development through disrupting major redox regulatory pathways. Proc Natl Acad Sci USA. (2018) 115:E812–21. doi: 10.1073/pnas.1717520115
35. Moghaddam SJ, Li H, Cho SN, Dishop MK, Wistuba II, Ji L, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. (2009) 40:443–53. doi: 10.1165/rcmb.2008-0198OC
36. Karin M. Inflammation and cancer: the long reach of Ras. Nat Med. (2005) 11:20–1. doi: 10.1038/nm0105-20
37. Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKβ - and JNK1-dependent inflammation. Cancer Cell. (2010) 17:89–97. doi: 10.1016/j.ccr.2009.12.008
38. Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, et al. Requirement for NF-κB signalling in a mouse model of lung adenocarcinoma. Nature. (2009) 462:104–7. doi: 10.1038/nature08462
39. Keith RL. Chemoprevention of lung cancer. Proc Am Thoracic Soc. (2009) 6:187–93. doi: 10.1513/pats.200807-067LC
40. Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity. (2019) 50:778–95. doi: 10.1016/j.immuni.2019.03.012
41. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. (2010) 3:cm1. doi: 10.1126/scisignal.3105cm1
42. Jokhi PP, King A, Loke YW. Cytokine production and cytokine receptor expression by cells of the human first trimester placental-uterine interface. Cytokine. (1997) 9:126–37. doi: 10.1006/cyto.1996.0146
43. Mallardo M, Giordano V, Dragonetti E, Scala G, Quinto I. DNA damaging agents increase the stability of interleukin-1 alpha, interleukin-1β, and interleukin-6 transcripts and the production of the relative proteins. J Biol Chem. (1994) 269:14899–904.
44. Warner SJ, Auger KR, Libby P. Interleukin 1 induces interleukin 1. II. Recombinant human interleukin 1 induces interleukin 1 production by adult human vascular endothelial cells. J Immunol. (1987) 139:1911–7. doi: 10.1084/jem.165.5.1316
45. Malyak M, Smith MF Jr, Abel AA, Arend WP. Peripheral blood neutrophil production of interleukin-1 receptor antagonist and interleukin-1β. J Clin Immunol. (1994) 14:20–30. doi: 10.1007/BF01541172
46. Sporri B, Bickel M, Limat A, Waelti ER, Hunziker T, Wiesmann UN. Juxtacrine stimulation of cytokine production in cocultures of human dermal fibroblasts and T cells. Cytokine. (1996) 8:631–5. doi: 10.1006/cyto.1996.0084
47. Santarlasci V, Cosmi L, Maggi L, Liotta F, Annunziato F. IL-1 and T helper immune responses. Front Immunol. (2013) 4:182. doi: 10.3389/fimmu.2013.00182
48. Barrera L, Montes-Servin E, Barrera A, Ramirez-Tirado LA, Salinas-Parra F, Banales-Mendez JL, et al. Cytokine profile determined by data-mining analysis set into clusters of non-small-cell lung cancer patients according to prognosis. Ann Oncol. (2015) 26:428–35. doi: 10.1093/annonc/mdu549
49. Wang S, Chen J, Haken RKT, Stanton P, Kong FM. Prognostic value of cytokine profile on survival in non-small cell lung cancer patients treated with radiotherapy. J Clin Oncol. (2015) 33:7525–7525. doi: 10.1200/jco.2015.33.15_suppl.7525
50. Gottschlich A, Endres S, Kobold S. Can we use interleukin-1β blockade for lung cancer treatment? Transl Lung Cancer Res. (2018) 7:S160–4. doi: 10.21037/tlcr.2018.03.15
51. Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci USA. (2003) 100:2645–50. doi: 10.1073/pnas.0437939100
52. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci USA. (2019) 116:1361–9. doi: 10.1073/pnas.1812266115
53. Kumar MS, Hancock DC, Molina-Arcas M, Steckel M, East P, Diefenbacher M, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. (2012) 149:642–55. doi: 10.1016/j.cell.2012.02.059
54. McLoed AG, Sherrill TP, Cheng DS, Han W, Saxon JA, Gleaves LA, et al. Neutrophil-derived IL-1β impairs the efficacy of NF-kB inhibitors against lung cancer. Cell Rep. (2016) 16:120–32. doi: 10.1016/j.celrep.2016.05.085
55. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1833–42. doi: 10.1016/S0140-6736(17)32247-X
56. Jiang R, Wang H, Deng L, Hou J, Shi R, Yao M, et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer. (2013) 13:59. doi: 10.1186/1471-2407-13-59
57. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harbor Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
58. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. (2003) 374:1–20. doi: 10.1042/bj20030407
59. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Sem Immunol. (2014) 26:38–47. doi: 10.1016/j.smim.2014.01.008
60. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. (2016) 37:11553–72. doi: 10.1007/s13277-016-5098-7
61. Lee SO, Yang X, Duan S, Tsai Y, Strojny LR, Keng P, et al. IL-6 promotes growth and epithelial-mesenchymal transition of CD133+ cells of non-small cell lung cancer. Oncotarget. (2016) 7:6626–8. doi: 10.18632/oncotarget.6570
62. Caetano MS, Zhang H, Cumpian AM, Gong L, Unver N, Ostrin EJ, et al. IL6 blockade reprograms the lung tumor microenvironment to limit the development and progression of K-ras–mutant lung cancer. Cancer Res. (2016) 76:3189–99. doi: 10.1158/0008-5472.CAN-15-2840
63. Ochoa CE, Mirabolfathinejad SG, Ruiz VA, Evans SE, Gagea M, Evans CM, et al. Interleukin 6, but not T helper 2 cytokines, promotes lung carcinogenesis. Cancer Prevent Res. (2011) 4:51–64. doi: 10.1158/1940-6207.CAPR-10-0180
64. Karakasheva TA, Lin EW, Tang Q, Qiao E, Waldron TJ, Soni M, et al. IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. (2018) 78:4957–70. doi: 10.1158/0008-5472.CAN-17-2268
65. Chen MF, Lin PY, Wu CF, Chen WC, Wu CT. IL-6 expression regulates tumorigenicity and correlates with prognosis in bladder cancer. PLoS ONE. (2013) 8:e61901. doi: 10.1371/journal.pone.0061901
66. Belluco C, Nitti D, Frantz M, Toppan P, Basso D, Plebani M, et al. Interleukin-6 blood level is associated with circulating carcinoembryonic antigen and prognosis in patients with colorectal cancer. Ann Surg Oncol. (2000) 7:133–8. doi: 10.1007/s10434-000-0133-7
67. Chung YC, Chang YF. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J Surg Oncol. (2003) 83:222–6. doi: 10.1002/jso.10269
68. De Vita F, Orditura M, Auriemma A, Infusino S, Roscigno A, Catalano G. Serum levels of interleukin-6 as a prognostic factor in advanced non-small cell lung cancer. Oncol Reports. (1998) 5:649–701. doi: 10.3892/or.5.3.649
69. Silva EM, Mariano VS, Pastrez PRA, Pinto MC, Castro AG, Syrjanen KJ, et al. High systemic IL-6 is associated with worse prognosis in patients with non-small cell lung cancer. PLoS ONE. (2017) 12:e0181125. doi: 10.1371/journal.pone.0181125
70. Qu Z, Sun F, Zhou J, Li L, Shapiro SD, Xiao G. Interleukin-6 prevents the initiation but enhances the progression of lung cancer. Cancer Res. (2015) 75:3209–15. doi: 10.1158/0008-5472.CAN-14-3042
71. Kim NH, Kim SK, Kim DS, Zhang D, Park JA, Yi H, et al. Anti-proliferative action of IL-6R-targeted antibody tocilizumab for non-small cell lung cancer cells. Oncol Lett. (2015) 9:2283–8. doi: 10.3892/ol.2015.3019
72. Yuquan B, Hexiao T, Laiyi W, Gaofeng P, Xuefeng Z, Ming X, et al. Interaction between epidermal growth factor receptor and interleukin-6 receptor in NSCLC progression. J Cell Biochem. (2019) 120:872–81. doi: 10.1002/jcb.27448
73. Gupta S, Bi R, Kim C, Chiplunkar S, Yel L, Gollapudi S. Role of NF-kB signaling pathway in increased tumor necrosis factor-alpha-induced apoptosis of lymphocytes in aged humans. Cell Death Diff. (2005) 12:177–83. doi: 10.1038/sj.cdd.4401557
74. Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol. (2014) 10:593–619. doi: 10.1586/1744666X.2014.894886
75. Roebuck KA. Regulation of interleukin-8 gene expression. J Interferon Cytokine Res. (1999) 19:429–38. doi: 10.1089/107999099313866
