
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 December 2019
Sec. Cancer Metabolism
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01404
This article is part of the Research Topic Tumor Cell Metabolism and Autophagy as Therapeutic Targets View all 17 articles
 Abraham Pedroza-Torres1*†
Abraham Pedroza-Torres1*† Sandra L. Romero-Córdoba2†
Sandra L. Romero-Córdoba2† Montserrat Justo-Garrido3
Montserrat Justo-Garrido3 Iván Salido-Guadarrama4
Iván Salido-Guadarrama4 Rubén Rodríguez-Bautista5
Rubén Rodríguez-Bautista5 Sarita Montaño6Rodolfo Muñiz-Mendoza7
Sarita Montaño6Rodolfo Muñiz-Mendoza7 Cristian Arriaga-Canon3Verónica Fragoso-Ontiveros7Rosa María Álvarez-Gómez7
Cristian Arriaga-Canon3Verónica Fragoso-Ontiveros7Rosa María Álvarez-Gómez7 Greco Hernández8Luis A. Herrera3*
Greco Hernández8Luis A. Herrera3*Dysregulated metabolism is a common feature of cancer cells and is considered a hallmark of cancer. Altered tumor-metabolism confers an adaptive advantage to cancer cells to fulfill the high energetic requirements for the maintenance of high proliferation rates, similarly, reprogramming metabolism confers the ability to grow at low oxygen concentrations and to use alternative carbon sources. These phenomena result from the dysregulated expression of diverse genes, including those encoding microRNAs (miRNAs) which are involved in several metabolic and tumorigenic pathways through its post-transcriptional-regulatory activity. Further, the identification of key actionable altered miRNA has allowed to propose novel targeted therapies to modulated tumor-metabolism. In this review, we discussed the different roles of miRNAs in cancer cell metabolism and novel miRNA-based strategies designed to target the metabolic machinery in human cancer.
Ever since their discovery in 1993 (1), microRNAs emerged as a new class of small RNAs with a critical role in the regulation of gene expression. MicroRNAs (miRNAs or miRs) are endogenous small non-coding RNAs from 18-25 nucleotides in length that regulate gene expression via base complementarity between the seed region of the microRNA and the 3′-untranslated region (UTR) of the target mRNA (1, 2). Depending on the degree of complementarity, miRNAs binding can induce mRNA degradation, translational repression, or both (3–5). For the considerable relevance of miRNAs in gene expression, these tiny RNA molecules have recently been called “master regulators of gene expression” (6).
The biogenesis of miRNAs has been extensively studied (7–9). For instance, genes encoding miRNAs show distinct genomic locations, such as intergenic, intronic, exonic, or mirtronic (a type of miRNA that is located in the introns of the mRNAs). Genes encoding miRNAs are transcribed in the nucleus in the form of long primary transcripts (pri-miRNAs) typically, although not exclusively, by RNA Pol II (10). Afterward, pri-miRNAs are processed into a small stem-loop transcript of approximately 55-70 nucleotides by the RNA-binding protein DiGeorge Syndrome Critical Region 8 (DGCR8) and Drosha (a ribonuclease III enzyme) (11, 12). This new structure, termed pre-miRNA, is recognized by Exportin 5 (Exp 5) and is exported from the nucleus to the cytoplasm (13). Once in the cytoplasm, pre-miRNA hairpins are cleaved by the Dicer RNase III enzyme and TRBP (TAR RNA-binding protein), resulting in a ~22 nucleotide mature miRNA-miRNA* duplex (14–16).
Finally, the mature miRNA is loaded onto Argonaute 2 protein (AGO2) and the RNA-induced silencing complex (RISC) to catalyze site-specific cleavage or translational repression of their mRNA targets (17, 18). The post-transcriptional regulation of gene expression by miRNAs is of paramount importance, thus, it is estimated that miRNAs could regulate nearly 60% of all human protein-coding genes (19). miRNAs are involved in several cellular processes, such as proliferation, development, differentiation, apoptosis, carcinogenesis, and energy metabolism (20–26).
During tumorigenesis, dysregulated metabolism represents an adaptive advantage of cancer cell that promote uncontrolled cell division, cell growth, and survival (27, 28). One of the best characterized metabolic disorders during cancer development is the Warburg effect, that increase glucose uptake and lactate production. During the Warburg effect, miRNAs activity contributes to keeping high levels of glycolysis. miRNAs also control other crucial steps of the energy metabolism, including glucose transport, glycolysis, tricarboxylic acid cycle, glutaminolysis, altered lipid metabolism, insulin production, cholesterol, and lipid homeostasis, as well as amino acid and nucleotide biogenesis (29–33).
In this review, we focus on the different roles of miRNAs in cancer metabolism and discuss novel miRNA-based strategies designed to target different processes in human cancer. We also explore the links between microbiota and miRNA networks and cancer, with a particular focus on genotoxicity and tumor-metabolism.
Upon cancer onset and progression, cells exhibit various growth, proliferation, and survival phenotypes. These cancer hallmarks are supported by a catabolic and anabolic metabolism reprogramming. Increasing evidence has shown that metabolic changes are the result of complex processes, and several cellular pathways are implicated (34–36). Recent findings have led to a significant shift in our understanding of altered metabolic states, which now are seen as a central transformative force in cancer development (37–39).
The Warburg effect is thought to be an early event in cancer that promotes rapid adaptation to higher bioenergetic demands, such as, excessive proliferation and hypoxic microenvironments. Warburg effect is characterized by: (a) supports the demand for ATP synthesis and promotes its flux into biosynthetic pathways to achieve an uncontrolled proliferation; (b) maintains an acidic microenvironment via the accumulation of lactate; and (c) allows for ROS signaling homeostasis (40–43). Moreover, reprogramming energy metabolism promotes tumor cells to use alternative carbon sources such as glutamine, considered to be the second source of nutrients after glucose. Glutamine is the most abundant amino acid in cells, and its catabolism results in several amounts of cellular precursors, including glutamate, aspartate, pyruvate, lactate, alanine, and citrate (44–46).
For many years, the Warburg effect was considered as a synonym for metabolic reprogramming. However, it is clear that this phenotype alone cannot explain all the metabolic alterations that enhance the formation of primary tumors and their development throughout invasion and metastasis. Recent publications have also reported the metabolic interactions between tumors and the microenvironment involving cancer-associated fibroblasts, immune cells, and microbiota, which allows us to expand our understanding of the metabolic reprogramming and reveals the complex interaction networks required to establish the tumor phenotype (47–49). Most of the aforementioned metabolic features are a consequence of the deregulation of several cell pathways and often involve altered oncogenes, tumor suppressors, and miRNA.
In the last decade, a growing volume of evidence has revealed the role of miRNAs in the regulation of energy metabolism, directly, through the regulation of glucose transporters (GLUT family), enzymes (hexokinase 1/2, Aldolase A), and protein kinases (AMPK, PI3K), or indirectly, through inhibition of several transcriptional factors (p53, c-Myc) (50–52). In any case, the role of miRNAs in the regulation of energy metabolism has gained much interest by their nature to modulate cellular metabolism and the possibility to use miRNAs-targets genes circuits as cancer therapies. Therefore, we review the main pathways of energy metabolism, the genes involved in each metabolic signaling and their transcriptional landscapes articulated by the miRNAs in cancer programs.
Glucose represents the main source of cellular energy. In cancer, tumor cells increase their glucose consumption to maintain the high energy requirements. However, due to the hydrophilic composition of glucose, it is not able to cross the plasma membrane by its own. To overcome this situation, tumor cells induce the expression of several members of the glucose transporters family (GLUTs, also named SLC2A proteins). Glucose transporters are membrane-associated carrier proteins responsible for facilitating the transport of glucose across the plasma membrane. In the human genome, 14 GLUT proteins have been found. Among different members of the GLUTs family, the expression of GLUT1, GLUT2, and GLUT3 has been reported to be upregulated in different types of tumors, whereas GLUT4 and GLUT5 are downregulated (53, 54). miRNAs control glucose uptake by regulating the GLUTs expression; for example, miR-144 and miR-132 are two miRNAs that have been associated with the regulation of GLUT1, one of the most broadly expressed isoforms in various cell types. Lui et al. reported that the downregulation of miR-144 induces an increase in glucose uptake in lung cancer (55). Moreover, Qu et al. demonstrated that the decrease in miR-132 expression altered glucose metabolism in prostate cancer (56). Additionally, miR-150 has been reported as a GLUT1 regulator in CD4+ cells (57). In renal cell carcinoma, miR-138, miR-150, miR-199a-3p, and miR-532-5p overexpression are associated with a decreased expression of GLUT 1, whereas miR-19a, miR-19b, miR-130b, and miR-301a decrease are directly associated with an over-expression of GLUT 1 (58).
GLUT3, another member of the glucose transport proteins family, is also regulated by miRNAs. Fei et al. demonstrated that miR-195-5p directly regulates the expression of GLUT3, and consequently decreases glucose uptake and inhibits cell growth in T24 bladder cancer cells (59). Similar results were reported by Dai DW in U251 and LN229 glioblastoma cells through the activity of miR-106a over GLUT3. Additionally, the authors indicated that miR-106a down-regulation is associated with glioblastoma patients survival (60).
Other examples of miRNAs that regulate glucose uptake are miR-233 and miR-133, which directly regulate the expression of GLUT4 (26, 61). Interestingly, miR-21 and miR-23a indirectly regulate the expression of GLUT4, as a result of their regulation over two GLUT4 translocators: PTEN and SMAD4 (62, 63). An exhaustive work published by Esteves et al., highlight the role of miR-21a-5p, miR-29a-3p, miR-29c-3p, miR-93-5p, miR-106b-5p, miR-133a-3p, miR-133b-3p, miR-222-3p, and miR-223-3p that directly or indirectly regulate the expression of GLUT4 (64). To our knowledge, there are no reports describing other members of the GLUT family regulated by miRNAs, although miRNA target prediction analysis identifies a set of miRNAs capable to silence them; however, further studies are needed to determine their contribution to aberrant tumor cell metabolism.
Unlike tumor cells, normal cells obtain energy in the form of ATP through the glucose-derived pyruvate by the mitochondrial oxidative phosphorylation. Conversely, regardless of oxygen conditions, tumor cells prefer anaerobic glycolysis, a less efficient process for obtaining ATP that produces large amounts of lactate. To compensate for this apparent decrease in energy flow, tumor cells increase glucose uptake and trigger alternative pathways to metabolize alternative carbon sources, such as glutamine, and some amino acids, such as arginine and glycine. This change in the energy metabolism confers several advantages to tumor cells, in addition it also provides necessary biomolecules for the high rates of cell division (65, 66).
During the first step of glycolysis, glucose is transformed into glucose-6 phosphate through the phosphorylation of the 6-hydroxyl group of glucose by the enzyme hexokinase (HK). The hexokinase family of enzymes comprises four isoforms (HK1–HK4) (67–69). Isoform 2 (HK2) has been reported to be upregulated in a wide variety of tumors (70–72).
One of the first works that demonstrated the regulation of miRNAs on the HK2 enzyme was published by Fang et al. Interestingly, they demonstrated that miR-125a and miR-143 regulate HK2, which modifies glucose metabolism and cell proliferation in lung cancer cells (73). This finding was confirmed by Peschiaroli et al. in head and neck squamous cell carcinoma (HNSCC)-derived cell lines (74), and by Gregersen et al. in colon cancer cells (75). Another miRNA, miR-199a-5p, regulates HK2 expression and has been reported to be under-expressed in liver cancer cells. Remarkably, overexpression of HIF1α decreased miR-199a-5p expression, which promotes glycolysis and lactate production (30). In stomach cancer cells, miR-181b directly inhibits the expression of HK2 and causes a decrease in glucose uptake and lactate production (76). In addition, miR-155 has also been reported as a regulator of the expression of HK2. Jiang et al. demonstrated that miR-155 regulates the expression of HK2 by two different mechanisms. First, miR-155 promotes the indirect transcription of HK2 through the activation of STAT3, a transcriptional activator of HK2. Second, miR-155 regulates the expression of C/EBPβ, a transcriptional activator of miR-143, whose overexpression is related to the inhibition of HK2 (77).
A couple of works showed that the enzyme responsible for catalyzing the second reaction of glycolysis, glucose-6-phosphate isomerase (GPI), is regulated by miR-200 in breast cancer cells (78) and by miR-302b and miR-17-5p in chicken primordial germ cells (79). Another glycolytic enzyme regulated by miRNAs is phosphofructokinase 1 (PFK1). PFK1 is the main regulatory enzyme for glycolysis; it catalyzes the phosphorylation reaction of fructose-6-phosphate to convert it into fructose-1,6-bisphosphate. In this sense, Yang et al. demonstrated that miR-135 targets PFK1, inhibits aerobic glycolysis, and suppresses tumor growth (31).
Similarly, Aldolase A, a glycolytic enzyme that catalyzes the conversion of fructose-1,6-bisphosphate to glyceraldehyde 3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP), is targeted by several miRNAs. Among the miRNAs that have been reported to regulate Aldolase A expression are the following: miR-122 in liver cells (80), miR-15a and miR-16-1 in leukemia (81), and miR-31 and miR-200a in Y79 retinoblastoma cells (82).
The expression level of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) has been widely used for normalizing quantitative gene expression experiments. GAPDH catalyzes the sixth reaction of glycolysis, where a molecule of NADH is released. Like other enzymes in glycolysis, GAPDH is targeted by some miRNAs such as miR-644a (83) and miR-155 (84).
The last reaction of the glycolysis pathway is catalyzed by pyruvate kinase 2 (PKM2) enzyme. PKM2 dephosphorylates phosphoenolpyruvate to produce pyruvate regardless of oxygen concentration. PKM2 has been reported to be over-expressed in many tumors due to the dysregulation of various miRNAs that down-modulate it. Some of the miRNAs reported to directly regulate the expression of PKM2 are miR-133a, miR-133b, miR-326, and miR-122 (85–87) whereas those that indirectly regulate it are miR-99a, miR-124, miR-137, and miR-340 (88, 89).
In tumors, after the glycolysis phase, pyruvate is converted into lactate by the lactate dehydrogenase enzyme (LDH). Some works have reported increased levels of LDH and its correlation with tumor aggressiveness (90–92). Interestingly, LDH expression is also regulated by miRNAs. For instance, miR-375 regulates the subunit B of LDH (LDHB) in maxillary sinus and esophageal anaplasias (93). In addition, subunit A of LHA (LDHA) has been reported to be regulated by miR-34a, miR-34c, miR-369-3p, miR-374, miR4524a/b, miR-323a-3p, miR-200c, miR-30d-5p, and miR-30a-5p in breast cancer cells and osteosarcoma tissues, which induces a decrease in glycolysis, lactate production, ATP generation, and cell proliferation (94–99).
Lactate fluxes are mainly maintained by monocarboxylate transporter (MCTs). MCTs are membrane proteins acting as carriers for lactate, pyruvate, and ketone bodies. Up to now, four MCT isoforms (MCT1, MCT2, MCT2, and MCT4) have been described in humans, and each of them exhibits a distinct cellular distribution (100). In the same way as LDH enzymes, lactate carriers (MCT proteins) are regulated by diverse miRNAs. For example, MCT1 is targeted by miR-29a, miR-29b, miR-124, and miR-495 in pancreatic β cells (101, 102). Another MCT1-regulatory miRNA is miR-342-3p, which promotes alterations in lactate and glucose flows. In addition, miR-342-3p over-expression significantly decreased cell proliferation, viability, and migration in breast cancer cell lines (103). MCT4, another member of the family of lactate transporters, is regulated by miR-145, which causes the accumulation of lactate within tumor cells in hepatocellular carcinoma cells (HCC) (104).
Glutamine metabolism (glutaminolysis) represents the second source of nutrients in cancer cells. Actually, high rates of glutaminolysis are necessary for metabolic reprogramming as it provides substrates for increased lipogenesis and nucleic acid biosynthesis that are critical to preserve the high proliferation rates of tumor cells (105, 106). Glutaminolysis converts glutamine into TCA cycle metabolites through the activity of multiple enzymes. First, glutamine is transported into the cells by solute transporters SLC1A5 and SLC7A5. Once inside the cell, glutamine is converted into glutamate and later into alpha-ketoglutarate (α-KG) by glutaminase (GLS), glutamate dehydrogenase (GDH, and other enzymes, such as glutamate pyruvate transaminase (GPT) for alanine production and glutamate oxaloacetate transaminase (GOT) for aspartate production. In addition, glutaminolysis produces considerable amounts of succinate, fumarate, malate, NADH, and ATP molecules. The transport of glutamine into the cell is strictly regulated by the membrane protein SLC1A5 (also called ASCT2 protein). SLC1A5 and other members of the ASC solute transporters family have been reported to be overexpressed in a wide variety of tumors. Dong J et al. showed that the exogenous expression of miR-137 and miR-122 markedly inhibited the SLC1A5 expression in a dose-dependent manner therefore altering tumor glutamine metabolism (107).
