 Lv Lv1
Lv Lv1 Liang He2Shaohua Chen3Yaqun Yu4Guosong Che1Xuan Tao1Shengtao Wang1Zhiyuan Jian5*Xuemei Zhang6*
Liang He2Shaohua Chen3Yaqun Yu4Guosong Che1Xuan Tao1Shengtao Wang1Zhiyuan Jian5*Xuemei Zhang6*- 1Department of Emergency and Trauma Surgery, Affiliated Hospital of Guilin Medical University, Guilin, China
- 2Department of Anesthesiology, Affiliated Hospital of Guilin Medical University, Guilin, China
- 3Department of Breast and Thyroid Surgery, Affiliated Hospital of Guilin Medical University, Guilin, China
- 4Department of Hepatobiliary and Pancreatic Surgery, Affiliated Hospital of Guilin Medical University, Guilin, China
- 5Gastrointestinal Surgery Department, Affiliated Hospital of Guilin Medical University, Guilin, China
- 6Department of Pathology, Affiliated Hospital of Guilin Medical University, Guilin, China
This study aimed to identify the roles of the long non-coding RNA LINC00114 in colorectal cancer (CRC) development. The expression levels of LINC00114 and miR-133b in CRC were determined by reverse transcription (RT)-polymerase chain reaction (PCR) and the functions of LINC00114 in CRC were evaluated in vitro and in vivo. Methylation-specific PCR assay was performed to detect the miR-133b promoter methylation in CRC cells. Bioinformatics analysis, RNA immunoprecipitation, dual luciferase assay, RNA pull-down, co-immunoprecipitation (IP), and chromatin IP (ChIP) assays were used to elucidate whether LINC00114 could recruit EZH2/DNMT1 and bind to the miR-133b promoter region, leading to dysregulated methylation and the depression of miR-133b. The expression levels of DNA methyltransferases (DNMTs), EZH2, and nucleoporin 214(NUP214) were analyzed by western blotting. Data showed that LINC00114 was highly expressed, whereas miR-133b was downregulated in the CRC tissues and cells. In vitro, silencing LINC00114 inhibited cell proliferation and impeded cell cycle at the G1/S phase by upregulating miR-133b. In vivo, LINC00114 knockdown reduced tumor growth. Further analysis showed that the methylation in miR-133b promoter region was increased in the CRC and silencing LINC00114 increased miR-133b expression through depressing methylation of its promoter region. ChIP-PCR experiments demonstrated that EZH2 and DNMT1 could bind to the miR-133b promoter region and it was abolished by LINC00114 knockdown. sh-EZH2 reversed the overexpression of DNMTs and CRC cell cycle progression induced by the LINC00114 upregulation. LINC00114 could regulate the NUP214 protein expression by sponging miR-133b. These results demonstrated that LINC00114 suppressed miR-133b expression via EZH2/DNMT1-mediated methylation of its promoter region, indicating that LINC00114 might be a potential novel target for CRC diagnosis and treatment.
Introduction
Colorectal cancer (CRC) is the third leading cause of cancer-related deaths worldwide (1). Importantly, CRC incidence rates have been increasing rapidly in several countries that have been historically at low risk for CRC (2), and relative survival rates indicate poor 5-year CRC prognosis (1). Although recently developed medical techniques have improved diagnosis and treatment, unfortunately, CRC is usually detected at advanced stages, which significantly impedes its treatment (3). Therefore, elucidating early diagnostic markers of CRC and discovering the pathogenesis of CRC are critical.
Long non-coding RNAs (lncRNAs) are a class of nucleotides, approximately 200 bp in length with no protein-coding capacity. Accumulated evidences indicate that lncRNAs play vital roles in tumor growth and metastasis (4). lncRNAs are categorized into several types and exert their functions through different molecular mechanisms. Notably, lncRNAs were reported to be intimately related to the development and progression of CRC. The lncRNA MAPKAPK5 antisense RNA 1, which is upregulated in CRC, can promote the development of CRC by binding to p21 (5). Moreover, nuclear enriched abundant transcript 1, another lncRNA, facilitates the progression of CRC by binding to DEAD-box helicase 5 (DDX5) via activation of the Wnt/β-catenin signaling pathway (6). In addition, lncRNAs regulate the transcription of downstream molecules by regulating DNA methylation or hydroxymethylation as well as histone acetylation or methylation. Hypermethylation often occurs in the promoter region near the tumor suppressor genes transcription factor 21 (TCF21) and TCF21 antisense RNA inducing promoter demethylation (TARID), resulting in gene silencing. The LncRNA Chaer (cardiac hypertrophy associated epigenetic regulator) can directly bind to the catalytic domain of polycomb repressive complex 2 (PRC2) and interfere with PRC2 binding to target gene sites. This binding suppresses histone H3K27 methylation in the promoter region of cardiac hypertrophy-related genes, thereby affecting their expression (7). However, the roles of lncRNAs in CRC tumorigenesis remain to be elucidated.
miR-133b was previously identified as a tumor suppressor and was shown to inhibit cell proliferation and tumorigenesis in several cancers, such as prostate cancer (8), breast cancer (9), ovarian cancer (10), and CRC (11). Additionally, a variety of validated miR-133b targets including nuclear pore protein nucleoporin 214 (NUP214) (12), CXC chemokine receptor-4 (CXCR4) (13), and B cell lymphoma-2 (BCL-2) (14) are involved in mitosis, tumorigenesis, and apoptosis. Although these studies ascribe an antitumor role for miR-133b, no studies to date have investigated lncRNA-mediated epigenetic mechanisms that regulate miR-133b expression in CRC.
In the current study, we elucidated the mechanism underlying the modulation of miR-133b expression by LINC00114, which was determined by bioinformatics analysis. We determined whether LINC00114 suppressed miR-133b expression via DNA methylation in CRC and demonstrated that LINC00114 could directly inhibit CRC progression via miRNA sponging, providing a potential mechanism that might be utilized in CRC diagnosis and treatment.
Materials and Methods
Tissues and Cells
The human colon epithelial cell line NCM460 and the CRC cell lines HT-29, HCT116, SW620, and LoVo were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai). NCM460 cells were incubated in McCoy's 5a medium with 10% fetal bovine serum (FBS; Gibco, California, USA). The CRC cell lines were cultured in DMEM medium (Hyclone, Logan, UT, USA) containing 10% FBS. All cells were cultured with 5% CO2 at 37°C.
CRC specimens were obtained from Affiliated Hospital of Guilin Medical University, and adjacent normal tissues at least 3 cm away from the tumor border were isolated for analyses. Tissue samples were preserved in liquid nitrogen for transportation and stored at −70°C. The use of the specimens was approved by the Institutional Review Board of Affiliated Hospital of Guilin Medical University.
In situ Hybridization
For in situ hybridization (ISH), CRC, and adjacent normal tissue specimens were fixed with 4% para-formaldehyde, dehydrated, and embedded in paraffin. The specimens were sliced into 4-μm-thick sections and mounted onto charged slides. After dewaxing and hydration, the sections were air-dried and immersed in distilled water containing 3% hydrogen peroxide, followed by immersion in pepsin solution for 30 min at 37°C. The sections were treated with hybridization buffer for 2 h at 37°C. Next, hybridization was performed by incubating the sections with the target DIG-labeled probe (AAGAAGCTGCTGAAGAACCCA) at 42°C overnight, followed by washing with 2 ×, 0.5 ×, and 0.2 × SSC solution, respectively. The sections were then blocked with biotinylated digoxin (Boster Biological Technology, Wuhan) for 1 h at 37°C and the streptavidin-biotin-peroxidase complex (Boster) for 20 min at 37°C. Hybridization signals were detected using DAB (3,3′-diaminobenzidine; P013IH, Auragene), and the sections were counterstained with hematoxylin. After dehydration, the sections were mounted with neutral gum and observed under the microscope.
