 Wenhui Zheng1
Wenhui Zheng1 Shuo Wang
Shuo Wang Ming Gu
Ming Gu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 12 December 2019
Sec. Cancer Molecular Targets and Therapeutics
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01403
Methyltransferase-like 3 (METTL3), a predominantly catalytic enzyme in the N6-methyladenosine (m6A) methyltransferase system, is dysregulated and plays a dual role (oncogene or tumor suppressor) in different human cancers. The expression and pro- or anticancer role of METTL3 in different cancers remain controversial. METTL3 is implicated in many aspects of tumor progression, including tumorigenesis, proliferation, invasion, migration, cell cycle, differentiation, and viability. Most underlying mechanisms involve multiple signaling pathways that rely on m6A-dependent modification. However, METTL3 can also modulate the cancer process by directly promoting the translation of oncogenes via interaction with the translation initiation machinery through recruitment of eukaryotic translation initiation factor 3 subunit h (eIF3h). In this review, we summarized the current evidence on METTL3 in diverse human malignancies and its potential as a prognostic/ therapeutic target.
Chemical modifications of nucleobases are critical for controlling gene expression at different levels, which subsequently induces changes in protein translation and modulates signaling pathways. N6-methyladenosine (m6A) modification of various RNAs, including eukaryotic messenger RNAs (mRNAs) (1), microRNAs (miRNAs) (2), and long non-coding RNAs (lncRNAs) (3) is enriched in near stop codon and 3′ untranslated terminal region (UTR) (4) and translated near 5′ UTR in a cap-independent manner (5) and has been considered one of the most ubiquitous, reversible and abundant internal modifications on RNA molecules.
In recent years, substantial progress has been made in understanding m6A modifications in various metabolic and infectious diseases, as well as cancer (6). In addition, a variety of studies have shown that m6A modification plays promotive or inhibitory roles in various cancers (7). Upon alteration of m6A regulatory genes or a change in the expression of proteins related to m6A methylation, m6A impacts the initiation and progression of various human malignancies through diverse mechanisms and is involved in different biological processes, including viral infection (8), immune responses, tissue renewal (8–10), stem cell differentiation, and motility (11), and therefore exerts a profound impact on cancer development (8).
m6A deposition is accomplished by a methyltransferase (MTase) complex referred to as “writers.” In mammalian cells, m6A MTase activity on mRNA requires at least two separate proteins: MT-A (200 KDa) and MT-B (800 KDa). MT-A is a multimeric protein that contains a 70-KDa S-adenosylmethionine-binding subunit referred to as MT-A70, which is also known as METTL3 (12). METTL3 is a predominantly catalytic enzyme of m6A MTase systems and belongs to the class I MTase family. It usually forms a stable heterodimeric complex with METTL14 (13). Along with the m6A-METTL associated complex composed of Wilms tumor 1-associated protein (WTAP), RNA binding motif protein 15 (RBM15), zinc finger CCCH-type containing 13 (ZC3H13), vir like m6A MTase associated (VIRMA), and HAKAI, they catalyze the formation of m6A. The fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5) are categorized as “erasers” which selectively remove the methyl code from target RNAs and reverse the methylation. YT521-B homology (YTH) domain-containing protein, eukaryotic translation initiation factor 3 (eIF3), IGF2 mRNA binding proteins (IGF2BP) families, and heterogeneous nuclear ribonucleoprotein (HNRNP) protein families are categorized as “readers” which decode m6A methylation and generate a functional signal (6, 14). Thus, m6A may affect RNAs splicing, stability, transcription, and translation (15) as shown in Figure 1.
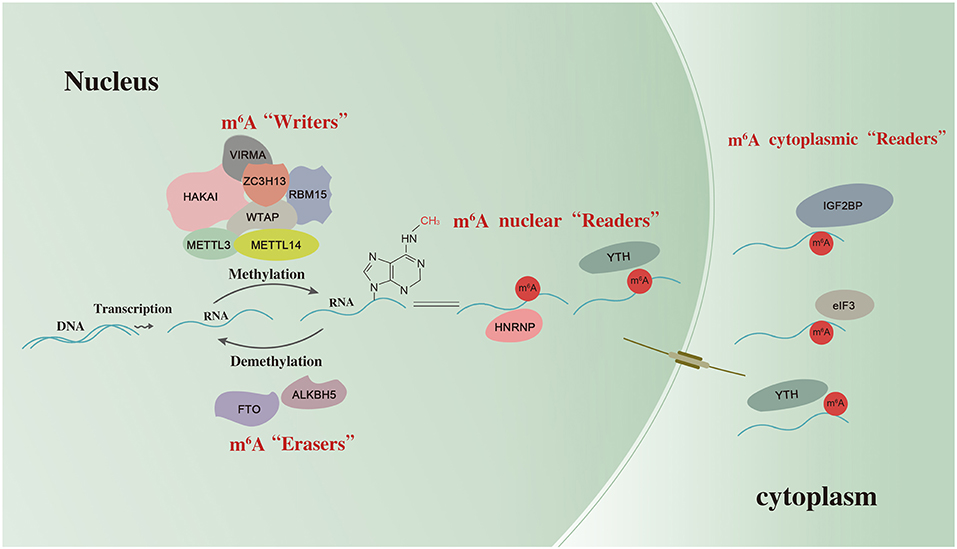
Figure 1. The scheme of m6A RNA methylation. m6A RNA methylation is regulated by “writers”, “erasers,” and “readers”. METTL3, methyltransferase-like 3; METTL14, methyltransferase like 14; WTAP, Wilms tumor 1-associated protein; RBM15, RNA binding motif protein 15; ZC3H13, zinc finger CCCH-type containing 13; VIRMA, vir like m6A methyltransferase associated; FTO, the fat mass and obesity-associated protein; ALKBH5, alkB homolog 5; YTH, YT521-B homology domain-containing protein; eIF3, eukaryotic translation initiation factor 3; IGF2BP, IGF2 mRNA binding proteins; HNRNP, heterogeneous nuclear ribonucleoprotein.