76. Bickel M. The role of interleukin-8 in inflammation and mechanisms of regulation. J Periodontol. (1993) 64:456–60.
77. Kosmopoulos M, Christofides A, Drekolias D, Zavras PD, Gargalionis AN, Piperi C. Critical role of IL-8 targeting in gliomas. Curr Med Chem. (2018) 25:1954–67. doi: 10.2174/0929867325666171129125712
78. Jia L, Li F, Shao M, Zhang W, Zhang C, Zhao X, et al. IL-8 is upregulated in cervical cancer tissues and is associated with the proliferation and migration of HeLa cervical cancer cells. Oncol Lett. (2018) 15:1350–6. doi: 10.3892/ol.2017.7391
79. Liu F, Yu C. IL-8 promote carcinogenesis of primary epithelial cells from familial adenomatous polyposis. Cell Biochem Biophys. (2014) 70:1765–71. doi: 10.1007/s12013-014-0126-y
80. Dominguez C, McCampbell KK, David JM, Palena C. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight. (2017) 2:94296. doi: 10.1172/jci.insight.94296
81. Tan PHS, Chia SS, Toh SL, Goh JCH, Nathan SS. The dominant role of IL-8 as an angiogenic driver in a three-dimensional physiological tumor construct for drug testing. Tissue Eng Part A. (2014) 20:1758–66. doi: 10.1089/ten.tea.2013.0245
82. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-Induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. (2012) 21:836–47. doi: 10.1016/j.ccr.2012.04.024
83. Zhu YM, Webster SJ, Flower D, Woll PJ. Interleukin-8/CXCL8 is a growth factor for human lung cancer cells. Br J Cancer. (2004) 91:1970–6. doi: 10.1038/sj.bjc.6602227
84. Luppi F, Longo AM, de Boer WI, Rabe KF, Hiemstra PS. Interleukin-8 stimulates cell proliferation in non-small cell lung cancer through epidermal growth factor receptor transactivation. Lung Cancer. (2007) 56:25–33. doi: 10.1016/j.lungcan.2006.11.014
85. Sunaga N, Kaira K, Tomizawa Y, Shimizu K, Imai H, Takahashi G, et al. Clinicopathological and prognostic significance of interleukin-8 expression and its relationship to KRAS mutation in lung adenocarcinoma. Br J Cancer. (2014) 110:2047–53. doi: 10.1038/bjc.2014.110
86. Sunaga N, Imai H, Shimizu K, Shames DS, Kakegawa S, Girard L, et al. Oncogenic KRAS-induced interleukin-8 overexpression promotes cell growth and migration and contributes to aggressive phenotypes of non-small cell lung cancer. Int J Cancer. (2012) 130:1733–44. doi: 10.1002/ijc.26164
87. Wislez M, Fujimoto N, Izzo JG, Hanna AE, Cody DD, Langley RR, et al. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res. (2006) 66:4198–207. doi: 10.1158/0008-5472.CAN-05-3842
88. Gong L, Cumpian AM, Caetano MS, Ochoa CE, De la Garza MM, Lapid DJ, et al. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer. (2013) 12:154. doi: 10.1186/1476-4598-12-154
89. Liu YN, Chang TH, Tsai MF, Wu SG, Tsai TH, Chen HY, et al. IL-8 confers resistance to EGFR inhibitors by inducing stem cell properties in lung cancer. Oncotarget. (2015) 6:10415–31. doi: 10.18632/oncotarget.3389
90. Waugh DJJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. (2008) 14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843
91. Sanmamed MF, Perez-Gracia JL, Schalper KA, Fusco JP, Gonzalez A, Rodriguez-Ruiz ME, et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann Oncol. (2017) 28:1988–95. doi: 10.1093/annonc/mdx190
92. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
93. Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. (1999) 162:1246–51.
94. Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. (2006) 177:4662–9. doi: 10.4049/jimmunol.177.7.4662
95. Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. (2003) 170:2106–12. doi: 10.4049/jimmunol.170.4.2106
96. Anipindi VC, Bagri P, Dizzell SE, Jiménez-Saiz R, Jordana M, Snider DP, et al. IL-17 production by γδ+ T cells is critical for inducing Th17 responses in the female genital tract and regulated by estradiol and microbiota. ImmunoHorizons. (2019) 3:317–30. doi: 10.4049/immunohorizons.1900040
97. Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. (2003) 14:155–74. doi: 10.1016/S1359-6101(03)00002-9
98. Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, et al. T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci USA. (2014) 111:5664–9. doi: 10.1073/pnas.1319051111
99. Rezalotfi A, Ahmadian E, Aazami H, Solgi G, Ebrahimi M. Gastric cancer stem cells effect on Th17/Treg balance; a bench to beside perspective. Front Oncol. (2019) 9:226. doi: 10.3389/fonc.2019.00226
100. Cunningham D, Zhang Q, Liu S, Parajuli KR, Nie Q, Ma L, et al. Interleukin-17 promotes metastasis in an immunocompetent orthotopic mouse model of prostate cancer. Am J Clin Exp Urol. (2018) 6:114–22.
101. Guan X, Liu Z, Zhang J, Jin X. Myeloid-derived suppressor cell accumulation in renal cell carcinoma is correlated with CCL2, IL-17 and IL-18 expression in blood and tumors. Adv Clin Exp Med. (2018) 27:947–53. doi: 10.17219/acem/70065