Transfection
LINC00114 interfering plasmid (pGMLV-hU6-MCS-CMV-ZsGreen1-WPRE) and sequences are GCCGATTAAGGTCTGAGAAGT, GCCAACCACACAAGAAATAGG, and GCACATCATCATTGTGCTTCT), LINC00114 overexpression plasmid (pcDNA3.1) and enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) interfering plasmid (sequences are GCTCCTCTAACCATGTTTACA, GCCAACCACACAAGAAATAGG, and GCTCCTCTAACCATGTTTACA) were obtained from Auragene (Changsha, China). For transfection experiments, cells were plated in six-well plates at a density of 4 × 105 cells per well and transfected using Lipofectamine ®2000 (ThermoFisher Scientific, CA, USA), according to the manufacturer's protocol. After 48 h of incubation, the cells were collected for further analyses.
MTT Assay
Cells were plated into 96-well plates at a density of 5,000 cells/well and were treat with or without miR-133b inhibitor 12 h later. After a another incubation for 24, 48, and 72 h, 10 μl MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution (Sangon Biotech, China) was added to each well, the cells were cultured for 4 h at 37°C, and the medium was removed. Next, the cells were incubated with 150 μl dimethyl sulfoxide solution (MP Biomedicals, USA) for 10 min for cell lysis. The absorbance was measured at 570 nm using the Multiskan MK microplate reader (ThermoFisher Scientific, USA). All experiments were repeated three times.
TUNEL Assay
For the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay, cells were fixed in 4% paraformaldehyde, incubated with permeabilization buffer containing Triton X-100 (Sigma), fluorescence-labeled reagent (45 μl) and terminal deoxynucleotidyl transferase (5 μl) (cat. #, 24529300; Sigma) for 1 h at 37°C. The nuclei were labeled with 4′,6-diamidino-2-phenylindole (Solarbio) for visualization. A fluorescence microscope system (Leica, Germany) was used to observe and capture images of the cells.
Flow Cytometry
Forty-eight hours after transfection, the cells were collected, washed three times with cold phosphate-buffered saline (PBS) and fixed with 70% ethanol overnight. Next, the cells were stained with propidium iodide (PI; Keygentec, China) for 30 min at 4°C in the dark for cell cycle analysis. The cell cycle status was evaluated using a flow cytometer (Beckman Coulter, USA). Apoptosis of cells was assessed 48 h after transfection using an Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit. Briefly, the cells were collected, washed twice with cold PBS, resuspended in 100 μL binding buffer, and incubated at room temperature in the dark for 30 min.
Animals
The right flanks of BALB/c nude mice (aged 4–5 weeks) were subcutaneously injected with 1 × 106 stable expression of LINC00114 -sh HCT116 cells (n = 4 per group). After 4 weeks, the animals were sacrificed and the weight and volume of tumors were measured. Next, the tumor was cut into two pieces. One piece was fixed with 4% paraformaldehyde and sliced into 5-μm-thick sections for immunohistochemistry assay. The other was stored in liquid nitrogen. All animal experiments were approved by the Animal Care Committee of Affiliated Hospital of Guilin Medical University. Immunohistochemistry Staining was performed using 5-μm-thick sections that were blocked with 10% FBS. After washing with PBS, the sections were incubated in TBST containing Ki67 (3174S, Cell Signaling Technologies, Danvers, MA, USA) overnight at 4°C. Next, the sections were washed with PBS and incubated with the secondary antibody for 30 min, followed by incubation with DAB and hematoxylin for 30 s to visualize the signal.
Reverse Transcription-Polymerase Chain Reaction
Total RNA was extracted from tissues and cells using TRIZOL® (Invitrogen). The PrimeScript RT reagent kit (Takara, Dalian, China) was used for reverse transcription, and SYBR® Premix DimerEraser (Takara) and StepOnePlus real-time polymerase chain reaction (PCR) system (Applied Biosystems, Foster City, CA, USA) were utilized for RT-PCR. Primer sequences (Generay Biotechnology, Shanghai) are presented as follows: LINC00114 (forward: TTGTGGCATCATAGCAGT, reverse: CTAAAGCATATTTGGGTGA), NUP214(forward: CGAGCGGGAGATGAAGGATTT, reverse: ACCAGCGAAGACCAGACCATA). TaqMan® 2 × universal master mix (Applied Biosystems) was used for quantitative PCR. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the endogenous control. The relative expression of targets was calculated using the 2−ΔΔCt method (15).
Western Blotting
Cells were lysed using RIPA lysis buffer (Auragene) with a protease inhibitor cocktail (Roche, USA) and phenylmethylsulfonyl fluoride (Auragene). Equal amounts (25 μg) of protein were loaded on 10–12% sodium dodecyl sulfate-polyacrylamide gels, and the separated proteins were transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore, USA). The membranes were blocked in TBST containing 3% BSA with gentle shaking for 90 min at room temperature and incubated with primary antibodies overnight at 4°C. After washing with TBST, the membranes were incubated with specific secondary antibodies 1 h at room temperature. The following primary antibodies from Abcam (Massachusetts, USA) were used: β-actin antibody (ab8226, 1:5,000), EZH2 (ab191250, 1:1,000), DNA methyltransferase 1 (DNMT1, ab92314, 1:1,000), H3K27me3 (ab6147, 1:500), DNMT3B (ab2851, 1:200), and NUP214 (ab70497, 1:2,000).
RNA Immunoprecipitation
RNA immunoprecipitation (RIP) experiments were performed using EZMagna RIP kit (Millipore, Billerica, MA, USA), according to the manufacturer's instructions. Briefly, HCT116 and LoVo cells were lysed using the RIP lysis buffer, and the lysates were incubated with magnetic beads containing specific antibodies (Ago, EZH2) or control IgG (Millipore). The antibody against EZH2 (Abcam) was used at a 1:200 dilution. Next, the beads were washed and incubated with Proteinase K for protein removal. Finally, RT-qPCR assay was used to measure the RNAs.
RNA Pull-Down
Briefly, T7 RNA polymerase (Promega, Madison, WI, USA) and biotinylated RNA-tagged mixtures (Roche, Basel, Switzerland) were used to label the transcriptional LINC00114 in vitro. Biotinylated LINC00114 was then mixed and incubated with cell lysates. Streptavidin magnetic beads (Life Technologies) were added to each binding reaction and further incubated at room temperature for 1 h. Beads were then washed briefly three times and boiled in SDS buffer, and the retrieved protein was detected by standard western blotting.
Chromatin Immunoprecipitation
For chromatin immunoprecipitation (ChIP), the Magna ChIP Kit (Millipore, Bedford, MA) was used in accordance with the manufacturer's instructions. Briefly, CRC cells were treated with 4% formaldehyde, and chromatin fragments with sizes ranging from 200 to 300 bp were generated by sonicating the cell lysates. Next, chromatin was immunoprecipitated with anti-EZH2, anti-DNMT1, anti-DNMT3B, and anti-He3mek27 antibodies, all from Abcam. IgG was used as control. RT-PCR was performed to recover and measure the precipitated chromatin DNA. The forward primer sequences are TCAATGCTATCCCCTTGTTAAACC and the reverse are ACCAA TACCCATGAACAACG. The forward primer sequences of negative control (around −7,000) are GCCAAGTGACACACCACATC and the reverse GCATAGGGCAAGGGCATCTA. The amount of immunoprecipitated DNA was calculated and normalized to the amount of input DNA.
Luciferase Reporter Assay
The predicted miR-133b combining site of LINC00114 was mutated and sequences were generated by chemical synthesis. The Wt-LINC0014 and Mut-LINC0014 overexpression constructs were provided by genepharma (Suzhou, China), with a psi-CHECK2 vector. HCT116 and LoVo cells were maintained and co-transfected with miR-133 mimics, inhibitors, or negative control (NC), respectively, using the Cellfectin®II reagent. After incubation for 48 h, the firefly luciferase activity in cells was determined using the dual luciferase reporter assay system (E1910; Promega).
Methylation-Specific PCR Assay
For the methylation-specific PCR (MSP) assay, genomic DNA was processed with EZ DNA Methylation-Gold™ kit (Zymo Research, USA). PCR was performed with HotStar Taq polymerase (Qiagen, Germany) using the following conditions: initial incubation of 94°C for 4 min; 35 cycles of 94°C for 30 s, 62°C for 30 s, and 72°C for 30 s; and one cycle of 72°C for 5 min. PCR products were electrophoresed in 3% agarose gels and visualized by ultraviolet illumination. The miR-133b MSP primer sequences were as follows: forward, 3′-TTTATTTAAAATATAAAAATA GCGG-5′; reverse, 3′-TCACCCAAACTAAAATACAAT AACG-5′; unmethylated miR-133b forward, 3′-TTTTTA TTTAAAATATAAAAATAGTGG-5′; unmethylated miR-133b reverse, 3′-ACC CAAACTAAAATACAATAACACT-5′.