METTL3 plays a pivotal role in all stages of the RNA life cycle involving m6A, including pre-mRNA splicing (16), nuclear export (17), translation regulation (18), mRNA decay (19), and miRNA processing (20). With the help of other components of “writers,” the METTL3 catalytic process modifies most m6A sites and has been considered the most common m6A pathway, especially in mRNA (21). METTL3 has been demonstrated to be dysregulated in diverse human malignancies and involved in many aspects of carcinogenesis. Zhou et al. showed that METTL3 and METTL14 exhibited higher expression in clear cell renal cell carcinoma than in normal samples and that patients with deletion of METTL3 had poorer overall survival (OS) and disease-free survival (DFS) (22). Liu et al. revealed that the proliferation, viability, and migration of gastric cancer cells with METTL3 silencing in vitro were significantly inhibited compared with those of control cells (23). This research group also noted that METTL3 expression was associated with biological processes, including adipogenesis, the mTOR pathway, and reactive oxygen species (24). Taketo et al. found that METTL3-deleted cells showed higher sensitivity to anticancer reagents, indicating that METTL3 may promote drug resistance in pancreatic cancer (25). The abovementioned study suggested that METTL3 plays a role in oncogenesis. High expression of METTL3 may predict poor survival and drug resistance in patients.
In contrast, Deng et al. showed that high expression of METTL3 was significantly associated with longer survival time and METTL3 played a tumor-suppressive role in colorectal cancer (26).
The role of METTL3 in cancer cells is controversial (27). The contradictory conclusions reached in previous studies must be related to the differences in the mechanisms of origin of different cancers. In this review, we summarized the recent advances made in relation to METTL3 dysregulation and its dual role coupled with the underlying mechanisms in various human cancers.
In the majority of cancer research, METTL3 has been found to be upregulated and to play an oncogenic role accompanied by increased m6A levels compared with those in normal tissues or cell lines. However, some pieces of research have yielded opposite results even in the same cancer type, as shown in Table 1.
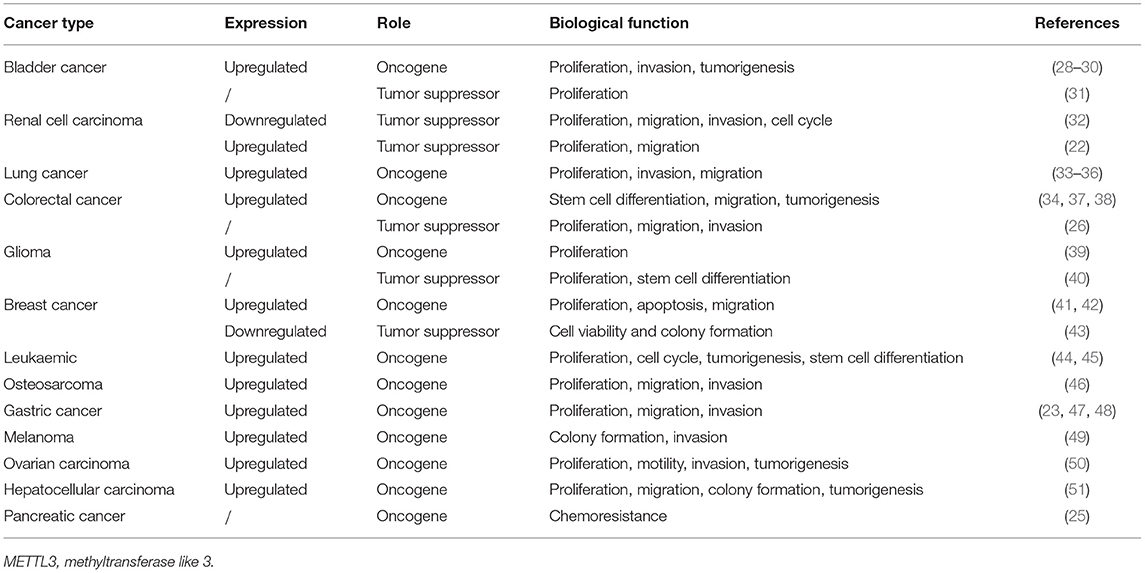
Table 1. Expression, clinical significance, and biological function of METTL3 in various cancers.
Recent studies noted that METTL3 was drastically upregulated in bladder cancer tissues and was related to tumor histological grade. Patients with high expression of METTL3 had poor prognosis and reduced survival time (28–30). Knockdown of METTL3 significantly reduced bladder cancer cell invasion, proliferation, and survival in vitro and tumorigenicity in vivo (31). The above studies proved that METTL3 acts as an oncogene in bladder cancer.
However, Zhao et al. showed that deletion of METTL3 significantly increased the proliferation of bladder cancer cell line 5637. Wild-type METTL3 successfully restored the normal growth rate and somatic mutations in METTL3 may disrupt the m6A methylation process and promote cancer cell growth. METTL3 acts as a tumor suppressor gene in bladder cancer (31). Similarly, Li et al. showed that METTL3 expression was lower in renal cell carcinoma samples compared with adjacent non-tumor samples. Negative METTL3 expression was significantly associated with larger tumor sizes and higher histological grade. Patients with high METTL3 expression had obviously extended survival time. Moreover, knockdown of METTL3 appreciably promoted cell proliferation, migration, and invasion and induced G0/G1 arrest, suggesting that METTL3 may act as a tumor suppressor in renal cell carcinoma (32).
