 Celine Chen
Celine Chen Heathcliff Dorado Garcia
Heathcliff Dorado Garcia Monika Scheer2
Monika Scheer2 Anton G. Henssen
Anton G. Henssen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 20 December 2019
Sec. Pediatric Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01458
This article is part of the Research Topic Novel Molecular Targets and Therapies for Pediatric Solid Tumors View all 14 articles
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children, and can be subcategorized histologically and/or based on PAX-FOXO1 fusion gene status. Over the last four decades, there have been no significant improvements in clinical outcomes for advanced and metastatic RMS patients, underscoring a need for new treatment options for these groups. Despite significant advancements in our understanding of the genomic landscape and underlying biological mechanisms governing RMS that have informed the identification of novel therapeutic targets, development of these therapies in clinical trials has lagged far behind. In this review, we summarize the current frontline multi-modality therapy for RMS according to pediatric protocols, highlight emerging targeted therapies and immunotherapies identified by preclinical studies, and discuss early clinical trial data and the implications they hold for future clinical development.
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children, comprising 4.5% of all childhood cancer with an annual incidence of 4.5 cases per 1 million children (1, 2). RMS cells resemble skeletal muscle progenitor cells, though they can arise from non-skeletal tissue origins (3). RMS is historically classified based on histopathologic features into distinct clinical subtypes— embryonal RMS (ERMS), alveolar RMS (ARMS), pleomorphic, and spindle cell and sclerosing RMS (ssRMS) (4, 5). ERMS represents the majority of cases and is associated with a favorable prognosis, while ARMS is more clinically aggressive due to a propensity for metastasis and recurrence (6–8). Eighty percentage of ERMS tumors are characterized by a loss of heterozygosity at the 11p15 locus, and they generally represent a more biologically heterogeneous group of tumors compared to ARMS (1). The recently recognized subtype, spindle cell and sclerosing RMS is a rare variant of RMS characterized by recurring fusions of VGLL2 or NOCA2 and has a favorable prognosis, so it is treated without the aggressive multimodal regimens used to treat ARMS and ERMS (5). In this review, we focus on the ARMS and ERMS subtypes.
The majority of ARMS tumors harbor a recurrent chromosomal translocation, t(2;13)(q35;q14) or t(1;13)(p36;q14). The 2;13 and 1;14 translocations encode for a chimeric transcription factor (TF), consisting of the N-terminal DNA binding domain of PAX3 or PAX7 fused to the C-terminal transactivation domain of FOXO1 (9, 10). Of all ARMS patients, approximately 60% express PAX3-FOXO1, 20% express PAX7-FOXO1, 20% are fusion negative (11, 12), and a small subset express rare variants such as PAX3-FOXO4 or PAX3-NOXA1 (12). Patients with the PAX7-FOXO1 rearrangement have superior overall survival (82%) compared to patients with the PAX3-FOXO1 rearrangement (61%) (12). Notably, chromosomal amplification was reported in the majority (93%) of PAX7-FOXO1 cases compared to PAX3-FOXO1 (9%) (13), raising the question of whether fusion gene amplification is linked to more favorable outcomes. At this point in time, it is unknown whether the PAX7 fusion partner or gene amplification is the main determinant of favorable outcome, but prospective tracking of fusion gene amplification in COG study ARST1431 is expected to clarify if gene amplifications contribute toward the observed difference. The remaining 20% of fusion-negative ARMS tumors present a similar molecular profile and clinical outcome to the ERMS subtype (14–16). The PAX-FOXO1 chimeric protein behaves as a highly active transcription factor to drive aberrant gene expression, encoding for downstream gene targets required for oncogenic transformation. The oncogenic capacity of the PAX-FOXO1 fusion proteins has been well characterized by multiple studies and has been shown to act as a dominant-acting oncogene in driving tumorigenesis in fusion-positive RMS (FP RMS) (4, 17). On the other hand, fusion-negative RMS (FN RMS) is characterized by higher rates of aneuploidy and single-nucleotide variations, with the RAS pathway most commonly activated in the majority of FN tumors (18–20). There is a known link between RMS and cancer predisposition syndromes, such as Li-Frameni syndrome, neurofibromatosis, Beckwith-Wiedemann syndrome, and Costello syndrome (19).
Over 90% of patients with low-risk localized disease can be cured with multi-modal therapy, but overall survival rates of patients with metastatic or recurrent disease remain dismal at 21% and 30%, respectively (21, 22). Because RMS is a rare disease, cooperative trials in Europe (European pediatric Soft Tissue Sarcoma Study Group (23), Cooperative Weichteilsarkom Studiengruppe der Gesellschaft für pädiatrische Onkologie und Hämatologie (CWS) (21, 23) and North America (Children's Oncology Group) (24) have been crucial for clinical study of this disease. Given that no significant improvements in the survival outcomes of metastatic and recurrent RMS patients in the last 30 years have been reached, there is an unmet need for novel treatment paradigms. Moreover, RMS patients could benefit from molecularly targeted and immunotherapeutic approaches, which could reduce the treatment-associated toxicities caused by current chemotherapy and radiation therapy (RT). Because funding of drug development for a rare childhood cancer such as RMS is limited, preclinical studies have focused on molecularly actionable targets that have been studied in other human cancers, many of which have clinically approved therapies. Here, we review the current frontline therapies, followed by an overview of emerging targeted therapies and immunotherapies in RMS (Figure 1).
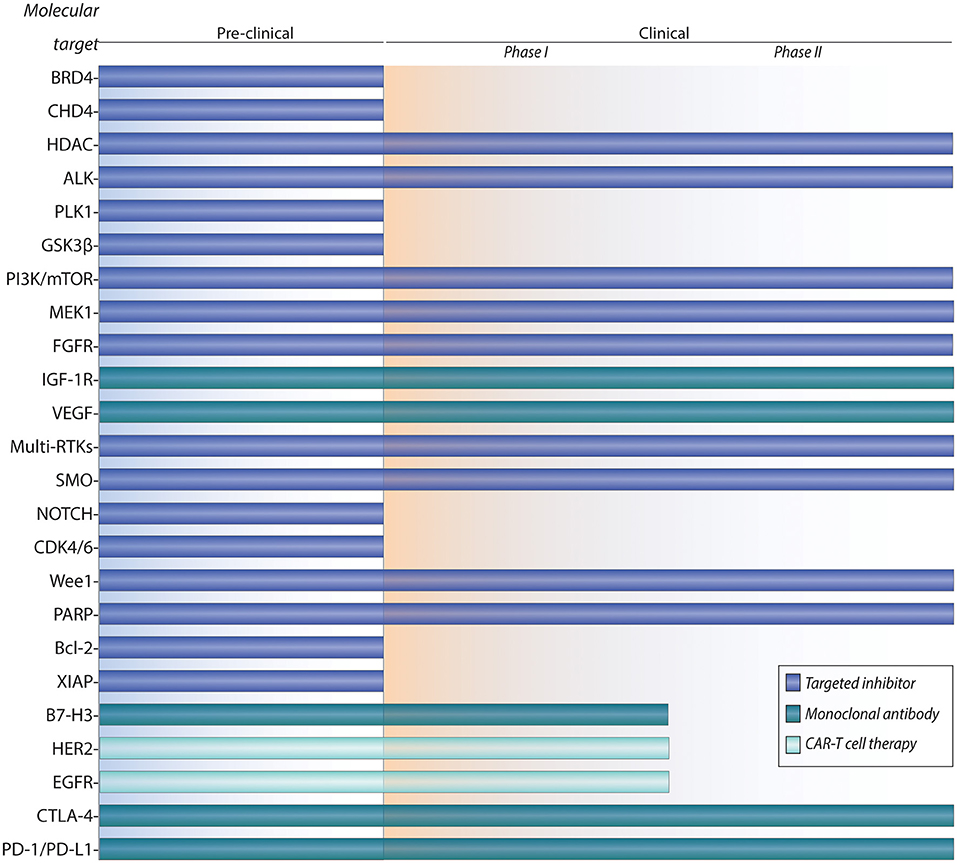
Figure 1. Pipeline of preclinical and clinical development for targeted therapies and immunotherapies of rhabdomyosarcoma.
The last five decades of cooperative group trials for RMS have improved the 5-year overall survival of patients with pediatric RMS, which now exceeds 70% (25–28). These improvements follow collaborative group clinical trial efforts, which have enabled improvements in chemotherapeutic dosing regimens, local control, and management of treatment-related toxicities. However, improvements in cure rate have generally been limited to patients with low- and intermediate-risk RMS, while no significant progress has been reached in cure rates for patients with advanced or metastatic RMS. Both European and American cooperative group studies have developed more sophisticated risk stratification systems to include more comprehensive prognostic features [patient age, tumor size and site, lymph node involvement, and/ or metastases and surgical group classification (IRS)] that allow more personalized and effective treatment approaches (29, 30).
The support for inclusion of fusion status for risk stratification in clinical trials has been disputed by conflicting studies. A study by Missiaglia et al. reported that positive fusion status correlated with an inferior clinical outcome, while Stegmaier et al. reported there to be no correlation between fusion status and clinical outcome (31, 32). The inconsistent results from these retrospective studies can be partly explained by methodological biases of convenience sampling, where samples are not truly representative of the whole population but rather chosen based on archival sample availability (33–35). Support for the inclusion of fusion status in future clinical trials comes from retrospective analyses such as one by Selfe et al., which argued that re-assignment based on fusion status could spare a significant number of patients from treatment-associated toxicities caused by unnecessary intensive therapy (36). Prompted by conflicting results from previous studies, in 2019 the COG re-examined the prognostic importance of fusion status and determined it was the second most important prognostic factor, after metastatic status (37). Taken together with additional supporting evidence for the inclusion of fusion status as a significant prognostic marker (31, 38) and evidence that FN ARMS and ERMS are molecularly indistinguishable (16), ARST1431 was the first COG trial to use fusion status instead of histopathological status (39). On the European side, fusion gene status will now be used instead of histological status for risk stratification in EpSSG trials (40). It should be mentioned that currently in Europe and North America, high-risk stratification is assigned based on metastatic status, irrespective of fusion status and histology. Whereas, previous studies focused on fusion status as an important prognostic marker in low- and intermediate-risk RMS (38, 41, 42), a review of high-risk RMS cases found that fusion status does not offer the same level of predictive value for metastatic patients. In cases of metastatic RMS, clinical risk factors remain the major predictors of outcome.
The current frontline treatment for all risk-groups of RMS is a multi-modal approach, comprising chemotherapy, surgical resection, and/or radiation therapy. In North America, the standard chemotherapy backbone includes vincristine, actinomycin D, and cyclophosphamide (VAC) (43, 44) and in Europe, the backbone consists of isofasfamide, vincristine, and actinomycin D (IVA) (45). A randomized trial confirmed there to be no significant difference in patient outcomes between the two treatment combinations, so VAC and IVA have continued to be used in their respective regions (25). Since the VAC/IVA regimen was first established four decades ago, the chemotherapy backbone has remained the same besides changes in duration, dosage, and route of administration. After three decades of controversy regarding the inclusion of doxorubicin in the chemotherapy regimen (43, 46–48), an open-label phase 3 trial (EpSSG RMS 2005) conclusively showed that addition of doxorubicin to the standard IVA backbone did not improve patient outcomes in high-risk rhabdomyosarcoma (49). Because of doxorubicin's known risk of cardiotoxicity (especially in younger patients), there is a lack of justification for its continued inclusion in the chemotherapy regimen.
Today, children with low-risk RMS (localized to favorable anatomical sites, grossly resected ERMS) treated with frontline multi-modality therapy have excellent outcomes (90% relapse-free survival). Recent clinical research on low-risk RMS has focused on reducing toxicity of treatment by decreasing dosage and duration of alkylating agent, without compromising the ability to prevent disease recurrence. The alkylating agent, cyclophosphamide used in the VAC chemotherapy regimen is known to cause acute and late effects, including severe myelosuppression, infectious complications, and infertility (50). Aiming to minimize treatment-related toxicities, the COG ARST0331 study showed that shorter duration therapy (which included lower-dose cyclophosphamide) and radiation therapy (RT) did not compromise failure-free survival for the majority of patients (51).
Eradication of gross primary tumor is achieved by a combination of surgery and/or RT, in addition to the standard systemic chemotherapy backbone. RT is included in the frontline treatment for nearly all RMS patients, although long-term toxicity poses a significant concern in younger patients (52). Strategies to reduce radiation-related toxicities include incorporation of intensity-modulated RT (IMRT) or proton bean RT and the use of brachytherapy for specific sites e.g., bladder or vagina, both of which are assumed to reduce late toxicities (skeletal muscle/soft tissue changes, joint stiffness, skeletal growth problems, and secondary malignancy) (53–56). It is worth noting a difference in treatment philosophies between North American and European studies. European studies use overall survival as the study end-point, preferring less aggressive local treatment (omission of RT if possible) to mitigate late toxicities, while tolerating a greater risk of relapsed disease. Conversely, North American studies tend to focus on event-free survival as the study end-point, so treatment strategies favor more aggressive local treatment with radiation therapy (1).