Statistical Analysis
Data were presented as means ± standard error. All experiments were repeated at least three times. All data was analyzed using GraphPad Prism 7.0 (CA, USA). Multiple comparisons were analyzed using one-way analysis of variance, and differences between groups were compared using Dunnett's multiple comparisons test. P < 0.05 was considered statistically significant.
Results
Expression Levels of LINC00114 and mIR-133b in CRC Tissues and Cell Lines
We previously demonstrated that miR-133b expression was reduced in CRC, although the upstream regulatory mechanism remained unclear (11). Therefore, we first analyzed the TCGA database to see if there was any lncRNAs regulating miR-133b as a upstream regulator and found that there were 1,046 lncRNAs upregulated in CRC. We next investigated the potential targets of miR-133b using the miRanda software and LINC00114 was identified as a potential target of miR-133b. The relatively high expression level of LINC00114 in CRC was also verified in the GEPIA database (http://gepia.cancer-pku.cn/) (Figure 1A). To verify whether LINC00114 and miR-133b were both dysregulation in CRC, we next performed RT-PCR and ISH assays and found that LINC00114 was upregulated while miR-133b was downregulated in the CRC specimens compared with the adjacent tissues (Figures 1B–D), suggesting a negative correlation between LINC00114 and miR-133b (Figure 1E). Additionally, the expression trend of LINC00114 and miR-133b in CRC cells was same with it in tissues (Figures 1F,G).
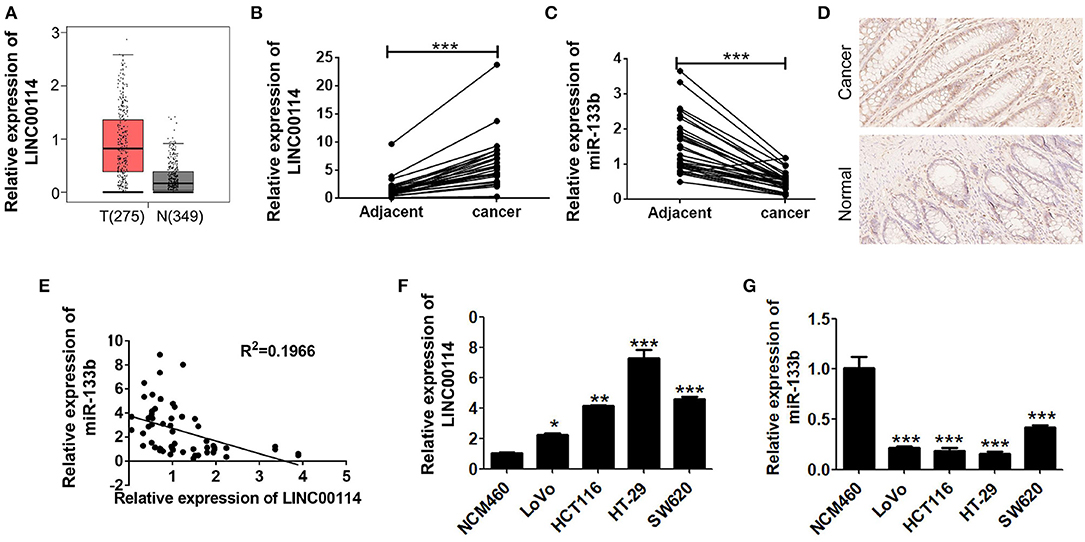
Figure 1. LINC00114 is upregulated in CRC. (A) Highly expressed LINC00114 is predicted in the GEPIA database. (B) LINC00114 is upregulated in the CRC samples compared to the adjacent normal tissue samples, determined via RT-PCR assay. (C) RT-PCR result shows that expression of miR-133b expression is reduced in the CRC samples. (D) LINC00114 expression is higher in the CRC as measured by in situ hybridization (200X). (E) There is an inverse correlation between LINC00114 and miR-133b expression in the CRC tissue samples. (F,G) The expression levels of LINC00114 and miR-133b in the CRC cells are determined by RT-PCR. *P < 0.05; **P < 0.01; ***P < 0.001.
LINC00114 Regulates CRC Progression Through Inhibiting miR-133b Expression in vitro and in vivo
To determine whether LINC00114 promoted the progression of CRC, we transfected sh-LINC00114 plasmids (LINC00114-sh1 and LINC00114-sh2) into HCT116 and LoVo cells (Figure S1). The results showed that LINC00114 knockdown inhibited the proliferation of these cells (Figures 2A,B) and the result was further confirmed by flow cytometry and TUNEL assays (Figures 2C,D). What's more, silencing LINC00114 disrupted the transition of G1/S in HCT116 and LoVo cells (Figure 2E). We also performed a nude mouse xenograft model to explore the effects of LINC00114 in vivo and found that, consistent with the findings above, LINC00114 suppression significantly reduced the tumor growth, compared with the control group (Figures 3A–C). RT-PCR and immunohistochemistry analysis revealed that the expression of miR-133b was promoted and the signal of Ki67 in the tumors of LINC00114 suppression group was weaker than that in the control group (Figures 3D,E). All these data firmly demonstrated that LINC00114 played vital roles in the growth of CRC in vivo and in vitro.
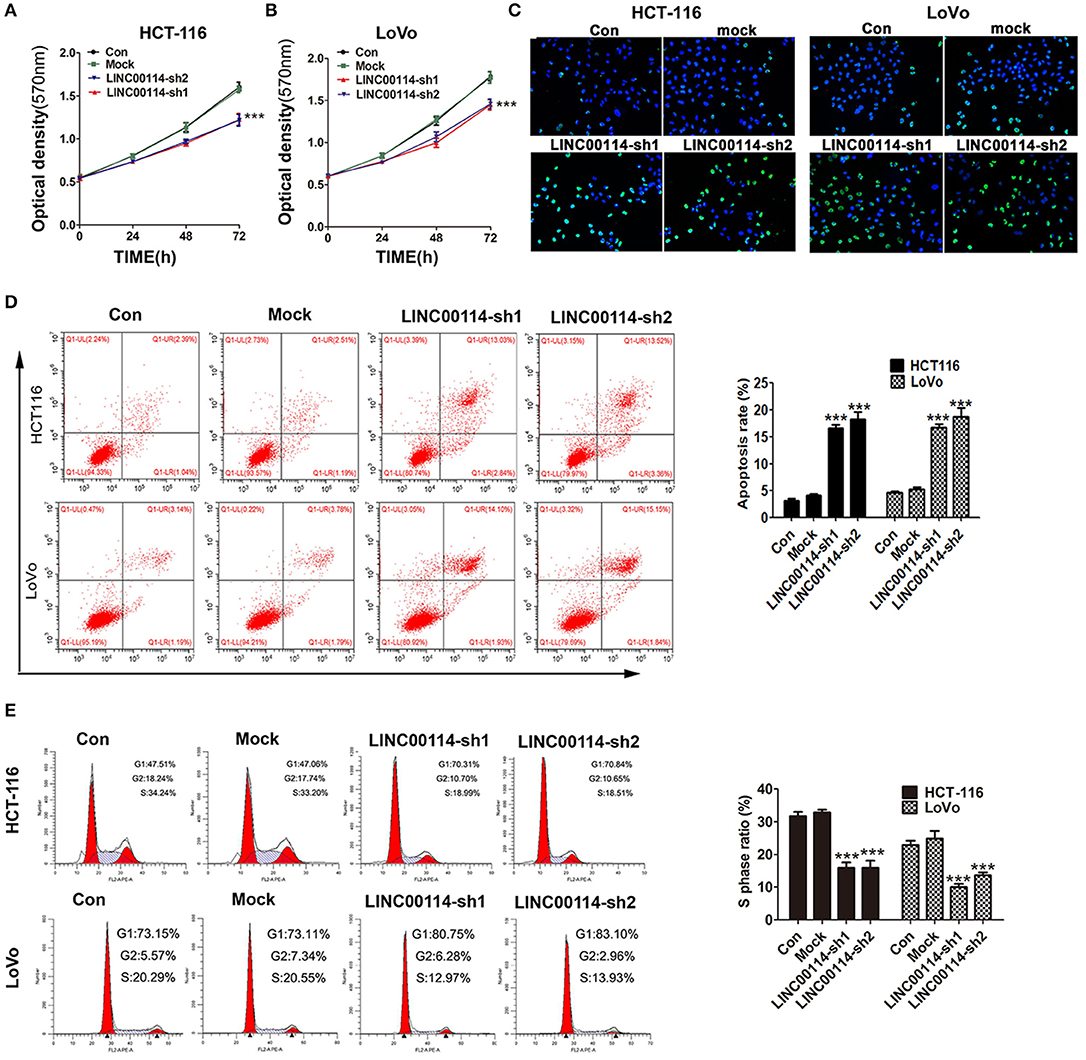
Figure 2. LINC00114 interference facilitates apoptosis and represses cell cycle progression in vitro. (A–C) sh-LINC00114 inhibits cells proliferation. (D) Apoptosis of CRC cells transfected with the sh-LINC00114 or the control plasmid for 48 h, analyzed by flow cytometry. (E) Cell cycle analysis of HCT116 and LoVo cells transfected with the LINC00114 or the control plasmid for 48 h using flow cytometry. ***P < 0.001. Con, the group only treated with PBS; Mock, the cells transfected with the empty vector plasmid.