METTL3 was upregulated in primary human lung adenocarcinomas compared with adjacent normal tissues, and METTL3 depletion suppressed the growth of lung cancer xenografts in vivo (33, 34). In addition, Du et al. revealed that METTL3 expression was higher in non-small cell lung carcinoma tissues than in adjacent tissues (35). METTL3 promotes the proliferation, survival, migration, and invasion of human lung cancer cells (34, 36). Collectively, these studies on METTL3 in lung cancer suggest the oncogenic role of METTL3.
Liu and colleagues compared the m6A-related genes in colorectal cancer and found that most m6A-related genes, including METTL3, were upregulated, except METTL14, YTHDF3, and ALKBH5 (37). METTL3 expression was consistently elevated in recurrent colorectal cancer, matched lymph node, and metastatic liver tissues. Colorectal cancer patients with high METTL3 expression had reduced OS and DFS times (37, 38). Knockdown of METTL3 in colorectal cancer cells significantly inhibited tumorigenesis and metastasis, cell self-renewal, and the frequency and migration of stem cells in vitro and in vivo (34), suggesting the oncogenic role of METTL3 in colorectal cancer.
However, Deng et al. noted that positive expression of METTL3 inhibits cell proliferation, migration, and invasion in colorectal cancer (26). Negative expression of METTL3 was significantly correlated with larger tumor size and metastasis. Multivariate analysis indicated that METTL3 expression status is an independent prognostic factor for DFS (26). In addition, knockdown of METTL3 promoted tumor proliferation and metastasis. The tumor suppressor role of METTL3 in this research was related to the P38/ERK pathway (26).
METTL3 expression was found to be elevated in glioma stem-like cells and attenuated during differentiation (39). Glioblastoma tumors exhibited elevated levels of METTL3 transcripts, and silencing METTL3 inhibited tumor growth coupled with prolonged survival of mice in vivo (39), suggesting the oncogenic role of METTL3 in glioblastoma. However, in another study on the function of m6A in glioblastoma, METTL3 overexpression inhibited stem cell growth and self-renewal, accompanied by suppressed tumor progression (40).
Recent studies showed that METTL3 was upregulated in breast cancer tissue and cells. Knockdown of METTL3 reduced cell proliferation and accelerated apoptosis and migration by targeting Bcl-2, suggesting the oncogenic role of METTL3 in breast cancer (41, 42). However, another study that explored key m6A-related enzymes via combined analysis of data from the ONCOMINE and The Cancer Genome Atlas databases with 36 pairs of breast cancer and adjacent non-cancerous tissues, revealed that the expression of all m6A methylases, including METTL3, was reduced in breast cancer. These researchers also noted that METTL3 and METTL14 were upregulated in the normal breast-like and luminal breast cancer subtypes compared with the basal-like and HER2-overexpressing types. MTase overexpression induced m6A expression and inhibited tumor cell viability and colony formation. Thus, METTL3 acts as a tumor suppressor (43).
Vu et al. showed that METTL3 mRNA and protein are expressed more abundantly in acute myeloid leukemia cells than in healthy human hematopoietic stem/progenitor cells or other types of tumor cells. Knockdown of METTL3 promoted cell differentiation accompanied by reduced cell proliferation (44). Moreover, another study revealed that knockdown of METTL3 results in differentiation of leukemic cells, failure to establish leukemia in immunodeficient mice, coupled with cell cycle arrest (45). Collectively, research in leukemia suggests that METTL3 is upregulated and significantly related to tumor cell differentiation, tumor formation, the cell cycle, and proliferation.
METTL3 and m6A were upregulated in human osteosarcoma (46), gastric cancer (23, 47, 48), melanoma (49), ovarian carcinoma (50), and hepatocellular carcinoma (51). Its expression level gradually increased with the increasing tumor stage and grade. METTL3 was an indicator of poor prognosis and METTL3 silencing inhibited the proliferation, migration, and invasion abilities of cells, colony formation, and motility (24, 47–52), demonstrating that METTL3 plays an oncogenic role in these cancers. Taketo et al. reported that a METTL3-knockdown pancreatic cancer cell line showed higher sensitivity to anticancer agents such as gemcitabine, 5-fluorouracil, cisplatin, and irradiation, suggesting that METTL3 is a potential target for the enhancement of therapeutic efficacy (25).
As mentioned above, METTL3 is implicated in many aspects of human cancer cell progression, which has prompted many researchers to explore its possible molecular mechanism. Next, we summarize the results of studies on the main pathways of METTL3 in various cancers as shown in Figure 2.