In localized high-risk RMS, the benefit of additional maintenance therapy is currently investigated by the two European groups EpSSG and CWS in two international trials. The EpSSG reported an improved overall survival with cyclophosphamide/vinorelbine in the first preliminary assessment at the end of the recruitment period of EpSSG RMS2005 (2008–2013) (57). The ongoing CWS-2007HR trial is a randomized study of whether the addition of an oral maintenance regimen of O-TIE (etoposide, idarubicin, tofosfamide) can benefit patients with localized high-risk RMS. The EpSSG RMS 2005 randomized phase III trial reported that addition of maintenance chemotherapy improved survival for patients with non-metastatic high-risk RMS (58, 59). Since this study was published in 2019, vinorelbine/cyclophosphamide maintenance chemotherapy has been established as the new standard-of-care for treatment of localized high-risk RMS.
Survival outcomes for patients with metastatic disease remain dismal (event free survival <20%, excluding patients <10 years old diagnosed with ERMS), and the frontline treatment has not advanced significantly over the last 30 years (22, 29, 60). Today in Europe and the United States, the Oberlin score is used for risk stratification and classification of metastatic RMS, assigning a risk score based on patient age, primary tumor site, number of metastases, histology, and bone marrow involvement (29). Unlike for localized disease, for metastatic RMS multimodal therapy frequently fails due to lack of a proper local therapy to treat metastatic sites such as the bone marrow and lungs. A European trial for metastatic RMS reported that high-dose chemotherapy (HD-CT) did not significantly improve survival outcomes compared to standard chemotherapy, despite increased treatment-associated toxicities (61). This report is consistent with a retrospective analysis of 389 patients, which found no significant improvements in survival after HD-CT with hematopoietic stem cell rescue in the treatment of metastatic rhabdomyosarcoma (62). Results from the COG ARST0431 trial for patients with high-risk RMS found that high-dose chemotherapy (dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation) did not produce meaningful benefit for most patients, except for a minority of patients with embryonal histology and limited metastatic disease (restricted to lungs) (63). Notably, the inefficacy of HD-CT has also been documented in trials for other metastatic childhood solid tumors, including Ewing sarcoma and osteosarcoma (64–66). Observations from a trial conducted on pediatric neuroblastoma patients treated with HD-CT and stem cell transplantation rescue found there were long-term health consequences (hearing loss, gonadal insufficiency) associated with treatment (67). An important recent study by Merker et al. reported that haploidentical allogeneic hematopoietic stem cell transplantation (HSCT) has inferior outcomes compared to standard maintenance therapies, such as O-TIE (68). Here, the authors argue against the use of allogeneic HSCT, which has more severe side effects than standard maintenance therapy. Taken together, HD-CT and allogeneic HSCT should be discontinued because they failed to achieve curative effects in metastatic RMS, and similar outcomes can be achieved with the less toxic maintenance therapies.
Ongoing European studies investigating the role of maintenance therapy are based off a report by Klingebiel et al. for the HD CWS-96 study. The HD CWS-96 trial was a non-randomized trial comparing the efficacy of high dose therapy (HDT) vs. oral maintenance therapy (OMT) in patients with stage IV soft tissue sarcoma (69). While high dose therapy failed to improve survival, oral maintenance therapy was a promising alternative, since the oral administration can provide long lasting exposure to chemotherapy without increasing toxic side effects. As such, an OMT regimen of O-TIE is the current standard of care within the CWS, while an OMT combination of cyclophosphamide/vinorelbine is used within the EpSSG for metastatic RMS. In North America, the COG does not currently regard maintenance therapy as the standard of care for metastatic RMS; however, COG study protocols include much longer absolute durations of therapy. There are considerations as to whether the concept of maintenance therapy or absolute duration of therapy is the more relevant metric for treatment.
In cases of metastatic disease with distant spread of disease from the primary tumor site, the value of localized treatment of the primary tumor is often overlooked. A European retrospective study demonstrated that aggressive localized treatment of the primary tumor (combined surgical resection and RT compared to either alone) led to improved outcomes in patients with metastatic RMS (70). Another retrospective study found that local treatment to all metastatic sites in stage IV RMS was associated with an improved progression-free survival (PFS) and overall survival (OS) at 5 years (71). These lines of evidence support the importance of strong localized therapy at both primary and metastatic sites. However, extended local therapy (RT or chemotherapy) is not always feasible in patients. Other directions that are currently being considered include targeting genetically quiescent cells with the administration of oral maintenance therapy (69) and efforts to design therapeutic agents specifically targeted toward the metastatic phenotype (72, 73).
Approximately one-third of pediatric RMS patients will experience progressive disease or relapse, with a median time to relapse/progression of 13 months from initial diagnosis (74). Based on multivariate statistical modeling, it is now possible to predict the chance of salvage following first relapse of localized RMS based on a number of factors associated with worse outcome: metastatic status, prior exposure to RT and chemotherapy, unfavorable size and site of the tumor, lymph node involvement, alveolar histology, and shorter time to relapse (75). In general, most relapsed RMS patients are treated with chemotherapy and local control (surgery and/or RT). Knowing the chance of salvage on a case-by-case basis is important for deciding treatment options for each patient. For instance, patients with a very low chance of cure will not respond effectively to salvage therapy, so these patients should be prioritized for enrollment in experimental trials (75). Meanwhile, patients who demonstrate relapse after low-risk disease may benefit from salvage chemotherapy, such as irinotecan/vincristine or alternating vincristine/doxorubicin/cyclophosphamide, and etoposide/ofosfamide (76, 77). Disappointingly, phase II trials for children with relapsed RMS have not demonstrated meaningful, single-agent activity of targeted inhibitors, such as a monoclonal antibody against IGF-1R (R1507) and a multi-kinase inhibitor, sorafenib (78, 79). Clearly, there is a need to understand why these therapeutic agents which show promise in the pre-clinical stage fail to translate into meaningful outcomes in patients, and to identify strategies targeting resistance mechanisms hindering their clinical efficacy. At any rate, novel therapeutic targets (Table 1) that are backed by supportive clinical evidence should also be explored as experimental options for patients with relapsed RMS.
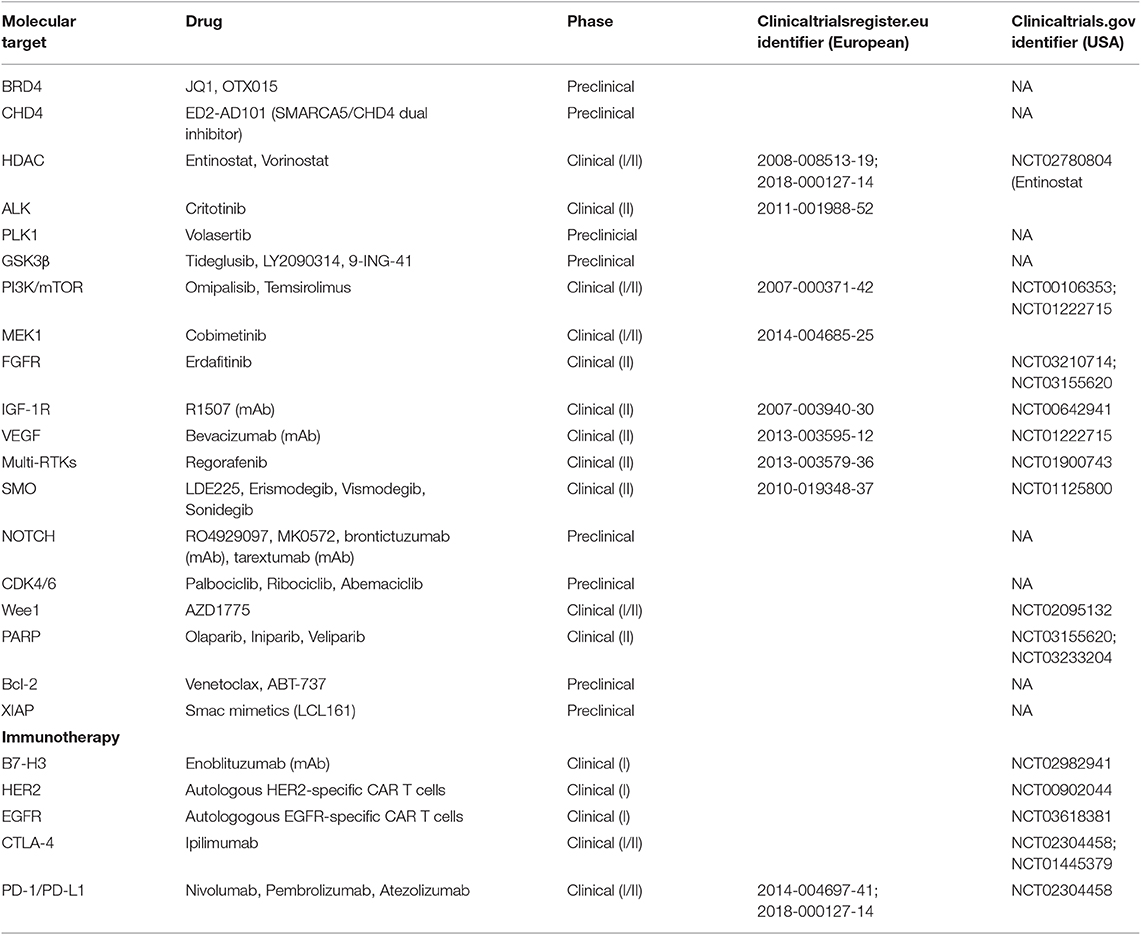
Table 1. Current targeted therapies and immunotherapies targets under evaluation in preclinical and/or clinical development in North America and Europe for rhabdomyosarcoma.
Given the inherent limitations of therapeutic options available for metastatic and recurrent RMS, experimental trials should prioritize patients with metastatic or recurrent disease, including emerging targeted therapy and immunotherapy strategies. Below, we summarize the key preclinical and clinical findings on novel targeted therapy and immunotherapy options in RMS (Figure 2).
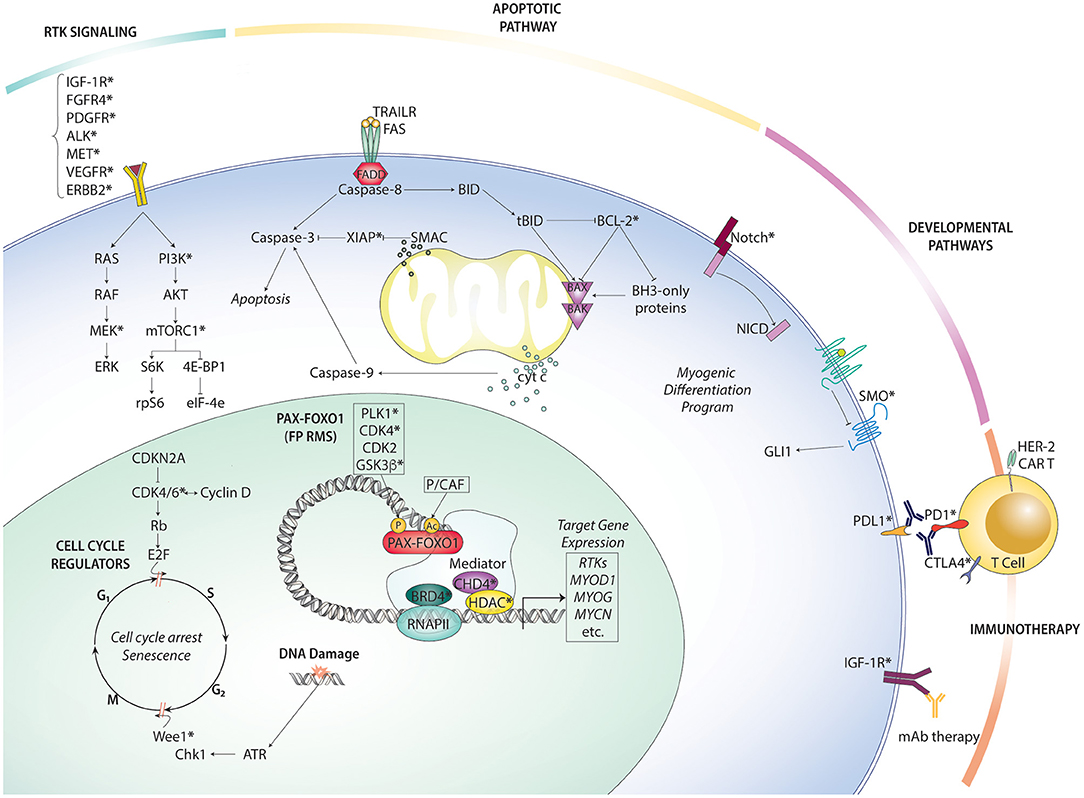
Figure 2. Overview of rhabdomyosarcoma targeted therapies organized by pathway. Therapeutically actionable targets (at least one existing small molecule inhibitor or antibody) are indicated with an asterisk (*).
In FP RMS, the chimeric transcription factor, PAX-FOXO1 presents the most direct and promising target. Conceptually, it is more effective to target one upstream transcription factor than multiple downstream signaling cascades and hundreds of target genes. Moreover, because PAX-FOXO1 fusion protein is uniquely expressed in tumor cells but not in normal cells, it is an attractive target. Small molecule inhibitors against oncogenic fusion proteins have achieved remarkable clinical success in some human cancers, such as the targeting of BCR-ABL in leukemia and EML4-ALK1 in lung carcinoma (80, 81). Until recently, TFs were considered to be an “undruggable” class of proteins due to an absence of deep hydrophobic pockets, large protein-protein interaction interfaces, and nuclear localization (82). Today, the opinion on drugging transcription factors is beginning to shift [reviewed by (83)], as approaches to inhibit transcription factors have demonstrated some success in preclinical and clinical studies. Novel approaches to drug transcription factors are currently being investigated in other disease contexts, with the possibility of adapting these strategies for targeting PAX-FOXO1. One experimental approach is the use of nanoparticle carriers to deliver naked siRNA or antisense oligonucleotides (ASO) into tumor cells to silence specific genes. The advantage of this approach is that any gene can theoretically be targeted by simply knowing the complementary base pairing for the gene of interest. A recent publication demonstrated that liposome-protamine-siRNA (LRP) particles targeting PAX3-FOXO1 were efficiently delivered to ARMS cell lines and downregulated PAX3-FOXO1 and its target genes in vitro, leading to delayed tumor growth and inhibition of tumor initiation in ARMS xenograft models (84). However, safety concerns over off-target effects by the RNAi transcripts and the toxicity of delivery systems remain significant obstacles to translation of this approach into the clinic.