102. Majumder S, Amatya N, Revu S, Jawale CV, Wu D, Rittenhouse N. IL-17 metabolically reprograms activated fibroblastic reticular cells for proliferation and survival. Nat Immunol. (2019) 20:534–45. doi: 10.1038/s41590-019-0367-4
103. Gaffen SL. An overview of IL-17 function and signaling. Cytokine. (2008) 43:402–7. doi: 10.1016/j.cyto.2008.07.017
104. Amatya N, Garg AV, Gaffen SL. IL-17 Signaling: the yin and the yang. Trends Immunol. (2017) 38:310–22. doi: 10.1016/j.it.2017.01.006
105. Asadzadeh Z, Mohammadi H, Safarzadeh E, Hemmatzadeh M, Mahdian-Shakib A, Jadidi-Niaragh F, et al. The paradox of Th17 cell functions in tumor immunity. Cell Immunol. (2017) 322:15–25. doi: 10.1016/j.cellimm.2017.10.015
106. Li Q, Han Y, Fei G, Guo Z, Ren T, Liu Z. IL-17 promoted metastasis of non-small-cell lung cancer cells. Immunol Lett. (2012) 148:144–50. doi: 10.1016/j.imlet.2012.10.011
107. Akbay EA, Koyama S, Liu Y, Dries R, Bufe LE, Silkes M, et al. Interleukin-17A promotes lung tumor progression through neutrophil attraction to tumor sites and mediating resistance to PD-1 blockade. J Thorac Oncol. (2017) 12:1268–79. doi: 10.1016/j.jtho.2017.04.017
108. Chung Y, Yang X, Chang SH, Ma L, Tian Q, Dong C. Expression and regulation of IL-22 in the IL-17-producing CD4+ T lymphocytes. Cell Res. (2006) 16:902–7. doi: 10.1038/sj.cr.7310106
109. Honda K. IL-22 from T cells: better late than never. Immunity. (2012) 37:952–4. doi: 10.1016/j.immuni.2012.11.006
110. Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. (2011) 141:237–248:248.e231. doi: 10.1053/j.gastro.2011.04.007
111. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem. (2002) 277:33676–82. doi: 10.1074/jbc.M204204200
112. Parks OB, Pociask DA, Hodzic Z, Kolls JK, Good M. Interleukin-22 signaling in the regulation of intestinal health and disease. Front Cell Devel Biol. (2015) 3:85. doi: 10.3389/fcell.2015.00085
113. Khosravi N, Caetano MS, Cumpian AM, Unver N, De la Garza Ramos C, Noble O, et al. IL22 promotes kras-mutant lung cancer by induction of a protumor immune response and protection of stemness properties. Cancer Immunol Res. (2018) 6:788–97. doi: 10.1158/2326-6066.CIR-17-0655
114. Cobleigh MA, Robek MD. Protective and pathological properties of IL-22 in liver disease: implications for viral hepatitis. Am J Pathol. (2013) 182:21–8. doi: 10.1016/j.ajpath.2012.08.043
115. Tufman A, Huber RM, Völk S, Aigner F, Edelmann M, Gamarra F, et al. Interleukin-22 is elevated in lavage from patients with lung cancer and other pulmonary diseases. BMC Cancer. (2016) 16:409. doi: 10.1186/s12885-016-2471-2
116. Bi Y, Cao J, Jin S, Lv L, Qi L, Liu F, et al. Interleukin-22 promotes lung cancer cell proliferation and migration via the IL-22R1/STAT3 and IL-22R1/AKT signaling pathways. Mol Cell Biochem. (2016) 415:1–11. doi: 10.1007/s11010-016-2663-8
117. Guillon A, Gueugnon F, Mavridis K, Dalloneau E, Jouan Y, Diot P, et al. Interleukin-22 receptor is overexpressed in nonsmall cell lung cancer and portends a poor prognosis. Eur Resp J. (2016) 47:1277–80. doi: 10.1183/13993003.01580-2015
118. Kobold S, Völk S, Clauditz T, Küpper NJ, Minner S, Tufman A, et al. Interleukin-22 is frequently expressed in small- and large-cell lung cancer and promotes growth in chemotherapy-resistant cancer cells. J Thorac Oncol. (2013) 8:1032–42. doi: 10.1097/JTO.0b013e31829923c8
119. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. (2001) 104:487–501. doi: 10.1016/S0092-8674(01)00237-9
120. Pfeffer K. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. (2003) 14:185–91. doi: 10.1016/S1359-6101(03)00022-4
121. Sedger LM, McDermott MF. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev. (2014) 25:453–72. doi: 10.1016/j.cytogfr.2014.07.016
122. Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Sem Immunol. (2014) 26:237–45. doi: 10.1016/j.smim.2014.02.009
123. Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryotic Gene Exp. (2010) 20:87–103. doi: 10.1615/CritRevEukarGeneExpr.v20.i2.10
124. Patel HJ, Patel BM. TNF-α and cancer cachexia: molecular insights and clinical implications. Life sciences. (2017) 170:56–63. doi: 10.1016/j.lfs.2016.11.033
125. Enewold L, Mechanic LE, Bowman ED, Zheng YL, Yu Z, Trivers G, et al. C. Serum concentrations of cytokines and lung cancer survival in African Americans and Caucasians. Cancer Epidemiol Biomark Prevent. (2009) 18:215–22. doi: 10.1158/1055-9965.EPI-08-0705
126. Zhang YW, Chen QQ, Cao J, Xu LQ, Tang X, Wang J, et al. Expression of tumor necrosis factor receptor 2 in human non-small cell lung cancer and its role as a potential prognostic biomarker. Thoracic Cancer. (2019) 10:437–44. doi: 10.1111/1759-7714.12948