In a well-conducted work, Gao et al. demonstrated that the repression of miR-23a and miR-24b by the oncogenic transcription factor c-Myc resulted in a greater expression of GLS proteins and led to the upregulation of glutamine catabolism (108). Another miRNA reported to regulate GLS protein expression is miR-203, which additionally sensitizes malignant melanoma cells to temozolomide chemotherapy (109). Expression of glutamate cysteine ligase, the rate limiting enzyme of glutathione (GSH) synthesis, is attenuated by miR-18a in liver cancer (110) and by miRNA-153 in glioblastoma (111). Additionally, miR-450a limits the metastatic potential of ovarian cancer cells by targeting a set of mitochondrial mRNAs to reduce glycolysis and glutaminolysis (112).
Oxidative phosphorylation (OXPHOS) is a metabolic pathway combining two cellular processes to generate energy in the form of ATP. First, in an oxidative stage, the electron donors such as NADH and FADH2 are oxidized by the electron transport chain that turns the released energy into a proton gradient across the mitochondrial inner membrane. In the second stage, phosphorylation, ATP synthase uses the proton gradient to phosphorylate ADP to ATP. OXPHOS involves a system of protein complexes with oxidoreductase functions (complex I–IV) and ATP synthase (complex V). Even though OXPHOS is the most efficient way to produce cellular energy, tumor cells prefer to metabolize glucose via aerobic glycolysis. Several studies have recently indicated that, contrary to what is generally accepted, tumor cells could alternate between these two processes, OXPHOS and aerobic glycolysis, depending on the tumor microenvironment (113–115).
Interestingly, it has been proposed that several miRNAs regulate OXPHOS by inducing the inhibition of many components of the electron transport chain. For instance, miR-210 regulates the activity of the mitochondrial complex I (NADH: ubiquinone oxidoreductase) via the iron-sulfur cluster assembly enzyme (ISCU) by reducing the availability of iron and sulfur ions (116). Another study published by Muralimanoharan et al. revealed that miR-210 overexpression significantly reduces the complex III expression of the electron transport chain (ubiquinone:cytochrome c oxidoreductase) (117). Cytochrome c oxidase (complex IV), another enzyme of the electron transport chain, is also regulated by miRNAs. The following miRNAs have been reported to regulate cytochrome c oxidase: miR-181c (118), miR-338 (119), and miR-210 (117).
Finally, ATP synthase (complex V), a transmembrane enzyme that catalyzes ATP synthesis from an ADP molecule, is also regulated by miRNAs. Willers et al. reported that miR-127-5p reduces the expression of the catalytic subunit of ATP synthase (β-F1 subunit) in BT-549 cells in breast cancer (120). Another miRNA, miR-141, reduces the activity of ATP synthase by reducing SLC25A3 proteins (121).
miRNAs are also capable of modulating metabolic reprogramming through regulating various transcription factors relevant in metabolic pathways (122). The metabolic shift of tumor cells may be a potential strategy to evade programmed cell death and triggers cell survival and growth by activating oncogenes, such as RAS, MYC, and p53 (51, 123–126). Tumor metabolic reprogramming seems to be influenced by oncogenes and tumor suppressor networks. For example, phosphatidylinositol 3-kinase (PI3K), a lipid kinase that regulates the levels of phosphorylated phosphatidylinositol at the plasma membrane and enhances glucose uptake and glycolysis in cancer cell metabolism, is targeted by miR-320, miR-123a, miR-422, miR-506, and miR-136 (127). Catanzaro et al. showed evidence that downregulation of miR-139-5p in pediatric low-grade gliomas drives cell proliferation by regulating PI3K/AKT signaling (128). Furthermore, miR-33a/b, targets metabolic enzymes involved in fatty acid metabolism and the AMPK pathway, whereas miR-29b targets amino acid catabolism, which regulates cancer cell metabolism and biogenesis to support tumor growth and proliferation (61, 129–131). Like PI3K, AKT, and mTORC1, the MYC transcription factor has important metabolic roles beyond enhancing glycolysis. MYC promotes mitochondrial gene expression and mitochondrial biogenesis. MYC mainly depends on glutamine as a carbon source for mitochondrial metabolism (132). The oncogene MYC can bind to the promoter region of other oncogenes such as some miRNAs; for example, miR-9 is frequently upregulated in glioma specimens and cells, and it could significantly enhance proliferation, migration, and invasion of glioma cells (133).
On the other hand, miRNAs regulate important signaling pathways in mitochondria by triggering adaptive mechanisms to optimize the oxidative phosphorylation concerning the substrate supply and energy demands. For example, exogenous exosomes carrying miRNAs can induce metabolic reprogramming by restoring respiration in cancer cells and thus suppressing tumor growth. The exosomal-miRNAs involved in the modulation of cancer metabolism may be used for better diagnoses and therapies (134, 135).
Hypoxia-inducible factor 1 (HIF-1), another pathway related to tumor metabolism, is also regulated by miRNAs. HIF-1 activation can stablished oncogenic signaling by promoting glycolysis of cancer cells; but also, an alternative mechanisms over the glucose carbon mitochondrial metabolism confers HIF-1 a tumor suppressor role in some types of cancer (136, 137). In this way, miR-125-5p, miR-33-5p, and miR-190-5p are known to target the master regulator of oxygen deprivation response, HIF-1 (138). On the other hand, HIF-1 is a key molecule in adapting cancer cells to the reduced oxygen availability in the microenvironment (139–141). HIF-1 induces metabolic reprogramming as it upregulates genes such as HK1, HK2, LDHA, PDK1, GLUT1, and GLUT3, which enhance lactate production through the glycolytic pathway (142, 143). HIF1 also influences the activity of the pentose phosphate pathway, nucleotide biogenesis, angiogenesis, and suppresses the mitochondrial function (144, 145).
Finally, the oncogene c-MYC regulates HIF1 expression regardless of oxygen levels, and both act in concert to “fine-tune” adaptive responses during tumor growth (146–149). Moreover, it has been reported that more than 50% of tumors have mutations in the tumor suppressor p53, which leads to metabolic changes and contributes to the Warburg effect through the upregulation of c-MYC, HIF-1, and a broad range of genes involved in other aspects of cancer biology, including tumor cell survival and proliferation, migration, drug resistance, and immune evasion (51, 150, 151). The advance in molecular biology techniques has allowed us to detect how a diversity of miRNAs regulate tumors metabolism, as we show in Table 1 and Figure 1.
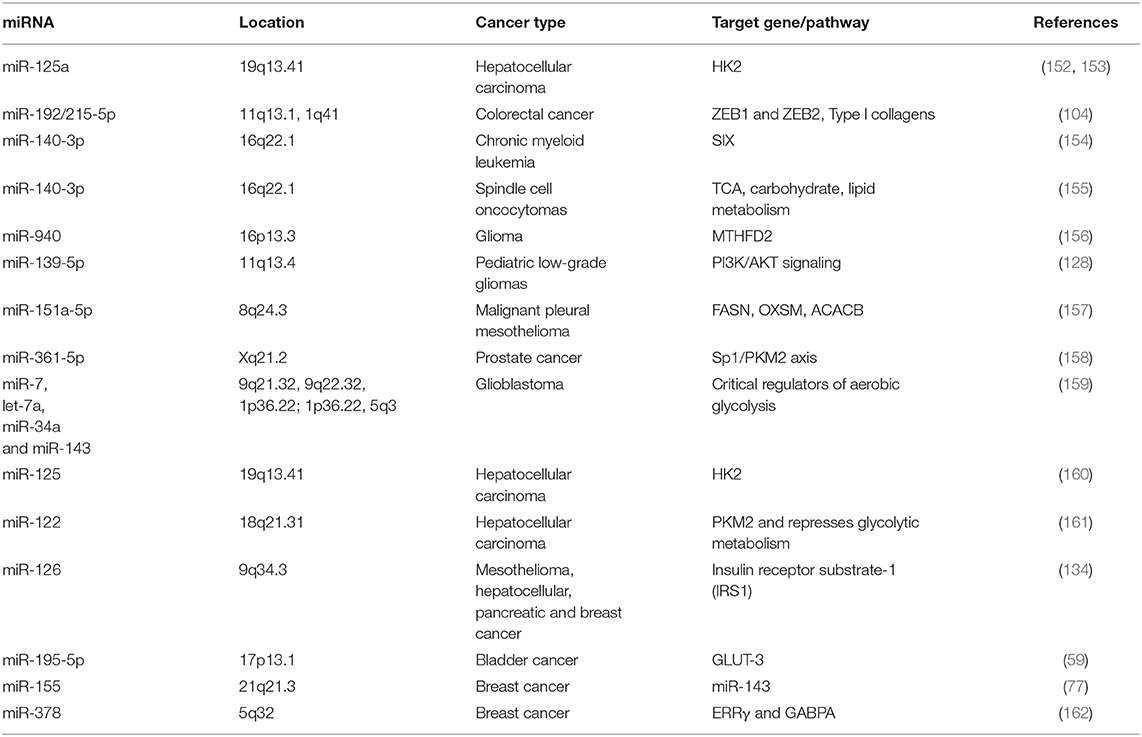
Table 1. Main miRNAs that regulate cellular metabolism in different types of cancer.
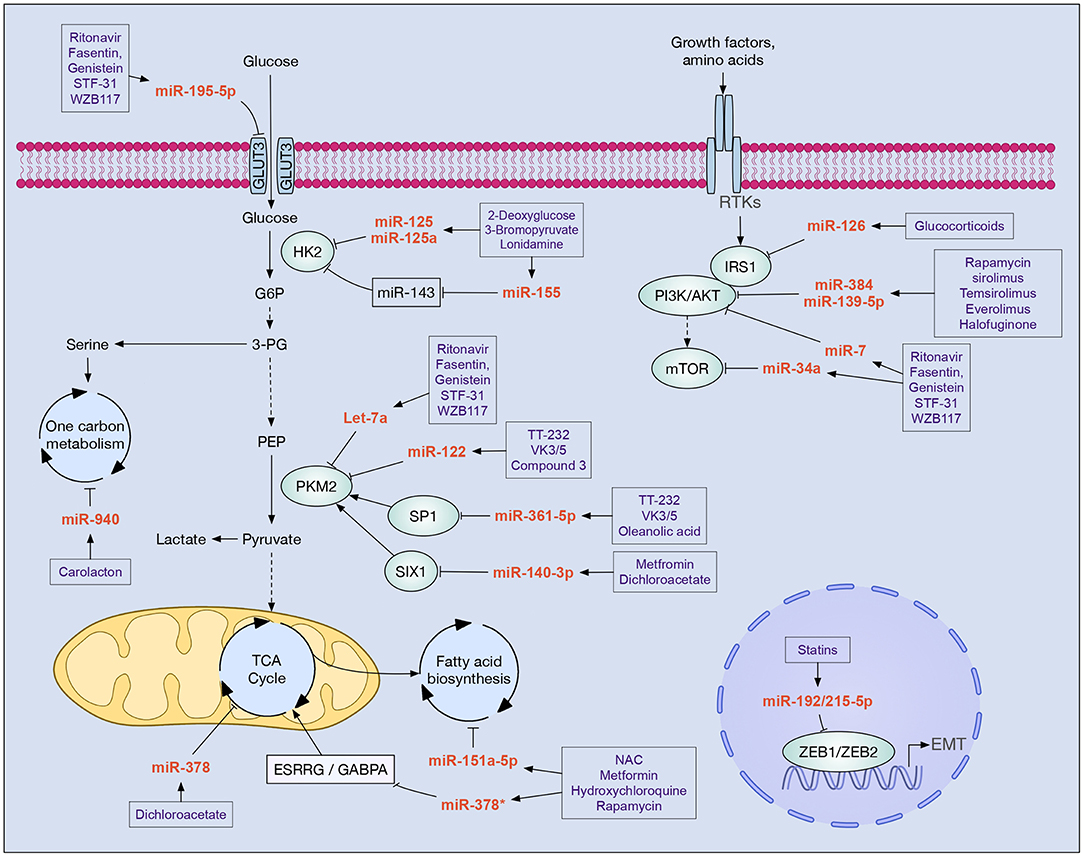
Figure 1. Drugs with clinical potential in cancer that modulate miRNAs implicated in cell metabolism. In boxes are shown drugs that potentially modulate the main miRNAs involved in the metabolic reprogramming of tumor cells. Increased glycolysis flow, alteration of the PI3K/AKT/mTOR pathway, and epithelial-mesenchymal transition (EMT) are key processes that allow tumor cells to reprogram their metabolism in order to survive, proliferate, migrate, and evade new niches. Different miRNAs participate in these processes inhibiting the expression of enzymes (e.g., HK2, PKM2, IRS1, PI3K, AKT, mTOR), transcription factors (e.g., SP1, SIX1, ZEB1, ZEB2, GABPA), and cellular receptors (e.g., GLUT3, ESRRG).
The unveiled connection between cancer profiles and metabolic reprogramming shed light on the reassessment of metabolism-targeting pharmacologic therapies as potential opportunities in cancer. Alterations in key miRNA regulatory networks contribute to the oncogenic transformation of cancer cells through genes involved in the metabolic switch (163). New insights into the altered tumor metabolism have provided novel therapeutic strategies that are being evaluated in preclinical models or clinical trials as effective therapies for many human cancers (164). Pharmacological targeting of altered miRNAs may have therapeutic effects by suppressing relevant cancer signaling pathways without affecting normal cells (165). Furthermore, pleuritic effects of metabolic drugs include miRNAs modulation that impairs signaling pathways and regulates cell energy production, which reveal miRNAs as potential drug targets.
Numerous studies now suggest that drug repurposing, which is the discovery of new therapeutic indications for known drugs, represents an attractive route in drug harnessing in cancer. Unlike the development of new molecules, drug repurposing identifies new uses for existing drugs that already have clinical and safety descriptions (166). Repurposing drugs with an oncological and non-oncological primary purpose, such as metabolic-based drugs, might be an attractive strategy to offer more effective treatment options to cancer patients and faster translate the research knowledge into the clinics (167). Interestingly, a growing body of evidence has shown that many of the antineoplastic effects and improved responses to these metabolic-based drugs may be mediated through induction of tumor suppressor miRNAs and suppression of oncogenic miRNAs.
In this section, we describe existing evidence of molecules with biochemical mechanisms impairing tumor metabolism. These molecules appear as the most promising repurposing and de-novo pharmacological interventions as shown by preclinical and clinical studies. Particular emphasis was put on chemo-resistance, which is recognized as a critical cause of treatment failure. It is reported that dysregulations of miRNAs contribute to therapy resistance via drug efflux mechanisms, alterations in drug targets, energy metabolism, DNA repair pathways, evasion of apoptosis, cell cycle control, among others (6, 168, 169). We briefly described below some pharmacologic therapies employed in different metabolic-related diseases and how they could selectively target metabolic pathways in cancer cells and modulate miRNAs networks, we will also comment some of the most relevant evidence of each of the metabolic therapeutically intervention and its anti-carcinogenic properties via miRNA activity. A more extensive over-view of miRNA expression portraits modulated by pharmacological treatment, as well as cooperative or resistance phenotypes toward drug activity is listed in Table 2 and Figure 2.