To date, there have not been significant efforts to design inhibitors which directly bind PAX-FOXO1. Yet, emerging strategies to directly drug transcription factors are currently being explored in other human cancers. These new approaches include modulation of the auto-inhibitory state of transcription factors, proteolysis targeting chimeras (PROTACs), use of cysteine reactive inhibitors, and targeting intrinsically disordered regions [reviewed in (83)]. Notably, PROTAC uses a bifunctional molecule simultaneously targeting the protein of interest and engaging an E3 ubiquitin ligase to promote proteasomal degradation of the target. So far, it has not been explored in RMS, but the documented efficacy in other studies support its consideration for targeting PAX-FOXO1 (85–87). The advantage of the PROTAC approach over traditional pharmacological inhibition is that a single molecule could be used for multiple rounds of proteasome-targeted degradation. The first step would be to identify ligands capable of binding PAX-FOXO1 with sufficient specificity and affinity. This is more feasible than other inhibitory approaches, since the ligand only needs to bind to a tractable surface, rather than a specific functional site which is much harder to target.
An alternative approach to disrupting PAX-FOXO1 activity is to target essential protein-protein interactions with co-regulators and chromatin-remodeling proteins required for oncogenic transcriptional activity. PAX-FOXO1 acts as a pioneering TF to establish looped super-enhancers, recruiting chromatin remodeling proteins, coactivators, and other TFs to aberrantly drive transcription at target genes. Only a small subset of TFs form the core regulatory circuit of TFs, which cancer cells are uniquely dependent on (88, 89). In FP ARMS, PAX-FOXO1 orchestrates the formation of super-enhancers, which drive the transcription of core regulatory TFs in a strong autoregulatory loop. A promising therapeutic approach is to disassemble the super-enhancer with small molecule inhibitors, thereby disrupting the oncogenic core regulatory circuit (90).
The selective disruption of super-enhancers by small molecule inhibitors can specifically suppress transcription at key oncogenic drivers (91). Gryder et al. was first to demonstrate a mechanistic link between the chromatin reader, BET bromodomain-containing protein (BRD4) and PAX3-FOXO1. The authors show that BRD4 small molecule inhibitor, JQ1 selectively disrupts the interaction between BRD4 and PAX3-FOXO1, leading to rapid degradation of the fusion gene and abrogation of transcriptional output (89). Another study reported that the antitumor activity of JQ1 is mediated by a decrease in angiogenic activity (92), which is consistent with the hypothesis that disruption of the super-enhancer ablates transcriptional output of gene targets, one of which is vascular endothelial growth factor (VEGF). Among the five structurally diverse BET bromodomain inhibitors tested in this study, OTX015 was reported to be most potent across a range of FP RMS cell lines, but its clinical efficacy has not been evaluated. A related therapeutic strategy targets chromatin helix DNA binding protein 4 (CHD4), an ATP-dependent chromatin remodeling protein which plays an integral role in the Nucleosome Remodeling Deacetylase (NuRD) complex. CHD4 is required for the recruitment of the transcriptional machinery, and its role in nucleosome eviction is required for transcription to proceed. (93) Upon knockdown of CHD4 in vitro, gene expression profiling showed that CHD4 activity is essential for the expression of a subset of PAX3-FOXO1 target genes, and that the observed effect was specific to FP RMS (93). Independently, another group found that CHD4 acts as a crucial coregulator of PAX3-FOXO1 (identified as a top candidate from a siRNA screen of 60 candidate interactors), suggesting the role of CHD4 as a therapeutic target in FP RMS (93). A first-in-class inhibitor (ED2-AD101) of SMARCA5/CHD4 was recently shown to suppress cell growth in acute myeloid leukemia (AML) cells, but no inhibitors specifically targeting CHD4 are currently available for clinical use (94).
Other studies have implicated that inhibition of another epigenetic regulator, histone deacetylase (HDAC) has antitumor effects in preclinical RMS models. A recent study designed a screen for epigenetic chemical probes to differentiate between super-enhancer driven transcription and constitutive transcription, revealing that the acetylation-axis is more important for the core regulatory TF circuit than the methylation-axis (90). Independent studies have reported the ability of HDAC inhibitors, entinostat, panobinostat, and vorinostat to delay tumor growth in xenograft models of RMS (95, 96). Several HDAC inhibitors are already approved for treatment of other cancers, but early clinical data show that HDAC inhibitors against solid tumors are far less effective than against hematological diseases, likely due to the pharmacokinetic differences in these two different tumor contexts (97). An ongoing clinical trial (NCT02780804) of the HDAC1/2/3 inhibitor entinostat in pediatric patients with advanced solid tumors is expected to shed new insight on HDAC inhibitors in RMS. Beyond disrupting transcriptional complexes to suppress the expression of key oncogenic genes, perturbation with HDAC inhibitors has also been shown to induce transcriptional chaos in cancer cells, driving cells into terminal differentiation or apoptotic cell death (98).
Taken together, these studies illustrate that FP RMS cells are differentially sensitive to the targeted disruption of super-enhancer complexes. Importantly, the knowledge that FP RMS cells are selectively dependent on epigenetic readers, writers, and erasers of the histone acetylation-axis can be exploited in the pharmacological disruption of these complexes (90). One important limitation to this approach is that epigenetic regulators also play a role in normal cellular transcriptional programs, meaning that there is a narrow therapeutic window and an increased risk of dose toxicity compared to an approach which directly targets the chimeric transcription factor.
A third approach is to target regulatory post-translational networks regulating the activity and stability of PAX-FOXO1. Many of these post-translational modification sites (phosphorylation, acetylation, methylation) in the fusion protein have been identified through high throughput mass spectrometry experiments or in vitro enzymatic screens performed in wildtype FOXO1 TF (99). However, only some of these sites have been functionally validated. Of these, most are phosphorylation sites which are targeted by common kinases, and many of these kinase are druggable by known kinase inhibitors. In general, there are two approaches for targeting the regulatory networks of PAX-FOXO1; (1) targeting the regulatory kinases that influence protein stability, and (2) targeting the regulatory kinases required for activation of the fusion protein.
Several phosphorylation sites are known to influence protein stability of the fusion product. In one study, a two-armed screening approach of kinome siRNA and small molecules identified that the kinase PLK1 stabilizes the fusion protein by phosphorylating S503. Inhibition of PLK1 directly led to ubiquitination of the fusion protein, followed by rapid proteasomal degradation (100). This evidence is consistent with the known function of PLK1 in the cell cycle, which is to coordinate entry into mitosis at the G2/M checkpoint. The expression levels of PAX3-FOXO1 and PLK1 both peak in G2 in a cell-cycle dependent manner. Upregulation of PAX3-FOXO1 transcripts and its stabilization by PLK1 phosphorylation permit the cell to progress past the G2/M checkpoint (101). Another upstream enzyme, acetyltransferase KAT2B (P/CAF) is known to modulate fusion protein stability by acetylating residues K426 and K429 (102). Other phosphorylation sites are known to control the transcriptional activity of PAX3-FOXO1, including the residues S201 (phosphorylated by the kinase GSK3β) (103), S205/S209 (by CK2) (104), and S430 (by CDK4) (105). Notably, several GSK3β inhibitors significantly suppressed transcriptional activity of PAX3-FOXO1, leading to inhibition of cellular proliferation and induction of apoptosis in ARMS cell lines (106). Given the availability of kinase inhibitors that have been studied in other human cancers, further functional validation of post-translational modifications of PAX3-FOXO1 and characterization of their respective kinases is a promising therapeutic strategy.
Receptor tyrosine kinases are a family of membrane-bound cell surface receptors which are aberrantly activated in many human malignancies. Constitutive activation of RTK signaling can reprogram numerous intracellular signaling pathways (metabolism, differentiation, apoptosis, growth) to promote tumor progression (Figure 2). In FP RMS, overexpressed PAX-FOXO1 targets include RTKs e.g., FGFR4, CXCR4, IGFR1, MET, and PDGFRA. In FN RMS, activating mutations in RTKs caused by molecular lesions can lead to hyperactive RTK signaling. Taken together, both FP and FN RMS could benefit from targeting RTK signaling. Generally speaking, the two strategies for targeting RTKs include small molecule kinase inhibitors and immunotherapy (monoclonal antibodies, CAR T). Several studies have shown the that RTK inhibitors can induce tumor regression in preclinical models (summarized in Table 1). Despite that many of these candidate targets are currently being evaluated in early phase I/II trials which recruit RMS patients, there has only been one clinical trial opened specifically for RMS patients. This trial (NCT03041701) is open to patients with relapsed or refractory RMS and its aim is to study the combination of the IFG-1R monoclonal antibody, ganitumab in combination with the SRC family kinase inhibitor, dasatinib. This treatment combination was based on preclinical evidence which reported that IGF-1R inhibition promotes a bypass resistance pathway through other kinases, such as the SRC family kinase YES (107) and ALK (108), suggesting that targeting multiple RTKs in combination is likely necessary to overcome resistance.
Despite the remarkable genetic and molecular differences between FP and FN RMS, the RTK/RAS/PI3K axis is commonly hijacked by both, suggesting that targeting this axis presents a more general therapeutic approach which could benefit a wide range of patients. A comprehensive genomic analysis of 147 RMS tumor samples by Shern et al. reported that the RTK/RAS/phosphoinositide 3-kinase (PI3K) pathway is altered in 93% of the tumors profiled, mostly dependent on the FGF and IGF receptor pathways (19). Fortunately, many of these pathway components (Table 1) can be targeted by clinically available therapeutics (19). The receptor tyrosine kinase, FGFR4 is frequently mutated and/or overactivated in RMS tumors, and recent work has implicated the role of FGF signaling in the evasion of apoptosis (109). Khan et al. identified potaninib from a panel of five tyrosine kinase inhibitors as a potent FGFR4 inhibitor that inhibits tumor growth in a RMS mouse model (110). The receptor tyrosine kinase, IGF2 is another potential target, given that either loss of imprinting or PAX-FOXO1-driven gene expression can induce the overexpression of IGF2 in rhabdomyosarcoma tumors (111). However, a recent clinical trial evaluating a monoclonal antibody against IGF-1R, R1507 in advanced sarcoma patients failed to achieve meaningful clinical responses to the therapy (79). Future studies should focus on elucidating potential resistance mechanisms to IGF-1R inhibition and identifying predictive biomarkers for IGF-1R inhibition sensitivity. A recent publication investigating a MEK inhibitor, trametinib in combination with IGF-1R inhibition showed a potent decrease in RMS cell viability and slowed tumor growth in xenograft models (112). Finally, a randomized phase II trial of bevacizumab (VEGF-A inhibitor) or temsirolimus (mTOR inhibitor) in combination with chemotherapy reported that the relapsed RMS patients who received temsirolimus achieved a better response (39). Although the RTK/RAS/PI3K axis is a common platform for therapeutic intervention in both FP and FN RMS, there is still a need to identify predictive biomarkers of response. Due to the extensive cross-talk across RTK signaling axes, combination therapies are likely needed to derive therapeutic benefit from this approach. Most of the data on targeting the RTK/RAS/PI3K axis in RMS comes from scattered preclinical reports, and none have demonstrated significant single agent activity in early phase clinical trials.
The knowledge that key developmental pathways, such as Hedgehog and Notch are commonly hijacked in a subset of RMS tumors can be exploited for therapeutic intervention. The Hedgehog (Hh) pathway is a highly conserved developmental pathway, which plays crucial roles in embryonic development, stem cell biology, and tissue homeostasis (113). In the canonical Hh pathway, repressive binding of Smoothened (Smo) to the transmembrane receptor Patched1 (PTCH1) maintains Hh signaling in an inactive state. Hh ligand binding to PTCH1 releases Smo, which becomes free to activate the Gli family of transcription factors (114). The link between Hh signaling and RMS was first described by Hahn et al. in a study of a Patched knockout mouse model that showed an ERMS phenotype. The relationship between Hh signaling dysregulation and RMS has subsequently been supported by several studies (115–118). Aberrant Hh signaling can be attributed to various germline mutations— loss of chromosomal region 9q22 containing PTCH in 33% of ERMS tumors (119, 120), loss of SUFU in 18% ERMS tumors (121), and/or genomic amplification of 12q13-15 containing the GLI1 gene in a small subset of ARMS tumors (116). Several studies have demonstrated that targeting the Hh pathway can inhibit tumor growth and impair tumor initiation in xenografted RMS models (118, 122–124). So far, preclinical evaluation of Smo inhibitors has been difficult to interpret due to the heterogeneity of response in preclinical models, depending on the RMS cell line and Smo inhibitor assessed (125, 126). Recent FDA approval of the Smo inhibitors, vismodegib, and sonidegib for the treatment of advanced basal cell carcinoma (BCC) and entry of other Smo inhibitors into clinical trials for pediatric medulloblastoma raise the possibility of expanding these inhibitors into clinical trials for pediatric RMS (127). However, because only a subset of RMS tumors appear to be sensitive to Smo inhibitors (such as ERMS tumors with a germline PTCH mutation), more robust predictive biomarkers for this therapy need to be established (126). Concerns over the side effects of Smo inhibitors, such as premature closure of bone growth plates may limit their use to only skeletally mature patients (128).