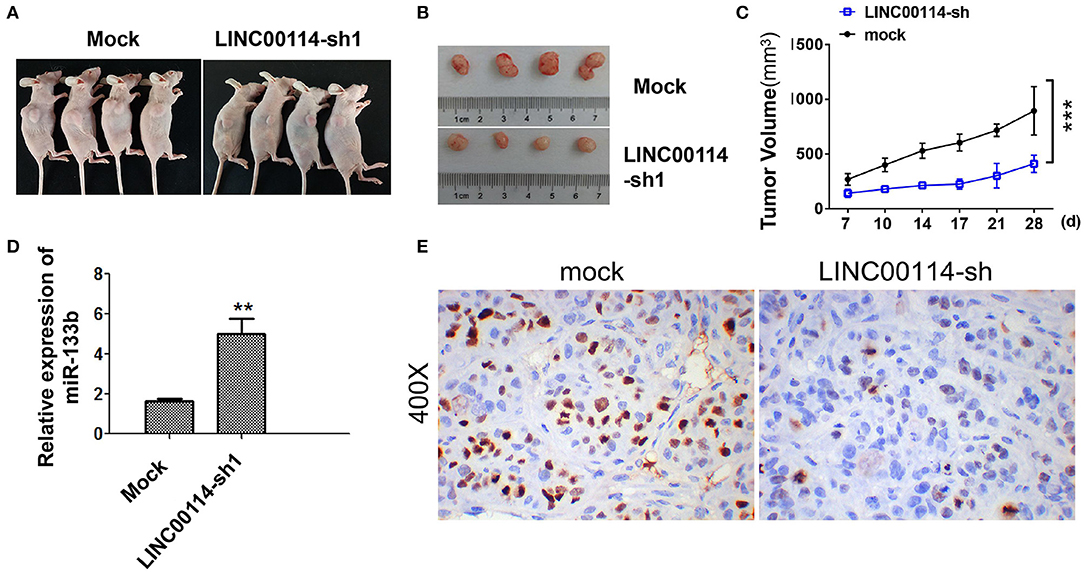
Figure 3. LINC00114 interference inhibits the development of CRC in vivo. (A) Tumor formation in nude mice. (B) Tumor body in nude mice. (C) Tumor growth curve. (D) Expression of miR-133b in tumor tissues. (E) Ki67 expression determined by immunohistochemistry. **P < 0.01; ***P < 0.001.
LINC00114 Regulates the Methylation of miR-133b Promoter Region
To explore the mechanism underlying LINC00114-mediated CRC cell proliferation, we then investigated the localization of LINC00114 (http://www.rna-society.org/rnalocate/) and found that LINC00114 was localized in both the cytoplasm and the nucleus (60 and 40%, respectively) (Figure 1D). These were further confirmed by RT-PCR assay (Figure 4A). We next used the dual luciferase reporter and RIP assays to verify that miR-133b and LINC00114 could bind to each other in a direct or indirect manner (Figures 4B–D). Together, these data identified that LINC00114 could bind to miR-133b. Previous studies demonstrated that the methylation level of miR-133b promoter region was high in CRC (11). We further confirmed these using MSP assay, as shown in Figure 4E. The miR-133b methylation level was low in NCM460 while high in CRC cells (LoVo, HCT116, HT-29, and SW620). Considering that LINC00114 depletion increases the expression of miR-133b (Figure 3D), we were urged to explore if LINC00114 regulates the expression of miR-133b by regulating methylation levels in its promoter region. To this end, we transfected LoVo and HCT116 cells with LINC00114-sh1 or treated with RG108, a DNA methyltransferase inhibitor, and found that LINC00114 knockdown and RG108 treatment reduced the methylation level of the miR-133b promoter region both (Figure 4F), as we expected. The treatments also increased the expression of miR-133b in CRC cells (Figure 4G), indicating that LINC00114 might regulate the methylation level in miR-133b promoter region.
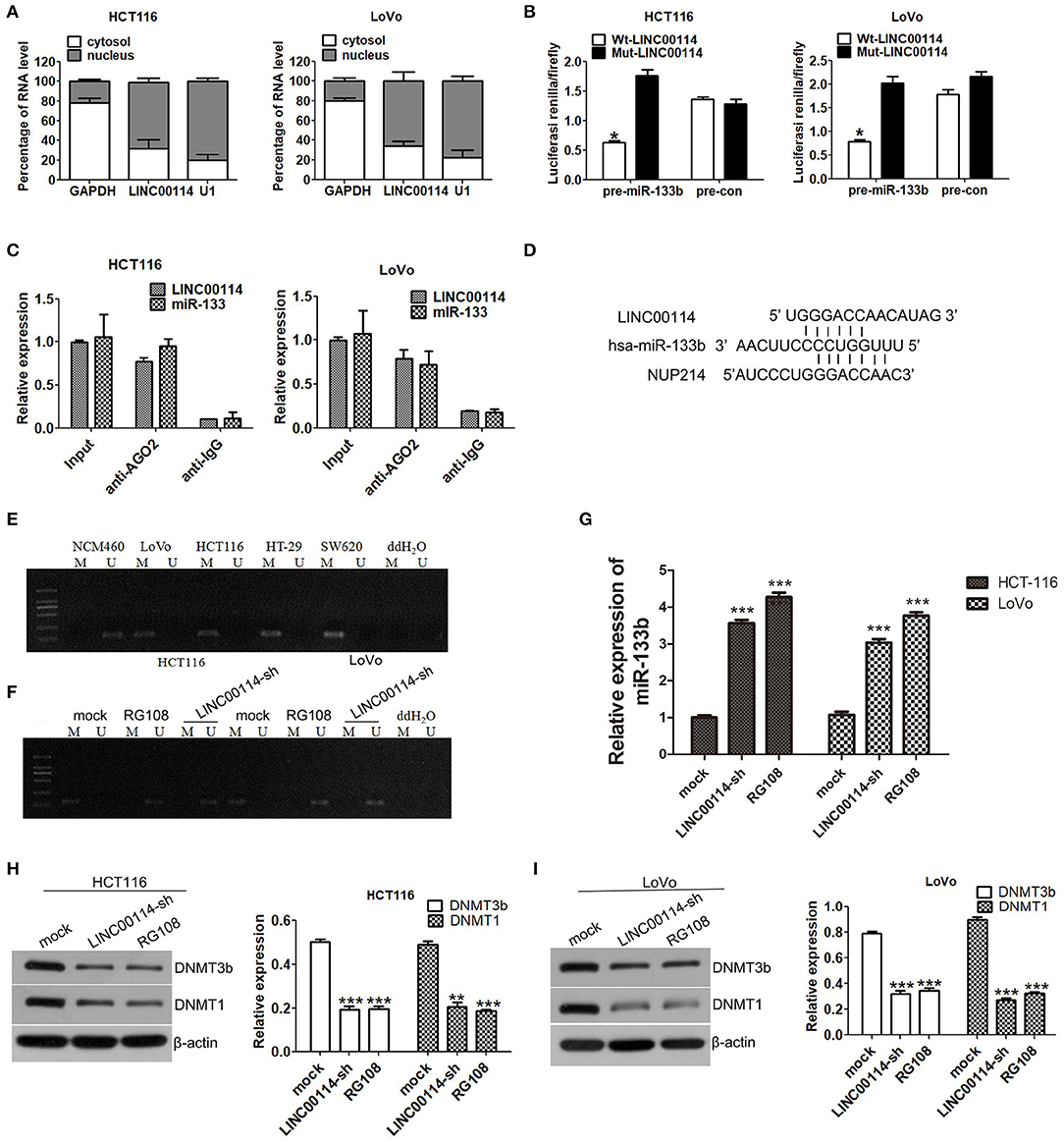
Figure 4. LINC00114 regulates DNA methylation of the miR-133b promoter region. (A) Expression levels of LINC00114 in the nucleus and cytoplasm of HCT116 and LoVo cells. (B) Luciferase activity of cells cotransfected with miR-133b overexpression and control plasmids after LINC00114 overexpression or mutation. (C) RIP assay was used to analyse the binding of LINC00114 to miR-133b. (D) The schematic diagram showed the binding site of miR-133b with LINC00114 and NUP214 mRNA. (E) Methylation detection of miR-133b promoter region in cells. (F) LINC00114 interference and RG108 treatment reduced the methylation levels of miR-133b promoter region in HCT116 and LoVo cells. (G) miR-133b levels after LINC00114 interference and RG108 treatment reduced the expression of miR-133b in cells. (H,I) Western blotting result showed that DNMT1 and DNMT3B were reduced in LINC00114-silenced or RG108-treated cells. *P < 0.05; **P < 0.01; ***P < 0.001. M, methylation. U, non-methylation.