127. Gupta M, Babic A, Beck AH, Terry K. TNF-α expression, risk factors, and inflammatory exposures in ovarian cancer: evidence for an inflammatory pathway of ovarian carcinogenesis? Hum Pathol. (2016) 54:82–91. doi: 10.1016/j.humpath.2016.03.006
128. Mikami S, Mizuno R, Kosaka T, Saya H, Oya M, Okada Y. Expression of TNF-α and CD44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. Int J Cancer. (2015) 136:1504–14. doi: 10.1002/ijc.29137
129. Gong L, da Silva Caetano M, Cumpian AM, Daliri S, Garza Flores A, Chang SH, et al. Tumor necrosis factor links chronic obstructive pulmonary disease and K-ras mutant lung cancer through induction of an immunosuppressive pro-tumor microenvironment. Oncoimmunology. (2016) 5:e1229724. doi: 10.1080/2162402X.2016.1229724
130. Tomita Y, Yang X, Ishida Y, Nemoto-Sasaki Y, Kondo T, Oda M, et al. Spontaneous regression of lung metastasis in the absence of tumor necrosis factor receptor p55. Int J Cancer. (2004) 112:927–33. doi: 10.1002/ijc.20493
131. Karabela SP, Kairi CA, Magkouta S, Psallidas I, Moschos C, Stathopoulos I, et al. Neutralization of tumor necrosis factor bioactivity ameliorates urethane-induced pulmonary oncogenesis in mice. Neoplasia. (2011) 13:1143–51. doi: 10.1593/neo.111224
132. Wang X, Ju W, Renouard J, Aden J, Belinsky SA, Lin Y. 17-allylamino-17-demethoxygeldanamycin synergistically potentiates tumor necrosis factor-induced lung cancer cell death by blocking the nuclear factor-kB pathway. Cancer Res. (2006) 66:1089–95. doi: 10.1158/0008-5472.CAN-05-2698
133. Gong K, Guo G, Gerber DE, Gao B, Peyton M, Huang C, et al. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J Clin Invest. (2018) 128:2500–18. doi: 10.1172/JCI96148
134. Madhusudan S, Muthuramalingam SR, Braybrooke JP, Wilner S, Kaur K, Han C, et al. Study of etanercept, a tumor necrosis factor-alpha inhibitor, in recurrent ovarian cancer. J Clin Oncol. (2005) 23:5950–59. doi: 10.1200/JCO.2005.04.127
135. Madhusudan S, Foster M, Muthuramalingam SR, Braybrooke JP, Wilner S, Kaur K, et al. A phase II study of etanercept (Enbrel), a tumor necrosis factor alpha inhibitor in patients with metastatic breast cancer. Clin Cancer Res. (2004) 10:6528–34. doi: 10.1158/1078-0432.CCR-04-0730
136. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
137. Arenberg DA, Keane MP, DiGiovine B, Kunkel SL, Strom SRB, Burdick MD, et al. Macrophage infiltration in human non-small-cell lung cancer: the role of CC chemokines. Cancer Immunol Immunother. (2000) 49:63–70. doi: 10.1007/s002620050603
138. Chen JJ, Yao PL, Yuan A, Hong TM, Shun CT, Kuo ML, et al. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin Cancer Res. (2003) 9:729–37.
139. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
140. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. (2006) 66:605. doi: 10.1158/0008-5472.CAN-05-4005
141. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. (2002) 196:254–65. doi: 10.1002/path.1027
142. Zaynagetdinov R, Sherrill TP, Polosukhin VV, Han W, Ausborn JA, McLoed AG, et al. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J Immunol. (2011) 187:5703–11. doi: 10.4049/jimmunol.1100558
143. Seth P, Csizmadia E, Hedblom A, Vuerich M, Xie H, Li M, et al. Deletion of lactate dehydrogenase-A in myeloid cells triggers antitumor immunity. Cancer Res. (2017) 77:3632–43. doi: 10.1158/0008-5472.CAN-16-2938
144. Belmont L, Rabbe N, Antoine M, Cathelin D, Guignabert C, Kurie J, et al. Expression of TLR9 in tumor-infiltrating mononuclear cells enhances angiogenesis and is associated with a worse survival in lung cancer. Int J Cancer. (2014) 134:765–77. doi: 10.1002/ijc.28413
145. Schmall A, Al-tamari HM, Herold S, Kampschulte M, Weigert A, Wietelmann A, et al. Macrophage and cancer cell cross-talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am J Res Crit Care Med. (2015) 191:437–47. doi: 10.1164/rccm.201406-1137OC
146. Zaynagetdinov R, Sherrill TP, Gleaves LA, Hunt P, Han W, McLoed AG, et al. Chronic NF-κB activation links COPD and lung cancer through generation of an immunosuppressive microenvironment in the lungs. Oncotarget. (2016) 7:5470–82. doi: 10.18632/oncotarget.6562
147. Conway EM, Pikor LA, Kung SH, Hamilton MJ, Lam S, Lam WL, et al. L. macrophages, inflammation, and lung cancer. Am J Respir Crit Care Med. (2016) 193:116–30. doi: 10.1164/rccm.201508-1545CI
148. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
149. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. (2012) 33:949–55. doi: 10.1093/carcin/bgs123
150. Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin JF, et al. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol. (1998) 152:83–92.