The Notch pathway regulates cell fate determination and stem cell differentiation during tissue development and maintenance. A recent publication used a zebrafish transgenic model of ERMS to identify intracellular NOTCH1 (ICN1) as an important regulator of balancing self-renewal and differentiation in ERMS (129). This work highlights the mechanistic underpinnings of the NOTCH1/SNAI1 pathway in driving self-renewal and blocking MEF2C regulated myogenic differentiation in RMS, describing a rationale for targeting the NOTCH1/SNAI1/MEF2C axis in ERMS. Another study highlighted that downregulation of Notch3 is sufficient to induce RMS cells into a terminal myogenic differentiation program, suggesting Notch3 as another potential therapeutic target (130). One group reported that the oncogenic signaling circuit between the Notch and YAP pathways drives stemness and tumorigenesis in ERMS, suggesting a rationale for co-targeting Notch and YAP (131). Given that there are clinically available gamma-secretase/Notch signaling pathway inhibitors (RO4929097), Notch1 inhibitors (MK0752), and Notch1 monoclonal antibodies (brontictuzumab, tarextumab), these drugs should be expanded into clinical trials for pediatric RMS.
In order to sustain chronic proliferation, cancer cells must meet the demanding needs imposed by energy metabolism and cellular division. The family of cyclin dependent kinases (CDK) which tightly control the cell cycle are frequently overactive in cancer cells, and have been extensively investigated as a molecular vulnerability in various human cancers. Several selective CDK4/6 inhibitors have already been approved for treatment in advanced stage breast cancer (132), and have been investigated in the context of RMS, as CDK4 is overexpressed in a subset of FP RMS tumors through the amplification of chromosomal region 12q13-q14 (133). CDK4 and its binding partner Cyclin D are required for progression through the G1/S checkpoint, and its overexpression allows cancer cells to adapt to the high proliferation rates needed to sustain tumorigenesis. One study showed that treatment of rhabdomyosarcoma cell lines and xenograft models with the clinically available CDK4/6 inhibitor, palbociclib is sufficient to inhibit proliferation by inducing cell cycle arrest at G1.
The DNA damage response (DDR) pathway plays a critical role in normal cellular homeostasis and its dysregulation is closely linked to increased mutation rates which drive oncogenesis. The poly(ADP-ribose) polymerases (PARP) belong to a family of DNA damage sensors which target the poly(ADP-ribose) polymerase by binding to single strand DNA breaks, recruiting other components of the homologous recombination (HR) repair machinery (134). PARP inhibitors (PARPi) are a well-established class of compounds capable of abrogating single strand break repair, which are converted into double strand breaks, subsequently leading deficient DNA repair and cell death (135). The understanding that inhibition of the DDR can be exploited in cancer cells to sensitize them to DNA lesions induced by chemotherapy or RT has been well-established in other cancers. However, the role of PARP inhibitors in RMS has not been extensively studied. A recent study showed that PARP inhibitors can sensitize RMS cell lines to ionizing radiation (IR), resulting in more potent cytotoxic effects compared to either modality alone (136). A related study reported that pretreatment with three PARP1 inhibitors (olaparib, iniparib, veliparib) was able to sensitize soft tissue sarcoma cells to radiation by inducing cell cycle arrest at the G2/M checkpoint (137). Taken together, these data point to the effective approach of combining PARP inhibitors with radiotherapy, sensitizing cancer cells to the ionizing radiation and tolerating lower doses of radiation. A recent preclinical study reported that the combination of olaparib and temozolomide (DNA-damaging agent) is a potent therapy for elimination of tumor cells in a human xenografted tumor zebrafish model of RMS. Both ERMS and ARMS were sensitive to combination treatment, suggesting the broad therapeutic potential of PARP inhibition in RMS (138). While combination therapy of olaparib and temozolomide is currently being investigated in phase II trials for Ewing's sarcoma, there are no open trials for this combination treatment in rhabdomyosarcoma. The strong preclinical evidence for the combination therapy of olaparib and temozolomide (138) warrants further investigation in clinical studies focused on pediatric RMS.
The Wee1 kinase arrests the cell cycle at the G2/M checkpoint for necessary DNA repair before entry into mitosis. Because cancer cells are more reliant on the G2/M checkpoint for DNA repair than normal cells due to G1/S DNA repair deficiencies, Wee1 inhibition can halt progression through the G2/M checkpoint and selectively induce apoptosis in cancer cells. One preclinical study reported that Wee1 kinase inhibitor AZD1775 possessed single-agent activity and synergized with conventional cytotoxic therapy (139). Wee1 inhibition against a background of cytotoxic drug-induced DNA damage results in mitotic catastrophe in tumor cells. A recent study used an integrated transcriptomic, epigenomic, and proteomic approach based on orthotopic patient-derived xenografts to validate and prioritize specific molecular vulnerabilities for high-risk RMS. The authors identified Wee1 kinase to be the most significant target for high-risk RMS, which led them to propose that patients with high-risk and recurrent RMS may benefit from combination therapy that includes AZD1775, irinotecan, and vincristine (140). Based on comprehensive preclinical testing data, patients with high-risk pediatric RMS were included in a phase I/II clinical trial (NCT02095132) of AZD1775 in combination with the chemotherapy agent irinotecan. A recent consensus article by American and European RMS leaders argued for the prioritization of the WEE1 inhibitor AZD1775 in combination with vincristine/irinotecan for the next clinical trial for patients with initially metastatic or recurrent RMS (141).
Direct modulation of apoptotic machinery has been exploited therapeutically in many human cancers, as most cancer cells are more sensitive to apoptotic induction than normal cells (142, 143). Because the Bcl-2 family of antiapoptotic proteins is required for cancer cell survival, inhibiting its function is one potential therapeutic approach. The development of BH3 mimetics, small molecule inhibitors which mimic the function BH3-only proteins by antagonizing the pro-survival function of anti-apoptotic Bcl-2 family, has recently gained traction as a therapeutic intervention in a number of human cancers. The Bcl-2 inhibitor, venetoclax is currently under evaluation in a trial for pediatric neuroblastoma and hematological malignancies (NCT03236857). Preuss et al. showed that ABT-737 (BH3 mimetic) and AZD8055 (mTOR inhibitor) cooperate synergistically to induce the mitochondrial apoptotic pathway in ERMS and ARMS cell lines (144). Another study showed that venetolax sensitized RMS cells to JNJ, an HDAC inhibitor (145). Taken together, these findings suggest that BH3 mimetics synergize with other targeted therapies by priming cancer cells to be sensitive to apoptotic induction.
A related strategy is the inhibition of X-linked Inhibitor of Apoptosis Proteins (XIAP), a family of proteins which block apoptosis by directly binding and inhibiting caspases, and which are frequently overexpressed in cancer cells. Smac mimetics are a class of molecules designed to mimic the endogenous antagonist of XIAPs, second mitochondrial activator of caspases (Smac). Upon activation of the mitochondrial apoptotic pathway, Smac is released into the cytosol, where it binds and neutralizes XIAPs, thereby allowing the caspase cascade to proceed. One study reported that Smac mimetics sensitized two RMS cell lines toward natural killer (NK) cell-mediated killing in an apoptotic-dependent manner (146). Currently, several Smac mimetics are being evaluated in early clinical trials for other human cancers but there are no open trials for RMS (147).
The recurrent 2;13 and 1;13 translocations in FP RMS encode for the PAX3-FOXO1 and PAX7-FOXO1 chimeric transcription factors, which are uniquely expressed in malignant cells but not in normal cells. This raises the possibility that the PAX-FOXO1 chimeric proteins can be leveraged as novel tumor-associated antigens in immunotherapy. The unique translocation breakpoint region may be processed, displayed on the tumor cell surface by major histocompatibility complex Class I (MHC-I) molecules, and targeted for killing by cytotoxic T cell lymphocytes (CTL) (148, 149). While data from preclinical mouse studies highlighted the vaccine-based approach as a promising strategy (149), a pilot clinical trial that generated vaccines by pulsing immature dendritic cells from breakpoint region peptides failed to improve patient outcomes (150). A separate study generated a human CTL line capable of lysing HLA-B7 rhabdomyosarcoma tumor cells (151). However, due to the limitation that this vaccine would only be applicable to the minority of the population who express the HLA-B7 allele, its clinical potential is limited. Attempts to generate vaccines targeted against other class I molecules including HLA-A1, HLA-A2, HLA-A3 are unlikely to be successful, as predicted by MHC-peptide binding algorithims (152). A pilot trial of consolidative immunotherapy (integration of immunotherapy into a multi-modal chemotherapeutic regimen), which administered vaccines of dendritic cells pulsed with breakpoint peptides reported positive outcomes in patients with high-risk pediatric ARMS, highlighting that vaccine-based approaches targeting the fusion protein could still be a valuable strategy. However, there is still room for improvement in refining this approach, such as using alternative approaches for generating more potent dendritic cells and identification of immunogenic peptides (153). New models that predict the immunogenicity of MHC-binding peptides from tumor transcriptomes can be leveraged to identify novel immunogenic peptides (154).
In the last two decades, monoclonal antibodies (mAbs) have become standard-of-care treatment of several human malignancies, but its role in RMS is not well established. Monocolonal antibodies can directly target cancer cells through a number of mechanisms, including inhibition of oncogenic signaling pathways, delivery of cytotoxic moieties to malignant cells, or induction of antibody-dependent cellular toxicity (155). One strategy has been the development of an Fc-enhanced anti-B7-H3 monoclonal antibody to target the B7-H3 protein upregulated on the surfaces of many pediatric solid tumor cells (156). Based on support from preclinical testing, a Phase I study was opened to evaluate enoblituzumab, an Fc-enhanced, humanized IgG1 monoclonal antibody specific for B7-H3 and engineered with an Fc domain with increased affinity for the activating receptor CD16A, thereby enhancing antibody dependent cellular cytotoxicity (ADCC) (157). Compared to classical mAb therapies which are less effective due to the existence of natural polymorphisms of FcγR, the strategy of fine tuning the Fc domain to optimize effector cell function should be considered in mAb-based approaches for RMS.
Early phase clinical studies of CAR T cell therapy for patients with pediatric solid tumors has demonstrated that while it can be safely administered, antitumor activity is limited (158). Targeting the human epidermal growth factor receptor 2 (HER2) expressed on tumor cells with the anti-HER2 antibody trastuzumab is an established therapy for the treatment of HER2-positive breast cancers (159, 160). Given that a subset of sarcomas express HER2 on tumor cell surfaces, targeting this receptor by immunotherapy is one potential strategy. Because HER2 expression levels are too low in sarcoma cells for a monoclonal antibody-based approach to be therapeutically actionable, HER2-positive sarcoma patients may be more sensitive to HER2-directed CAR T cell therapy (161). Following preclinical evaluation of a HER2-specific CAR containing a CD28.ζ signaling domain, Navai et al. (162) conducted a small phase I study (NCT00902044) evaluating the efficacy of HER2-targeted CAR T-cell therapy in combination with lymphodepletion chemotherapy in patients with advanced HER2-positive sarcoma. One pediatric RMS patient treated achieved a complete response for 12 months, but relapsed later (163). Another recent preclinical study of CAR T cells targeting B7-H3 (an immune checkpoint antigen) in xenograft models of various pediatric solid tumors, including RMS demonstrated that they could induce tumor regression in xenograft models (164). Other potential cell surface immune targets (FGFR4, SLC19A1, ACVR2A, EPHB4) were identified by Khan et al., in a study which used gene expression datasets to rank potential immune targets by their differential expression between 12 pediatric cancer tissues and normal tissue (165). Such gene expression approaches can be a useful strategy to generate a list of possible immune targets, but validation that these targets are actually expressed at the protein level on tumor cells (and not expressed on normal cells) is required before they are considered for CAR T therapy.
Compared to the success of CAR T cell therapy for treating B-cell-derived malignancies, the clinical efficacy of CAR T cell therapy to pediatric solid tumors has so far been limited. As reviewed by DeRenzo et al., treatment of solid pediatric tumors presents a unique set of challenges that must be carefully taken into consideration. Challenges of designing CAR T cell therapy for solid tumor malignancies include: heterogeneous antigen expression, limited migration of T cells to tumor sites, and an immunosuppressive, hostile microenvironment (158). In order to advance the field of CAR T cell therapy in pediatric solid tumors, there is a need for further optimization of CAR T cells at the preclinical stage, identification of immunogenic targets, and a technique to non-invasively monitor CAR T activity in patients in the clinical trial stage (158).