It is known that DNMT3A and DNMT3B enzymes are important for the DNA methylation (16, 17), and DNMT1 also is a designated maintenance DNA methyltransferase, in a replication-dependent manner DNMTs (18, 19). To investigate the roles of these DNMTs in the LINC00114-regulated change of miR-133b promoter methylation, we performed western blotting assays and found that the expression of DNMT1 and DNMT3B were decreased in the cells with LINC00114 silenced or RG108 treatment (Figures 4H,I), suggesting that LINC00114 might regulate the methylation of the miR-133b promoter through methyltransferases.
LINC00114 Regulates Methylation Level of miR-133b Promoter by Binding to EZH2
Several studies had been reported that the function of some lncRNAs was EZH2-depended and participated in tumor progression by recruiting PRC2 (20, 21). Therefore, we first explored whether LINC00114 could bind to DNA methyltransferase using online RNA-protein interaction prediction software (http://pridb.gdcb.iastate.edu/RPISeq/). As expected, it showed that LINC00114 might bind to EZH2 and DNMT1 (with RF and SVM scores of >0.5), which did bind to miR-133b (Table S1). We also confirmed that LINC00114 expression was positively correlated with EZH2 using GEPIA (http://gepia.cancer-pku.cn/index.html) (Figure 5A). Further, we found that the expression levels of EZH2, DNMT1, and H3K27me3 were reduced when EZH2 knocked down (Figure 5B). Also, EZH2 depression promoted the expression of miR-133b (Figure 5C) and EZH2 indeed binds with DNMT1 and H3K27me3 as shown in the result of Co-IP assay (Figure 5D). Moreover, the RNA pull-down assay revealed that biotinylated LINC00114 could enrich the EZH2 and DNMT1 proteins in cells (Figure 5E) and LINC00114 was enriched in the RNA-protein complexes immunoprecipitated by EHZ2 and DNMT1. These results confirmed the interplay between LINC00114, EZH2, and DNMT1.
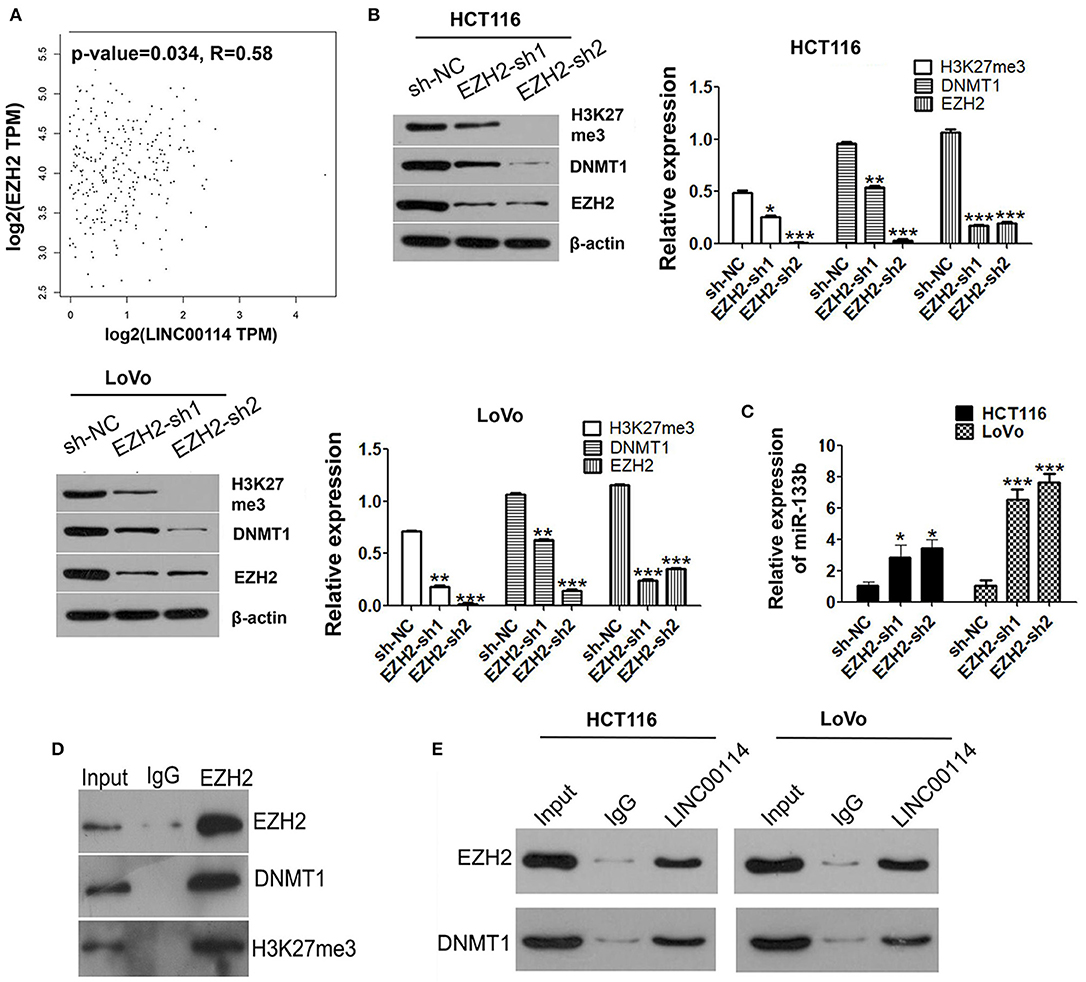
Figure 5. LINC00114 binds to EZH2 protein. (A) Correlation analysis of LINC00114 and EZH2 using the GEPIA database, also see it in method section. (B) EZH2 interference plasmids were transfected into HCT116 and LoVo cells and then H3K27me3, DNMT1, and EZH2 were detected using western blotting assay. (C) miR-133b levels were detected by RT-PCR in cells transfected with sh-EZH2. (D) Co-IP analysis showed that EZH2 could bind to DNMT1 and H3K27me3. (E) RNA pull-down assay was performed to detect the binding of LINC00114 with EZH2 and DNMT1. *P < 0.05; **P < 0.01; ***P < 0.001.