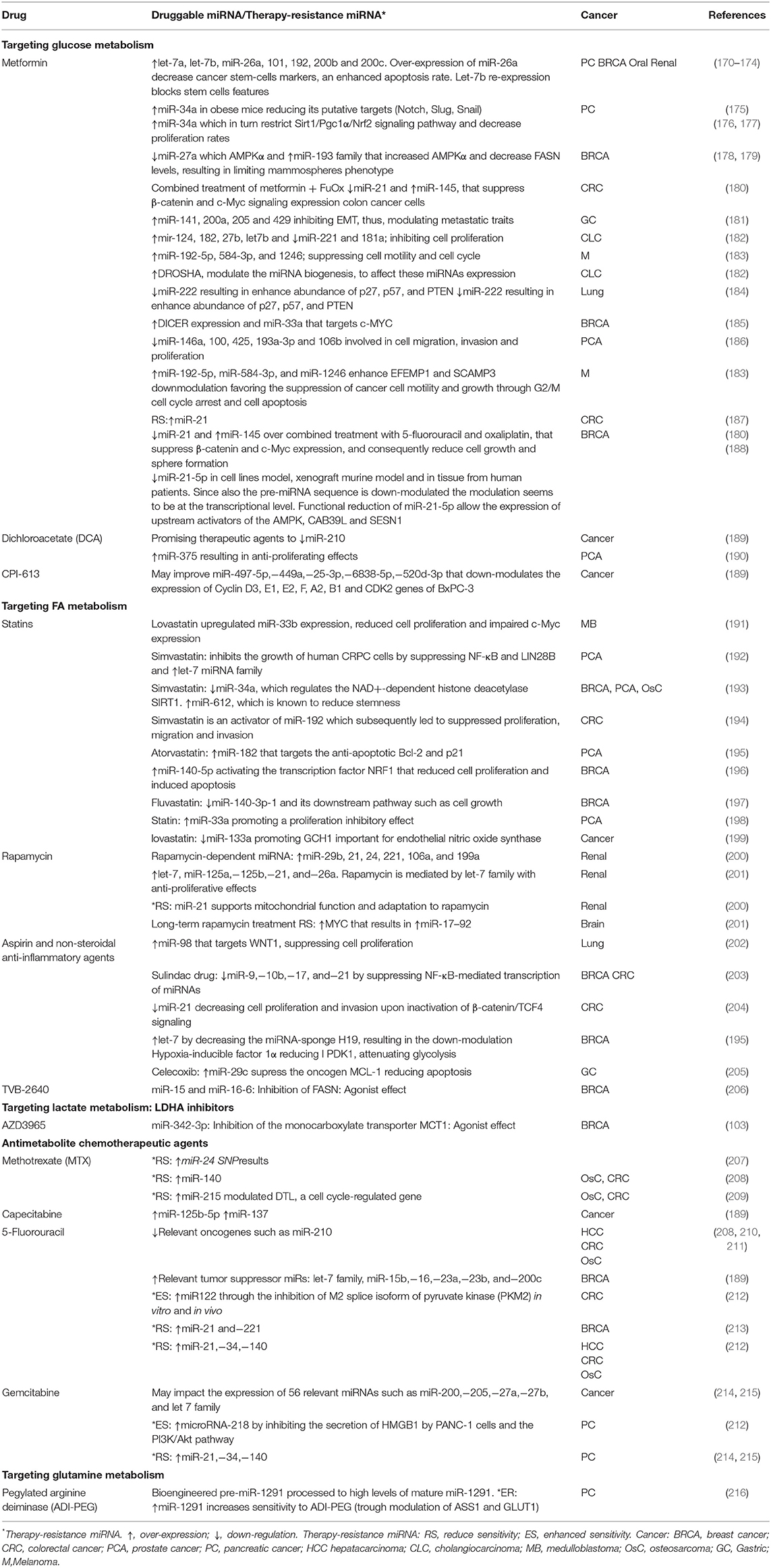
Table 2. miRNAs target by metabolic-drugs or miRNAs related to therapy resistance.
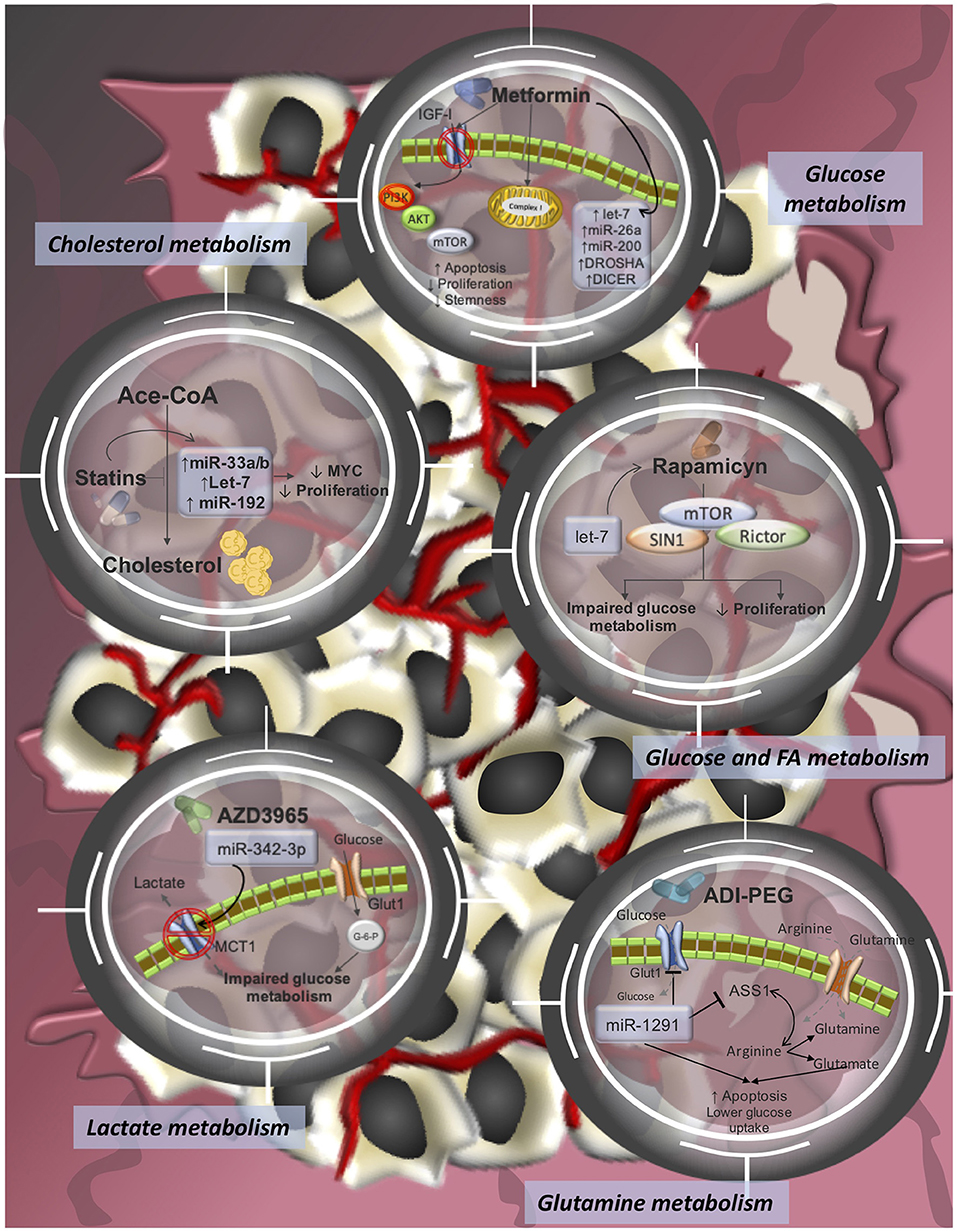
Figure 2. Pharmacological-targeting of tumor metabolism and miRNA-modulating networks of drugs tested in clinical trials or already approved FDA drugs for cancer treatments. It is reported that dysregulations of miRNAs contribute to therapy resistance via drug efflux mechanisms, alterations in drug targets, energy metabolism, Glutamine metabolism, lactate metabolism, cholesterol metabolism, among others.
Metformin, a commonly prescribed drug for treating type 2 diabetes, inhibits the mitochondrial complex I that impairs respiration, which results in a systemic impede of glucose uptake and neoglucogenesis (217–220) that reduces blood glycemia and insulinemia in hyperglycemic/diabetic patients. The tumor-suppressing effect of metformin has been reported in epidemiological studies describing a statistical association between metformin use and improved clinical outcomes in cancer (221–224). One striking example of this onco-suppressive feature is the cooperative effect of metformin and neoadjuvant chemotherapy to achieve complete tumor regression in some breast cancer patients (225). Although the precise anti-tumorigenic mechanism of action is not well-described, recent studies have shown that metformin can partially direct mitochondrial complex I inhibition, reduce NADH oxidation, and increase AMP/ATP ratio in tumors, with the consequent inhibition of mTOR signaling and decrease of fatty acid and cholesterol synthesis (218, 220, 226). Thus, metformin favors a catabolic process over an anabolic one in tumor cells. Overall, this metabolic pressure causes proliferation decline and triggers apoptosis in cancer cell lines [(227); Table 2 and Figure 2].
A variety of evidence, both in-vitro and in-vivo along with epidemiological studies, supported the protective effect of metformin against cancer development (228–231). Even more, the role of metformin on cancer not only fall in limiting its incidence, but also as a novel therapeutically intervention as shown by the 335 registered clinical trials that have evaluated patients benefit of incorporate Metformin in their treatment. The underlying mechanism of the anticancer activity of Metformin can be partially explained through its ability to modulate miRNA expression, activity and biogenesis in a variety of tumor types (Table 2 and Figure 2). For instance, overexpression of the tumor suppressors let-7, miR-26, and miR-200 family members has been reported in the literature as a pleuritic effect of Metformin molecular activity in breast, colorectal, pancreatic, oral and renal cancer. Briefly, Metformin up-modulates let-7a, that epigenetically inhibits the oncomiR miRNA-181a, which actively participated in the epithelial-to-mesenchymal transition, thus, abrogating this aggressive phenotype in BRCA (170). In CRC, the metabolic drug overexpress let-7, miR-200b/c, and miR-26a that limit the stem-like phenotype, which has been linked to poor clinical outcomes (171). Consistently, in pancreatic tumors Metformin induces the expression of miR-26a and let-7c miRNAs reducing cell proliferation, invasion, and migration. Particularly, miR-26a down-regulates the oncogene HMGA1 contributing to the observed phenotype (172). Studies in oral cancer cell models reveal that Metformin significantly increases miR-26a levels which directly decreases Mcl-1 expression that enhances apoptotic rates and reduces tumor-cell viability (173). Finally, in renal carcinoma Metformin treatment limits cell proliferation by miR-26a up-modulation that in turn down-regulates Bcl-2, cyclin D1 and upregulates the tumor suppressor PTEN, which all together influence cell cycle and cell death (174).
Dichloroacetate (DCA, PDK inhibitor) is a small molecule that inhibits the pyruvate dehydrogenase kinase (PDK) and regulates mitochondrial pyruvate dehydrogenase complex that catalyzes the irreversible decarboxylation of pyruvate into acetyl-CoA (232). PDK is overexpressed in several tumors and favors pyruvate conversion into lactate (233). Inhibition of PDK by DCA in cancer cells prompts glucose oxidation, reverses mitochondrial apoptosis, and suppresses tumor growth (234). CPI-613 is a novel anticancer agent (lipoic acid analog) that inhibits PDK through targeting lipoyl-binding pockets and selectively target the altered mitochondrial energy metabolism in tumor cells and produces changes in mitochondrial and redox status, which leads to tumor cells death (232, 235, 236). One of the main clinical challenges in colorectal cancer management is the development of chemoresistance. Interestingly, DCA treatment improve chemosensitivity to 5-fluorouracil. The evidence pointed out that the DCA over-express miR-149-3p which consequently enhanced 5-FU-induced apoptosis. Importantly, miR-149-3p is a post-transcriptional regulator of PDK2 transcript. Thus, DCA treatment overcome chemoresistant phenotype by modulating miR-149-3p/PDK2 axis (237).
Several pieces of evidence propose that targeting de novo fatty acid synthesis might be effective in the treatment of some cancers. For example, statins, cholesterol-lowering drugs, have been recently related to antitumor, cytostatic, and cytotoxic activity in diverse clinical trials of advanced malignancies (238); however, the studies are still inconclusive. Epidemiological studies have shown that statins lower the risk of presenting lung, breast, bowel, and prostate cancer (239, 240). Furthermore, different preclinical in-vitro studies show that statins may produce a variety of antineoplastic responses in cancer cells, including a cytostatic effect (cell cycle G1/S phase arrest), pro-apoptotic activity by downmodulating BCL-2 (241, 242), anti-metastatic properties through NF-kB and matrix metalloproteinase inactivation (243, 244) and anti-angiogenic properties. Different studies have provided novel evidence of the pleiotropic effects of statins independent to its cholesterol signaling modulation in cancer. For instance, in-vitro assays have shown that more than 400 miRNAs are altered by statins interventions. Including, some well-known tumor suppressor miRNAs such as miR-612, which is up-modulated after statins treatment promoting cancer cell differentiation and enhancing cancer cells response to chemotherapy (193). In another study, miR33a (198), and miR-33b (191) resulted up-modulated and participates in the anti-oncogenic properties of statins by promoting proliferation inhibitory effects and down-regulating the oncogene c-Myc. Another statin-regulated miRNA is miR-182, which down-regulates the antiapoptotic Bcl-2 transcript and consequently favors cell apoptosis (195). In a more complex regulatory circuit, simvastatin reduces NF-κB and LIN28B expression and subsequently increased let-7 levels, that in summary significantly inhibited cell viability and clonal proliferation [(192); Table 2 and Figure 2].
In a different fashion, rapalogs that inhibit mTOR (e.g., rapamycin and its derivatives, everolimus, and temsirolimus) exhibit anti-tumor effects by targeting PI3K/Akt/mTOR axis and cell proliferation. A wide spectrum of tumors is being evaluated in monotherapy or in combination. Temsirolimus and everolimus have been recently approved for the treatment of patients with advanced renal cell carcinoma (245, 246). Since mTOR is also involved in glucose metabolism by stimulating GLUT1, it is reasonable to propose a combinatory therapy with metformin to synergistically kill tumor cells [(247); Table 2]. Once again, let-7 family is one of the most reported miRNAs related with Rapamycin mechanism, playing a dual role. In one hand, in short-term treatments the inhibitory effect of rapamycin over cancer cells is mediated by increased expression of let-7 members that regulates c-MYC post-transcriptionally regulates c-MYC. On the other hand, re-expression of let-7 restore rapamycin sensitivity in resistant tumor cells (201). Long-term rapamycin treatment up-modulates miR-17–92 cluster that is related to rapamycin resistance, probably by its positive regulation over c-Myc (201). From a combinatory point of view rapamycin and metformin are able to synergize their activities against cancer cells, since this last one inhibits miR-21-5p which induces signaling of mTOR, a rapamycin-target (188).
Finally, TVB- 2640 compound is one of the first bioavailable fatty acid synthase (FASN) inhibitor to enter clinical trials for breast, colon, and astrocytic tumors, in combination with chemotherapy with the aim of enhancing clinical responses and prolonging stable disease times (NIH). Its antineoplastic activity leads to reduced cell signaling, induces tumor cell apoptosis, and inhibits cell proliferation in tumor cells by restricting lipid signaling, mainly fatty acid production, which is necessary to satisfy tumors metabolic needs [(248–250); Table 2 and Figure 2].
Aspirin, a non-steroidal anti-inflammatory drug (NSAIDs), has shown metabolic and antitumor properties (251). Aspirin may impair tumor cell migration and metastasis through preventing platelet clot formation (252). Aspirin also activates AMPK and inhibits mTOR and FA synthesis in cancer cell lines (253). Recently, aspirin has been demonstrated to have effective anti-tumor effects against RAS/RAF-mutant cells in colorectal cancer by simultaneously affecting BRAF/CRAF dimerization and hyper-activating the AMPK and ERK pathway [(254); Table 2 and Figure 2]. Besides the well-described cardioprotective effects of NSAIDs, there are substantial preclinical, clinical, and observational data that supports its activity in preventing cancer, with strong evidence in colorectal (255), lung (256), and ovarian cancer (257, 258). In preclinical studies NSAIDs administration confer a chemopreventive effect in different cancer cell models and in-vivo assays, probably via miRNA modulation. Recently, a novel mechanism of action of aspirin has been reported, in which the drug induces the expression of well-known tumor suppressors miRNAs, such as miR-98 that in turn suppress WNT1 and consequently limits cell proliferation in lung cancer (202). Moreover, NSAIDs favor let-7 expression by decreasing the abundance of one of its ncRNA-sponge, attenuating in this way glycolysis in breast cancer [(195); Table 2]. Anti-inflammatory drugs are also able to abrogate the oncogene miR-21, that results in low cell proliferation and invasion rates in BRCA and CRC (203, 204).
Several clinical trials evaluating LDHA inhibitors in different solid cancers are currently underway. One mechanism of action of LDHA inhibitors is to limit lactate export from cancer cells into the extracellular space. Accumulating intracellular lactate moves LDHA catalyzed-reaction to produce pyruvate, which prevents NAD+ regeneration and affects the energy source that established a fine competition between cancer cells that resulted in cell death. AZD3965, a drug affecting lactate metabolism, inhibits lactate transporter MCT1, which is overexpressed in several tumors and is associated with poor outcomes (259–262). MCT1 inhibitors probably synergize with the exogenous restauration of miR-342-3p that should provide a more effective inhibition of lactate transportation, which result in loss of cancer cell metabolism homeostasis [(103); Table 2 and Figure 2].
Antimetabolites as chemotherapeutic agents (e.g., methotrexate, capecitabine, 5-fluorouracil, and gemcitabine) are small molecules that resemble nucleotide metabolites; they inhibit the activity of enzymes involved in nucleotide synthesis by preventing cell division and triggering cell death. They are widely used in clinics to treat cancer since neoplastic cells have an increased metabolic demand that requires a huge nucleotide biosynthesis and DNA replication (263). More in detail, methotrexate is a folate analog that inhibits carbon transfer reactions required for de novo nucleotide synthesis. Fluorouracil (5-FU) is a synthetic analog of uracil that inhibits thymidylate synthase by limiting the availability of thymidine nucleotides for DNA synthesis (264) and has been reported that enhances the expression of relevant tumor suppressors such as let-7 family, miR-15b,−16,−23a,−23b, and−200c, some of them well-describe metabolic modulators (189). Moreover, 5-FU represses miR-210 (208, 210, 211), that down-modulates GPD1L, a negative regulator of HIF, restricting HIF-1α stability [(265, 266); Table 2]. Similarly, capecitabine is widely used in chemotherapies for gastrointestinal cancers. It halts tumor cells by inhibiting DNA and RNA synthesis and limiting the precursor of thymidine triphosphate (267, 268). Gemcitabine, another nucleoside analog, is intercalated into the DNA molecule and blocks DNA polymerases (269). Notably, the literature reports its effect over several miRNAs such as miR-200,−205,−27a,−27b, and let 7 family [(215, 269); Table 2]. All these agents can achieve important clinical responses and lead to complete remission in many cases.