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) belong to a class of inhibitory receptors, which are negative regulators of T-cell immune function. In a tumor context, cancer cells have evolved mechanisms to co-opt this system, enabling cancer cells to evade immune surveillance. For example, when PD-1 receptor on T cells is engaged by its native ligand, PD-L1, T cell effector function is inhibited. Thus, tumor cells have evolved to express PD-L1 on their surfaces to deactivate T cell effector function, enabling them to evade destruction by the immune system. Release of the negative regulators at these checkpoints with checkpoint blockade therapy can induce a latent anti-tumor immune response (166). Given the clinical success of immune checkpoint inhibitors in metastatic melanoma and metastatic squamous non-small cell lung cancer, early-phase clinical trials are currently investigating their clinical efficacy in pediatric solid tumors (167–169).
Ipilimumab is a first-in-class anti-CTLA-4 immune checkpoint inhibitor approved for treatment of metastatic melanoma and was recently evaluated in a phase I clinical trial for the treatment of pediatric advanced solid tumors. Even though ipilimumab is safely tolerated in these patients, its efficacy as a monotherapy is limited. The authors of this study recommend future investigation of anti-CTLA-4 therapy in combination with other checkpoint inhibitors and/or immune-modifying agents (170). Targeting the PD-1/PD-L1 axis by mAb therapy (nivolumab, pembrolizumab) has a similar mechanism to CTLA-4 therapy, in which brakes are released on the anti-tumor activity of T cells. A pilot phase I trial (NCT01445379) of ipilimumab in children with advanced refractory solid tumors showed that no objective tumor regressions were achieved (170). Combination therapy of both CTLA-4 and PD-L1 inhibitors have demonstrated clinical efficacy in advanced melanoma, suggesting its consideration in pediatric cancers (171, 172). Initial data from an ongoing phase I/II trial (NCT02304458) evaluating nivolumab with/without ipilimumab in children with recurrent or refractory solid tumors or sarcomas showed that single-agent nivolumab has no activity, but in combination with ipilimumab demonstrated efficacy in certain sarcoma subtypes (173).
It is unlikely that immune checkpoint blockade in pediatric patients will achieve the same levels of response seen in adult patients. In adult cancers, a high mutational burden (more neoantigens) is correlated with a strong response to immune checkpoint inhibitors. Pediatric cancers are characterized by a low mutational burden, but it may be interesting to study whether RMS patients with higher mutational burdens (ERMS subtype) are more responsive to immune checkpoint therapy. Moreover, children do not yet have a fully developed immune system, which is required for optimal response to immunotherapy. Finally, the highly immunosuppressive microenvironment of pediatric sarcomas due to the presence of regulatory T cells and myeloid-derived suppressor cells limits the efficacy of immunotherapy.
In an ideal world with unlimited financial resources and time, drug development efforts would be focused on developing pediatric cancer-specific drugs, such as a direct inhibitor of the PAX-FOXO1 fusion protein uniquely expressed in FP RMS. In reality, few drugs have been developed specifically for childhood cancers due to a small market for a rare childhood diseases, and the ability to directly target PAX-FOXO1 does not appear to be within reach for some time. For now, most clinical trials opened for RMS exploit known drugs targeting common pathways which are dysregulated in other human cancers (Figure 2). Careful review of how targeted therapies have been successful in clinical trials for other human malignancies [e.g., immune checkpoint blockade in metastatic melanoma (168, 169, 172, 174, 175)] and systematic analysis of clinical trials of related families of childhood cancers [e.g., PARP inhibition in Ewing's sarcoma (138)] provide valuable insight into translating these therapies into a RMS tumor context.
In FP RMS, one strategy to target PAX-FOXO1 has been the selective disruption co-regulatory and post-translational networks of PAX-FOXO1 with clinically approved inhibitors. While such approaches have shown encouraging responses in preclinical studies, these targets have normal physiological functions unrelated to the fusion protein, so careful consideration must be given to off-target effects. On the other hand, FN RMS (lacking the chimeric transcription factor) harbors recurrent genetic alterations (RAS, FGFR4, IGFR1, CDK4, PI3KCA, etc.), conferring these tumors molecular dependencies which can be targeted by clinically available drugs (19).
Based on data from early clinical trials enrolling pediatric rhabdomyosarcoma patients, single-agent therapies do not appear to achieve durable responses. As with most targeted therapies, almost all cancers treated with a single-agent therapy will eventually acquire resistance and reduced sensitivity to subsequent lines of treatment. Of equal importance to identifying novel therapeutic targets, is the unequivocal need to better understand how RMS tumors develop resistance to these therapies. Given the recent surge of research completed to characterize the genomic, epigenomic, transcriptomic, and proteomic landscape of rhabdomyosarcoma (117, 140), we now have a comprehensive list of therapeutic vulnerabilities, which are currently under preclinical and clinical evaluation. However, narrowly focusing on identifying these targets is inadequate, and a commensurate amount of effort ought to be given to studying mechanisms of resistance to targeted therapy. For patients diagnosed with metastatic rhabdomyosarcoma, insufficient local therapy options and incomplete eradication of occult microscopic residual disease are the most common causes of treatment failure (176). These new targeted therapies and immunotherapies hold promise for patients with metastatic or recurrent RMS, but only insofar as we concurrently advance our understanding of how to overcome inevitable drug resistance. While the identification of novel therapeutic vulnerabilities for RMS is gaining significant traction, it is equally important for clinicians to remain one step ahead by being able to anticipate resistance mechanisms and to identify strategies to overcome resistance accordingly.
Even the most successful targeted therapies that have been approved for the treatment of human cancers fail to completely eliminate residual disease in patients, leading to eventual relapse despite an initial response. A bulk solid tumor, such as rhabdomyosarcoma is composed of a heterogeneous population of cells which evolves to be more genetically unstable and complex as selective pressure is applied during drug treatment (177). Pediatric cancers are characterized by dynamic chromosomal instability, which can result in loss of chromosomal segments or copy-number alterations, contributing to the genetic heterogeneity of the tumor mass. A small subpopulation of drug-resistant tumor cells (harboring a genetic alteration conferring a survival advantage) present at initial treatment may persist and expand, resulting in eventual failure to eliminate residual tumor mass. Generally speaking, the two known strategies for overcoming drug resistance are intermittent dosing schedules and combination therapies. Intermittent dosing relies on the principle that periods of interspersed drug-withdrawal between drug-treatments can restore drug sensitivity by allowing drug-sensitive subpopulations to repopulate the tumor mass. Mathematical modeling can be used to predict the optimal dosing schedules which can sustain drug sensitivity, as demonstrated by a study which used an algorithm to design a tyrosine kinase inhibitor (TKI) dosing schedule for treatment of non-small cell lung cancers (178). These modeling platforms should be integrated into the design of future clinical trials for TKIs in RMS, where TKIs such as IGF-1R have demonstrated limited efficacy so far in early phase clinical trials (79, 179). A second approach is to anticipate which pre-existing subclonal populations will be drug-resistant, identify molecular vulnerabilities for these drug-resistant subclones, and design combination therapies focused on eradicating the maximal percentage of the heterogeneous tumor mass during first-line therapy. As an example, one study in non-small lung cell cancers showed that drug-resistant tumor cells with acquired resistance conferred by the EGFRT790M mutation could be re-sensitized to EGFR TKIs by co-targeting them with navitoclax, an inhibitor of BCL-2 and BCL-XL (180). With the expansion of CRISPR-Cas9-based gene editing systems, we now have the ability to conduct unbiased genome-wide screens for therapeutic vulnerabilities in RMS to identify synthetic lethal combinations (181, 182).
Initial efforts to bring immunotherapies designed for adults into rhabdomyosarcoma pediatric trials has been met with limited success. These immunotherapies fail to translate because pediatric solid tumors are characterized by a lower mutational burden and a non-inflammatory tumor microenvironment (defined by very few infiltrating T cells and low levels of chemokines/cytokines) (183). Although further study is needed to elucidate the landscape of the RMS tumor microenvironment, pediatric solid tumors are generally regarded as less “inflamed” than adult tumors (184). As recently reviewed here (183), patients with “hot” tumor microenvironments (immune infiltrated) respond better to immune checkpoint blockade, whereas patients with “cold” tumor microenvironments (immune non-infiltrated) are better suited for adoptive T cell therapy. Further characterization of the tumor microenvironment of rhabdomyosarcoma would help guide the choice of an immunotherapeutic approach.
As whole-genome and transcriptome sequencing of rhabdomyosarcoma tumors has revealed, the genomic diversity of this disease requires a personalized (genotype-guided) approach to therapy. The presence or absence of the PAX-FOXO1 fusion gene reflects the vast genetic and molecular differences between FP and FN tumors, and this prognostic marker should guide the design of therapies specific to each subclass (37). Multi-region sampling of the tumor, single-cell sequencing, autopsy analysis, and longitudinal analysis of liquid biopsy samples can be used to reconstruct a holistic picture of tumor evolution in response to drug treatment, which can guide clinical decisions to counteract potential drug resistance (177). The gap between the observed efficacy of targeted therapies in preclinical models of RMS and the marginal improvements in patient outcomes observed in clinical trials has forced a reconsideration of our approach. Only when the mechanisms of drug resistance are understood will these new treatments be effective for children with metastatic or recurrent RMS, for which intensive chemotherapeutic regimens have already been exhausted.
Our current understanding of the genomic and molecular landscape of rhabdomyosarcoma has equipped us with a valuable list of potential targets for targeted therapy and immunotherapy based approaches. Effective clinical translation of these agents remains an ongoing challenge, underscoring the need to elucidate why tumors eventually acquire resistance to targeted therapy. Given the small number of patients available for enrollment in clinical trials, it is necessary to prioritize which therapeutic targets and combination therapies will have the most clinical benefit for the greatest number of patients. The marked heterogeneity across RMS subtypes (and specifically within the ERMS subtype) means more personalized, genotype-guided approaches are needed to inform treatment decisions. As such, there is a need to identify reliable and objective biomarkers to determine the most effective therapy for each patient. With the inadequate outcomes observed in early RMS clinical trials of targeted therapies and immunotherapy, the conversation must shift toward how we can close the gap between the preclinical and clinical efficacy of these therapies.