To investigate whether DNMTs and EZH2 were involved in the LINC00114 -mediated methylation of miR-133b promoter, we performed ChIP assay and it showed that the knockdown of LINC00114 reduced the binding of the DNMTs and EZH2 to the miR-133b promoter region (Figure 6A). This result indicated that LINC00114 altered the EZH2-mediated H3K27me3 trimethylation, thereby leading to the miR-133b repression in hypermethylation-depended manner. In addition, sh-EZH2 neutralized the miR-133b depression-mediated by LINC00114 overexpression (Figure 6B). The TUNEL and MTT assays revealed that LINC00114 overexpression failed to promote the proliferation of CRC cells, owing to EZH2 depression (Figures 6C,D). Rescue experiments demonstrated that sh-EZH2 attenuated the effect of LINC00114 overexpression on DNMT1 expression (Figure 6E). These results indicated that LINC00114 promoted miR-133b promoter methylation by inducing the EZH2/DNMT1 binding to the miR-133b promoter region, thereby facilitating the development of CRC.
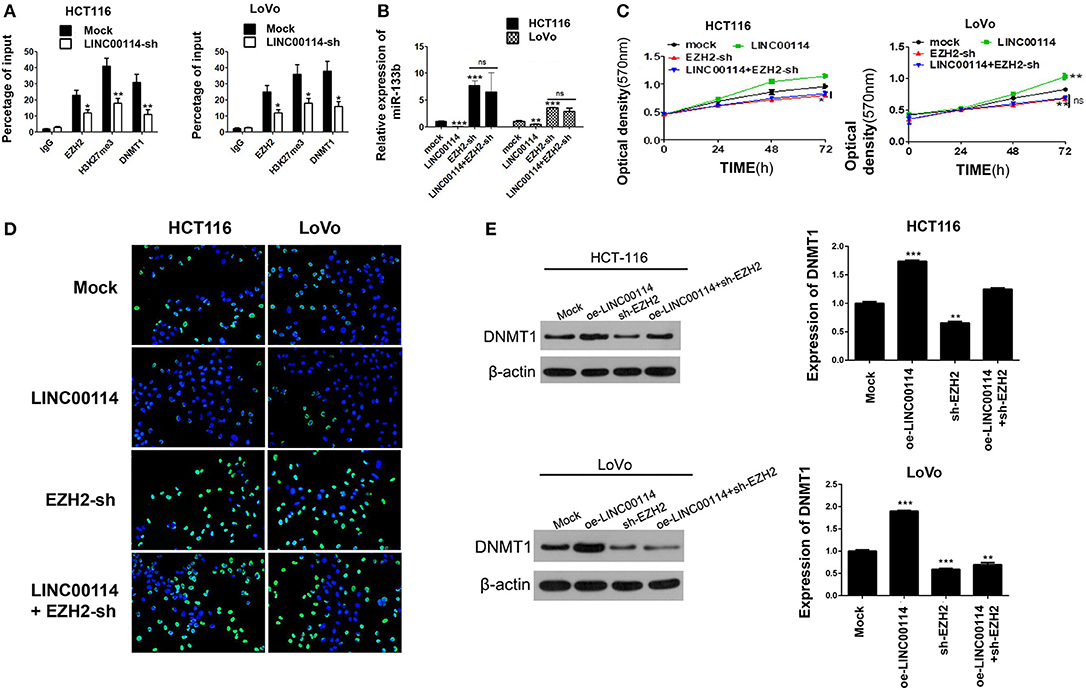
Figure 6. LINC00114 promotes the methylation of miR-133b promoter region by recruiting EZH2 and DNMT1. (A) LINC00114 affects EZH2, DNMT1, and H3K27me3 binding with the miR-133b promoter region in CRC cells, as detected by ChIP assay. (B) EZH2-sh reversed the LINC00114-induced inhibition of miR-133b in cells. (C,D) MTT and TUNEL assays result showed that EZH2-sh reversed the LINC00114-incduced proliferation promotion of cells. (E) EZH2-sh reversed the upregulation of DNMT1 induced by LINC00114 overexpression. oe, overexpression. *P < 0.05; **P < 0.01; ***P < 0.001.
LINC00114 Regulates CRC Cell Proliferation by Sponging miR-133b
Previous study reported that miR-133b could target NUP214, a member of the massive nuclear pore complex, illustrating that the miR-133b-mediated NUP214 downregulation arrested cells in the early mitotic phase and led to aberrant chromosome segregation and cell death (12). Thus, we assessed whether LINC00114 regulated cell cycle progression through NUP214. We next found that both NUP214 mRNA and LINC00114 shared the binding site for miR-133b (Figure 4D). Furthermore, LINC00114 knockdown facilitated miR-133b expression while depressed NUP214 expression in cells (Figures 7A–C). In addition, the assays revealed that miR-133b silencing promoted the CRC cell growth and that sh-LINC00114 neutralized the proliferation promotion mediated by miR-133b inhibitor (Figures 7D,E), indicating that LINC00114 regulated cell proliferation via sponging miR-133b.
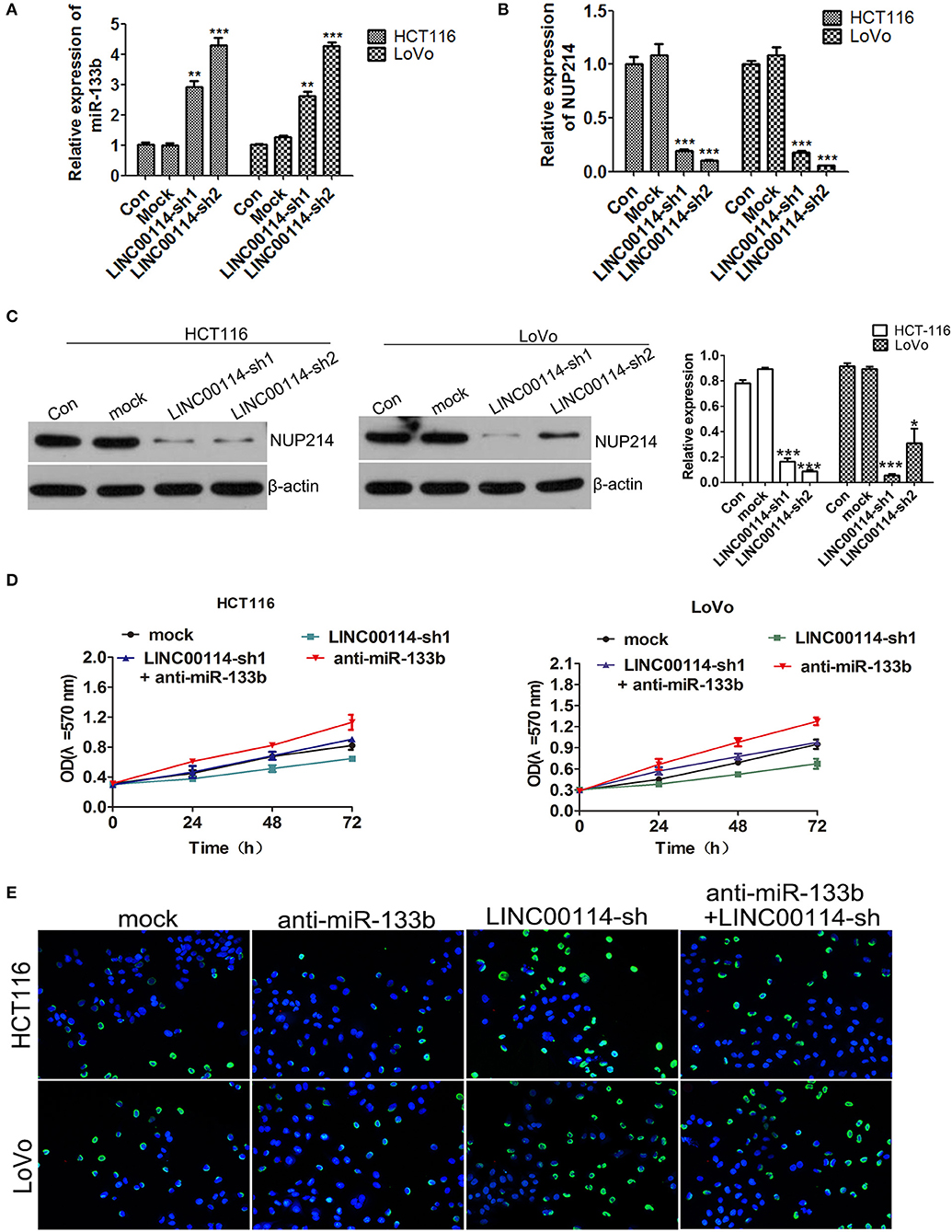
Figure 7. LINC00114 regulates cell proliferation via sponging miR-133b. (A) Expression of miR-133b detection in LINC00114 silenced cells. (B,C) The expression of NUP214 was determined by RT-PCR and western blotting, respectively. (D,E) Inhibition of miR-133b maintained proliferation promotion of cells in LINC00114 silenced cells, as shown in MTT and TUNEL assays, respectively. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Although radiotherapy chemotherapy and primary treatment approaches for CRC can prolong life expectancy and reduce patient burden, the CRC prognosis remains poor (22, 23). LncRNAs were recently demonstrated to play vital roles as major regulators of gene expression and cancer cell proliferation (24) in multiple cancers. Several lncRNAs have been identified as regulators in CRC cell proliferation and metastasis (25). In current study, we focused on a cancer-related lncRNA, LINC00114, which we found to be upregulated in CRC. Moreover, we demonstrated that LINC00114 negatively regulated miR-133b expression via DNA methylation, suggesting that LINC00114 contributed to the CRC development as an oncogene. We further found that LINC00114 served as an miR-133b sponge to regulate NUP214 expression, thus participating in the progression of CRC.