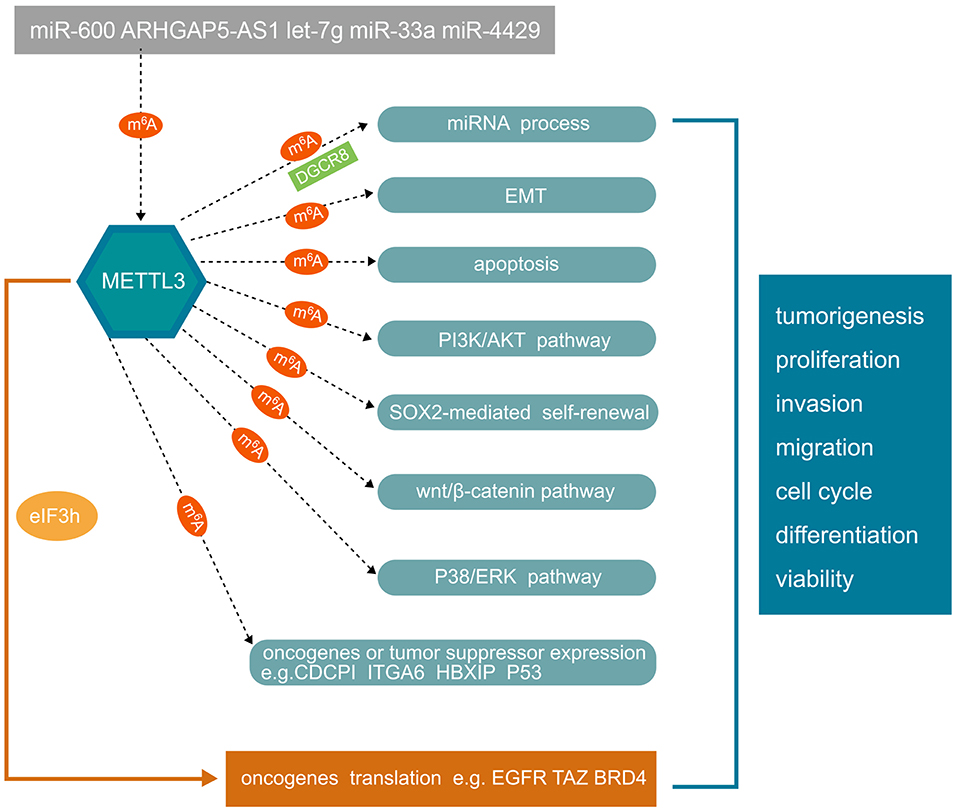
Figure 2. The mechanisms of METTL3 involved in human cancer progression. Non-coding RNAs can regulate METTL3 expression. Conversely, METTL3 can regulate miRNA process in m6A manner. METTL3 plays an important role in tumorigenesis, tumor cell proliferation, invasion, migration, cell cycle, stem cell differentiation, and viability in an m6A-dependent manner. The underlying mechanisms involve EMT, apoptosis, PI3K/AKT, SOX2-mediated self-renewal, wnt/β-catenin, and P38/ERK pathways. In addition, METTL3 can also regulate the expression of oncogenes or tumor suppressors in an m6A-dependent manner. METTL3 can also directly regulate oncogene translation independently of m6A by recruiting eIF3h. METTL3, methyltransferase-like 3; EMT, epithelial-mesenchymal transition; PI3K, phosphatidylinositol 3-kinase; SOX2, SRY-box transcription factor 2; ERK, extracellular regulated MAP kinase; CDCP1, CUB domain containing protein 1; ITGA6, integrin subunit alpha 6; HBXIP, mammalian hepatitis B X-interacting protein; EGFR, epidermal growth factor receptor; BRD4, bromodomain containing 4.
miRNAs or lncRNAs can modulate the expression of oncogenes or tumor suppressors by targeting the 3′UTR of METTL3 in an m6A-dependent manner. Wei et al. showed that miR-600 can reverse the effect of METTL3 overexpression, as well as overexpression of the related genes in the PI3K/AKT and β-catenin/stat3 signaling pathways (36). The lncRNA ARHGAP5-AS1 can stimulate m6A modification of ARHGAP5 mRNA to stabilize ARHGAP5 mRNA, which is upregulated in gastric cancer, in the cytoplasm to promote chemoresistance by recruiting METTL3 (52). Hepatitis B X-interacting protein (HBXIP) upregulates METTL3 in breast cancer cells by inhibiting the miRNA let-7g, which downregulates the expression of METTL3 by targeting its 3′UTR. Conversely, METTL3 promotes the expression of HBXIP through m6A modification (42). Du et al. showed that miR-33a can attenuate non-small cell lung cancer cell proliferation by targeting the 3′UTR of METTL3 mRNA (35). Moreover, miR4429 inhibits gastric cancer progression by targeting METTL3 to hinder m6A-induced stabilization of SEC62 (53).
Alarcon et al. found that METTL3 promotes the maturation of miRNAs by interacting with the microprocessor protein DGCR8 (2, 20). Han et al. found that METTL3 positively modulates pri-miR221/222 processing in an m6A-dependent manner by interacting with DGCR8, resulting in a reduction in the PTEN level, which ultimately results in proliferation of bladder cancer (29). Wang et al. explored the function and mechanism of METTL3 and m6A in colistin-induced kidney injury and found that METTL3 interacts with DGCR8 and positively modulates the processing of mature miR-873-5p in an m6A-dependent manner (54). Collectively, this evidence suggests that DGCR8 plays an important role in the m6A-dependent regulation of miRNA processing by METTL3. METTL3 can modulate the expression of oncogenes or tumor suppressors by influencing miRNA maturation and processing via interaction with DGCR8.
The ability of epithelial cells to undergo transition to a mesenchymal phenotype during malignant progression, termed epithelial-mesenchymal transition (EMT), is now widely accepted as a core biological process (55). Liu et al. demonstrated that after knockdown of METTL3 in gastric cancer cells, the level of α-smooth muscle actin (α-SMA) was significantly reduced, while the expressions of the mesenchymal markers N-cadherin and vimentin were not markedly changed, suggesting that METTL3 silencing partially impairs EMT progression in gastric cancer cells (23). Yue et al. showed that METTL3 mediated m6A modification of zinc finger MYM-type containing 1 (ZMYM1) which mediated the repression of E-cadherin promoter and facilitated EMT and metastasis in gastric cancer (48). Relying on the reader ELAVL1, Lin et al. showed that m6A modification regulates EMT and that knockdown of METTL3 impairs migration, invasion and EMT both in vivo and in vitro (56). METTL3 overexpression promoted the accumulation of MMP2 and N-cadherin in melanoma cells (49). Hua et al. showed that METTL3 promotes EMT by upregulating the receptor tyrosine kinase AXL in ovarian carcinoma (50). The above mentioned research suggests that METTL3 plays an oncogenic role by promoting the EMT process.