151. Keizman D, Ish-Shalom M, Huang P, Eisenberger MA, Pili R, Hammers H, et al. The association of pre-treatment neutrophil to lymphocyte ratio with response rate, progression free survival and overall survival of patients treated with sunitinib for metastatic renal cell carcinoma. Eur J Cancer. (2012) 48:202–8. doi: 10.1016/j.ejca.2011.09.001
152. Donskov F. Immunomonitoring and prognostic relevance of neutrophils in clinical trials. Sem Cancer Biol. (2013) 23:200–7. doi: 10.1016/j.semcancer.2013.02.001
153. Shaul ME, Fridlender ZG. Neutrophils as active regulators of the immune system in the tumor microenvironment. J Leukocyte Biol. (2017) 102:343–9. doi: 10.1189/jlb.5MR1216-508R
154. Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. (2010) 22:238–44. doi: 10.1016/j.coi.2010.01.021
155. Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, et al. J. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. (2012) 122:3127–44. doi: 10.1172/JCI61067
156. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF- β: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
157. Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. (2013) 24:631–44. doi: 10.1016/j.ccr.2013.10.009
158. Kargl J, Busch SE, Yang GHY, Kim KH, Hanke ML, Metz HE, et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat Commun. (2017) 8:14381. doi: 10.1038/ncomms14381
159. Acuff HB, Carter KJ, Fingleton B, Gorden DL, Matrisian LM. Matrix metalloproteinase-9 from bone marrow–derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res. (2006) 66:259–66. doi: 10.1158/0008-5472.CAN-05-2502
160. Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase–mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. (2010) 16:219–23. doi: 10.1038/nm.2084
161. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA. (2006) 103:12493–8. doi: 10.1073/pnas.0601807103
162. Sato T, Takahashi S, Mizumoto T, Harao M, Akizuki M, Takasugi M, et al. Neutrophil elastase and cancer. Surg Oncol. (2006) 15:217–22. doi: 10.1016/j.suronc.2007.01.003
163. Welch DR, Schissel DJ, Howrey RP, Aeed PA. Tumor-elicited polymorphonuclear cells, in contrast to “normal” circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proc Natl Acad Sci USA. (1989) 86:5859–63. doi: 10.1073/pnas.86.15.5859
164. Faget J, Groeneveld S, Boivin G, Sankar M, Zangger N, Garcia M, et al. Neutrophils and snail orchestrate the establishment of a pro-tumor microenvironment in lung cancer. Cell Rep. (2017) 21:3190–204. doi: 10.1016/j.celrep.2017.11.052
165. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
166. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
167. Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. (1999) 162:5728–37.
168. Chen MF, Kuan FC, Yen TC, Lu MS, Lin PY, Chung YH, et al. IL-6-stimulated CD11b+ CD14+ HLA-DR– myeloid-derived suppressor cells, are associated with progression and poor prognosis in squamous cell carcinoma of the esophagus. Oncotarget. (2014) 5:8716–28. doi: 10.18632/oncotarget.2368
169. Elkabets M, Ribeiro VS, Dinarello CA, Ostrand–Rosenberg S, Di Santo JP, Apte RN, et al. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol. (2010) 40:3347–57. doi: 10.1002/eji.201041037
170. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. (2012) 122:4094–104. doi: 10.1172/JCI64115
171. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. (2009) 124:2621–33. doi: 10.1002/ijc.24249
172. Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. (2008) 222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x
173. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. (2005) 5:641–54. doi: 10.1038/nri1668
174. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. (2004) 172:989–99. doi: 10.4049/jimmunol.172.2.989
175. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. (2007) 13:828–35. doi: 10.1038/nm1609
176. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
177. Hu CE, Gan J, Zhang RD, Cheng YR, Huang GJ. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. (2011) 46:156–64. doi: 10.3109/00365521.2010.516450
178. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. (2007) 179:977–83. doi: 10.4049/jimmunol.179.2.977
179. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+ CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
180. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
181. Sfanos KS, Bruno TC, Maris CH, Xu L, Thoburn CJ, DeMarzo AM, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res. (2008) 14:3254–61. doi: 10.1158/1078-0432.CCR-07-5164
182. Koyama K, Kagamu H, Miura S, Hiura T, Miyabayashi T, Itoh R, et al. Reciprocal CD4+ T-cell balance of effector CD62Llow CD4+ and CD62LhighCD25+ CD4+ regulatory T cells in small cell lung cancer reflects disease stage. Clin Cancer Res. (2008) 14:6770–9. doi: 10.1158/1078-0432.CCR-08-1156
183. Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. (2010) 51:154–64. doi: 10.1002/hep.23291
184. Amedei A, Niccolai E, Benagiano M, Della Bella C, Cianchi F, Bechi P, et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother. (2013) 62:1249–60. doi: 10.1007/s00262-013-1429-3