In recent years, there has been substantial attention to the role of miRNAs in regulating metabolic reprogramming. Researchers have tried to reveal the mechanisms that regulate metabolic alterations in tumor cells and identify the interactions (miRNA-mRNA, miRNA-transcription factor, and miRNA-metabolic pathway) that are susceptible of being therapeutically actionable. Although studies are still incipient, robust data have been generated, describing how miRNAs directly or indirectly regulate the dysregulated metabolism of tumor cells. Based on the evidence described in this work, it is appropriate to hypothesize that there are miRNA interactions susceptible of being modulated by therapeutic interventions to reverse the metabolic alterations that allow tumor cells to uncontrollably proliferate. In addition, it is necessary to emphasize the usefulness of miRNAs-based gene therapies to enhance the regulatory activity of those identified miRNAs. However, more studies need to be conducted in a broader spectrum of components of the energetic metabolism of tumor cells, such as enzymes, transcription factors, positive regulators, and enhancers to provide more evidence on the impact of regulation mediated by miRNAs and their signaling networks on oncogenic processes.
In the last section we discussed how metabolic-target drugs and chemotherapy can modulate miRNA signaling programs as a beneficial pleiotropic effect. But it is also necessary to emphasize the usefulness of miRNAs-based therapies to improve or moderate their regulatory activity. Recent advances have permitted to study the effects of directly manipulating cellular miRNA levels by suppressing the expression of oncomiRs, that somehow enhance cancer metabolism, and which are frequently overexpressed in human cancers. Or on the contrary, by reestablishing the expression of tumor suppressor miRNAs that in many cases collaborate to restrict cancer energetics programs (270). Evidences obtained from these studies have prompted the designing and refinement of dedicated technology aimed at, either, inhibiting miRNAs (i.e., antisense oligonucleotides, locked nucleic acid, antagomiRs, miRNA sponges, and small molecule inhibitors that inactivate mature miRNA sequence in the RISC complex) (271–273) or restoring their levels by mimic sequences that can be recognized by Dicer and Ago2 proteins to be functional. Notable, miRNA delivery systems have been improved during the last years, resulting in robust and more specific devices such as liposomes, adenovirus, adeno-associated virus, EDV nanocell, and nano-particles accompanied with conjugate antibodies (274–276).
Below we briefly describe some examples of clinical trials that have been evaluated the therapeutic impact of targeting miRNAs involved in the regulation of emerging hallmarks of cancer like tumor metabolism, already described in previous sections. For instance, MRX34 was the first miRNA-based therapy undergoing in a clinical trial for cancer treatment, its aim was to re-express miR-34, that regulates LDHA, by introducing a mimic sequence through the lipid carrier NOV40 to treat patients with lymphoma, melanoma, multiple myeloma, liver, small cell lung, and renal carcinoma. Unfortunately, although promising results were observed, the trial was terminated due to severe immune-related reactions developed by some patients (277).
The first completed phase 1 trial evaluated the TargomiR technology, intended for delivering miRNA mimic sequences in vehicles containing bacterially derived minicells and a targeting moiety antibody against EGFR to treat non-small cell lung cancer. A similar example is the MesomiR-1 drug, which reintroduces miR-16, a miRNA that regulates Aldolase A in glycolysis process (278, 279). Another, drug delivery system being evaluated in stage 1 clinical trial is the locked oligonucleotide acid-modified inhibitor for miR-155 (MRG-106), as part of the clinical intervention for cutaneous T-cell lymphoma patients (280, 281). This therapeutic intervention re-expresses miR-155 targets such as miR-143, that negatively regulates HK2 and consequently limits the active glycolytic phenotype (77). Other examples include the new miRNA delivery system from Regulus company named RGLS5579, an anti-miRNA against miR-10b, for patients diagnosed with glioblastoma multiforme (282). Interestingly, under hypoxic conditions, HIF1 upregulates the transcription factor TWIST that results in the induction of the oncomiR miR-10b (283).
A further candidate of miRNA-based therapeutic currently under evaluation by Regulus and Sanofi companies, although not for cancer patient's treatment, is RG-012 which silences miR-21 in patients with Alport syndrome (284). Along the text we widely discussed miR-21 activity as a promoter of the tumoral-metabolism and its role in resistance against metabolic-based drugs. Miragen, another company, maintains also an active phase 1 study for miR-29 mimic (MRG-201) to treat keloid, fibrosis and scar tissue formation (ClinicalTrials: NCT03601052). Importantly, miR-29 is frequently lost in cancer and has been reported to negatively regulates MCT1, a lactate transporter (101, 102).
Lastly, combinatorial therapy strategies have provided successful results to treat cancer since this approach can target several tumor cell survival pathways and establish molecular landscapes to overcome resistance, offering a holistic way to reduce tumor development and evolution (285). Taking advantage of the technological advances, chemotherapeutic agents can be coordinately administrated with miRNA-based therapeutics to provide synergistic effects and enhance patient response. Since, these examples represent the first generation of miRNA-based therapeutics, there are some challenges and limitations. As an illustration, preclinical experiments in in-vivo models have shown low RNA stability, numerous mRNAs targets can be regulated by a single miRNA and different biological effects can be achieved by a miRNA in different tissues (286). Thus, it is important to guarantee tumor-specific delivery and local retention of miRNAs, for example by nanoparticle which facilitates target-specific shipment of miRNAs (286, 287).
In addition to tumoral-intracellular metabolic reprogramming, tumor cells encounter a variety of systemic factors that can influence tumorigenesis and cell metabolism (27, 34, 38, 41, 164, 288). For instance, obesity is a metabolic disorder that promote tumor growth and a connection between obesity and certain cancers, including colorectal, renal, breast cancer, esophageal, adenocarcinoma, thyroid, endometrial, prostate, and leukemia, have been reported in numerous cohort studies (289–292). In recent years, there has been substantial attention to miRNA roles in obesity-linked cancer (293). miRNA regulation programs can modulate adipogenic differentiation by controlling signaling pathways related to its biogenesis, additionally, several miRNAs associated with obesity also have well-described roles in carcinogenesis, thus, their deregulated expression portrait may act as a functional link between obesity and cancer (294–296).
Furthermore, over the last decade, a huge advent of next-generation sequencing occurred, allowing to deeply characterize the diversity of microorganisms that colonize human epitheliums, known as microbiota. Human microbiome produces small molecules and metabolites through a complex community network with relevant biological effects both at local and systemic levels and its dysregulation contributes to cancer establishment, progression and therapy response (297–300). Carcinogenesis is a complex process on which exogenous, as well as, endogenous factors could impact in different ways on malignant transformation. Among endogenous factors, metabolites generated as byproducts of metabolic activity can either act as carcinogen compounds (i.e., nitrosamines, conversion of alcohol to acetaldehyde, and tumor-promoting secondary bile acids) or as anticarcinogens (i.e., activation of dietary phytochemicals and inactivation of hormones that stimulate tumor cells growth). Even more, metabolism of different substances within the body can be affected by different health conditions like diabetes or obesity, which is characterized by chronic inflammation. In this context, bacterial metagenome has revealed to be an important player in fine-tuning tumor metabolic function, as is enriched in genes that participates in nutrients, bile acid and xenobiotic metabolism, as well as biosynthesis of vitamins and isoprenoids, therefore has become an emergent factor that affects tumor development (301–303). Based on these novel data, the gut microbiome is increasingly being recognized as a dynamic ecosystem influenced by environmental conditions such as diet and drug therapy with relevant effects on tumoral biology and metabolism (304, 305). As an open system, gut microbes elicit their effects on cancer cells via their capacity to induce pro-inflammatory responses (306–308) or more indirectly by the production of secondary metabolites (309–311). Recent evidence showed that short chain fatty acids (SCFAs), hydrogen sulfide (H2S), bile acids, and some other metabolites are produced by gut microbiota and impact the genome and epigenome of cancer cells, including miRNAs. Thus, the gut microbiome is an important regulator of host transcriptional dynamics in part through the establishment of inter-communications via miRNA signaling (312).
Host microbiome has pointed out as a potential modulator of cancer metabolism and could be a future target for precision medicine. While there is less evidence of how microbiota affects most of the miRNA landscapes in human tumors, there are growing data that explain how the microbiota confers some effect on cancer pathways in colorectal cancer (CRC). Under physiological conditions, the microbiota promotes a metabolic niche that produces a huge amount of the energy required by the intestinal epithelial cells (313) through the production of butyrate, a SCFAs, as a result of complex carbohydrates fermentation. CRC cells preferentially use glucose over butyrate as the major source of energy, resulting in a gut microbiome related dysbiosis (314). In the tumoral context, low butyrate concentrations enhance MYC expression, which in turn up-modulates the levels of the oncogenic miR-17-92 cluster (315). The overexpression of miR-17-92a cluster has been shown to enhance cell proliferation, metastasis, and angiogenesis (316–318). This data demonstrates an antitumor mechanism of butyrate over the MYC /miR-17-92a axis in CRC cells. As exemplified, miRNAs activity is a relevant feature in mediating metabolic changes and modulating the interaction of host transcriptional portrait and microbiota. Some other evidences are described in Table 3. Results from numerous studies now suggest an additionally level in the complex interplay between miRNAs and gut microbiome, including data describing the influence of miRNAs in controlling gut composition and growth rates by improving selectively pressure on the surrounding microenvironment (Table 3).

Table 3. miRNA portrait and gut microbiota in cancer.
Furthermore, results from numerous studies suggest that intestinal miRNAs come from two main sources: host and food (55, 324). The intestinal epithelial cells are the main contributors of host-derived miRNAs, but miRNAs contained in food can as well be absorbed by the host and regulate gene expression in a cross-species regulation manner (325, 326). Recently it has been showed that Ginger derived exosome-like vesicles, containing RNA, are taken up by the gut microbiota and can alter microbiome composition and host physiology. Briefly, the exosomal particles are preferentially engulfed by Lactobacillus rhamnosus and the exosomal microRNAs-cargo target various genes in the bacteria, such as Ginger miR7267-3p that mediate the production of IL-22, favoring an improvement in the colitis via IL-22-dependent mechanisms (324). These findings reveal how plant products and their effects on the microbiome may be exploited to specially target host processes to modify tumor growth through specific diet interventions (Figure 3).
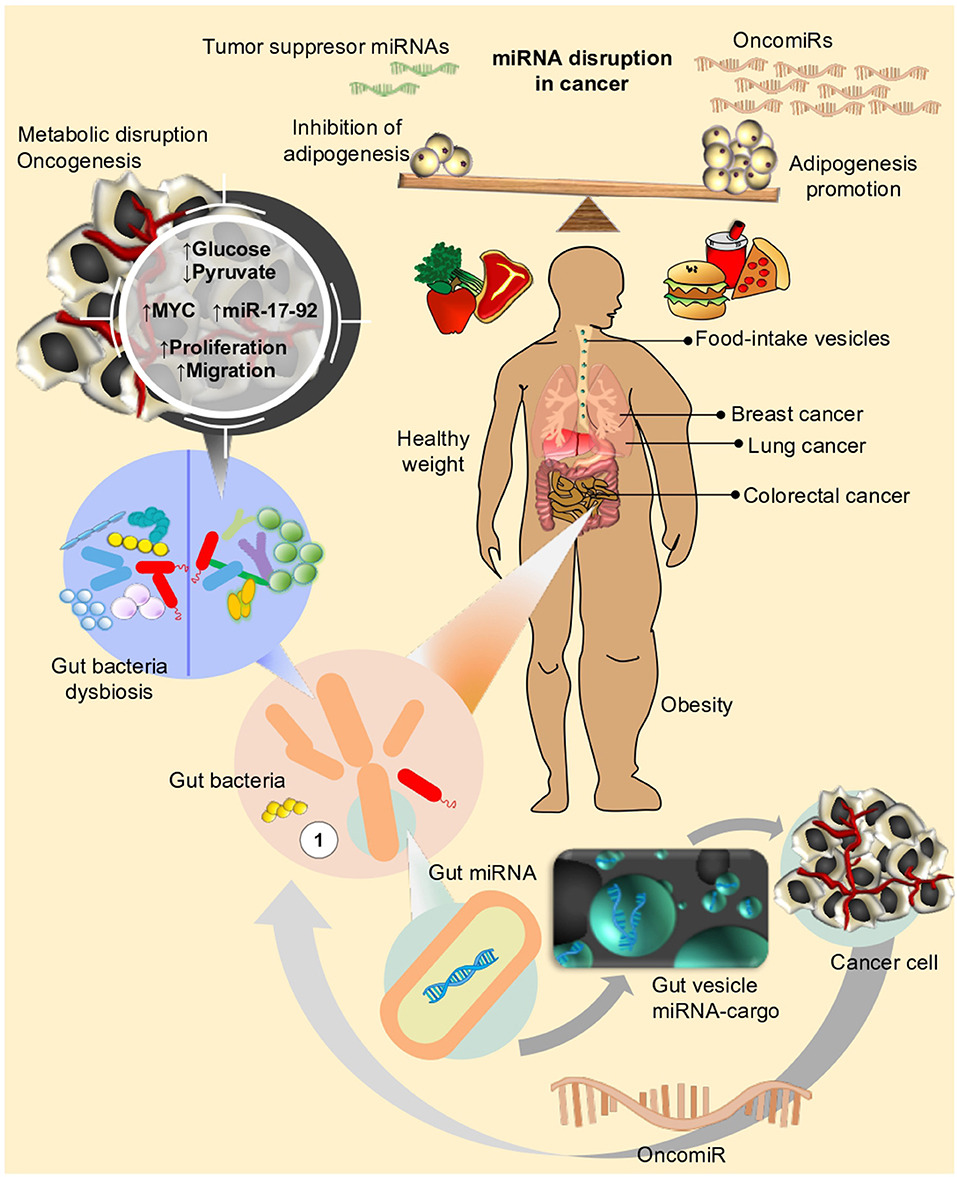
Figure 3. Life style and diet has an impact on different metabolic mechanisms in human cells. Disruption of metabolic fluxes, might particularly affect expression of genes and miRNAs related to control of cell proliferation, cell cycle, and adhesion, eventually leading or favoring neoplastic processes to take place in different organs (i.e., Breast, Prostate, Lung, Colon, etc.). Microbiota, on the other side, the new star player in the complex interaction between environment and human organism, can also influence the effect of nutrients or drug intake within host. In an unhealthy weight scenario (i.e., obesity), disequilibrium in adipogenesis leads to chronic inflammation and triggering of signals for over-expression of oncomiRs. Under this condition, dysbiosis (e.g., loss of balance in gut bacteria composition) could further concur to sustain or even enhance the metabolic perturbations favoring neoplastic transformations.
Although studies are still incipient, robust data have been generated, describing how microRNAs serve as important communication factors between the gut microbiome and the host. On the basis of these evidences, it's appropriate to hypothesize there is an open bi-directional communication between host cells and microbes, potentially mediated through miRNA activity. However, more studies are required to be conducted in a broader spectra of cancers, to provide more evidence on the impact of gut microbiota and their miRNAs signaling networks on oncogenic and metabolic processes (300), which finally will allow us take advantage of these changes and devise new strategies to translate the modulation of metabolic alterations into patient management.
AP-T, GH, SR-C, MJ-G, IS-G, RR-B, and SM conceived, designed, and wrote most of the manuscript. RM-M, CA-C, VF-O, RÁ-G, and LH contributed to the writing as well as gathered information for the manuscript. SR-C, MJ-G, IS-G, and RR-B assembled figures and tables.
This work was supported by Cátedra-CONACyT Program (Mexico), Grant No. 2425, Genómica integral del cancer. VF-O, RM-M, and RÁ-G were supported by an internal funding of the National Institute of Cancer (INCAN, Mexico). GH was supported by CONACyT (Mexico), Grant No. 273116, Identification of new mutations in the Androgen Receptor gene specific of Mexican men and their clinic impact on prostate cancer, and by an internal funding of the National Institute of Cancer (INCAN, Mexico).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
SR-C was supported by Cátedra Salvador Zubirán, UNAM/INCMNSZ.
1. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-y
2. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. (1993) 75:855–62. doi: 10.1016/0092-8674(93)90530-4
3. Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. (2006) 20:1885–98. doi: 10.1101/gad.1424106
4. Behm-Ansmant I, Rehwinkel J, Izaurralde E. MicroRNAs silence gene expression by repressing protein expression and/or by promoting mRNA decay. Cold Spring Harb Symp Quant Biol. (2006) 71:523–30. doi: 10.1101/sqb.2006.71.013
5. Vella MC, Choi E-Y, Lin S-Y, Reinert K, Slack FJ. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3'UTR. Genes Dev. (2004) 18:132–7. doi: 10.1101/gad.1165404
6. Garofalo M, Croce CM. microRNAs: master regulators as potential therapeutics in cancer. Annu Rev Pharmacol Toxicol. (2011) 51:25–43. doi: 10.1146/annurev-pharmtox-010510-100517
7. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. (2009) 10:126–39. doi: 10.1038/nrm2632
8. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/s0092-8674(04)00045-5
9. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. (2014) 15:509–24. doi: 10.1038/nrm3838
10. Lee Y, Kim M, Han J, Yeom K-H, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. (2004) 23:4051–60. doi: 10.1038/sj.emboj.7600385
11. Han J, Lee Y, Yeom K-H, Kim Y-K, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. (2004) 18:3016–27. doi: 10.1101/gad.1262504
12. Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. (2007) 39:380–5. doi: 10.1038/ng1969
13. Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. (2003) 17:3011–6. doi: 10.1101/gad.1158803
14. Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. (2004) 118:57–68. doi: 10.1016/j.cell.2004.06.017
15. Koscianska E, Starega-Roslan J, Krzyzosiak WJ. The role of Dicer protein partners in the processing of microRNA precursors. PLoS ONE. (2011) 6:e28548. doi: 10.1371/journal.pone.0028548
16. Fareh M, Yeom K-H, Haagsma AC, Chauhan S, Heo I, Joo C. TRBP ensures efficient Dicer processing of precursor microRNA in RNA-crowded environments. Nat Commun. (2016) 7:13694. doi: 10.1038/ncomms13694
17. Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. (2005) 436:740–4. doi: 10.1038/nature03868
18. Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. (2004) 15:185–97. doi: 10.1016/j.molcel.2004.07.007
19. Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. (2009) 19:92–105. doi: 10.1101/gr.082701.108
20. Roccaro AM, Sacco A, Thompson B, Leleu X, Azab AK, Azab F, et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood. (2009) 113:6669–80. doi: 10.1182/blood-2009-01-198408
21. Chen J-F, Tao Y, Li J, Deng Z, Yan Z, Xiao X, et al. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J Cell Biol. (2010) 190:867–79. doi: 10.1083/jcb.200911036
22. Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. (2008) 105:13421–6. doi: 10.1073/pnas.0801613105
23. Schickel R, Park S-M, Murmann AE, Peter ME. miR-200c regulates induction of apoptosis through CD95 by targeting FAP-1. Mol Cell. (2010) 38:908–15. doi: 10.1016/j.molcel.2010.05.018
24. Otsubo T, Akiyama Y, Hashimoto Y, Shimada S, Goto K, Yuasa Y. MicroRNA-126 inhibits SOX2 expression and contributes to gastric carcinogenesis. PLoS ONE. (2011) 6:e16617. doi: 10.1371/journal.pone.0016617
25. Hu Q, Du K, Mao X, Ning S. miR-197 is downregulated in cervical carcinogenesis and suppresses cell proliferation and invasion through targeting forkhead box M1. Oncol Lett. (2018) 15:10063–9. doi: 10.3892/ol.2018.8565
26. Lu H, Buchan RJ, Cook SA. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc Res. (2010) 86:410–20. doi: 10.1093/cvr/cvq010
27. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
28. Oermann EK, Wu J, Guan K-L, Xiong Y. Alterations of metabolic genes and metabolites in cancer. Semin Cell Dev Biol. (2012) 23:370–80. doi: 10.1016/j.semcdb.2012.01.013
29. Chen D, Wang H, Chen J, Li Z, Li S, Hu Z, et al. MicroRNA-129-5p Regulates glycolysis and cell proliferation by targeting the glucose transporter SLC2A3 in gastric cancer cells. Front Pharmacol. (2018) 9:502. doi: 10.3389/fphar.2018.00502
30. Guo W, Qiu Z, Wang Z, Wang Q, Tan N, Chen T, et al. MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatol Baltim Md. (2015) 62:1132–44. doi: 10.1002/hep.27929
31. Yang Y, Ishak Gabra MB, Hanse EA, Lowman XH, Tran TQ, Li H, et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat Commun. (2019) 10:809. doi: 10.1038/s41467-019-08759-0
32. Zhang X, Li Z, Xuan Z, Xu P, Wang W, Chen Z, et al. Novel role of miR-133a-3p in repressing gastric cancer growth and metastasis via blocking autophagy-mediated glutaminolysis. J Exp Clin Cancer Res CR. (2018) 37:320. doi: 10.1186/s13046-018-0993-y
33. Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. (2006) 3:87–98. doi: 10.1016/j.cmet.2006.01.005
34. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
35. Dang CV. Links between metabolism and cancer. Genes Dev. (2012) 26:877–90. doi: 10.1101/gad.189365.112
36. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. (2010) 330:1340–4. doi: 10.1126/science.1193494
37. Vaupel P, Schmidberger H, Mayer A. The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol. (2019) 95:912–9. doi: 10.1080/09553002.2019.1589653
38. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
39. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
40. Lebelo MT, Joubert AM, Visagie MH. Warburg effect and its role in tumourigenesis. Arch Pharm Res. (2019) 42:833–47. doi: 10.1007/s12272-019-01185-2
41. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. (2011) 11:85–95. doi: 10.1038/nrc2981
42. Wu W, Zhao S. Metabolic changes in cancer: beyond the Warburg effect. Acta Biochim Biophys Sin. (2013) 45:18–26. doi: 10.1093/abbs/gms104
43. Soga T. Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. (2013) 104:275–81. doi: 10.1111/cas.12085
44. Kato Y, Maeda T, Suzuki A, Baba Y. Cancer metabolism: new insights into classic characteristics. Jpn Dent Sci Rev. (2018) 54:8–21. doi: 10.1016/j.jdsr.2017.08.003
45. Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci CMLS. (2016) 73:377–92. doi: 10.1007/s00018-015-2070-4
46. Li C, Zhang G, Zhao L, Ma Z, Chen H. Metabolic reprogramming in cancer cells: glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer. World J Surg Oncol. (2016) 14:15. doi: 10.1186/s12957-016-0769-9
47. Schwörer S, Vardhana SA, Thompson CB. Cancer metabolism drives a stromal regenerative response. Cell Metab. (2019) 29:576–91. doi: 10.1016/j.cmet.2019.01.015
48. Ippolito L, Morandi A, Taddei ML, Parri M, Comito G, Iscaro A, et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene. (2019) 38:5339–55. doi: 10.1038/s41388-019-0805-7
49. Netea-Maier RT, Smit JWA, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: a mutual relationship. Cancer Lett. (2018) 413:102–9. doi: 10.1016/j.canlet.2017.10.037
50. Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. (2019) 38:2223–40. doi: 10.1038/s41388-018-0582-8
51. Yeung SJ, Pan J, Lee M-H. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci CMLS. (2008) 65:3981–99. doi: 10.1007/s00018-008-8224-x
52. Zwaans BMM, Lombard DB. Interplay between sirtuins, MYC and hypoxia-inducible factor in cancer-associated metabolic reprogramming. Dis Model Mech. (2014) 7:1023–32. doi: 10.1242/dmm.016287
53. Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. (2013) 34:121–38. doi: 10.1016/j.mam.2012.07.001
54. Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. (2010) 298:E141–5. doi: 10.1152/ajpendo.00712.2009
55. Liu M, Gao J, Huang Q, Jin Y, Wei Z. Downregulating microRNA-144 mediates a metabolic shift in lung cancer cells by regulating GLUT1 expression. Oncol Lett. (2016) 11:3772–6. doi: 10.3892/ol.2016.4468
56. Qu W, Ding S-M, Cao G, Wang S-J, Zheng X-H, Li G-H. miR-132 mediates a metabolic shift in prostate cancer cells by targeting Glut1. FEBS Open Bio. (2016) 6:735–41. doi: 10.1002/2211-5463.12086
57. King BC, Esguerra JLS, Golec E, Eliasson L, Kemper C, Blom AM. CD46 activation regulates miR-150–mediated control of GLUT1 expression and cytokine secretion in human CD4 + T Cells. J Immunol. (2016) 196:1636–45. doi: 10.4049/jimmunol.1500516
58. Chow T-FF, Mankaruos M, Scorilas A, Youssef Y, Girgis A, Mossad S, et al. The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J Urol. (2010) 183:743–51. doi: 10.1016/j.juro.2009.09.086
59. Fei X, Qi M, Wu B, Song Y, Wang Y, Li T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. (2012) 586:392–7. doi: 10.1016/j.febslet.2012.01.006
60. Dai D-W, Lu Q, Wang L-X, Zhao W-Y, Cao Y-Q, Li Y-N, et al. Decreased miR-106a inhibits glioma cell glucose uptake and proliferation by targeting SLC2A3 in GBM. BMC Cancer. (2013) 13:478. doi: 10.1186/1471-2407-13-478
61. Horie T, Ono K, Nishi H, Iwanaga Y, Nagao K, Kinoshita M, et al. MicroRNA-133 regulates the expression of GLUT4 by targeting KLF15 and is involved in metabolic control in cardiac myocytes. Biochem Biophys Res Commun. (2009) 389:315–20. doi: 10.1016/j.bbrc.2009.08.136
62. Ling H-Y, Hu B, Hu X-B, Zhong J, Feng S-D, Qin L, et al. MiRNA-21 reverses high glucose and high insulin induced insulin resistance in 3T3-L1 adipocytes through targeting phosphatase and tensin homologue. Exp Clin Endocrinol Diabetes Off J Ger Soc Endocrinol Ger Diabetes Assoc. (2012) 120:553–9. doi: 10.1055/s-0032-1311644
63. Raychaudhuri S. MicroRNAs overexpressed in growth-restricted rat skeletal muscles regulate the glucose transport in cell culture targeting central TGF-β factor SMAD4. PLoS ONE. (2012) 7:e34596. doi: 10.1371/journal.pone.0034596
64. Esteves JV, Enguita FJ, Machado UF. MicroRNAs-mediated regulation of skeletal muscle GLUT4 expression and translocation in insulin resistance. J Diabetes Res. (2017) 2017:7267910. doi: 10.1155/2017/7267910
65. Yin C, Qie S, Sang N. Carbon source metabolism and its regulation in cancer cells. Crit Rev Eukaryot Gene Expr. (2012) 22:17–35. doi: 10.1615/critreveukargeneexpr.v22.i1.20
66. Dang CV. Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle Georget Tex. (2010) 9:3884–6. doi: 10.4161/cc.9.19.13302
67. John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE. (2011) 6:e17674. doi: 10.1371/journal.pone.0017674
68. Mandarino LJ, Printz RL, Cusi KA, Kinchington P, O'Doherty RM, Osawa H, et al. Regulation of hexokinase II and glycogen synthase mRNA, protein, and activity in human muscle. Am J Physiol. (1995) 269:E701–8. doi: 10.1152/ajpendo.1995.269.4.E701
69. Wyatt E, Wu R, Rabeh W, Park H-W, Ghanefar M, Ardehali H. Regulation and cytoprotective role of hexokinase III. PLoS ONE. (2010) 5:e13823. doi: 10.1371/journal.pone.0013823
70. Yasuda S, Arii S, Mori A, Isobe N, Yang W, Oe H, et al. Hexokinase II and VEGF expression in liver tumors: correlation with hypoxia-inducible factor-1α and its significance. J Hepatol. (2004) 40:117–23. doi: 10.1016/S0168-8278(03)00503-8
71. Jiao L, Zhang H-L, Li D-D, Yang K-L, Tang J, Li X, et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy. (2018) 14:671–84. doi: 10.1080/15548627.2017.1381804
72. Palmieri D, Fitzgerald D, Shreeve SM, Hua E, Bronder JL, Weil RJ, et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol Cancer Res. (2009) 7:1438–45. doi: 10.1158/1541-7786.MCR-09-0234
73. Fang R, Xiao T, Fang Z, Sun Y, Li F, Gao Y, et al. MicroRNA-143 (miR-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J Biol Chem. (2012) 287:23227–35. doi: 10.1074/jbc.M112.373084
74. Peschiaroli A, Giacobbe A, Formosa A, Markert EK, Bongiorno-Borbone L, Levine AJ, et al. miR-143 regulates hexokinase 2 expression in cancer cells. Oncogene. (2013) 32:797–802. doi: 10.1038/onc.2012.100
75. Gregersen LH, Jacobsen A, Frankel LB, Wen J, Krogh A, Lund AH. MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer. (2012) 12:232. doi: 10.1186/1471-2407-12-232
76. Li L-Q, Yang Y, Chen H, Zhang L, Pan D, Xie W-J. MicroRNA-181b inhibits glycolysis in gastric cancer cells via targeting hexokinase 2 gene. Cancer Biomark Sect Dis Markers. (2016) 17:75–81. doi: 10.3233/CBM-160619
77. Jiang S, Zhang L-F, Zhang H-W, Hu S, Lu M-H, Liang S, et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. (2012) 31:1985–98. doi: 10.1038/emboj.2012.45
78. Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W, et al. Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res. (2011) 71:3400–9. doi: 10.1158/0008-5472.CAN-10-0965
79. Rengaraj D, Park TS, Lee SI, Lee BR, Han BK, Song G, et al. Regulation of glucose phosphate isomerase by the 3'UTR-specific miRNAs miR-302b and miR-17-5p in chicken primordial germ cells. Biol Reprod. (2013) 89:33. doi: 10.1095/biolreprod.112.105692
80. Fabani MM, Gait MJ. miR-122 targeting with LNA/2'-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA NYN. (2008) 14:336–46. doi: 10.1261/rna.844108
81. Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci USA. (2008) 105:5166–71. doi: 10.1073/pnas.0800121105
82. Montoya V, Fan H, Bryar PJ, Weinstein JL, Mets MB, Feng G, et al. Novel miRNA-31 and miRNA-200a-mediated regulation of retinoblastoma proliferation. PLoS ONE. (2015) 10:e0138366. doi: 10.1371/journal.pone.0138366
83. Sikand K, Singh J, Ebron JS, Shukla GC. Housekeeping gene selection advisory: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin are targets of miR-644a. PLoS ONE. (2012) 7:e47510. doi: 10.1371/journal.pone.0047510
84. Khanna M, Saini S, Shariff M, Ronsard L, Singh JK, Kumar H. Data highlighting miR-155 and GAPDH correlation. Data Brief. (2019) 24:103945. doi: 10.1016/j.dib.2019.103945
85. Wong T-S, Liu X-B, Chung-Wai Ho A, Po-Wing Yuen A, Wai-Man Ng R, Ignace Wei W. Identification of pyruvate kinase type M2 as potential oncoprotein in squamous cell carcinoma of tongue through microRNA profiling. Int J Cancer. (2008) 123:251–7. doi: 10.1002/ijc.23583
86. Kefas B, Comeau L, Erdle N, Montgomery E, Amos S, Purow B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro-Oncol. (2010) 12:1102–12. doi: 10.1093/neuonc/noq080
87. Liu AM, Xu Z, Shek FH, Wong K-F, Lee NP, Poon RT, et al. miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS ONE. (2014) 9:e86872. doi: 10.1371/journal.pone.0086872
88. Sun Y, Zhao X, Zhou Y, Hu Y. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol Rep. (2012) 28:1346–52. doi: 10.3892/or.2012.1958
89. Li W, Wang J, Chen Q-D, Qian X, Li Q, Yin Y, et al. Insulin promotes glucose consumption via regulation of miR-99a/mTOR/PKM2 pathway. PLoS ONE. (2013) 8:e64924. doi: 10.1371/journal.pone.0064924
90. Feng Y, Xiong Y, Qiao T, Li X, Jia L, Han Y. Lactate dehydrogenase A: a key player in carcinogenesis and potential target in cancer therapy. Cancer Med. (2018) 7:6124–36. doi: 10.1002/cam4.1820
91. Li J, Zhu S, Tong J, Hao H, Yang J, Liu Z, et al. Suppression of lactate dehydrogenase A compromises tumor progression by downregulation of the Warburg effect in glioblastoma. Neuroreport. (2016) 27:110–5. doi: 10.1097/WNR.0000000000000506
92. Dong T, Liu Z, Xuan Q, Wang Z, Ma W, Zhang Q. Tumor LDH-A expression and serum LDH status are two metabolic predictors for triple negative breast cancer brain metastasis. Sci Rep. (2017) 7:6069. doi: 10.1038/s41598-017-06378-7