METTL3 inhibition can induce cancer cell apoptosis by regulating the expression of apoptosis-related genes in an m6A-dependent manner. Lin et al. showed that downregulation of METTL3 increases the levels of the positive regulators Bax and active caspase-3 but decreases the expression of Bcl2, a negative regulator of apoptosis, suggesting that downregulation of METTL3 activates this apoptosis-related pathway (47). Wei et al. showed that nuclear METTL3 increases the Bax/Bcl2 ratio in lung cancer cells and that METTL3 knockdown strongly increases the levels of cleaved caspase3 and PARP, implying that knockdown of METTL3 induces the mitochondrial apoptotic pathways in lung cancer cells (36).
Knockdown of METTL3 results in a reduction in m6A and subsequently promotes cancer cell proliferation and invasion by activating PI3K-AKT signaling. Several studies noted that knockdown of METTL3 inhibited PI3K expression and reduced the levels of the phosphorylated form of AKT, ribosomal protein S6 kinase B1 (p70S6K, an AKT downstream effector), β-catenin, and cyclin D1 (36, 44, 47, 57). METTL3 knockdown significantly suppressed cell proliferation, and METTL3 acted as an oncogene in acute myeloid leukemia cells by suppressing Myc expression via a reduction in the m6A level in Myc mRNA (44, 58). Also, Cheng et al. revealed that the major role of m6A modification in hematopoietic stem cell differentiation arises from its ability to regulate symmetric commitment by controlling Myc mRNA stability (59).
In addition, PH domain leucine-rich-repeat protein phosphatase 2 (PHLPP2), a tumor suppressor that inhibits cancer cell proliferation and invasion, has been proven to be a negative regulator of AKT (60). METTL3 can promote miR-25 maturation and subsequently target PHLPP2 and AKT (61). Further, Liu et al. showed that decreased expression of METTL3 results in a reduction in m6A methylation, which, in turn, leads to decreased expression of PHLPP2 and increased expression of the positive AKT regulator mTORC2 (62). These findings revealed that knockdown of METTL3 can activate the PI3K-AKT pathway by promoting AKT phosphorylation via modulation of the expression of AKT regulators such as PHLPP2 and mTORC2 in an m6A-dependent manner, as shown in Figure 3.
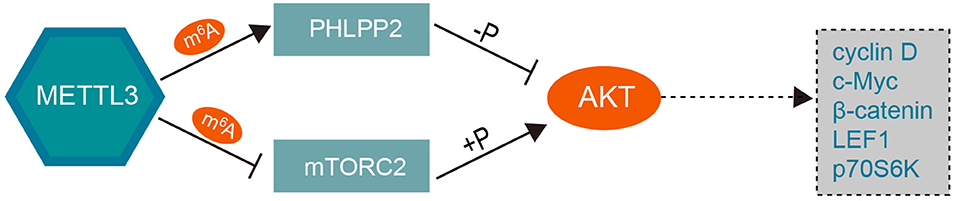
Figure 3. The mechanism underlying the role of METTL3 in the PI3K/AKT pathway. Overexpression of METTL3 coupled with increased m6A level promotes the negative AKT regulator PHLPP2 and inhibits the expression of the positive AKT regulator mTORC2, which subsequently decrease the AKT phosphorylation level and downstream effector. Knockdown of METTL3 can activate the PI3K/AKT pathway by positively regulating the phosphorylation of AKT. METTL3, methyltransferase-like 3; PHLPP2, PH domain leucine-rich-repeat protein phosphatase 2; LEF1, lymphoid enhancer binding factor 1; p70S6K, ribosomal protein S6 kinase B1.
Sex-determining region Y (SRY)-box transcription factor 2 (SOX2) is a transcription factor whose activity is associated with cancer stem cell differentiation. High SOX2 levels are usually associated with poor outcomes. SOX2 is an important marker for the promotion of tumor initiation and proliferation and participates in tumor metastasis (63–65). Li and her colleagues found that after knockdown of METTL3 in colorectal cancer cells, the genes with the greatest expression changes were enriched in the stem cell differentiation pathway. Among these genes, SOX2 exhibited the most consistently decreased m6A level in METTL3-knockdown colorectal cancer cells compared with control cells (39). Li et al. showed that the m6A reader IGF2BP2 bound to the SOX2 coding sequence (CDS) region to enhance SOX2 mRNA stability in an m6A-dependent manner (38). After METTL3 inhibition, the expression levels of cancer stem cell surface antigens such as CD133, CD44, and epithelial cell adhesion molecule were markedly reduced. In addition, decreased sphere numbers and sizes as well as a markedly reduced frequency of stem cells were observed. Moreover, the expressions of downstream SOX2 genes, including those of cyclin D1 and Myc, were consistently suppressed. Exogenous overexpression of a SOX2 mutant without the 3′UTR reversed the inhibitory effect of neurosphere formation in METTL3-knockdown glioma stem-like cells (39). Collectively, the research on METTL3 and SOX2 suggests that METTL3-mediated m6A modification regulates SOX2-associated stem cell self-renewal and tumor progression.
METTL3 can regulate tumor cell progression by modulating the expression of oncogenes such as CUB domain-containing protein 1 (CDCP1) (30), integrin subunit alpha 6 (ITGA6) (66), and mammalian HBXIP (42) or tumor suppressors [such as P53 (67)] in an m6A-dependent manner. Miao et al. demonstrated that METTL3 silencing decreased the expression of lymphoid enhancer-binding factor 1 (LEF1) because knockdown of METTL3 reduced the m6A level and shortened the half-life of LEF1 mRNA transcripts and subsequently inhibited the wnt/β-catenin pathway in human osteosarcoma (46). Deng et al. showed that in colorectal cancer cells, knockdown of METTL3 promotes cell proliferation and migration via activation of p-p38 and p-ERK, possibly indicating that METTL3 inhibits colorectal cancer cell proliferation and migration by modulating the P38/ERK pathway (26). Wang et al. showed that METTL3-mediated m6A modification of HDGF promotes tumor angiogenesis and that there was a correlation between nuclear HDGF level and glycolysis in gastric cancer cells, both of which were correlated with subsequent tumor growth and liver metastasis (68).