On one hand, lncRNAs can competitively bind to miRNAs, regulating the proliferation and metastasis of cancer cells (26). Gastric cancer associated transcript 3, an lncRNA, is upregulated in colon cancer and it combines with miR-3127 via ELAV like RNA binding protein 1 to promote CRC (26). Moreover, nuclear enriched abundant transcript 1 is another lncRNA that can compete with sirtuin 1 to bind miR-34a and regulate the Wnt/β-catenin signaling pathway and promote the development of CRC (27). In the current study, we identified that knockdown of LINC00114 significantly increased the expression of miR-133b, and LINC00114 was inversely correlated with miR-133b in CRC. Numerous studies demonstrate that miR-133b plays an important role in CRC progression by inhibiting cell proliferation and metastasis (11, 12, 28). Our results showed that the knockdown of LINC00114 inhibited CRC cell proliferation by negatively regulating miR-133b expression, indicating that LINC00114 might promote the development of CRC by suppressing miR-133b.
Conversely, lncRNAs can regulate gene expression through various mechanisms, such as chromatin modification and pre- and post-transcriptional regulation. Growing evidences indicate that some lncRNAs regulate the expression of downstream target genes by DNA methylation (29, 30). In this study, we showed that silencing LINC00114 decreased DNA methylation in the miR-133b promoter region. Previous study reported that lncRNAs were associated with several RNA-binding proteins including EZH2, SUZ12, LSD1, staufen double-stranded RNA binding protein 1, and DNMT1 (31, 32). EZH2 is a component of the methyltransferase in PRC2 and regulated by lncRNAs at the transcriptional level. EZH2 induces H3K27 methylation; it also induces LSD1 to regulate H3K4 demethylation (33). EZH2 usually mediates the methylation of the second lysine of histone H3 and is associated with tumor progression, angiogenesis, inflammation (34). Moreover, EZH2 may also recruit DNMT1 to the promoter region of its target genes. DNMTs and methyl-CpG binding protein 2 were reported to play an important role in methylation modification (35–37). These studies raise the possibility that lncRNAs may have a primary role in recruiting polycomb proteins to their target genes (32, 36). In our study, we found that LINC00114, together with EZH2 and DNMT1, played a direct role by online RIP-seq data. We hypothesized that LINC00114 recruited EZH2 and DNMT1 to the miR-133b promoter region for its hypermethylation and repression of miR-133b expression. To validate the relationship among EZH2, LINC00114, and DNMTs, we performed an RNA pull-down assay and confirmed that EZH2 and DNMT1 were binding partners of LINC00114. RT-PCR analysis confirmed that knockdown of LINC00114 affected the expression of DNMTs and miR-133b. In agreement with previous studies which reported that EZH2 could regulate DNA methylation through DNMTs (38), we determined that silencing EZH2 resulted in downregulation of DNMT1 and upregulation of miR-133b expression. Furthermore, knockdown of EZH2 reversed the LINC00114-induced DNMT1 upregulation and cell cycle progression. Additionally, knockdown of LINC00114 reduced the containing of EZH2 and DNMT1 that bind to the promoter region of miR-133b. These results indicated that LINC00114 might regulate the expression of miR-133b by binding to EZH2 and DNMT1.
It was reported that miR-133b impacted the development of CRC by regulating NUP214. Nucleopore is essential for many species to enter the nucleus, and NUP214 regulates mitosis and promotes carcinogenesis (39). NUP214 is involved in impeding mitotic progression and is a reported target of miR-133b; it is also highly expressed in HCT116 cells (12). In the current study, we demonstrated that LINC00114 could regulate NUP214 protein expression by sponging miR-133b. In addition, miR-133b and NUP214 act as key factors in the pathogenesis of CRC. These findings also suggest that LINC00114 affects the development of CRC by competitively binding to miR-133b through NUP214.
In conclusion, the current study demonstrated that LINC00114 expression was increased in CRC, which might represent an unfavorable prognostic factor for CRC. In addition, LINC00114 depletion inhibited cancer cell proliferation both in vitro and in vivo. Moreover, the oncogenic effects of LINC00114 were facilitated by its binding to EZH2 and DNMT1 and consequent induction of miR-133b promoter methylation which led to the downregulation of miR-133b. Our results also revealed that LINC00114 served as a molecular sponge for miR-133b, reversing its ability to repress NUP214 protein. Overall, these findings reveal novel therapeutic targets that should be considered for CRC.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: http://gepia.cancer-pku.cn/.
Ethics Statement
The studies involving human participants were reviewed and approved by The Ethics Committee of the Guilin Medical University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by The Ethics Committee of the Guilin Medical University. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
ZJ and XZ designed the experiments. LL and LH performed the most of the experiments. SC and YY analyzed the data. GC and XT wrote the paper. SW revised the paper.
Funding
This research was supported by Guangxi Natural Science Foundation under Grant No. 2018GXNSFBA050062, Guangxi Natural Science Foundation under Grant No. 2018GXNSFDA281003, Guangxi Natural Science Foundation under Grant No. 2017GXNSFAA198131, and National Natural Science Foundation of China; Grant No. 81802478.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.01383/full#supplementary-material
Figure S1. (A,B) Verification of the efficiency of LINC00114 interference plasmid in HCT116 and LoVo cells. ***P < 0.001.
Table S1. Prediction of the binding ability of LINC00114 and mir-133b to genes.
References
1. Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. (2017) 67:177–93. doi: 10.3322/caac.21395
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. (2011) 61:69–90. doi: 10.3322/caac.20107
3. Dienstmann R, Salazar R, Tabernero J. Molecular subtypes and the evolution of treatment decisions in metastatic colorectal cancer. Am Soc Clin Oncol Educ Book. 38:231–8. doi: 10.1200/EDBK_200929
4. Gugnoni M, Ciarrocchi A. Long noncoding RNA and epithelial mesenchymal transition in cancer. Int J Mol Sci. (2019) 20:1924. doi: 10.3390/ijms20081924
5. Ji H, Hui B, Wang J, Zhu Y, Tang L, Peng P, et al. Long noncoding RNA MAPKAPK5-AS1 promotes colorectal cancer proliferation by partly silencing p21 expression. Cancer Sci. (2019) 110:72–85. doi: 10.1111/cas.13838
6. Meng Z, Weiwei W, Qiongyan Z, Yong W, Shujuan N, Cong T, et al. The lncRNA NEAT1 activates Wnt/β-catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J Hematol Oncol. (2018) 11:113. doi: 10.1186/s13045-018-0656-7
7. Wang Z, Zhang X-J, Ji Y-X, Zhang P, Deng K-Q, Gong J, et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. (2016) 22:1131. doi: 10.1038/nm.4179
8. Shuai H, Qingde W, Jincheng P, Xinsheng P, Dong R, Qiji L, et al. Transcriptional downregulation of miR-133b by REST promotes prostate cancer metastasis to bone via activating TGF-β signaling. Cell Death Dis. (2018) 9:779. doi: 10.1038/s41419-018-0807-3
9. Qiu-Yu W, Ci-Xiang Z, Meng-Na Z, Jun T, Chen-Long W, Cheng-Ning M, et al. MiR-133b targets Sox9 to control pathogenesis and metastasis of breast cancer. Cell Death Dis. (2018) 9:752. doi: 10.1038/s41419-018-0715-6
10. Yang L, Hou J, Cui XH, Suo LN, Lv YW. MiR-133b regulates the expression of CTGF in epithelial-mesenchymal transition of ovarian cancer. Eur Rev Med Pharmacol Sci. (2017) 21:5602–9.