CC, HD, MS, and AH contributed to the conception and design of the review. CC wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
AH was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)−398299703 and the Wilhelm Sander Stiftung. AH was a participant in the BIH-Charité Clinical Scientist Program funded by the Charité—Universitätsmedizin Berlin and the Berlin Institute of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Dasgupta R, Fuchs J, Rodeberg D. Rhabdomyosarcoma. Semin Pediatr Surg. (2016) 25:276–83. doi: 10.1053/j.sempedsurg.2016.09.011
2. Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer. (2009) 115:4218–26. doi: 10.1002/cncr.24465
3. Drummond CJ, Hatley ME. A Case of mistaken identity: rhabdomyosarcoma development from endothelial progenitor cells. Mol Cell Oncol. (2018) 5:e1448246. doi: 10.1080/23723556.2018.1448246
4. Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Therapy. (2002) 1:97–104. doi: 10.4161/cbt.51
5. Whittle SB, Hicks MJ, Roy A, Vasudevan SA, Reddy K, Venkatramani R. Congenital spindle cell rhabdomyosarcoma. Pediatr Blood Cancer. (2019) 66:e27935. doi: 10.1002/pbc.27935
6. Xia SJ, Rajput P, Strzelecki DM, Barr FG. Analysis of genetic events that modulate the oncogenic and growth suppressive activities of the PAX3-FKHR fusion oncoprotein. Lab Investig. (2007) 87:318. doi: 10.1038/labinvest.3700521
7. Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. (2001) 20:5736–46. doi: 10.1038/sj.onc.1204599
8. Mercado GE, Barr FG. Fusions involving PAX and FOX genes in the molecular pathogenesis of alveolar rhabdomyosarcoma: recent advances. Curr Mol Med. (2007) 7:47–61. doi: 10.2174/156652407779940440
9. Olanich ME, Barr FG. A call to ARMS: targeting the PAX3-FOXO1 gene in alveolar rhabdomyosarcoma. Expert Opin Ther Targets. (2013) 17:607–23. doi: 10.1517/14728222.2013.772136
10. Davis RJ, Bennicelli JL, Macina RA, Nycum LM, Biegel JA, Barr FG. Structural characterization of the FKHR gene and its rearrangement in alveolar rhabdomyosarcoma. Hum Mol Genet. (1995) 4:2355–62. doi: 10.1093/hmg/4.12.2355 Available online at: https://cancerres.aacrjournals.org/content/62/16/4704.long
11. Barr FG, Qualman SJ, Macris MH, Melnyk N, Lawlor ER, Strzelecki DM, et al. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res. (2002) 62:4704–10. Available online at: https://cancerres.aacrjournals.org/content/62/16/4704.long
12. Wachtel M, Dettling M, Koscielniak E, Stegmaier S, Treuner J, Simon-Klingenstein K, et al. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res. (2004) 64:5539–45. doi: 10.1158/0008-5472.CAN-04-0844
13. Duan F, Smith LM, Gustafson DM, Zhang C, Dunlevy MJ, Gastier-Foster JM, et al. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children's Oncology Group. Genes Chromosomes Cancer. (2012) 51:662–74. doi: 10.1002/gcc.21953
14. Davicioni E, Anderson MJ, Finckenstein FG, Lynch JC, Qualman SJ, Shimada H, et al. Molecular classification of rhabdomyosarcoma–genotypic and phenotypic determinants of diagnosis: a report from the Children's Oncology Group. Am J Pathol. (2009) 174:550–64. doi: 10.2353/ajpath.2009.080631
15. Davicioni E, Anderson JR, Buckley JD, Meyer WH, Triche TJ. Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: a report from the children's oncology group. J Clin Oncol. (2010) 28:1240–6. doi: 10.1200/JCO.2008.21.1268
16. Williamson D, Missiaglia E, de Reynies A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. (2010) 28:2151–8. doi: 10.1200/JCO.2009.26.3814
17. Fredericks WJ, Ayyanathan K, Herlyn M, Friedman JR, Rauscher FJ III An engineered PAX3-KRAB transcriptional repressor inhibits the malignant phenotype of alveolar rhabdomyosarcoma cells harboring the endogenous PAX3-FKHR oncogene. Mol Cell Biol. (2000) 20:5019–31. doi: 10.1128/MCB.20.14.5019-5031.2000
18. Chen L, Shern JF, Wei JS, Yohe ME, Song YK, Hurd L, et al. Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet. (2015) 11:e1005075. doi: 10.1371/journal.pgen.1005075
19. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. (2014) 4:216–31. doi: 10.1158/2159-8290.CD-13-0639
20. Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. Children's research Hospital–Washington University Pediatric Cancer Genome, targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. (2013) 24:710–24. doi: 10.1016/j.ccr.2013.11.002
21. Dantonello TM, Int-Veen C, Schuck A, Seitz G, Leuschner I, Nathrath M, et al. Survival following disease recurrence of primary localized alveolar rhabdomyosarcoma. Pediatr Blood Cancer. (2013) 60:1267–73. doi: 10.1002/pbc.24488
22. Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer. (2012) 59:5–10. doi: 10.1002/pbc.24118
23. Ferrari, Trama A, De Paoli A, Bergeron C, Merks JHM, Jenney M, et al. Access to clinical trials for adolescents with soft tissue sarcomas: enrollment in European pediatric Soft tissue sarcoma Study Group (EpSSG) protocols. Pediatr Blood Cancer. (2017) 64:e26348. doi: 10.1002/pbc.26348
24. Hawkins DS, Spunt SL, Skapek SX, COG Soft Tissue Sarcoma Committee. Children's oncology Group's 2013 blueprint for research: soft tissue sarcomas. Pediatr Blood Cancer. (2013) 60:1001–8. doi: 10.1002/pbc.24435
25. Crist WM, Anderson JR, Meza JL, Fryer C, Raney RB, Ruymann FB, et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol. (2001) 19:3091–102. doi: 10.1200/JCO.2001.19.12.3091
26. Arndt CA, Stoner JA, Hawkins DS, Rodeberg DA, Hayes-Jordan AA, Paidas CN, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children's oncology group study D9803. J Clin Oncol. (2009) 27:5182–8. doi: 10.1200/JCO.2009.22.3768
27. Stevens MC, Rey A, Bouvet N, Ellershaw C, Flamant F, Habrand JL, et al. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology–SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol. (2005) 23:2618–28. doi: 10.1200/JCO.2005.08.130
28. Oberlin O, Rey A, Sanchez de Toledo J, Martelli H, Jenney MEM, Scopinaro M, et al. Randomized comparison of intensified six-drug versus standard three-drug chemotherapy for high-risk nonmetastatic rhabdomyosarcoma and other chemotherapy-sensitive childhood soft tissue sarcomas: long-term results from the international society of pediatric oncology MMT95 Study. J Clin Oncol. (2012) 30:2457–65. doi: 10.1200/JCO.2011.40.3287
29. Oberlin O, Rey A, Lyden E, Bisogno GM, Stevens CG, Meyer WH, et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European Cooperative Groups. J Clin Oncol. (2008) 26:2384–9. doi: 10.1200/JCO.2007.14.7207
30. Meza JL, Anderson J, Pappo AS, Meyer WH. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The Children's Oncology Group. J Clin Oncol. (2006) 24:3844–51. doi: 10.1200/JCO.2005.05.3801
31. Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol. (2012) 30:1670–7. doi: 10.1200/JCO.2011.38.5591
32. Stegmaier S, Poremba C, Schaefer KL, Leuschner I, Kazanowska B, Bekassy AN, et al. Prognostic value of PAX-FKHR fusion status in alveolar rhabdomyosarcoma: a report from the cooperative soft tissue sarcoma study group (CWS). Pediatr Blood Cancer. (2011) 57:406–14. doi: 10.1002/pbc.22958
33. Barr FG, Smith LM, Lynch JC, Strzelecki D, Parham DM, Qualman SJ, et al. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Intergroup Rhabdomyosarcoma Study-III trial: a report from the Children's Oncology Group. J Mol Diagn. (2006) 8:202–8. doi: 10.2353/jmoldx.2006.050124
34. Rosenberg AR, Skapek SX, Hawkins DS. The inconvenience of convenience cohorts: rhabdomyosarcoma and the PAX-FOXO1 biomarker. Cancer Epidemiol Biomarkers Prev. (2012) 21:1012–8. doi: 10.1158/1055-9965.EPI-12-0207
35. Williamson D, Missiaglia E, Chisholm J, Shipley J. Inconvenience of convenience cohorts–letter. Cancer Epidemiol Biomarkers Prev. (2012) 21:1388. doi: 10.1158/1055-9965.EPI-12-0724
36. Selfe J, Olmos D, Al-Saadi R, Thway K, Chisholm J, Kelsey A, et al. Impact of fusion gene status versus histology on risk-stratification for rhabdomyosarcoma: retrospective analyses of patients on UK trials. Pediatr Blood Cancer. (2017) 64:e26386. doi: 10.1002/pbc.26386
37. Hibbitts E, Chi Y, Hawkins DS, Barr FG, Bradley JA, Dasgupta R, et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: a report from the Children's Oncology Group. Cancer Med. (2019) 8:6437–48. doi: 10.1002/cam4.2504
38. Skapek SX, Anderson J, Barr FG, Bridge JA, Gastier-Foster JM, Parham DM, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children's oncology group report. Pediatric Blood Cancer. (2013) 60:1411–7. doi: 10.1002/pbc.24532
39. Mascarenhas L, Meyer WH, Lyden E, Rodeberg DA, Indelicato DJ, Linardic CM, et al. Randomized phase II trial of bevacizumab and temsirolimus in combination with vinorelbine (V) and cyclophosphamide (C) for first relapse/disease progression of rhabdomyosarcoma (RMS): A report from the Children's Oncology Group (COG). J Clin Oncol. (2014) 32:10003. doi: 10.1200/jco.2014.32.15_suppl.10003
40. Gallego S, Zanetti I, Orbach D, Ranchere D, Shipley J, Zin A, et al. Fusion status in patients with lymph node-positive (N1) alveolar rhabdomyosarcoma is a powerful predictor of prognosis: Experience of the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG). Cancer. (2018) 124:3201–9. doi: 10.1002/cncr.31553
41. Rudzinski ER, Teot LA, Anderson JR, Moore J, Bridge JA, Barr FG, et al. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Am J Clin Pathol. (2013) 140:82–90. doi: 10.1309/AJCPA1WN7ARPCMKQ
42. Arnold MA, Anderson JR, Gastier-Foster JM, Barr FG, Skapek SX, Hawkins DS, et al. Histology, fusion status, and outcome in alveolar rhabdomyosarcoma with low-risk clinical features: a report from the Children's Oncology Group. Pediatr Blood Cancer. (2016) 63:634–9. doi: 10.1002/pbc.25862
43. Maurer HM, Gehan EA, Beltangady M, Crist W, Dickman PS, Donaldson SS, et al. The intergroup rhabdomyosarcoma study-II. Cancer. (1993) 71:1904–22.
44. Maurer HM, Crist W, Lawrence W, Ragab AH, Raney RB, Webber B, et al. The intergroup rhabdomyosarcoma study-I. A final report. Cancer. (1988) 61:209–20.
45. Koscielniak E, Harms D, Henze G, Jürgens H, Gadner H, Herbst M, et al. Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol. (1999) 17:3706–19. doi: 10.1200/JCO.1999.17.12.3706
46. Arndt CA. Role of doxorubicin in rhabdomyosarcoma: is the answer knowable? Am Soc Clin Oncol Educ Book. (2012) 621–23. doi: 10.14694/EdBook_AM.2012.32.621
47. Sandler E, Lyden E, Ruymann F, Maurer H, Wharam M, Parham D, et al. Efficacy of ifosfamide and doxorubicin given as a phase II “window” in children with newly diagnosed metastatic rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Study Group*. Med Pediatr Oncol. (2001) 37:442–8. doi: 10.1002/mpo.1227
48. Crist W, Gehan EA, Ragab AH, Dickman PS, Donaldson SS, Fryer C, et al. The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol. (1995) 13:610–30. doi: 10.1200/JCO.1995.13.3.610
49. Bisogno G, Jenney M, Bergeron C, Gallego Melcón S, Ferrari A, Oberlin O, et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): a multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. (2018) 19:1061–71. doi: 10.1016/S1470-2045(18)30337-1
50. Kenney LB, Laufer MR, Grant FD, Grier H, Diller L. High risk of infertility and long term gonadal damage in males treated with high dose cyclophosphamide for sarcoma during childhood. Cancer. (2001) 91:613–21. doi: 10.1002/1097-0142(20010201)91:3<613::AID-CNCR1042>3.0.CO;2-R
51. Walterhouse DO, Pappo AS, Meza JL, Breneman JC, Hayes-Jordan AA, Parham DM, et al. Shorter-duration therapy using vincristine, dactinomycin, and lower-dose cyclophosphamide with or without radiotherapy for patients with newly diagnosed low-risk rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J Clin Oncol. (2014) 32:3547–52. doi: 10.1200/JCO.2014.55.6787
52. Eaton BR, McDonald MW, Kim S, Marcus RB Jr, Sutter AL, Chen Z, et al. Radiation therapy target volume reduction in pediatric rhabdomyosarcoma. Cancer. (2013) 119:1578–85. doi: 10.1002/cncr.27934
53. Ladra MM, Edgington SK, Mahajan A, Grosshans D, Szymonifka J, Khan F, et al. A dosimetric comparison of proton and intensity modulated radiation therapy in pediatric rhabdomyosarcoma patients enrolled on a prospective phase II proton study. Radiother Oncol. (2014) 113:77–83. doi: 10.1016/j.radonc.2014.08.033
54. McDonald MW, Esiashvili N, George BA, Katzenstein HM, Olson TA, Rapkin LB, et al. Intensity-modulated radiotherapy with use of cone-down boost for pediatric head-and-neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys. (2008) 72:884–91. doi: 10.1016/j.ijrobp.2008.01.058
55. Saltzman AF, Cost NG. Current treatment of pediatric bladder and prostate rhabdomyosarcoma. Curr Urol Rep. (2018) 19:11. doi: 10.1007/s11934-018-0761-8
56. Terezakis SA, Wharam MD. Radiotherapy for rhabdomyosarcoma: indications and outcome. Clin Oncol. (2013) 25:27–35. doi: 10.1016/j.clon.2012.07.009
57. Bisogno G, De Salvo GL, Bergeron C, Jenney M, Merks HMJ, Minard-Colin V, et al. Maintenance low-dose chemotherapy in patients with high-risk (HR) rhabdomyosarcoma (RMS): a report from the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG). J Clin Oncol. (2018) 36:LBA2. doi: 10.1200/JCO.2018.36.18_suppl.LBA2