METTL3 itself participates in controlling the translation of some m6A-containing mRNAs, such as the epidermal growth factor receptor, and the expression of the Hippo pathway effector TAZ, thus affecting Myc and RAS levels in lung cancer independently of m6A reader proteins (34). Choe et al. showed that the METTL3-eIF3h complex enhances the translation of bromodomain containing 4 (BRD4), which is also modified by m6A in lung cancer cells when tethered to reporter mRNA at sites near the stop codon, supporting an mRNA looping mechanism for ribosome recycling and translational control (33, 69). METTL3 can promote cancer cell growth, survival, and invasion by recruiting eIF3h to the translation initiation complex and directly promotes oncogene translation independently of its MTase activity (34).
The data described in this review suggest that METTL3 is upregulated in most cancer tissues and cell lines and plays an oncogenic role in tumor formation and progression (29). However, other data suggest converse conclusions about the expression and role of METTL3 in bladder cancer, renal cell carcinoma, colorectal cancer, glioma, and breast cancer. The contradictory expression patterns and functions of METTL3 may be largely attributed to differences in the tumor tissue origin, extracellular microenvironment, upstream and downstream regulatory factors, and research methods.
As noted herein, m6A modification plays a dual and important role in human cancer progression (70). METTL3 is a predominant MTase for m6A modification, and its underlying mechanism must be complex, involving multiple molecules and pathways. The lncRNA ARHGAP5-AS1, miR-600, miRNA let-7g, and miR-33a can influence human cancer progression by targeting METTL3 (35, 36, 52, 53). Conversely, METTL3 can promote pri-miRNA processing by interacting with DGCR8 (2, 20, 54). Dysregulation of METTL3 in various cancers can also influence cancer cell EMT (23, 49, 50), apoptosis (36, 47), and stem cell self-renewal (38, 39), which have been identified to be important in cancer progression. In addition, METTL3-mediated m6A modification can directly regulate the transcription and translation of oncogenes and tumor suppressors coupled to the most of the important pathways involved in cancer cell progression, such as the PI3K/AKT (36, 44, 47, 57, 60, 62), wnt/β-catenin (46), and P38/ERK (26) pathways. METTL3 can also regulate cancer-related gene expression via m6A modification (30, 42, 66, 67). In conclusion, METTL3-related m6A regulatory genes involve multiple pathways and the opposing role of METTL3 in different cancer types may be associated with genes with opposing function, some of which we currently do not know.
In addition, METTL3 can promote tumor progression by regulating oncogene translation independently of m6A reader proteins and its MTase activity (34). METTL3 is tethered to reporter mRNA at sites near the stop codon by recruiting eIF3h (33, 34, 69). This observation may help to explain the controversy regarding the expression and role of METTL3 in cancer cells.
The role of m6A in disease occurrence and development has received more and more attention in recent years. Especially in the prevention and treatment of malignancies, m6A and its related factors are expected to become new prognostic indicators and therapeutic targets. The research pertaining to m6A-related enzyme inhibitors has focused mainly on “erasers” (71–73), although it is still at the preliminary stage.
As mentioned above, METTL3 appears to be a predominantly catalytic enzyme in the m6A process. The research about the role METTL3 in various cancers revealed that METTL3 has great potential for clinical application by serving as a new diagnostic/prognostic/treatment target. The mechanism underlying the effects of METTL3 is complex and implicated in multiple signaling pathways. The precise molecular mechanisms underlying the role of METTL3 in cancer initiation and progression are not thoroughly understood and require further systematic investigation. Further studies are needed to overcome the challenges in gaining a comprehensive understanding of the potential and limitations of METTL3 and m6A in cancer diagnosis and treatment.
METTL3 is dysregulated and plays a dual role in various types of human cancers. Through m6A modification, METTL3 modulates tumor cell proliferation, invasion, migration, tumor formation, and drug resistance. These effects are orchestrated through multiple pathways, such as the miRNA processing, EMT, apoptosis, stem cell self-renewal, and PI3K/AKT pathways. Non-coding RNAs can upregulate or downregulate METTL3 expression. In addition, METTL3 can promote oncogene translation independently of m6A readers by recruiting eIF3h. METTL3 has great potential for clinical application by serving as a new diagnostic/prognostic/treatment target. However, further studies are still needed to clarify the exact details of METTL3 expression, roles, and mechanisms in human cancers.