11. Lv L, Zhou J, Lin C, Hu G, Yi L, Du J, et al. DNA methylation is involved in the aberrant expression of miR-133b in colorectal cancer cells. Oncol Lett. (2015) 10:907. doi: 10.3892/ol.2015.3336
12. Bhattacharjya S, Roy K, Ganguly A, Sarkar S, Panda CK, Bhattacharyya D, et al. Inhibition of nucleoporin member Nup214 expression by miR-133b perturbs mitotic timing and leads to cell death. Mol Cancer. (2015) 14:42. doi: 10.1186/s12943-015-0299-z
13. Duan FT, Qian F, Fang K, Lin KY, Wang WT, Chen YQ. miR-133b, a muscle-specific microRNA, is a novel prognostic marker that participates in the progression of human colorectal cancer via regulation of CXCR4 expression. Mol Cancer. (2013) 12:164. doi: 10.1186/1476-4598-12-164
14. Crawford M, Batte K, Yu L, Wu X, Nuovo GJ, Marsh CB, et al. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem Biophys Res Commun. (2009) 388:483–9. doi: 10.1016/j.bbrc.2009.07.143
15. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
16. Kato Y, Kaneda M, Hata K, Kumaki K, Hisano M, Kohara Y, et al. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet. (2007) 16:2272–80. doi: 10.1093/hmg/ddm179
17. Li E, Okano FM, Haber DF, Haber DA, Bell JD. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. (1999) 99:247–57. doi: 10.1016/S0092-8674(00)81656-6
18. Chen T. Structure and function of eukaryotic DNA methyltransferases. Curr Topics Dev Biol. (2004) 60:55. doi: 10.1016/S0070-2153(04)60003-2
19. Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. (2005) 74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721
20. Kaneko S, Li G, Son J, Xu C-F, Margueron R, Neubert TA, et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. (2010) 24:2615. doi: 10.1101/gad.1983810
21. Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. (2010) 464:1071–6. doi: 10.1038/nature08975
22. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. Ca Cancer J Clin. (2018) 60:277–300. doi: 10.3322/caac.20073
23. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. (2015) 136:E359–86. doi: 10.1002/ijc.29210
24. He J-H, Zhang J-Z, Han Z-P, Wang L, Lv Y, Li Y-G. Reciprocal regulation ofPCGEM1andmiR-145promote proliferation of LNCaP prostate cancer cells. J Exp Clin Cancer Res. (2014) 33:72. doi: 10.1186/PREACCEPT-1983004361129219
25. Song F, Li L, Liang D, Zhuo Y, Wang X, Dai H. Knockdown of long noncoding RNA urothelial carcinoma associated 1 inhibits colorectal cancer cell proliferation and promotes apoptosis via modulating autophagy. J Cell Physiol. (2019) 234:7420–34.
26. Pan B, Zhao M, Xu L. Long noncoding RNA gastric cancer-associated transcript 3 plays oncogenic roles in glioma through sponging miR-3127–5p. J Cell Physiol. (2019) 234:8825–33.
27. Luo Y, Chen J-J, Lv Q, Qin J, Huang Y-Z, Yu M-H, et al. Long non-coding RNA NEAT1 promotes colorectal cancer progression by competitively binding miR-34a with SIRT1 and enhancing the Wnt/β-catenin signaling pathway. Cancer Lett. (2019) 440:11–22.
28. Wu D, Pan H, Zhou Y, Zhou J, Fan Y, Qu P. microRNA-133b downregulation and inhibition of cell proliferation, migration and invasion by targeting matrix metallopeptidase-9 in renal cell carcinoma. Mol Med Rep. (2014) 9:2491–8. doi: 10.3892/mmr.2014.2116
29. Li W, Zheng J, Deng J, You Y, Wu H, Li N, et al. Increased levels of the long intergenic non–protein coding RNA POU3F3 promote DNA methylation in esophageal squamous cell carcinoma cells. Gastroenterology. (2014) 146:1714–26.e5. doi: 10.1053/j.gastro.2014.03.002
30. Teschendorff AE, Lee SH, Jones A, Fiegl H, Kalwa M. HOTAIR and its surrogate DNA methylation signature indicate carboplatin resistance in ovarian cancer. Genome Med. (2015) 7:108. doi: 10.1186/s13073-015-0233-4
31. Leonidas B, Evangelos V, George V, Eleftheria H. Non-coding RNAs and EZH2 interactions in cancer: long and short tales from the transcriptome. Int J Cancer. (2013) 133:267–74. doi: 10.1002/ijc.27859
32. Huang M, Hou J, Wang Y, Xie M, Wei C, Nie F, et al. Long noncoding RNA LINC00673 is activated by SP1 and exerts oncogenic properties by interacting with LSD1 and EZH2 in gastric cancer. Mol Ther. (2017) 25:1014. doi: 10.1016/j.ymthe.2017.01.017
33. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. (2010) 329:689–93. doi: 10.1126/science.1192002
34. Xiong L, Chun L, Rendong Z, Wenjun X, Xia N, Xiajun Y, et al. The EZH2- H3K27me3-DNMT1 complex orchestrates epigenetic silencing of the wwc1 gene, a Hippo/YAP pathway upstream effector, in breast cancer epithelial cells. Cell Signal. (2018) 51:243–56.
35. Xu Y, Yao Y, Jiang X, Zhong X, Wang Z, Li C, et al. SP1-induced upregulation of lncRNA SPRY4-IT1 exerts oncogenic properties by scaffolding EZH2/LSD1/DNMT1 and sponging miR-101–3p in cholangiocarcinoma. J Exp Clin Cancer Res. (2018) 37:81. doi: 10.1186/s13046-018-0747-x
36. Xiang S, Zou P, Tang Q, Zheng F, Wu J, Chen Z, et al. HOTAIR-mediated reciprocal regulation of EZH2 and DNMT1contribute to polyphyllin I-inhibited growth of castration-resistant prostate cancer cells in vitro and in vivo. Biochim Biophys Acta Gen Subj. (2017) 1862:589–99. doi: 10.1016/j.bbagen.2017.12.001
37. Ba M-C, Long H, Cui S-Z, Gong Y-F, Yan Z-F, Wu Y-B, et al. Long noncoding RNA LINC00673 epigenetically suppresses KLF4 by interacting with EZH2 and DNMT1 in gastric cancer. Oncotarget. (2017) 8:95542–53. doi: 10.18632/oncotarget.20980
38. Viré E, Brenner C, Deplus R, Blanchon LC, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. (2007) 25:279–80. doi: 10.1016/j.urolonc.2007.02.005
Keywords: LINC00114, EZH2, DNMT1, miR-133b, methylation, colorectal cancer
Citation: Lv L, He L, Chen S, Yu Y, Che G, Tao X, Wang S, Jian Z and Zhang X (2019) Long Non-coding RNA LINC00114 Facilitates Colorectal Cancer Development Through EZH2/DNMT1-Induced miR-133b Suppression. Front. Oncol. 9:1383. doi: 10.3389/fonc.2019.01383
Received: 03 August 2019; Accepted: 22 November 2019;
Published: 17 December 2019.
Edited by:
Cecilia Ana Suarez, National Council for Scientific and Technical Research (CONICET), ArgentinaReviewed by:
Hua Zhang, Guangdong Medical University, ChinaPei Yan Liu, University of New South Wales, Australia
Copyright © 2019 Lv, He, Chen, Yu, Che, Tao, Wang, Jian and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyuan Jian, amlhbnpoaXl1YW4yMDAxQDE2My5jb20=; Xuemei Zhang, Z3VpbGludGcwNzczQDE2My5jb20=