58. Bisogno G, De Salvo GL, Bergeron C, Gallego Melcón S, Merks JH, Kelsey A, et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. (2019) 20:1566–75. doi: 10.1016/S1470-2045(19)30617-5
59. Meyer WH. Maintenance chemotherapy in rhabdomyosarcoma: the new standard of care. Lancet Oncol. (2019) 20:1476–77. doi: 10.1016/S1470-2045(19)30618-7
60. Breneman JC, Lyden E, Pappo AS, Link MP, Anderson JR, Parham DM, et al. Prognostic Factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma—a report from the intergroup rhabdomyosarcoma Study IV. J Clin Oncol. (2003) 21:78–84. doi: 10.1200/JCO.2003.06.129
61. Carli M, Colombatti R, Oberlin O, Bisogno G, Treuner J, Koscielniak E, et al. European Intergroup Studies (MMT4–89 and MMT4–91) on childhood metastatic rhabdomyosarcoma: final results and analysis of prognostic factors. J Clin Oncol. (2004) 22:4787–94. doi: 10.1200/JCO.2004.04.083
62. Weigel BJ, Breitfeld PP, Hawkins D, Crist WM, Baker KS. Role of high-dose chemotherapy with hematopoietic stem cell rescue in the treatment of metastatic or recurrent rhabdomyosarcoma. J Pediatr Hematol Oncol. (2001) 23:272–6. doi: 10.1097/00043426-200106000-00007
63. Weigel BJ, Lyden E, Anderson JR, Meyer WH, Parham DM, Rodeberg DA, et al. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: a report from the Children's Oncology Group. J Clin Oncol. (2016) 34:117–22. doi: 10.1200/JCO.2015.63.4048
64. Grier HE, Krailo MD, Tarbell NJ, Link MPC, Fryer JH, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's Sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. (2003) 348:694–701. doi: 10.1056/NEJMoa020890
65. Harris MB, Gieser P, Goorin AM, Ayala A, Shochat SJ, Ferguson WS, et al. Treatment of metastatic osteosarcoma at diagnosis: a Pediatric Oncology Group Study. J Clin Oncol. (1998) 16:3641–3648. doi: 10.1200/JCO.1998.16.11.3641
66. Marina NM, Pappo AS, Parham DM, Cain AM, Rao BN, Poquette CA, et al. Chemotherapy dose-intensification for pediatric patients with Ewing's family of tumors and desmoplastic small round-cell tumors: a feasibility study at St. Jude Children's Research Hospital. J Clin Oncol. (1999) 17:180. doi: 10.1200/JCO.1999.17.1.180
67. Haghiri S, Fayech C, Dufour C, Pasqualini C, Bolle S, Boumaraf A, et al. Long-term health status of high-risk neuroblastoma survivors treated with high-dose chemotherapy and hematopoietic stem cell transplantation. J Clin Oncol. (2019) 37:10054. doi: 10.1200/JCO.2019.37.15_suppl.10054
68. Merker M, Meister MT, Rettinger E, Jarisch A, Soerensen J, Willasch A, et al. Haploidentical allogeneic hematopoietic stem cell transplantation in patients with high-risk soft tissue sarcomas: results of a single-center prospective trial. Bone Marrow Transpl. (2018) 53:891–4. doi: 10.1038/s41409-018-0088-6
69. Klingebiel T, Boos J, Beske F, Hallmen E, Int-Veen C, Dantonello T, et al. Treatment of children with metastatic soft tissue sarcoma with oral maintenance compared to high dose chemotherapy: report of the HD CWS-96 trial. Pediatr Blood Cancer. (2008) 50:739–45. doi: 10.1002/pbc.21494
70. Ben Arush M, Minard-Colin V, Mosseri V, Defachelles AS, Bergeron C, Algret N, et al. Does aggressive local treatment have an impact on survival in children with metastatic rhabdomyosarcoma? Eur J Cancer. (2015) 51:193–201. doi: 10.1016/j.ejca.2014.11.009
71. Mohan AC, Venkatramani R, Okcu MF, Nuchtern JG, Vasudevan SA, Mahajan A, et al. Local therapy to distant metastatic sites in stage IV rhabdomyosarcoma. Pediatr Blood Cancer. (2018) 65:e26859. doi: 10.1002/pbc.26859
72. Khanna C, Fan TM, Gorlick R, Helman LJ, Kleinerman ES, Adamson PC, et al. Toward a drug development path that targets metastatic progression in osteosarcoma. Clin Cancer Res. (2014) 20:4200–9. doi: 10.1158/1078-0432.CCR-13-2574
73. Hoshino, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. (2015) 527:329–35. doi: 10.1038/nature15756
74. Pappo AS, Anderson JR, Crist WM, Wharam MD, Breitfeld PP, Hawkins D, et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: a report from the Intergroup rhabdomyosarcoma Study Group. J Clin Oncol. (1999) 17:3487–93. doi: 10.1200/JCO.1999.17.11.3487
75. Chisholm JC, Marandet J, Rey A, Scopinaro M, de Toledo JS, Merks JHM, et al. Prognostic factors after relapse in nonmetastatic rhabdomyosarcoma: a nomogram to better define patients who can be salvaged with further therapy. J Clin Oncol. (2011) 29:1319–25. doi: 10.1200/JCO.2010.32.1984
76. Mascarenhas L, Lyden ER, Breitfeld PP, Walterhouse DO, Donaldson SS, Paidas CN, et al. Randomized phase II window trial of two schedules of irinotecan with vincristine in patients with first relapse or progression of rhabdomyosarcoma: a report from the Children's Oncology Group. J Clin Oncol. (2010) 28:4658–63. doi: 10.1200/JCO.2010.29.7390
77. Arndt CAS, Hawkins DS, Meyer WH, Sencer SF, Neglia JP, Anderson JR. Comparison of results of a pilot study of alternating vincristine/doxorubicin/cyclophosphamide and etoposide/ifosfamide with IRS-IV in intermediate risk rhabdomyosarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer. (2008) 50:33–6. doi: 10.1002/pbc.21093
78. Kim, Widemann BC, Krailo M, Jayaprakash N, Fox E, Weigel B, et al. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: a report from the Children's Oncology Group. Pediatr Blood Cancer. (2015) 62:1562–6. doi: 10.1002/pbc.25548
79. Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn CW, Chugh R, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: Results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. (2014) 120:2448–56. doi: 10.1002/cncr.28728
80. Salesse S, Verfaillie CM. BCR/ABL: from molecular mechanisms of leukemia induction to treatment of chronic myelogenous leukemia. Oncogene. (2002) 21:8547–859. doi: 10.1038/sj.onc.1206082
81. Sasaki T, Rodig SJ, Chirieac LR, Jänne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. (2010) 46:1773–80. doi: 10.1016/j.ejca.2010.04.002
82. Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the “undruggable” cancer targets. Nat Rev Cancer. (2017) 17:502. doi: 10.1038/nrc.2017.36
83. Bushweller JH Targeting transcription factors in cancer — from undruggable to reality. Nat Rev Cancer. (2019) 19:611–24. doi: 10.1038/s41568-019-0196-7
84. Rengaswamy V, Zimmer D, Süss R, Rössler J, RGD liposome-protamine-siRNA (LPR) nanoparticles targeting PAX3-FOXO1 for alveolar rhabdomyosarcoma therapy. J Control Release. (2016) 235:319–27. doi: 10.1016/j.jconrel.2016.05.063
85. Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew Chem Int Ed Engl. (2016) 55:807–10. doi: 10.1002/anie.201507634
86. Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. (2015) 11:611–7. doi: 10.1038/nchembio.1858
87. Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. (2015) 348:1376–81. doi: 10.1126/science.aab1433
88. Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet. (2018) 50:515–23. doi: 10.1038/s41588-018-0044-9
89. Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, et al. PAX3–FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. (2017) 7:884. doi: 10.1158/2159-8290.CD-16-1297
90. Gryder BE, Wu L, Woldemichael GM, Pomella S, Quinn TR, Park PMC, et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat Commun. (2019) 10:3004. doi: 10.1038/s41467-019-11046-7
91. Lovén J, Heather Hoke A, Charles Lin Y, Lau A, David Orlando A, Christopher Vakoc R, et al. selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. (2013) 153:320–34. doi: 10.1016/j.cell.2013.03.036
92. Matheson CJ, Backos DS, Reigan P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol Sci. (2016) 37:872–81. doi: 10.1016/j.tips.2016.06.006
93. Böhm M, Wachtel M, Marques JG, Streiff N, Laubscher D, Nanni P, et al. Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J Clin Investig. (2016) 126:4237–49. doi: 10.1172/JCI85057
94. Kishtagari Ng KP, Jarman C, Tiwari AD, Phillips JG, Schuerger C, et al. A first-in-class inhibitor of ISWI-Mediated (ATP-Dependent) transcription repression releases terminal-differentiation in AML cells while sparing normal hematopoiesis. Blood. (2018) 132:216. doi: 10.1182/blood-2018-99-119311
95. Abraham J, Nuñez-Álvarez Y, Hettmer S, Carrió E, Chen HIH, Nishijo K, et al. Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Devel. (2014) 28:1578–1591. doi: 10.1101/gad.238733.114
96. Hedrick E, Crose L, Linardic CM, Safe S. Histone deacetylase inhibitors inhibit rhabdomyosarcoma by reactive oxygen species–dependent targeting of specificity protein transcription factors. Mol Cancer Therap. (2015) 14:2143. doi: 10.1158/1535-7163.MCT-15-0148
97. McClure JJ, Li X, Chou CJ. Chapter Six: Advances and challenges of HDAC inhibitors in cancer therapeutics. In: Tew KD, Fisher PB, editors. Advances in Cancer Research. Cambridge, MA: Academic Press (2018). p. 183–211. doi: 10.1016/bs.acr.2018.02.006
98. Gonda TJ, Ramsay RG. Directly targeting transcriptional dysregulation in cancer. Nat Rev Cancer. (2015) 15:686. doi: 10.1038/nrc4018
99. Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. (2015) 43:D512–20. doi: 10.1093/nar/gku1267
100. Thalhammer V, Lopez-Garcia LA, Herrero-Martin D, Hecker R, Laubscher D, Gierisch ME, et al. PLK1 phosphorylates PAX3-FOXO1, the Inhibition of Which Triggers Regression of Alveolar Rhabdomyosarcoma. Cancer Res. (2015) 75:98. doi: 10.1158/0008-5472.CAN-14-1246
101. Kikuchi K, Hettmer S, Aslam MI, Michalek JE, Laub W, Wilky BA, et al. Cell-Cycle dependent expression of a translocation-mediated fusion oncogene mediates checkpoint adaptation in rhabdomyosarcoma. PLOS Genetics. (2014) 10:e1004107. doi: 10.1371/journal.pgen.1004107
102. Bharathy N, Suriyamurthy S, Rao VK, Ow JR, Lim HJ, Chakraborty P, et al. P/CAF mediates PAX3–FOXO1-dependent oncogenesis in alveolar rhabdomyosarcoma. J Pathol. (2016) 240:269–81. doi: 10.1002/path.4773
103. Loupe JM, Miller PJ, Ruffin DR, Stark MW, Hollenbach AD. Inhibiting phosphorylation of the oncogenic PAX3-FOXO1 reduces alveolar rhabdomyosarcoma phenotypes identifying novel therapy options. Oncogenesis. (2015) 4:e145. doi: 10.1038/oncsis.2015.2
104. Dietz KN, Miller PJ, Iyengar AS, Loupe JM, Hollenbach AD. Identification of serines 201 and 209 as sites of Pax3 phosphorylation and the altered phosphorylation status of Pax3-FOXO1 during early myogenic differentiation. Int J Biochem Cell Biol. (2011) 43:936–45. doi: 10.1016/j.biocel.2011.03.010
105. Liu L, Wu J, Ong SS, Chen T. Cyclin-dependent kinase 4 phosphorylates and positively regulates PAX3-FOXO1 in human alveolar rhabdomyosarcoma cells. PLoS ONE. (2013) 8:e58193. doi: 10.1371/journal.pone.0058193
106. Zeng FY, Dong H, Cui J, Liu L, Chen T. Glycogen synthase kinase 3 regulates PAX3–FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells. Biochem Biophys Res Commun. (2010) 391:1049–55. doi: 10.1016/j.bbrc.2009.12.017
107. Wan X, Yeung C, Heske C, Mendoza A, Helman LJ. IGF-1R inhibition activates a YES/SFK bypass resistance pathway: rational basis for co-targeting IGF-1R and Yes/SFK kinase in rhabdomyosarcoma. Neoplasia. (2015) 17:358–66. doi: 10.1016/j.neo.2015.03.001
108. van Gaal JC, Roeffen MHS, Flucke UE, van der Laak WM, van der Heijden G, de Bont JM, et al. Simultaneous targeting of insulin-like growth factor-1 receptor and anaplastic lymphoma kinase in embryonal and alveolar rhabdomyosarcoma: a rational choice. Eur J Cancer. (2013) 49:3462–70. doi: 10.1016/j.ejca.2013.06.022
109. Wachtel M, Rakic J, Okoniewski M, Bode P, Niggli F, et al. FGFR4 signaling couples to Bim and not Bmf to discriminate subsets of alveolar rhabdomyosarcoma cells. Int J Cancer. (2014) 135:1543–52. doi: 10.1002/ijc.28800
110. Li SQ, Cheuk AT, Shern JF, Song YK, Hurd L, Liao H, et al. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS ONE. (2013) 8:e76551. doi: 10.1371/journal.pone.0076551
111. Marshall AD, Grosveld GC. Alveolar rhabdomyosarcoma – The molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skeletal Muscle. (2012) 2:25. doi: 10.1186/2044-5040-2-25
112. Yohe ME, Gryder BE, Shern JF, Song YK, Chou HC, Sindiri S, et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci Transl Med. (2018) 10:eaan4470. doi: 10.1126/scitranslmed.aan4470
113. Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. (2013) 14:416. doi: 10.1038/nrm3598
114. Pak E, Segal RA. Hedgehog signal transduction: key players, oncogenic drivers, and cancer therapy. Dev Cell. (2016) 38:333–44. doi: 10.1016/j.devcel.2016.07.026
115. Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. (1998) 4:619–22. doi: 10.1038/nm0598-619
116. Pressey JG, Anderson JR, Crossman DK, Lynch JC, Barr FG. Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer. (2011) 57:930–8. doi: 10.1002/pbc.23174
117. Satheesha S, Manzella G, Bovay A, Casanova EA, Bode PK, Belle R, et al. Targeting hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene. (2016) 35:2020–30. doi: 10.1038/onc.2015.267
118. Zibat ME, Rosenberger A, Pritchard-Jones K, Shipley J, Hahn H, Fulda S. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene. (2010) 29:6323–30. doi: 10.1038/onc.2010.368