WZ and MG searched PubMed about METTL3 and m6A in human cancers and wrote the draft. XZ and MZ summarized the dyregulation and different functions in various human cancers. YZ and HJ searched the Kegg pathway and classified the complex mechanisms. XD and SW drawed the figures attached.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m6A RNA methylation in cancer. J Hematol Oncol. (2018) 11:48. doi: 10.1186/s13045-018-0590-8
2. Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell. (2015) 162:1299–308. doi: 10.1016/j.cell.2015.08.011
3. He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P, et al. ALKBH5 Inhibits pancreatic cancer motility by decreasing long non-coding RNA KCNK15-AS1 methylation. Cell Physiol Biochem. (2018) 48:838–46. doi: 10.1159/000491915
4. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
5. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5' UTR m(6)a promotes cap-independent translation. Cell. (2015) 163:999–1010. doi: 10.1016/j.cell.2015.10.012
6. Lence T, Paolantoni C, Worpenberg L, Roignant JY. Mechanistic insights into m6A RNA enzymes. Biochim Biophys Acta Gene Regul Mech. (2019) 1862:222–9. doi: 10.1016/j.bbagrm.2018.10.014
7. He L, Li J, Wang X, Ying Y, Xie H, Yan H, et al. The dual role of N6-methyladenosine modification of RNAs is involved in human cancers. J Cell Mol Med. (2018) 22:4630–9. doi: 10.1111/jcmm.13804
8. Chen B, Li Y, Song R, Xue C, Xu F. Functions of RNA N6-methyladenosine modification in cancer progression. Mol Biol Rep. (2019) 46:1383–91. doi: 10.1007/s11033-018-4471-6
9. Dang W, Xie Y, Cao P, Xin S, Wang J, Li S, et al. N(6)-methyladenosine and viral infection. Front Microbiol. (2019) 10:417. doi: 10.3389/fmicb.2019.00417
10. Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, et al. Publisher correction: m6A modification controls the innate immune response to infection by targeting type I interferons. Nat Immunol. (2019) 20:243. doi: 10.1038/s41590-019-0314-4
11. Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. (2015) 347:1002–6. doi: 10.1126/science.1261417
12. Bujnicki JM, Feder M, Radlinska M, Blumenthal RM. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m6A methyltransferase. J Mol Evol. (2002) 55:431–44. doi: 10.1007/s00239-002-2339-8
13. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. (2016) 534:575–8. doi: 10.1038/nature18298
14. Tong J, Flavell RA, Li HB. RNA m6A modification and its function in diseases. Front Med. (2018) 12:481–9. doi: 10.1007/s11684-018-0654-8
15. Chen XY, Zhang J, Zhu JS. The role of m6A RNA methylation in human cancer. Mol Cancer. (2019) 18:103. doi: 10.1186/s12943-019-1033-z
16. Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, et al. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. (2016) 540:301–4. doi: 10.1038/nature20577
17. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
18. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
19. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. (2014) 505:117–20. doi: 10.1038/nature12730
20. Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. (2015) 519:482–5. doi: 10.1038/nature14281
21. Meyer KD, Jaffrey SR. Rethinking m6A readers, writers, and erasers. Annu Rev Cell Dev Biol. (2017) 33:319–42. doi: 10.1146/annurev-cellbio-100616-060758
22. Zhou J, Wang J, Hong B, Ma K, Xie H, Li L, et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma - a retrospective study using TCGA database. Aging. (2019) 11:1633–47. doi: 10.18632/aging.101856
23. Liu T, Yang S, Sui J, Xu SY, Cheng YP, Shen B, et al. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. J Cell Physiol. (2020) 235:548–62. doi: 10.1002/jcp.28994
24. Kobayashi M, Ohsugi M, Sasako T, Awazawa M, Umehara T, Iwane A, et al. The RNA methyltransferase complex of WTAP, METTL3, and METTL14 regulates mitotic clonal expansion in adipogenesis. Mol Cell Biol. (2018) 38:e00116–18. doi: 10.1128/MCB.00116-18
25. Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. (2018) 52:621–9. doi: 10.3892/ijo.2017.4219
26. Deng R, Cheng Y, Ye S, Zhang J, Huang R, Li P, et al. m6A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. OncoTargets Ther. (2019) 12:4391–402. doi: 10.2147/OTT.S201052
27. Lobo J, Barros-Silva D, Henrique R, Jeronimo C. The emerging role of epitranscriptomics in cancer: focus on urological tumors. Genes. (2018) 9:E552. doi: 10.3390/genes9110552
28. Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, et al. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene. (2019) 38:3667–80. doi: 10.1038/s41388-019-0683-z
29. Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. (2019) 18:110. doi: 10.1186/s12943-019-1036-9
30. Yang F, Jin H, Que B, Chao Y, Zhang H, Ying X, et al. Dynamic m6A mRNA methylation reveals the role of METTL3-m6A-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. (2019) 38:4755–72. doi: 10.1038/s41388-019-0755-0
31. Zhao S, Liu J, Nanga P, Liu Y, Cicek AE, Knoblauch N, et al. Detailed modeling of positive selection improves detection of cancer driver genes. Nat Commun. (2019) 10:3399. doi: 10.1038/s41467-019-11284-9
32. Li X, Tang J, Huang W, Wang F, Li P, Qin C, et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. (2017) 8:96103–16. doi: 10.18632/oncotarget.21726
33. METTL3 Promotes mRNA translation to drive tumorigenesis. Cancer Discov. (2018) 8:1346. doi: 10.1158/2159-8290.CD-RW2018-167
34. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. (2016) 62:335–45. doi: 10.1016/j.molcel.2016.03.021
35. Du M, Zhang Y, Mao Y, Mou J, Zhao J, Xue Q, et al. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys Res Commun. (2017) 482:582–9. doi: 10.1016/j.bbrc.2016.11.077
36. Wei W, Huo B, Shi X. miR-600 inhibits lung cancer via downregulating the expression of METTL3. Cancer Manage Res. (2019) 11:1177–87. doi: 10.2147/CMAR.S181058
37. Liu X, Liu L, Dong Z, Li J, Yu Y, Chen X, et al. Expression patterns and prognostic value of m6A-related genes in colorectal cancer. Am J Transl Res. (2019) 11:3972–91.
38. Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. (2019) 18:112. doi: 10.1186/s12943-019-1038-7
39. Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. (2018) 37:522–33. doi: 10.1038/onc.2017.351
40. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. (2017) 18:2622–34. doi: 10.1016/j.celrep.2017.02.059
41. Wang H, Xu B, Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 722:144076. doi: 10.1016/j.gene.2019.144076
42. Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. (2018) 415:11–9. doi: 10.1016/j.canlet.2017.11.018
43. Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. (2019) 19:326. doi: 10.1186/s12885-019-5538-z
44. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. (2017) 23:1369–76. doi: 10.1038/nm.4416
45. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. (2017) 552:126–31. doi: 10.1038/nature24678
46. Miao W, Chen J, Jia L, Ma J, Song D. The m6A methyltransferase METTL3 promotes osteosarcoma progression by regulating the m6A level of LEF1. Biochem Biophys Res Commun. (2019) 516:719–25. doi: 10.1016/j.bbrc.2019.06.128
47. Lin S, Liu J, Jiang W, Wang P, Sun C, Wang X, et al. METTL3 promotes the proliferation and mobility of gastric cancer cells. Open Med. (2019) 14:25–31. doi: 10.1515/med-2019-0005
48. Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. (2019) 18:142. doi: 10.1186/s12943-019-1065-4
49. Dahal U, Le K, Gupta M. RNA m6A methyltransferase METTL3 regulates invasiveness of melanoma cells by matrix metallopeptidase 2. Melanoma Res. (2019) 29:382–9. doi: 10.1097/CMR.0000000000000580
50. Hua W, Zhao Y, Jin X, Yu D, He J, Xie D, et al. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. (2018) 151:356–65. doi: 10.1016/j.ygyno.2018.09.015
51. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. (2018) 67:2254–70. doi: 10.1002/hep.29683
52. Zhu L, Zhu Y, Han S, Chen M, Song P, Dai D, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. (2019) 10:383. doi: 10.1038/s41419-019-1585-2
53. He H, Wu W, Sun Z, Chai L. MiR-4429 prevented gastric cancer progression through targeting METTL3 to inhibit m6A-caused stabilization of SEC62. Biochem Biophys Res Commun. (2019) 517:581–7. doi: 10.1016/j.bbrc.2019.07.058
54. Wang J, Ishfaq M, Xu L, Xia C, Chen C, Li J. METTL3/m6A/miRNA-873–5p attenuated oxidative stress and apoptosis in colistin-induced kidney injury by modulating Keap1/Nrf2 pathway. Front Pharmacol. (2019) 10:517. doi: 10.3389/fphar.2019.00517
55. Li L, Li W. Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol Therap. (2015) 150:33–46. doi: 10.1016/j.pharmthera.2015.01.004
56. Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, et al. RNA m6A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat Commun. (2019) 10:2065. doi: 10.1038/s41467-019-09865-9
57. Zhang C, Zhang M, Ge S, Huang W, Lin X, Gao J, et al. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. (2019) 8:4766–81. doi: 10.1002/cam4.2360
58. Wang XS, He JR, Yu S, Yu J. Methyltransferase-like 3 promotes the proliferation of acute myeloid leukemia cells by regulating N6-methyladenosine Levels of MYC. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. (2018) 40:308–14. doi: 10.3881/j.issn.1000-503X.2018.03.002
59. Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, et al. m6A RNA methylation maintains hematopoietic stem cell identity and symmetric commitment. Cell Rep. (2019) 28:1703–16 e6. doi: 10.1016/j.celrep.2019.07.032
60. Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. (2007) 25:917–31. doi: 10.1016/j.molcel.2007.02.017
61. Zhang J, Bai R, Li M, Ye H, Wu C, Wang C, et al. Excessive miR-25–3p maturation via N(6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. (2019) 10:1858. doi: 10.1038/s41467-019-09712-x
62. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. (2018) 20:1074–83. doi: 10.1038/s41556-018-0174-4
63. Carrasco-Garcia E, Santos JC, Garcia I, Brianti M, Garcia-Puga M, Pedrazzoli J Jr, et al. Paradoxical role of SOX2 in gastric cancer. Am J Cancer Res. (2016) 6:701–13.
64. Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. (2009) 41:1238–42. doi: 10.1038/ng.465
65. Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. (2013) 12:15–30. doi: 10.1016/j.stem.2012.12.007
66. Jin H, Ying X, Que B, Wang X, Chao Y, Zhang H, et al. N(6)-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMed. (2019) 47:195–207. doi: 10.1016/j.ebiom.2019.07.068
67. Uddin MB, Roy KR, Hosain SB, Khiste SK, Hill RA, Jois SD, et al. An N(6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem Pharmacol. (2019) 160:134–45. doi: 10.1016/j.bcp.2018.12.014
68. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. (2019). doi: 10.1136/gutjnl-2019-319639. [Epub ahead of print].
69. Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. (2018) 561:556–60. doi: 10.1038/s41586-018-0538-8
70. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. (2018) 28:507–17. doi: 10.1038/s41422-018-0034-6
71. Wang R, Han Z, Liu B, Zhou B, Wang N, Jiang Q, et al. Identification of Natural Compound Radicicol as a Potent FTO Inhibitor. Mol Pharmaceutics. (2018) 15:4092–8. doi: 10.1021/acs.molpharmaceut.8b00522
72. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. (2015) 43:373–84. doi: 10.1093/nar/gku1276
Keywords: METTL3, m6A, cancer, mechanism, pathway
Citation: Zheng W, Dong X, Zhao Y, Wang S, Jiang H, Zhang M, Zheng X and Gu M (2019) Multiple Functions and Mechanisms Underlying the Role of METTL3 in Human Cancers. Front. Oncol. 9:1403. doi: 10.3389/fonc.2019.01403
Received: 16 September 2019; Accepted: 27 November 2019;
Published: 12 December 2019.
Edited by:
Matiullah Khan, AIMST University, MalaysiaReviewed by:
Paolo Armando Gagliardi, University of Bern, SwitzerlandCopyright © 2019 Zheng, Dong, Zhao, Wang, Jiang, Zhang, Zheng and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Gu, Z3VtaW5nXzIxQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.