93. Kinoshita T, Nohata N, Yoshino H, Hanazawa T, Kikkawa N, Fujimura L, et al. Tumor suppressive microRNA-375 regulates lactate dehydrogenase B in maxillary sinus squamous cell carcinoma. Int J Oncol. (2012) 40:185–93. doi: 10.3892/ijo.2011.1196
94. Xiao X, Huang X, Ye F, Chen B, Song C, Wen J, et al. The miR-34a-LDHA axis regulates glucose metabolism and tumor growth in breast cancer. Sci Rep. (2016) 6:21735. doi: 10.1038/srep21735
95. Chen H, Gao S, Cheng C. MiR-323a-3p suppressed the glycolysis of osteosarcoma via targeting LDHA. Hum Cell. (2018) 31:300–9. doi: 10.1007/s13577-018-0215-0
96. Li L, Kang L, Zhao W, Feng Y, Liu W, Wang T, et al. miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett. (2017) 400:89–98. doi: 10.1016/j.canlet.2017.04.034
97. Wang J, Wang H, Liu A, Fang C, Hao J, Wang Z. Lactate dehydrogenase A negatively regulated by miRNAs promotes aerobic glycolysis and is increased in colorectal cancer. Oncotarget. (2015) 6:19456–468. doi: 10.18632/oncotarget.3318
98. Yuan D, Zheng S, Wang L, Li J, Yang J, Wang B, et al. MiR-200c inhibits bladder cancer progression by targeting lactate dehydrogenase A. Oncotarget. (2017) 8:67663–9. doi: 10.18632/oncotarget.18801
99. He Y, Chen X, Yu Y, Li J, Hu Q, Xue C, et al. LDHA is a direct target of miR-30d-5p and contributes to aggressive progression of gallbladder carcinoma. Mol Carcinog. (2018) 57:772–83. doi: 10.1002/mc.22799
100. Halestrap AP. The SLC16 gene family - structure, role and regulation in health and disease. Mol Aspects Med. (2013) 34:337–49. doi: 10.1016/j.mam.2012.05.003
101. Pullen TJ, da Silva Xavier G, Kelsey G, Rutter GA. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol Cell Biol. (2011) 31:3182–94. doi: 10.1128/MCB.01433-10
102. Liang D, Zhang Y, Han J, Wang W, Liu Y, Li J, et al. Embryonic stem cell-derived pancreatic endoderm transplant with MCT1-suppressing miR-495 attenuates type II diabetes in mice. Endocr J. (2015) 62:907–20. doi: 10.1507/endocrj.EJ15-0186
103. Romero-Cordoba SL, Rodriguez-Cuevas S, Bautista-Pina V, Maffuz-Aziz A, D'Ippolito E, Cosentino G, et al. Loss of function of miR-342-3p results in MCT1 over-expression and contributes to oncogenic metabolic reprogramming in triple negative breast cancer. Sci Rep. (2018) 8:12252. doi: 10.1038/s41598-018-29708-9
104. Zhao Y, Li W, Li M, Hu Y, Zhang H, Song G, et al. Targeted inhibition of MCT4 disrupts intracellular pH homeostasis and confers self-regulated apoptosis on hepatocellular carcinoma. Exp Cell Res. (2019)111591. doi: 10.1016/j.yexcr.2019.111591
105. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. (2013) 123:3678–84. doi: 10.1172/JCI69600
106. Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin Cell Dev Biol. (2012) 23:362–9. doi: 10.1016/j.semcdb.2012.02.002
107. Dong J, Xiao D, Zhao Z, Ren P, Li C, Hu Y, et al. Epigenetic silencing of microRNA-137 enhances ASCT2 expression and tumor glutamine metabolism. Oncogenesis. (2017) 6:e356. doi: 10.1038/oncsis.2017.59
108. Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. (2009) 458:762–5. doi: 10.1038/nature07823
109. Chang X, Zhu W, Zhang H, Lian S. Sensitization of melanoma cells to temozolomide by overexpression of microRNA 203 through direct targeting of glutaminase-mediated glutamine metabolism. Clin Exp Dermatol. (2017) 42:614–21. doi: 10.1111/ced.13119
110. Anderton B, Camarda R, Balakrishnan S, Balakrishnan A, Kohnz RA, Lim L, et al. MYC-driven inhibition of the glutamate-cysteine ligase promotes glutathione depletion in liver cancer. EMBO Rep. (2017) 18:569–85. doi: 10.15252/embr.201643068
111. Liu Z, Wang J, Li Y, Fan J, Chen L, Xu R. MicroRNA-153 regulates glutamine metabolism in glioblastoma through targeting glutaminase. Tumour Biol J Int Soc Oncodevelopmental Biol Med. (2017) 39:1010428317691429. doi: 10.1177/1010428317691429
112. Muys BR, Sousa JF, Plaça JR, de Araújo LF, Sarshad AA, Anastasakis DG, et al. miR-450a acts as a tumor suppressor in ovarian cancer by regulating energy metabolism. Cancer Res. (2019) 79:3294–305. doi: 10.1158/0008-5472.CAN-19-0490
113. Zacksenhaus E, Shrestha M, Liu JC, Vorobieva I, Chung PED, Ju Y, et al. Mitochondrial OXPHOS induced by RB1 deficiency in breast cancer: implications for anabolic metabolism, stemness, and metastasis. Trends Cancer. (2017) 3:768–79. doi: 10.1016/j.trecan.2017.09.002
114. Marchiq I, Le Floch R, Roux D, Simon M-P, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. (2015) 75:171–80. doi: 10.1158/0008-5472.CAN-14-2260
115. Ippolito L, Marini A, Cavallini L, Morandi A, Pietrovito L, Pintus G, et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget. (2016) 7:61890–904. doi: 10.18632/oncotarget.11301
116. Patel B, Gove CD, Hutchinson WL, Mowbray J. Substantial quantities of the high energy derivative oligophosphoglyceroyl-ATP are located in mitochondria in rat liver. Biochim Biophys Acta. (1991) 1074:178–81. doi: 10.1016/0304-4165(91)90058-o
117. Muralimanoharan S, Maloyan A, Mele J, Guo C, Myatt LG, Myatt L. MIR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta. (2012) 33:816–23. doi: 10.1016/j.placenta.2012.07.002
118. Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res. (2012) 110:1596–603. doi: 10.1161/CIRCRESAHA.112.267732
119. Aschrafi A, Schwechter AD, Mameza MG, Natera-Naranjo O, Gioio AE, Kaplan BB. MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J Neurosci. (2008) 28:12581–90. doi: 10.1523/JNEUROSCI.3338-08.2008
120. Willers IM, Martínez-Reyes I, Martínez-Diez M, Cuezva JM. miR-127-5p targets the 3'UTR of human β-F1-ATPase mRNA and inhibits its translation. Biochim Biophys Acta. (2012) 1817:838–48. doi: 10.1016/j.bbabio.2012.03.005
121. Baseler WA, Thapa D, Jagannathan R, Dabkowski ER, Croston TL, Hollander JM. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am J Physiol Cell Physiol. (2012) 303:C1244–51. doi: 10.1152/ajpcell.00137.2012
122. Pucci S, Mazzarelli P. MicroRNA Dysregulation in colon cancer microenvironment interactions: the importance of small things in metastases. Cancer Microenviron. (2011) 4:155–62. doi: 10.1007/s12307-011-0062-y
123. Morton MJ, Pitel ML. Reflex sympathetic dystrophy syndrome complicating the management of TMJ symptoms. A case report. Cranio J Craniomandib Pract. (1989) 7:239–42.
124. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
125. Kimmelman AC. Metabolic dependencies in RAS-driven cancers. Clin Cancer Res Off J Am Assoc Cancer Res. (2015) 21:1828–34. doi: 10.1158/1078-0432.CCR-14-2425
126. Simabuco FM, Morale MG, Pavan ICB, Morelli AP, Silva FR, Tamura RE. p53 and metabolism: from mechanism to therapeutics. Oncotarget. (2018) 9:23780–823. doi: 10.18632/oncotarget.25267
127. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. (2009) 23:537–48. doi: 10.1101/gad.1756509
128. Catanzaro G, Besharat ZM, Miele E, Chiacchiarini M, Po A, Carai A, et al. The miR-139-5p regulates proliferation of supratentorial paediatric low-grade gliomas by targeting the PI3K/AKT/mTORC1 signalling. Neuropathol Appl Neurobiol. (2018) 44:687–706. doi: 10.1111/nan.12479
129. Koh H-J, Toyoda T, Fujii N, Jung MM, Rathod A, Middelbeek RJ-W, et al. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc Natl Acad Sci USA. (2010) 107:15541–6. doi: 10.1073/pnas.1008131107
130. Dávalos A, Goedeke L, Smibert P, Ramírez CM, Warrier NP, Andreo U, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci USA. (2011) 108:9232–7. doi: 10.1073/pnas.1102281108
131. Mersey BD, Jin P, Danner DJ. Human microRNA (miR29b) expression controls the amount of branched chain alpha-ketoacid dehydrogenase complex in a cell. Hum Mol Genet. (2005) 14:3371–7. doi: 10.1093/hmg/ddi368
132. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
133. Chen X, Yang F, Zhang T, Wang W, Xi W, Li Y, et al. MiR-9 promotes tumorigenesis and angiogenesis and is activated by MYC and OCT4 in human glioma. J Exp Clin Cancer Res CR. (2019) 38:99. doi: 10.1186/s13046-019-1078-2
134. Tomasetti M, Lee W, Santarelli L, Neuzil J. Exosome-derived microRNAs in cancer metabolism: possible implications in cancer diagnostics and therapy. Exp Mol Med. (2017) 49:e285. doi: 10.1038/emm.2016.153
135. Tomasetti M, Santarelli L, Neuzil J, Dong L. MicroRNA regulation of cancer metabolism: role in tumour suppression. Mitochondrion. (2014) 19(Pt A):29–38. doi: 10.1016/j.mito.2014.06.004
136. Shen G, Li X, Jia Y, Piazza GA, Xi Y. Hypoxia-regulated microRNAs in human cancer. Acta Pharmacol Sin. (2013) 34:336–41. doi: 10.1038/aps.2012.195
137. Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol. (2019) 7:4. doi: 10.3389/fcell.2019.00004
138. English SG, Hadj-Moussa H, Storey KB. MicroRNAs regulate survival in oxygen-deprived environments. J Exp Biol. (2018) 221(Pt 23):jeb190579. doi: 10.1242/jeb.190579
139. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. (2006) 3:187–97. doi: 10.1016/j.cmet.2006.01.012
140. Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
141. Hagen T. Oxygen versus reactive oxygen in the regulation of HIF-1α: the balance tips. Biochem Res Int. (2012) 2012:436981. doi: 10.1155/2012/436981
142. Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. (2009) 9:1084–101. doi: 10.2174/138955709788922610
143. Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. HIF-1-Dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int J Mol Sci. (2019) 20:238. doi: 10.3390/ijms20020238
144. Singh D, Arora R, Kaur P, Singh B, Mannan R, Arora S. Overexpression of hypoxia-inducible factor and metabolic pathways: possible targets of cancer. Cell Biosci. (2017) 7:62. doi: 10.1186/s13578-017-0190-2
145. Tokuda K, Baron B, Yamashiro C, Kuramitsu Y, Kitagawa T, Kobayashi M, et al. Up-regulation of the pentose phosphate pathway and HIF-1α expression during neural progenitor cell induction following glutamate treatment in rat ex vivo retina. Cell Biol Int. (2019) doi: 10.1002/cbin.11212. [Epub ahead of print].
146. Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. (2007) 12:108–13. doi: 10.1016/j.ccr.2007.07.006
147. Podar K, Anderson KC. A therapeutic role for targeting c-Myc/Hif-1-dependent signaling pathways. Cell Cycle Georget Tex. (2010) 9:1722–8. doi: 10.4161/cc.9.9.11358
148. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. (2007) 11:407–20. doi: 10.1016/j.ccr.2007.04.001
149. Kim J, Gao P, Liu Y-C, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. (2007) 27:7381–93. doi: 10.1128/MCB.00440-07
150. Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci USA. (2018) 115:E10869–78. doi: 10.1073/pnas.1808314115
151. Obacz J, Pastorekova S, Vojtesek B, Hrstka R. Cross-talk between HIF and p53 as mediators of molecular responses to physiological and genotoxic stresses. Mol Cancer. (2013) 12:93. doi: 10.1186/1476-4598-12-93
152. Jin F, Wang Y, Zhu Y, Li S, Liu Y, Chen C, et al. The miR-125a/HK2 axis regulates cancer cell energy metabolism reprogramming in hepatocellular carcinoma. Sci Rep. (2017) 7:3089. doi: 10.1038/s41598-017-03407-3
153. Lin YX, An FM, Zhan Q. [Research progress of microRNAs in regulation of lipid metabolism-related proteins for influencing the pathogenesis of hepatocellular carcinoma]. Zhonghua Gan Zang Bing Za Zhi Zhonghua Ganzangbing Zazhi Chin J Hepatol. (2019) 27:219–22. doi: 10.3760/cma.j.issn.1007-3418.2019.03.011
154. Nie Z-Y, Liu X-J, Zhan Y, Liu M-H, Zhang X-Y, Li Z-Y, et al. miR-140-5p induces cell apoptosis and decreases Warburg effect in chronic myeloid leukemia by targeting SIX1. Biosci Rep. (2019) 39:BSR20190150. doi: 10.1042/BSR20190150
155. Krokker L, Nyíro G, Reiniger L, Darvasi O, Szücs N, Czirják S, et al. Differentially expressed miRNAs influence metabolic processes in pituitary oncocytoma. Neurochem Res. (2019) 44:2360–71. doi: 10.1007/s11064-019-02789-2
156. Xu T, Zhang K, Shi J, Huang B, Wang X, Qian K, et al. MicroRNA-940 inhibits glioma progression by blocking mitochondrial folate metabolism through targeting of MTHFD2. Am J Cancer Res. (2019) 9:250–69.
157. De Santi C, Melaiu O, Bonotti A, Cascione L, Di Leva G, Foddis R, et al. Deregulation of miRNAs in malignant pleural mesothelioma is associated with prognosis and suggests an alteration of cell metabolism. Sci Rep. (2017) 7:3140. doi: 10.1038/s41598-017-02694-0
158. Ling Z, Liu D, Zhang G, Liang Q, Xiang P, Xu Y, et al. miR-361-5p modulates metabolism and autophagy via the Sp1-mediated regulation of PKM2 in prostate cancer. Oncol Rep. (2017) 38:1621–8. doi: 10.3892/or.2017.5852
159. Alfardus H, McIntyre A, Smith S. MicroRNA regulation of glycolytic metabolism in glioblastoma. BioMed Res Int. (2017) 2017:9157370. doi: 10.1155/2017/9157370
160. Guo H, Nan Y, Zhen Y, Zhang Y, Guo L, Yu K, et al. miRNA-451 inhibits glioma cell proliferation and invasion by downregulating glucose transporter 1. Tumour Biol. (2016) 37:13751–61. doi: 10.1007/s13277-016-5219-3
161. Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. (2015) 17:183–94. doi: 10.1038/ncb3094
162. Eichner LJ, Perry M-C, Dufour CR, Bertos N, Park M, St-Pierre J, et al. miR-378(*) mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. (2010) 12:352–61. doi: 10.1016/j.cmet.2010.09.002
163. Hatziapostolou M, Polytarchou C, Iliopoulos D. miRNAs link metabolic reprogramming to oncogenesis. Trends Endocrinol Metab. (2013) 24:361–73. doi: 10.1016/j.tem.2013.03.002
164. Vernieri C, Casola S, Foiani M, Pietrantonio F, de Braud F, Longo V. Targeting cancer metabolism: dietary and pharmacologic interventions. Cancer Discov. (2016) 6:1315–33. doi: 10.1158/2159-8290.CD-16-0615
165. Drakaki A, Hatziapostolou M, Iliopoulos D. Therapeutically targeting microRNAs in liver cancer. Curr Pharm Des. (2013) 19:1180–91. doi: 10.2174/138161213804805658
167. Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP, Vikas P. The repurposing drugs in oncology (ReDO) project. Ecancermedicalscience. (2014) 8:442. doi: 10.3332/ecancer.2014.442
168. Naidu S, Garofalo M. microRNAs: an emerging paradigm in lung cancer chemoresistance. Front Med. (2015) 2:77. doi: 10.3389/fmed.2015.00077
169. Ayers D, Vandesompele J. Influence of microRNAs and long non-coding RNAs in cancer chemoresistance. Genes. (2017) 8:95. doi: 10.3390/genes8030095
170. Oliveras-Ferraros C, Cufí S, Vazquez-Martin A, Torres-Garcia VZ, Del Barco S, Martin-Castillo B, et al. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: induction of the tumor suppressor miRNA let-7a and suppression of the TGFβ-induced oncomiR miRNA-181a. Cell Cycle. (2011) 10:1144–51. doi: 10.4161/cc.10.7.15210
171. Bao B, Wang Z, Ali S, Ahmad A, Azmi AS, Sarkar SH, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res. (2012) 5:355–64. doi: 10.1158/1940-6207.CAPR-11-0299
172. Li W, Yuan Y, Huang L, Qiao M, Zhang Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Res Clin Pract. (2012) 96:187–95. doi: 10.1016/j.diabres.2011.12.028
173. Wang F, Xu J, Liu H, Liu Z, Xia F. Metformin induces apoptosis by microRNA-26a-mediated downregulation of myeloid cell leukaemia-1 in human oral cancer cells. Mol Med Rep. (2016) 13:4671–6. doi: 10.3892/mmr.2016.5143
174. Yang F-Q, Wang J-J, Yan J-S, Huang J-H, Li W, Che J-P, et al. Metformin inhibits cell growth by upregulating microRNA-26a in renal cancer cells. Int J Clin Exp Med. (2014) 7:3289–96.