185. You R, DeMayo FJ, Liu J, Cho SN, Burt BM, Creighton CJ, et al. IL17A regulates tumor latency and metastasis in lung adeno and squamous SQ.2b and AD.1 cancer. Cancer Immunol Res. (2018) 6:645–57. doi: 10.1158/2326-6066.CIR-17-0554
186. Xu C, Hao K, Yu L, Zhang X. Serum interleukin-17 as a diagnostic and prognostic marker for non-small cell lung cancer. Biomarkers. (2014) 19:287–90. doi: 10.3109/1354750X.2014.908954
187. Ye ZJ, Zhou Q, Gu YY, Qin SM, Ma WL, Xin JB, et al. Generation and differentiation of IL-17–producing CD4+ T cells in malignant pleural effusion. J Immunol. (2010) 185:6348–54. doi: 10.4049/jimmunol.1001728
188. Santegoets SJAM, Turksma AW, Suhoski MM, Stam AGM, Albelda SM, Hooijberg E, et al. IL-21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J Transl Med. (2013) 11:37. doi: 10.1186/1479-5876-11-37
189. Laurence A, O'Shea JJ. TH-17 differentiation: of mice and men. Nat Immunol. (2007) 8:903. doi: 10.1038/ni0907-903
190. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. (2008) 28:454–67. doi: 10.1016/j.immuni.2008.03.004
191. Erfani N, Mehrabadi SM, Ghayumi MA, Haghshenas MR, Mojtahedi Z, Ghaderi A, et al. Increase of regulatory T cells in metastatic stage and CTLA-4 over expression in lymphocytes of patients with non-small cell lung cancer (NSCLC). Lung Cancer. (2012) 77:306–11. doi: 10.1016/j.lungcan.2012.04.011
192. Shimizu K, Nakata M, Hirami Y, Yukawa T, Maeda A, Tanemoto K. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thoracic Oncol. (2010) 5:585–90. doi: 10.1097/JTO.0b013e3181d60fd7
193. Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi MB, Harpole DH Jr, et al. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. (2006) 107:2866–72. doi: 10.1002/cncr.22282
194. Marshall EA, Ng KW, Kung SHY, Conway EM, Martinez VD, Halvorsen EC, et al. Emerging roles of T helper 17 and regulatory T cells in lung cancer progression and metastasis. Mol Cancer. (2016) 15:67. doi: 10.1186/s12943-016-0551-1
195. Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors in cancer therapy: a focus on T-regulatory cells. Immunol Cell Biol. (2018) 96:21–33. doi: 10.1111/imcb.1003
196. Ganesan AP, Johansson M, Ruffell B, Yagui-Beltrán A, Lau J, Jablons DM, et al. Tumor-infiltrating regulatory T cells inhibit endogenous cytotoxic T cell responses to lung adenocarcinoma. J Immunol. (2013) 191:2009–17. doi: 10.4049/jimmunol.1301317
197. Smyth MJ, Teng MW, Swann J, Kyparissoudis K, Godfrey DI, Hayakawa Y. CD4+ CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J Immunol. (2006) 176:1582–7. doi: 10.4049/jimmunol.176.3.1582
198. Zdanov S, Mandapathil M, Eid RA, Adamson-Fadeyi S, Wilson W, Qian J, et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol Res. (2016) 4:354–65. doi: 10.1158/2326-6066.CIR-15-0241
199. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. (2016) 30:147–60. doi: 10.1016/j.ccell.2016.05.016
200. Adeegbe DO, Liu S, Hattersley M, Bowden M, Zhou CW, Li S, et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in Kras-mutant non-small cell lung cancer. Cancer Immunol Res. (2018) 6:1234–45. doi: 10.1158/2326-6066.CIR-18-0077
201. Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell–dependent antitumor activity following radiation. J Clin Invest. (2018) 128:3926–40. doi: 10.1172/JCI96519
202. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. (2012) 482:400. doi: 10.1038/nature10755
203. Hanson HL, Donermeyer DL, Ikeda H, White JM, Shankaran V, Old LJ, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. (2000) 13:265–76. doi: 10.1016/S1074-7613(00)00026-1
204. Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol. (1998) 10:588–94. doi: 10.1016/S0952-7915(98)80228-8
205. Li J, Jie HB, Lei Y, Gildener-Leapman N, Trivedi S, Green T, et al. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. (2015) 75:508–18. doi: 10.1158/0008-5472.CAN-14-1215
206. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
208. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Disease. (2015) 6:e1792. doi: 10.1038/cddis.2015.162
209. Kontani K, Sawai S, Hanaoka J, Tezuka N, Inoue S, Fujino S. Involvement of granzyme B and perforin in suppressing nodal metastasis of cancer cells in breast and lung cancers. Eur J Surg Oncol. (2001) 27:180–6. doi: 10.1053/ejso.2000.1060
210. Mazzaschi G, Madeddu D, Falco A, Bocchialini G, Goldoni M, Sogni F, et al. Low PD-1 expression in cytotoxic CD8+ tumor-infiltrating lymphocytes confers an immune-privileged tissue microenvironment in NSCLC with a prognostic and predictive value. Clin Cancer Res. (2018) 24:407–19. doi: 10.1158/1078-0432.CCR-17-2156
211. Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. In: Alt FW, editor. Advances in Immunology. Oxford, UK: Academic Press (2014). p. 91–128. doi: 10.1016/B978-0-12-800267-4.000030-1
212. Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer Interdiscipl Int J Am Cancer Soc. (1997) 79:2320–28.
213. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer. (2000) 88:577–83. doi: 10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V
214. Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. (2006) 6:940–52. doi: 10.1038/nri1983
215. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
216. Martín-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-γ for T H 1 priming. Nat Immunol. (2004) 5:1260. doi: 10.1038/ni1138
217. O'Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med. (2012) 209:1869–82. doi: 10.1084/jem.20112738
218. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. (2016) 16:7–19. doi: 10.1038/nrc.2015.5
219. Cong J, Wang X, Zheng X, Wang D, Fu B, Sun R, et al. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metabol. (2018) 28:243–55.e245. doi: 10.1016/j.cmet.2018.06.021
220. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
221. Lai DM, Shu Q, Fan J. The origin and role of innate lymphoid cells in the lung. Military Med Res. (2016) 3:25. doi: 10.1186/s40779-016-0093-2
222. Bruchard M, Ghiringhelli F. Deciphering the roles of innate lymphoid cells in cancer. Front Immunol. (2019) 10:656. doi: 10.3389/fimmu.2019.00656
223. Mattner J, Wirtz S. Friend or Foe? The ambiguous role of innate lymphoid cells in cancer development. Trends Immunol. (2017) 38:29–38. doi: 10.1016/j.it.2016.10.004
224. Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, et al. NCR+ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun. (2015) 6:8280. doi: 10.1038/ncomms9280
225. Koh J, Kim HY, Lee Y, Park IK, Kang CH, Kim YT, et al. H. IL23-Producing human lung cancer cells promote tumor growth via conversion of innate lymphoid cell 1 (ILC1) into ILC3. Clin Cancer Res. (2019) 25:4026–37. doi: 10.1158/1078-0432.CCR-18-3458
226. Busser B, Sancey L, Brambilla E, Coll JL, Hurbin A. The multiple roles of amphiregulin in human cancer. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. (2011) 1816:119–31. doi: 10.1016/j.bbcan.2011.05.003
227. Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Gröne A, Sibilia M, et al. J. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. (2013) 38:275–84. doi: 10.1016/j.immuni.2012.09.023
228. Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, et al. Identification of innate IL-5–producing cells and their role in lung eosinophil regulation and antitumor immunity. J Immunol. (2012) 188:703–13. doi: 10.4049/jimmunol.1101270
229. Adderley H, Blackhall FH, Lindsay CR. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMed. (2019) 41:711–6. doi: 10.1016/j.ebiom.2019.02.049
230. Niyongere S, Saltos A, Gray JE. Immunotherapy combination strategies (non-chemotherapy) in non-small cell lung cancer. J Thoracic Disease. (2018) 10(Suppl. 3):S433–50. doi: 10.21037/jtd.2017.12.120
231. Miura Y, Sunaga N. Role of immunotherapy for oncogene-driven non-small cell lung cancer. Cancers. (2018) 10:245. doi: 10.3390/cancers10080245
232. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discovery. (2015) 14:561. doi: 10.1038/nrd4591
233. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. (2013) 39:38–48. doi: 10.1016/j.immuni.2013.07.004
234. Takahashi H, Okamoto M, Shimodaira S, Tsujitani Si, Nagaya M, Ishidao T, et al. Impact of dendritic cell vaccines pulsed with Wilms' tumour-1 peptide antigen on the survival of patients with advanced non-small cell lung cancers. Eur J Cancer. (2013) 49:852–9. doi: 10.1016/j.ejca.2012.11.005
235. Ge C, Li R, Song H, Geng T, Yang J, Tan Q, et al. Phase I clinical trial of a novel autologous modified-DC vaccine in patients with resected NSCLC. BMC Cancer. (2017) 17:884. doi: 10.1186/s12885-017-3859-3
236. Fischer J, Darjes H, Lahm H, Schindel M, Drings P, Krammer P. Constitutive secretion of bioactive transforming growth factor β 1 by small cell lung cancer cell lines. Lung Cancer. (1995) 3:270. doi: 10.1016/0169-5002(95)98719-Q
237. Masotti A, Morandini G, Ortolani R, Fumagalli L. Phase-II randomized study of pre-operative IL-2 administration in operable NSCLC. Lung Cancer. (1998) 20:191–202. doi: 10.1016/S0169-5002(98)00024-5
238. Albeituni SH, Ding C, Liu M, Hu X, Luo F, Kloecker G, et al. Yeast-derived particulate β-glucan treatment subverts the suppression of Myeloid-Derived Suppressor Cells (MDSC) by inducing polymorphonuclear MDSC apoptosis and monocytic MDSC differentiation to APC in cancer. J Immunol. (2016) 196:2167–80. doi: 10.4049/jimmunol.1501853
239. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase Ia study of FoxP3+ CD4 Treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res. (2015) 21:4327–36. doi: 10.1158/1078-0432.CCR-15-0357
240. Iversen TZ, Engell-Noerregaard L, Ellebaek E, Andersen R, Larsen SK, Bjoern J, et al. Long-lasting disease stabilization in the absence of toxicity in metastatic lung cancer patients vaccinated with an epitope derived from indoleamine 2,3 dioxygenase. Clin Cancer Res. (2014) 20:221–32. doi: 10.1158/1078-0432.CCR-13-1560
Keywords: lung cancer, inflammation, cytokine, tumor microenvironment, K-ras
Citation: Deng S, Clowers MJ, Velasco WV, Ramos-Castaneda M and Moghaddam SJ (2020) Understanding the Complexity of the Tumor Microenvironment in K-ras Mutant Lung Cancer: Finding an Alternative Path to Prevention and Treatment. Front. Oncol. 9:1556. doi: 10.3389/fonc.2019.01556
Received: 14 October 2019; Accepted: 23 December 2019;
Published: 22 January 2020.
Edited by:
Giovanna Schiavoni, Istituto Superiore di Sanità (ISS), ItalyReviewed by:
Zong Sheng Guo, University of Pittsburgh, United StatesAmedeo Amedei, University of Florence, Italy
Copyright © 2020 Deng, Clowers, Velasco, Ramos-Castaneda and Moghaddam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seyed Javad Moghaddam, c21vZ2hhZGRAbWRhbmRlcnNvbi5vcmc=