119. Bridge JA, Liu J, Qualman SJ, Suijkerbuijk R, Wenger G, Zhang J, et al. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes, Chromosomes Cancer. (2002) 33:310–21. doi: 10.1002/gcc.10026
120. Bridge JA, Liu J, Weibolt V, Baker KS, Perry D, Kruger R, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer. (2000) 27:337–44. doi: 10.1002/(sici)1098-2264(200004)27:4<337::aid-gcc1>3.0.co;2-1
121. Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgård R, Undén AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. (2006) 208:17–25. doi: 10.1002/path.1882
122. Eichenmüller M, Hemmerlein B, von Schweinitz D, Kappler R. Betulinic acid induces apoptosis and inhibits hedgehog signalling in rhabdomyosarcoma. Br J Cancer. (2010) 103:43–51. doi: 10.1038/sj.bjc.6605715
123. Tostar U, Toftgård R, Zaphiropoulos PG, Shimokawa T. Reduction of human embryonal rhabdomyosarcoma tumor growth by inhibition of the hedgehog signaling pathway. Genes Cancer. (2010) 1:941–51. doi: 10.1177/1947601910385449
124. Yamanaka H, Oue T, Uehara S, Fukuzawa M. Hedgehog signal inhibitor forskolin suppresses cell proliferation and tumor growth of human rhabdomyosarcoma xenograft. J Pediatr Surg. (2011) 46:320–5. doi: 10.1016/j.jpedsurg.2010.11.010
125. Geyer N, Ridzewski R, Bauer J, Kuzyakova M, Dittmann K, Dullin C, et al. Different response of Ptch mutant and Ptch wildtype rhabdomyosarcoma toward SMO and PI3K Inhibitors. Front Oncol. (2018) 8:396. doi: 10.3389/fonc.2018.00396
126. Ridzewski R, Rettberg D, Dittmann K, Cuvelier N, Fulda S, Hahn H. Hedgehog Inhibitors in Rhabdomyosarcoma: a comparison of four compounds and responsiveness of four cell lines. Front Oncol. (2015) 5:130. doi: 10.3389/fonc.2015.00130
127. Basset-Seguin N, Hauschild A, Grob JJ, Kunstfeld R, Dréno B, Mortier L, et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-planned interim analysis of an international, open-label trial. Lancet Oncol. (2015) 16:729–36. doi: 10.1016/S1470-2045(15)70198-1
128. Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget. (2017) 8:69295–302. doi: 10.18632/oncotarget.20619
129. Ignatius MS, Hayes MN, Lobbardi R, Chen EY, McCarthy KM, Sreenivas P, et al. The NOTCH1/SNAIL1/MEF2C pathway regulates growth and self-renewal in embryonal rhabdomyosarcoma. Cell Rep. (2017) 19:2304–18. doi: 10.1016/j.celrep.2017.05.061
130. Raimondi L, Ciarapica R, De Salvo M, Verginelli F, Gueguen M, Martini C, et al. Inhibition of Notch3 signalling induces rhabdomyosarcoma cell differentiation promoting p38 phosphorylation and p21(Cip1) expression and hampers tumour cell growth in vitro and in vivo. Cell Death Differ. (2012) 19:871–81. doi: 10.1038/cdd.2011.171
131. Slemmons KK, Crose LES, Riedel S, Sushnitha M, Belyea B, Linardic CM. A novel notch-YAP circuit drives stemness and tumorigenesis in embryonal rhabdomyosarcoma. Mol Cancer Res. (2017) 15:1777–91. doi: 10.1158/1541-7786.MCR-17-0004
132. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. (2016) 13:417. doi: 10.1038/nrclinonc.2016.26
133. Olanich ME, Sun W, Hewitt SM, Abdullaev Z, Pack SD, Barr FG. CDK4 Amplification reduces sensitivity to CDK4/6 inhibition in fusion-positive rhabdomyosarcoma. Clin Cancer Res. (2015) 21:4947–59. doi: 10.1158/1078-0432.CCR-14-2955
134. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. (2017) 355:1152–8. doi: 10.1126/science.aam7344
135. Wiltshire TD, Lovejoy CA, Wang T, Xia F, O'Connor MJ, Cortez D, et al. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J Biol Chem. (2010) 285:14565–71. doi: 10.1074/jbc.M110.104745
136. Camero S, Ceccarelli S, De Felice F, Marampon F, Mannarino O, Camicia L, et al. PARP inhibitors affect growth, survival and radiation susceptibility of human alveolar and embryonal rhabdomyosarcoma cell lines. J Cancer Res Clin Oncol. (2019) 145:137–52. doi: 10.1007/s00432-018-2774-6
137. Mangoni M, Sottili M, Salvatore G, Meattini I, Desideri I, Greto D, et al. Enhancement of soft tissue sarcoma cell radiosensitivity by poly(ADP-ribose) Polymerase-1 Inhibitors. Radiat Res. (2018) 190:464–472, 469. doi: 10.1667/RR15035.1
138. Yan C, Brunson DC, Tang Q, Do D, Iftimia NA, Moore JC, et al. Visualizing engrafted human cancer and therapy responses in immunodeficient Zebrafish. Cell. (2019) 177:1903–14.e1914,. doi: 10.1016/j.cell.2019.04.004
139. Kahen E, Yu D, Harrison DJ, Clark J, Hingorani P, Cubitt CL, et al. Identification of clinically achievable combination therapies in childhood rhabdomyosarcoma. Cancer Chemother Pharmacol. (2016) 78:313–23. doi: 10.1007/s00280-016-3077-8
140. Stewart E, McEvoy J, Wang H, Chen X, Honnell V, Ocarz M, et al. Children's Research Hospital – Washington University Pediatric Cancer Genome, identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell. (2018) 34:411–26.e19. doi: 10.1016/j.ccell.2018.07.012
141. Yohe ME, Heske CM, Stewart E, Adamson PC, Ahmed N, Antonescu CR, et al. Insights into pediatric rhabdomyosarcoma research: challenges and goals. Pediatr Blood Cancer. (2019). 66:e27869. doi: 10.1002/pbc.27869
142. Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. (2012) 151:344–55. doi: 10.1016/j.cell.2012.08.038
143. Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore VDG, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. (2011) 334:1129–33. doi: 10.1126/science.1206727
144. Preuss E, Hugle M, Reimann R, Schlecht M, Fulda S. Pan-mammalian target of rapamycin (mTOR) inhibitor AZD8055 primes rhabdomyosarcoma cells for ABT-737-induced apoptosis by down-regulating Mcl-1 protein. J Biol Chem. (2013) 288:35287–96. doi: 10.1074/jbc.M113.495986
145. Heinicke U, Haydn T, Kehr S, Vogler M, Fulda S. BCL-2 selective inhibitor ABT-199 primes rhabdomyosarcoma cells to histone deacetylase inhibitor-induced apoptosis. Oncogene. (2018) 37:5325–39. doi: 10.1038/s41388-018-0212-5
146. Dobson CC, Naing T, Beug ST, Faye MD, Chabot J, St-Jean M, et al. Oncolytic virus synergizes with Smac mimetic compounds to induce rhabdomyosarcoma cell death in a syngeneic murine model. Oncotarget. (2017) 8:3495–508. doi: 10.18632/oncotarget.13849
147. Fulda S. Promises and challenges of smac mimetics as cancer therapeutics. Clin Cancer Res. (2015) 21:5030. doi: 10.1158/1078-0432.CCR-15-0365
148. Goletz TJ, Mackall CL, Berzofsky JA, Helman LJ. Molecular alterations in pediatric sarcomas: potential targets for immunotherapy. Sarcoma. (1998) 2:77–87. doi: 10.1080/13577149878037
149. Mackall C, Berzofsky J, Helman LJ. Targeting tumor specific translocations in sarcomas in pediatric patients for immunotherapy. Clin Orthop Relat Res. (2000) 373:25–31. doi: 10.1097/00003086-200004000-00005
150. Dagher R, Long LM, Read EJ, Leitman SF, Carter CS, Tsokos M, et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion interleukin-2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: an inter-institute NIH study. Med Pediatr Oncol. (2002) 38:158–64. doi: 10.1002/mpo.1303
151. van den Broeke LT, Pendleton CD, Mackall C, Helman LJ, Berzofsky JA. Identification and epitope enhancement of a PAX-FKHR fusion protein breakpoint epitope in alveolar rhabdomyosarcoma cells created by a tumorigenic chromosomal translocation inducing CTL capable of lysing human tumors. Cancer Res. (2006) 66:1818. doi: 10.1158/0008-5472.CAN-05-2549
152. Rodeberg DA, Nuss RA, Heppelmann CJ, Celis E. Lack of effective T-lymphocyte response to the PAX3/FKHR translocation area in alveolar rhabdomyosarcoma. Cancer immunology, immunotherapy: Cancer Immunol Immunother. (2005) 54:526–34. doi: 10.1007/s00262-004-0625-6
153. Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res. (2008) 14:4850–8. doi: 10.1158/1078-0432.CCR-07-4065
154. Baldauf MC, Gerke JS, Kirschner A, Blaeschke F, Effenberger M, Schober K, et al. Systematic identification of cancer-specific MHC-binding peptides with RAVEN. Oncoimmunology. (2018) 7:e1481558. doi: 10.1080/2162402X.2018.1481558
155. Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. (2015) 15:361. doi: 10.1038/nrc3930
156. Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an Fc-Enhanced anti–B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. (2012) 18:3834. doi: 10.1158/1078-0432.CCR-12-0715
157. Desantes K, Maris JM, McDowell K, Mackall C, Shankar S, Vasselli J, et al. A phase 1, open-label, dose escalation study of enoblituzumab (MGA271) in pediatric patients with B7-H3-expressing relapsed or refractory solid tumors. J Clin Oncol. (2017) 35:TPS2596. doi: 10.1200/JCO.2017.35.15_suppl.TPS2596
158. DeRenzo C, Krenciute G, Gottschalk S. The landscape of CAR T cells beyond acute lymphoblastic leukemia for pediatric solid tumors. Am Soc Clin Oncol Educ Book. (2018) 830–7. doi: 10.1200/EDBK_200773
159. Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. (2002) 20:719–26. doi: 10.1200/JCO.2002.20.3.719
160. Mass RD, Press MF, Anderson S, Cobleigh MA, Vogel CL, Dybdal N, et al. Evaluation of clinical outcomes according to HER2 detection by fluorescence in situ hybridization in women with metastatic breast cancer treated with trastuzumab. Clin Breast Cancer. (2005) 6:240–6. doi: 10.3816/CBC.2005.n.026
161. Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of t cells overcomes low levels of tumor antigen expression. Mol Ther. (2009) 17:1779–87. doi: 10.1038/mt.2009.133
162. Navai SA, DeRenzo C, Joseph SK, Sanber K, Byrd T, Zhang H, et al. Administration of HER2-CAR T cells after lymphodepletion safely improves T cell expansion and induces clinical responses in patients with advanced sarcomas [abstract no. LB-147/4]. (2019). Available online at: https://www.abstractsonline.com/pp8/#!/6812/presentation/9413 (accessed May 3, 2019).
163. Hegde M, DeRenzo CC, Zhang H, Mata M, Gerken C, Shree A, et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. J Clin Oncol. (2017) 35:10508. doi: 10.1200/JCO.2017.35.15_suppl.10508
164. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. (2019) 25:2560. doi: 10.1158/1078-0432.CCR-18-0432
165. Orentas RJ, Yang JJ, Wen X, Wei JS, Mackall CL, Khan J. Identification of cell surface proteins as potential immunotherapy targets in 12 pediatric cancers. Front Oncol. (2012) 2:194. doi: 10.3389/fonc.2012.00194
166. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359:1350. doi: 10.1126/science.aar4060
167. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
168. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
169. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
170. Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin Cancer Res. (2016) 22:1364–70. doi: 10.1158/1078-0432.CCR-15-0491
171. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. (2015) 33:1974–82. doi: 10.1200/JCO.2014.59.4358
172. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
173. Davis KL, Fox E, Reid JM, Liu X, Minard CG, Weigel B, et al. ADVL1412: initial results of a phase I/II study of nivolumab and ipilimumab in pediatric patients with relapsed/refractory solid tumors—a COG study. J Clin Oncol. (2017) 35:10526. doi: 10.1200/JCO.2017.35.15_suppl.10526
174. Chicas-Sett R, Morales-Orue I, Rodriguez-Abreu D, Lara-Jimenez P. Combining radiotherapy and ipilimumab induces clinically relevant radiation-induced abscopal effects in metastatic melanoma patients: a systematic review. Clin Transl Radiat Oncol. (2017) 9:5–11. doi: 10.1016/j.ctro.2017.12.004
175. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. (2010) 11:155–64. doi: 10.1016/S1470-2045(09)70334-1
176. Wexler LH. Metastatic rhabdomyosarcoma: still room for improvement. J Clin Oncol. (2016) 34:105–6. doi: 10.1200/JCO.2015.64.3395
177. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2017) 15:81. doi: 10.1038/nrclinonc.2017.166
178. Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. (2011) 3:90ra59. doi: 10.1126/scitranslmed.3002356
179. Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. (2011) 29:4541–7. doi: 10.1200/JCO.2010.34.0000
180. Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. (2016) 22:262–9. doi: 10.1038/nm.4040
181. Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. (2015) 160:1246–60. doi: 10.1016/j.cell.2015.02.038
182. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. (2015) 517:583–8. doi: 10.1038/nature14136
183. Jones DTW, Banito A, Grünewald TGP, Haber M, Jäger N, Kool M, et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat Rev Cancer. (2019) 19:420–38. doi: 10.1038/s41568-019-0169-x
Keywords: rhabdomyosacoma, pediatric oncology, soft tissue sarcoma, targeted therapy, childhood cancer
Citation: Chen C, Dorado Garcia H, Scheer M and Henssen AG (2019) Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 9:1458. doi: 10.3389/fonc.2019.01458
Received: 04 October 2019; Accepted: 05 December 2019;
Published: 20 December 2019.
Edited by:
Karim Malik, University of Bristol, United KingdomReviewed by:
Javed Khan, National Cancer Institute (NCI), United StatesCopyright © 2019 Chen, Dorado Garcia, Scheer and Henssen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anton G. Henssen, aGVuc3NlbmxhYkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.