175. Cifarelli V, Lashinger LM, Devlin KL, Dunlap SM, Huang J, Kaaks R, et al. Metformin and rapamycin reduce pancreatic cancer growth in obese prediabetic mice by distinct microRNA-regulated mechanisms. Diabetes. (2015) 64:1632–42. doi: 10.2337/db14-1132
176. Do MT, Kim HG, Choi JH, Jeong HG. Metformin induces microRNA-34a to downregulate the Sirt1/Pgc-1α/Nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents. Free Radic Biol Med. (2014) 74:21–34. doi: 10.1016/j.freeradbiomed.2014.06.010
177. Xie W, Wang L, Sheng H, Qiu J, Zhang D, Zhang L, et al. Metformin induces growth inhibition and cell cycle arrest by upregulating microRNA34a in renal cancer cells. Med Sci Monit. (2017) 23:29–37. doi: 10.12659/msm.898710
178. Wahdan-Alaswad RS, Cochrane DR, Spoelstra NS, Howe EN, Edgerton SM, Anderson SM, et al. Metformin-induced killing of triple-negative breast cancer cells is mediated by reduction in fatty acid synthase via miRNA-193b. Horm Cancer. (2014) 5:374–89. doi: 10.1007/s12672-014-0188-8
179. Zhao W, Zhang X, Liu J, Sun B, Tang H, Zhang H. miR-27a-mediated antiproliferative effects of metformin on the breast cancer cell line MCF-7. Oncol Rep. (2016) 36:3691–9. doi: 10.3892/or.2016.5199
180. Nangia-Makker P, Yu Y, Vasudevan A, Farhana L, Rajendra SG, Levi E, et al. Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS ONE. (2014) 9:e84369. doi: 10.1371/journal.pone.0084369
181. Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Quirantes R, Segura-Carretero A, Micol V, et al. Metformin lowers the threshold for stress-induced senescence: a role for the microRNA-200 family and miR-205. Cell Cycle. (2012) 11:1235–46. doi: 10.4161/cc.11.6.19665
182. Jiang X, Ma N, Wang D, Li F, He R, Li D, et al. Metformin inhibits tumor growth by regulating multiple miRNAs in human cholangiocarcinoma. Oncotarget. (2015) 6:3178–94. doi: 10.18632/oncotarget.3063
183. Tseng H-W, Li S-C, Tsai K-W. Metformin treatment suppresses melanoma cell growth and motility through modulation of microRNA expression. Cancers. (2019) 11:209. doi: 10.3390/cancers11020209
184. Wang Y, Dai W, Chu X, Yang B, Zhao M, Sun Y. Metformin inhibits lung cancer cells proliferation through repressing microRNA-222. Biotechnol Lett. (2013) 35:2013–9. doi: 10.1007/s10529-013-1309-0
185. Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun. (2012) 3:865. doi: 10.1038/ncomms1859
186. Avci CB, Harman E, Dodurga Y, Susluer SY, Gunduz C. Therapeutic potential of an anti-diabetic drug, metformin: alteration of miRNA expression in prostate cancer cells. Asian Pac J Cancer Prev. (2013) 14:765–8. doi: 10.7314/APJCP.2013.14.2.765
187. Tsao C-J. microRNA-21-mediated regulation of Sprouty2 protein expression enhances the cytotoxic effect of 5-fluorouracil and metformin in colon cancer cells. Int J Mol Med. (2012) 29:920–6. doi: 10.3892/ijmm.2012.910
188. Pulito C, Mori F, Sacconi A, Goeman F, Ferraiuolo M, Pasanisi P, et al. Metformin-induced ablation of microRNA 21-5p releases Sestrin-1 and CAB39L antitumoral activities. Cell Discov. (2017) 3:17022. doi: 10.1038/celldisc.2017.22
189. Devlin C, Greco S, Martelli F, Ivan M. miR-210: more than a silent player in hypoxia. IUBMB Life. (2011) 63:94–100. doi: 10.1002/iub.427
190. Harting T, Stubbendorff M, Willenbrock S, Wagner S, Schadzek P, Ngezahayo A, et al. The effect of dichloroacetate in canine prostate adenocarcinomas and transitional cell carcinomas in vitro. Int J Oncol. (2016) 49:2341–50. doi: 10.3892/ijo.2016.3720
191. Takwi AAL, Li Y, Becker Buscaglia LE, Zhang J, Choudhury S, Park AK, et al. A statin-regulated microRNA represses human c-Myc expression and function. EMBO Mol Med. (2012) 4:896–909. doi: 10.1002/emmm.201101045
192. Kang M, Lee K-H, Lee HS, Jeong CW, Ku JH, Kim HH, et al. Concurrent treatment with simvastatin and NF-κB inhibitor in human castration-resistant prostate cancer cells exerts synergistic anti-cancer effects via control of the NF-κB/LIN28/let-7 miRNA signaling pathway. PLoS ONE. (2017) 12:e0184644. doi: 10.1371/journal.pone.0184644
193. Karlic H, Thaler R, Gerner C, Grunt T, Proestling K, Haider F, et al. Inhibition of the mevalonate pathway affects epigenetic regulation in cancer cells. Cancer Genet. (2015) 208:241–52. doi: 10.1016/j.cancergen.2015.03.008
194. Zheng X, Liu K, Wang X, Zhang R, Li X. MicroRNA-192 acts as a tumor suppressor in colon cancer and simvastatin activates miR-192 to inhibit cancer cell growth. Mol Med Rep. (2019) 19:1753–60. doi: 10.3892/mmr.2019.9808
195. Peng F, Wang J-H, Fan W-J, Meng Y-T, Li M-M, Li T-T, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene. (2018) 37:1062–74. doi: 10.1038/onc.2017.368
196. Bai F, Yu Z, Gao X, Gong J, Fan L, Liu F. Simvastatin induces breast cancer cell death through oxidative stress up-regulating miR-140-5p. Aging. (2019) 11:3198–219. doi: 10.18632/aging.101974
197. Bhardwaj A, Singh H, Trinidad CM, Albarracin CT, Hunt KK, Bedrosian I. The isomiR-140-3p-regulated mevalonic acid pathway as a potential target for prevention of triple negative breast cancer. Breast Cancer Res. (2018) 20:150. doi: 10.1186/s13058-018-1074-z
198. Karataş ÖF, Ittmann M. MiR-33a and statins collaboratively reduce the proliferative capacity of prostate cancer cells. Eur Res J. (2018) 4:266–74. doi: 10.18621/eurj.380619
199. Li P, Yin Y-L, Guo T, Sun X-Y, Ma H, Zhu M-L, et al. Inhibition of aberrant microRNA-133a expression in endothelial cells by statin prevents endothelial dysfunction by targeting GTP cyclohydrolase 1 in vivo. Circulation. (2016) 134:1752–65. doi: 10.1161/CIRCULATIONAHA.116.017949
200. Lam HC, Liu H-J, Baglini CV, Filippakis H, Alesi N, Nijmeh J, et al. Rapamycin-induced miR-21 promotes mitochondrial homeostasis and adaptation in mTORC1 activated cells. Oncotarget. (2017) 8:64714–27. doi: 10.18632/oncotarget.19947
201. Totary-Jain H, Sanoudou D, Ben-Dov IZ, Dautriche CN, Guarnieri P, Marx SO, et al. Reprogramming of the MicroRNA transcriptome mediates resistance to rapamycin. J Biol Chem. (2013) 288:6034–44. doi: 10.1074/jbc.M112.416446
202. Gan H, Lin L, Hu N, Yang Y, Gao Y, Pei Y, et al. Aspirin ameliorates lung cancer by targeting the miR-98/WNT1 axis. Thorac Cancer. (2019) 10:744–50. doi: 10.1111/1759-7714.12992
203. Li X, Gao L, Cui Q, Gary BD, Dyess DL, Taylor W, et al. Sulindac inhibits tumor cell invasion by suppressing NF-κB-mediated transcription of microRNAs. Oncogene. (2012) 31:4979–86. doi: 10.1038/onc.2011.655
204. Kang C. Genome-wide identification of TCF7L2/TCF4 target miRNAs reveals a role for miR-21 in Wnt-driven epithelial cancer. Int J Oncol. (2011) 40:519–26. doi: 10.3892/ijo.2011.1215
205. Saito Y, Suzuki H, Imaeda H, Matsuzaki J, Hirata K, Tsugawa H, et al. The tumor suppressor microRNA-29c is downregulated and restored by celecoxib in human gastric cancer cells. Int J Cancer. (2013) 132:1751–60. doi: 10.1002/ijc.27862
206. Wang J, Zhang X, Shi J, Cao P, Wan M, Zhang Q, et al. Fatty acid synthase is a primary target of MiR-15a and MiR-16-1 in breast cancer. Oncotarget. (2016) 7:78566–76. doi: 10.18632/oncotarget.12479
207. Mishra PJ, Humeniuk R, Mishra PJ, Longo-Sorbello GSA, Banerjee D, Bertino JR. A miR-24 microRNA binding-site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proc Natl Acad Sci USA. (2007) 104:13513–8. doi: 10.1073/pnas.0706217104
208. Song B, Wang Y, Xi Y, Kudo K, Bruheim S, Botchkina GI, et al. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene. (2009) 28:4065–74. doi: 10.1038/onc.2009.274
209. Song B, Wang Y, Titmus MA, Botchkina G, Formentini A, Kornmann M, et al. Molecular mechanism of chemoresistance by miR-215 in osteosarcoma and colon cancer cells. Mol Cancer. (2010) 9:96. doi: 10.1186/1476-4598-9-96
210. Valeri N, Gasparini P, Braconi C, Paone A, Lovat F, Fabbri M, et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2. (hMSH2). Proc Natl Acad Sci USA. (2010) 107:21098–103. doi: 10.1073/pnas.1015541107
211. Akao Y, Noguchi S, Iio A, Kojima K, Takagi T, Naoe T. Dysregulation of microRNA-34a expression causes drug-resistance to 5-FU in human colon cancer DLD-1 cells. Cancer Lett. (2011) 300:197–204. doi: 10.1016/j.canlet.2010.10.006
212. He J, Xie G, Tong J, Peng Y, Huang H, Li J, et al. Overexpression of microRNA-122 re-sensitizes 5-FU-resistant colon cancer cells to 5-FU through the inhibition of PKM2 in vitro and in vivo. Cell Biochem Biophys. (2014) 70:1343–50. doi: 10.1007/s12013-014-0062-x
213. Shah MY, Pan X, Fix LN, Farwell MA, Zhang B. 5-fluorouracil drug alters the microrna expression profiles in MCF-7 breast cancer cells. J Cell Physiol. (2011) 226:1868–78. doi: 10.1002/jcp.22517
214. Park J-K, Lee EJ, Esau C, Schmittgen TD. Antisense Inhibition of microRNA-21 or−221 arrests cell cycle, induces apoptosis, and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma: Pancreas. (2009) 38:e190–9. doi: 10.1097/MPA.0b013e3181ba82e1
215. Almeida MI, Reis RM, Calin GA. Decoy activity through microRNAs: the therapeutic implications. Expert Opin Biol Ther. (2012) 12:1153–9. doi: 10.1517/14712598.2012.693470
216. Kim EJ, Tu M-J, Duan Z, Gonzalez FJ, Bold RJ, Yu A. MicroRNA-1291 effects on pancreatic cancer (PC) cells sensitivity to arginine deprivation and chemotherapy through modulation of ASS1 and GLUT1. J Clin Oncol. (2018) 36:334. doi: 10.1200/JCO.2018.36.4_suppl.334
217. El-Mir M-Y, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
218. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/JCI13505
219. Bridges HR, Jones AJY, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. (2014) 462:475–87. doi: 10.1042/BJ20140620
220. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. (2014) 3:e02242. doi: 10.7554/eLife.02242
221. Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. (2005) 330:1304–5. doi: 10.1136/bmj.38415.708634.F7
222. Lee JH, Kim TI, Jeon SM, Hong SP, Cheon JH, Kim WH. The effects of metformin on the survival of colorectal cancer patients with diabetes mellitus. Int J Cancer. (2012) 131:752–9. doi: 10.1002/ijc.26421
223. Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS ONE. (2012) 7:e33411. doi: 10.1371/journal.pone.0033411
224. Gandini S, Puntoni M, Heckman-Stoddard BM, Dunn BK, Ford L, DeCensi A, et al. Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Prev Res. (2014) 7:867–85. doi: 10.1158/1940-6207.CAPR-13-0424
225. Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. (2009) 27:3297–302. doi: 10.1200/JCO.2009.19.6410
226. Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. (2016) 24:716–27. doi: 10.1016/j.cmet.2016.09.006
227. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. (2004) 101:3329–35. doi: 10.1073/pnas.0308061100
228. Kim HJ, Lee S, Chun KH, Jeon JY, Han SJ, Kim DJ, et al. Metformin reduces the risk of cancer in patients with type 2 diabetes: an analysis based on the Korean national diabetes program cohort. Medicine. (2018) 97:e0036. doi: 10.1097/MD.0000000000010036
229. Lee M-Y, Lin K-D, Hsiao P-J, Shin S-J. The association of diabetes mellitus with liver, colon, lung, and prostate cancer is independent of hypertension, hyperlipidemia, and gout in Taiwanese patients. Metabolism. (2012) 61:242–9. doi: 10.1016/j.metabol.2011.06.020
230. He K, Hu H, Ye S, Wang H, Cui R, Yi L. The effect of metformin therapy on incidence and prognosis in prostate cancer: a systematic review and meta-analysis. Sci Rep. (2019) 9:2218. doi: 10.1038/s41598-018-38285-w
231. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JMM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. (2009) 32:1620–5. doi: 10.2337/dc08-2175
232. Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: an old metabolic gatekeeper regulated by new pathways and pharmacological agents: pyruvate dehydrogenase complex in cancer. Int J Cancer. (2016) 138:809–17. doi: 10.1002/ijc.29564
233. Kim K, Kim JK, Han SH, Lim J-S, Kim KI, Cho DH, et al. Adiponectin is a negative regulator of NK cell cytotoxicity. J Immunol. (2006) 176:5958–64. doi: 10.4049/jimmunol.176.10.5958
234. Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. (2008) 99:989–94. doi: 10.1038/sj.bjc.6604554
235. Wen S, Zhu D, Huang P. Targeting cancer cell mitochondria as a therapeutic approach. Future Med Chem. (2013) 5:53–67. doi: 10.4155/fmc.12.190
236. Bingham PM, Stuart SD, Zachar Z. Lipoic acid and lipoic acid analogs in cancer metabolism and chemotherapy. Expert Rev Clin Pharmacol. (2014) 7:837–46. doi: 10.1586/17512433.2014.966816
237. Liang Y, Hou L, Li L, Li L, Zhu L, Wang Y, et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene. (2019) doi: 10.1038/s41388-019-1035-8
238. Bjarnadottir O, Romero Q, Bendahl P-O, Jirström K, Rydén L, Loman N, et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat. (2013) 138:499–508. doi: 10.1007/s10549-013-2473-6
239. Lin JJ, Ezer N, Sigel K, Mhango G, Wisnivesky JP. The effect of statins on survival in patients with stage IV lung cancer. Lung Cancer. (2016) 99:137–42. doi: 10.1016/j.lungcan.2016.07.006
240. Hung M-S, Chen I-C, Lee C-P, Huang R-J, Chen P-C, Tsai Y-H, et al. Statin improves survival in patients with EGFR-TKI lung cancer: a nationwide population-based study. PLoS ONE. (2017) 12:e0171137. doi: 10.1371/journal.pone.0171137
241. Newman A, Clutterbuck RD, Powles RL, Catovsky D, Millar JL. A comparison of the effect of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors simvastatin, lovastatin and pravastatin on leukaemic and normal bone marrow progenitors. Leuk Lymphoma. (1997) 24:533–7. doi: 10.3109/10428199709055590
242. Dimitroulakos J, Thai S, Wasfy GH, Hedley DW, Minden MD, Penn LZ. Lovastatin induces a pronounced differentiation response in acute myeloid leukemias. Leuk Lymphoma. (2000) 40:167–78. doi: 10.3109/10428190009054894
243. Denoyelle C. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis. (2001) 22:1139–48. doi: 10.1093/carcin/22.8.1139
244. Mannello F, Tonti GA. Statins and breast cancer: may matrix metalloproteinase be the missing link. Cancer Invest. (2009) 27:466–70. doi: 10.1080/07357900802491444
245. Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci. (2015) 36:124–35. doi: 10.1016/j.tips.2014.11.004
246. Tolcher AW, Bendell JC, Papadopoulos KP, Burris HA, Patnaik A, Jones SF, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol. (2015) 26:58–64. doi: 10.1093/annonc/mdu482
247. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
248. Zaytseva YY, Rychahou PG, Gao T, Lee EY, Weiss HL, Heuer TS, et al. Abstract 1010: evaluation of small-molecule FASN inhibitors in preclinical models of colorectal cancer. Cancer Res. (2016) 76:1010. doi: 10.1158/1538-7445.AM2016-1010
249. Brenner A, Falchook G, Patel M, Infante J, Arkenau H-T, Dean E, et al. Abstract P6-11-09: heavily pre-treated breast cancer patients show promising responses in the first in human study of the first-In-class fatty acid synthase (FASN) inhibitor, TVB-2640 in combination with paclitaxel. Cancer Res. (2017) 77(Suppl. 4). doi: 10.1158/1538-7445.SABCS16-P6-11-09
250. Zaytseva YY, Rychahou PG, Le A-T, Scott TL, Flight RM, Kim JT, et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget. (2018) 9: 24787–800. doi: 10.18632/oncotarget.25361
251. Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. (2002) 109:1321–6. doi: 10.1172/JCI0214955
252. Paez Espinosa EV, Murad JP, Khasawneh FT. Aspirin: pharmacology and clinical applications. Thrombosis. (2012) 2012:1–15. doi: 10.1155/2012/173124
253. Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. (2012) 336:918–22. doi: 10.1126/science.1215327
254. Hammerlindl H, Ravindran Menon D, Hammerlindl S, Emran AA, Torrano J, Sproesser K, et al. Acetylsalicylic acid governs the effect of sorafenib in RAS -mutant cancers. Clin Cancer Res. (2018) 24:1090–102. doi: 10.1158/1078-0432.CCR-16-2118
255. Drew DA, Cao Y, Chan AT. Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer. (2016) 16:173–86. doi: 10.1038/nrc.2016.4
256. Rothwell PM, Fowkes FGR, Belch JFF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet Lond Engl. (2011) 377:31–41. doi: 10.1016/S0140-6736(10)62110-1
257. Simon TG, Ma Y, Ludvigsson JF, Chong DQ, Giovannucci EL, Fuchs CS, et al. Association between aspirin use and risk of hepatocellular carcinoma. JAMA Oncol. (2018) 4:1683–90. doi: 10.1001/jamaoncol.2018.4154
258. Barnard ME, Poole EM, Curhan GC, Eliassen AH, Rosner BA, Terry KL, et al. Association of analgesic use with risk of ovarian cancer in the nurses' health studies. JAMA Oncol. (2018) 4:1675–82. doi: 10.1001/jamaoncol.2018.4149
259. Pinheiro C, Longatto-Filho A, Scapulatempo C, Ferreira L, Martins S, Pellerin L, et al. Increased expression of monocarboxylate transporters 1, 2, and 4 in colorectal carcinomas. Virchows Arch. (2008) 452:139–46. doi: 10.1007/s00428-007-0558-5
260. Pinheiro C, Albergaria A, Paredes J, Sousa B, Dufloth R, Vieira D, et al. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma: MCT1 in breast cancer. Histopathology. (2010) 56:860–7. doi: 10.1111/j.1365-2559.2010.03560.x
261. Bola BM, Chadwick AL, Michopoulos F, Blount KG, Telfer BA, Williams KJ, et al. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol Cancer Ther. (2014) 13:2805–16. doi: 10.1158/1535-7163.MCT-13-1091
262. Halford SER, Jones P, Wedge S, Hirschberg S, Katugampola S, Veal G, et al. A first-in-human first-in-class (FIC) trial of the monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with advanced solid tumours. J Clin Oncol. (2017) 35:2516. doi: 10.1200/JCO.2017.35.15_suppl.2516
263. Mehrmohamadi M, Jeong SH, Locasale JW. Molecular features that predict the response to antimetabolite chemotherapies. Cancer Metab. (2017) 5:8. doi: 10.1186/s40170-017-0170-3
264. Showalter SL, Showalter TN, Witkiewicz A, Havens R, Kennedy EP, Hucl T, et al. Evaluating the drug-target relationship between thymidylate synthase expression and tumor response to 5-fluorouracil: Is it time to move forward? Cancer Biol Ther. (2008) 7:986–94. doi: 10.4161/cbt.7.7.6181
265. Costales MG, Haga CL, Velagapudi SP, Childs-Disney JL, Phinney DG, Disney MD. Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J Am Chem Soc. (2017) 139:3446–55. doi: 10.1021/jacs.6b11273
266. Kelly TJ, Souza AL, Clish CB, Puigserver P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol Cell Biol. (2011) 31:2696–706. doi: 10.1128/MCB.01242-10
267. Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, et al. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. (1957) 179:663–6. doi: 10.1038/179663a0
268. Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE. Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol. (2006) 24:2903–9. doi: 10.1200/JCO.2005.05.0245
269. Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev. (2009) 109:2880–93. doi: 10.1021/cr900028p
270. Hanna J, Hossain GS, Kocerha J. The potential for microRNA therapeutics and clinical research. Front Genet. (2019) 10:478. doi: 10.3389/fgene.2019.00478
271. Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. (2010) 9:775–89. doi: 10.1038/nrd3179
272. van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Mol Med. (2014) 6:851–64. doi: 10.15252/emmm.201100899
273. Shah MY, Ferrajoli A, Sood AK, Lopez-Berestein G, Calin GA. microRNA therapeutics in cancer - an emerging concept. EBioMedicine. (2016) 12:34–42. doi: 10.1016/j.ebiom.2016.09.017
274. Yingchoncharoen P, Kalinowski DS, Richardson DR. Lipid-based drug delivery systems in cancer therapy: what is available and what is yet to come. Pharmacol Rev. (2016) 68:701–87. doi: 10.1124/pr.115.012070
275. Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang H-W, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. (2009) 137:1005–17. doi: 10.1016/j.cell.2009.04.021
276. MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP, et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell. (2007) 11:431–45. doi: 10.1016/j.ccr.2007.03.012
277. Beg MS, Brenner AJ, Sachdev J, Borad M, Kang Y-K, Stoudemire J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. (2017) 35:180–8. doi: 10.1007/s10637-016-0407-y
278. Reid G, Kao SC, Pavlakis N, Brahmbhatt H, MacDiarmid J, Clarke S, et al. Clinical development of TargomiRs, a miRNA mimic-based treatment for patients with recurrent thoracic cancer. Epigenomics. (2016) 8:1079–85. doi: 10.2217/epi-2016-0035
279. van Zandwijk N, Pavlakis N, Kao SC, Linton A, Boyer MJ, Clarke S, et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. (2017) 18:1386–96. doi: 10.1016/S1470-2045(17)30621-6
280. Seto AG, Beatty X, Lynch JM, Hermreck M, Tetzlaff M, Duvic M, et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br J Haematol. (2018) 183:428–44. doi: 10.1111/bjh.15547
281. Querfeld C, Pacheco T, Foss FM, Halwani AS, Porcu P, Seto AG, et al. Preliminary results of a phase 1 trial evaluating MRG-106, a synthetic microRNA antagonist (LNA antimiR) of microRNA-155, in patients with CTCL. Blood. (2016) 128:1829. doi: 10.1182/blood.V128.22.1829.1829
282. Ghosh D, Nandi S, Bhattacharjee S. Combination therapy to checkmate Glioblastoma: clinical challenges and advances. Clin Transl Med. (2018) 7:33. doi: 10.1186/s40169-018-0211-8
283. Haque I, Banerjee S, Mehta S, De A, Majumder M, Mayo MS, et al. Cysteine-rich 61-connective tissue growth factor-nephroblastoma-overexpressed 5 (CCN5)/Wnt-1-induced signaling protein-2 (WISP-2) regulates microRNA-10b via hypoxia-inducible factor-1α-TWIST signaling networks in human breast cancer cells. J Biol Chem. (2011) 286:43475–85. doi: 10.1074/jbc.M111.284158
284. Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. (2015) 125:141–56. doi: 10.1172/JCI75852
285. Steeg PS, Theodorescu D. Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol. (2008) 5:206–19. doi: 10.1038/ncponc1066
286. Chakraborty C, Wen Z-H, Agoramoorthy G, Lin C-S. Therapeutic microRNA delivery strategies with special emphasis on cancer therapy and tumorigenesis: current trends and future challenges. Curr Drug Metab. (2016) 17:469–77. doi: 10.2174/1389200217666160126142408
287. Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. (2004) 56:1649–59. doi: 10.1016/j.addr.2004.02.014
288. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. (2017) 168:657–69. doi: 10.1016/j.cell.2016.12.039
289. Larsson SC, Wolk A. Overweight and obesity and incidence of leukemia: a meta-analysis of cohort studies: overweight and obesity and incidence of leukemia. Int J Cancer. (2008) 122:1418–21. doi: 10.1002/ijc.23176
290. Roberts DL, Dive C, Renehan AG. Biological mechanisms linking obesity and cancer risk: new perspectives. Annu Rev Med. (2010) 61:301–16. doi: 10.1146/annurev.med.080708.082713
291. Castillo JJ, Reagan JL, Ingham RR, Furman M, Dalia S, Merhi B, et al. Obesity but not overweight increases the incidence and mortality of leukemia in adults: a meta-analysis of prospective cohort studies. Leuk Res. (2012) 36:868–75. doi: 10.1016/j.leukres.2011.12.020
292. Lehr S, Hartwig S, Sell H. Adipokines: a treasure trove for the discovery of biomarkers for metabolic disorders. Proteomics Clin Appl. (2012) 6:91–101. doi: 10.1002/prca.201100052
293. Arner P, Kulyté A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat Rev Endocrinol. (2015) 11:276–88. doi: 10.1038/nrendo.2015.25
294. Iacomino G, Siani A. Role of microRNAs in obesity and obesity-related diseases. Genes Nutr. (2017) 12:23. doi: 10.1186/s12263-017-0577-z
295. Kasiappan R, Rajarajan D. Role of microRNA regulation in obesity-associated breast cancer: nutritional perspectives. Adv Nutr Int Rev J. (2017) 8:868–88. doi: 10.3945/an.117.015800
296. Ayers D, Boughanem H, Macías-González M. Epigenetic influences in the obesity/colorectal cancer axis: a novel theragnostic avenue. J Oncol. (2019) 2019:7406078. doi: 10.1155/2019/7406078
298. Tsilimigras MCB, Fodor A, Jobin C. Carcinogenesis and therapeutics: the microbiota perspective. Nat Microbiol. (2017) 2:17008. doi: 10.1038/nmicrobiol.2017.8
299. Elinav E, Garrett WS, Trinchieri G, Wargo J. The cancer microbiome. Nat Rev Cancer. (2019) 19:371–6. doi: 10.1038/s41568-019-0155-3
300. Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, Wargo JA. The microbiome, cancer, and cancer therapy. Nat Med. (2019) 25:377–88. doi: 10.1038/s41591-019-0377-7
301. Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science. (2006) 312:1355–9. doi: 10.1126/science.1124234
302. Philipp B. Bacterial degradation of bile salts. Appl Microbiol Biotechnol. (2011) 89:903–15. doi: 10.1007/s00253-010-2998-0
303. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. (2013) 13:800–12. doi: 10.1038/nrc3610
304. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486:207–14. doi: 10.1038/nature11234
305. McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, et al. American gut: an open platform for citizen science microbiome research. mSystems. (2018) 3:e00031-18. doi: 10.1128/mSystems.00031-18
306. Francescone R, Hou V, Grivennikov SI. Microbiome, inflammation, and cancer. Cancer J. (2014) 20:181–9. doi: 10.1097/PPO.0000000000000048
307. Irrazábal T, Belcheva A, Girardin SE, Martin A, Philpott DJ. The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell. (2014) 54:309–20. doi: 10.1016/j.molcel.2014.03.039
308. Chen J, Pitmon E, Wang K. Microbiome, inflammation and colorectal cancer. Semin Immunol. (2017) 32:43–53. doi: 10.1016/j.smim.2017.09.006
309. Mischke M, Plösch T. The gut microbiota and their metabolites: potential implications for the host epigenome. In: A. Schwiertz, editor. Microbiota of the Human Body. Cham: Springer International Publishing (2016). p. 33–44. doi: 10.1007/978-3-319-31248-4_3
310. Bhat MI, Kapila R. Dietary metabolites derived from gut microbiota: critical modulators of epigenetic changes in mammals. Nutr Rev. (2017) 75:374–89. doi: 10.1093/nutrit/nux001
311. Ye J, Wu W, Li Y, Li L. Influences of the gut microbiota on DNA methylation and histone modification. Dig Dis Sci. (2017) 62:1155–64. doi: 10.1007/s10620-017-4538-6
312. Williams MR, Stedtfeld RD, Tiedje JM, Hashsham SA. MicroRNAs-based inter-domain communication between the host and members of the gut microbiome. Front Microbiol. (2017) 8:1896. doi: 10.3389/fmicb.2017.01896
313. Yuan C, Subramanian S. microRNA-mediated tumor–microbiota metabolic interactions in colorectal cancer. DNA Cell Biol. (2019) 38:281–5. doi: 10.1089/dna.2018.4579
314. Warburg O. On the Origin of Cancer Cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
315. Hu S, Liu L, Chang EB, Wang J-Y, Raufman J-P. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol Cancer. (2015) 14:180. doi: 10.1186/s12943-015-0450-x
316. Dews M, Fox JL, Hultine S, Sundaram P, Wang W, Liu YY, et al. The Myc-miR-17 92 axis blunts TGF signaling and production of multiple TGF -dependent antiangiogenic factors. Cancer Res. (2010) 70:8233–46. doi: 10.1158/0008-5472.CAN-10-2412
317. Zhang G, Zhou H, Xiao H, Liu Z, Tian H, Zhou T. MicroRNA-92a functions as an oncogene in colorectal cancer by targeting PTEN. Dig Dis Sci. (2014) 59:98–107. doi: 10.1007/s10620-013-2858-8
318. Ke T-W, Wei P-L, Yeh K-T, Chen WT-L, Cheng Y-W. MiR-92a promotes cell metastasis of colorectal cancer through PTEN-mediated PI3K/AKT pathway. Ann Surg Oncol. (2015) 22:2649–55. doi: 10.1245/s10434-014-4305-2
319. Yuan C, Burns MB, Subramanian S, Blekhman R. Interaction between host microRNAs and the gut microbiota in colorectal cancer. mSystems. (2018) 3:e00205-17. doi: 10.1128/mSystems.00205-17
320. Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor–κB, and up-regulating expression of microRNA-21. Gastroenterology. (2017) 152:851–66.e24. doi: 10.1053/j.gastro.2016.11.018
321. Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. (2014) 63:1932–42. doi: 10.1136/gutjnl-2013-305257
322. Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. (2014) 5:675–80. doi: 10.4161/19490976.2014.969989
323. Baek SH. A novel link between SUMO modification and cancer metastasis. Cell Cycle. (2006) 5:1492–5. doi: 10.4161/cc.5.14.3008
324. Teng Y, Ren Y, Sayed M, Hu X, Lei C, Kumar A, et al. Plant-derived exosomal microRNAs shape the gut microbiota. Cell Host Microbe. (2018) 24:637–52.e8. doi: 10.1016/j.chom.2018.10.001
325. Zhang L, Hou D, Chen X, Li D, Zhu L, Zhang Y, et al. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res. (2012) 22:107–26. doi: 10.1038/cr.2011.158
Keywords: microRNAs, reprogramming metabolism, regulation, therapeutic targets, tumor cell metabolism
Citation: Pedroza-Torres A, Romero-Córdoba SL, Justo-Garrido M, Salido-Guadarrama I, Rodríguez-Bautista R, Montaño S, Muñiz-Mendoza R, Arriaga-Canon C, Fragoso-Ontiveros V, Álvarez-Gómez RM, Hernández G and Herrera LA (2019) MicroRNAs in Tumor Cell Metabolism: Roles and Therapeutic Opportunities. Front. Oncol. 9:1404. doi: 10.3389/fonc.2019.01404
Received: 23 September 2019; Accepted: 27 November 2019;
Published: 11 December 2019.
Edited by:
Luis Enrique Gomez-Quiroz, Universidad Autónoma Metropolitana, MexicoReviewed by:
Barbara Marengo, University of Genoa, ItalyCopyright © 2019 Pedroza-Torres, Romero-Córdoba, Justo-Garrido, Salido-Guadarrama, Rodríguez-Bautista, Montaño, Muñiz-Mendoza, Arriaga-Canon, Fragoso-Ontiveros, Álvarez-Gómez, Hernández and Herrera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abraham Pedroza-Torres, YWJyYW5lZXRAZ21haWwuY29t; Luis A. Herrera, bWV0aWxAaG90bWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.