 Manqing Wu
Manqing Wu Jun Shen
Jun Shen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 22 November 2019
Sec. Molecular and Cellular Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01307
This article is part of the Research Topic Human and Oncoviral Noncoding RNAs as Modulators of Cancer Aggressiveness and Disease Progression View all 15 articles
Super-enhancers (SEs) are clusters of enhancers that play a key role in regulating genes that determine cell identity. Enhancer RNAs (eRNAs) are non-coding RNAs transcribed from enhancers that function to promote the enhancer's functions via multiple mechanisms, such as recruiting transcription factors to specific enhancers, promoting enhancer-promoter looping, directing chromatin accessibility, interacting with RNA polymerase II and facilitating histone acetylation. Understanding how super-enhancer RNAs (seRNAs) contribute to specific gene regulation has thus become an area of active interest. Immune checkpoint deregulation is one of the key characteristics of tumors and autoimmune diseases, and is also closely related to cell identity. Recent studies revealed a potential pathway for seRNA's involvement in regulating the expression of immune checkpoints. The present study reviews the current knowledge of eRNA function, immune checkpoint blockage mechanism, and its effect. In addition, for the first time, we explore the direct and indirect roles of seRNAs in regulating immune checkpoint expression in cancer and autoimmune diseases.
The identification of substantial amounts of non-protein coding transcripts and their versatile functions is one of the most striking findings of contemporary genomic research. Only ~2% of the transcribed human genome is accounted for by protein coding exons, thus non-coding RNAs constitute the majority of transcripts (1). Long non-coding RNAs (lncRNAs) are non-coding RNAs that are longer than 200 nucleotides, which play a significant role in the regulation of gene expression, splicing, translation, and epigenetic regulation.
Enhancers are DNA elements of a few hundred base pairs in length that are characterized by acetylation. Enhancers interact with transcription factors (TFs) and promoters. A mammalian cell contains thousands of active enhancers and ~1 million active enhancers have been found in all human cells (2). Super-enhancers (SEs), also known as stretch enhancers, are regions where multiple enhancers are clustered together. They exert more potent effects than typical enhancers and are associated with genes that are involved in determining cell identity in both the physiological and pathological state. Cell identity genes are a cluster of functionally interconnected genes that jointly establish the unique phenotype of a given cell type on epigenomic, transcriptomic, proteomic, and metabolomic level. For instance, NPC1L1, APOC3, and LCT in enterocytes, FOXP3, CTLA1, and IL2RA in T regulatory cells (3, 4). OCT4, NANOG, and PRDM14 are cell identity genes of ESC, they encode core transcription factors of embryonic stem cell (ESC) (5). These genes function together to enable expression of genes necessary to maintain its ESC pluripotency. The suppression of their expression leads to loss of pluripotency and self-renewal ability in ESC. Super-enhancers are required for cell type-specific processes and are linked with disease-associated genomic variations. Enhancer RNAs (eRNAs), another marker of active enhancers, are a novel species of non-coding RNA molecules that are transcribed from enhancer regions. Two types of eRNAs have been identified, comprising short, bi-directional and non-polyadenylated eRNAs and long, unidirectional, and polyadenylated eRNAs. The exact function of eRNAs is not clearly understood, and it has been hypothesized that eRNAs are transcription noise that do not contribute to gene expression (6). However, recent findings suggested that at least some eRNAs have a role in enhancer function by recruiting TFs to specific enhancers, promoting enhancer-promoter looping, directing chromatin accessibility, interacting with RNA polymerase II (RNAP II), and stimulating histone acetylation (7–12). Research into enhancers has expanded over the last decade and the biological function of enhancers has become increasingly clear. However, the exact function and mechanism of eRNAs are currently under investigation.
The immune system comprises innate and adaptive immunity. Immune checkpoints, consisting of co-stimulatory checkpoints and co-inhibitory checkpoints, are vital for the maintenance of self-response and prevention of autoimmunity. They are paired molecules that act as a double check before the stimulation or inhibition of an immune response. Immune checkpoints are expressed in a tissue or cell subset-specific manner. The application of immunotherapy in a wide variety of cancers has led to significant tumor shrinkage and improved clinical outcomes in patients by revitalizing the anti-tumor immune response (13, 14). The mostly widely studied inhibitory checkpoints are programmed cell death receptor-1 (PD-1), programmed cell death ligand (PD-L1), and cytotoxic T lymphocyte-associated molecule-4 (CTLA-4).
Recent studies have shown that SEs play key roles in determining cell identity in both healthy and pathological states. Over 25,000 enhancers were identified as deferentially activated in renal, breast, and prostate tumor cells, as compared with normal cells (15). This suggested a potential network between malignancy and enhancer activity. In addition, SEs are located at oncogenes and other genes that are essential for tumor pathogenesis in cancer cells, indicating their possible utility as biomarkers for tumor-specific pathologies (2). Considering the notion that evading immune destruction as a hallmark of malignancy, it is suspected that SEs in immune cells may be involved in the regulation of inhibitory checkpoint expression (16). In this review, we summarize the current understanding of eRNA function, their mechanism of action, and immune checkpoints. Then, we focus on the crosstalk between eRNA and immune checkpoints in pathological stages. A better understanding of the link between SEs, eRNAs, and immune checkpoints, may lead to eRNAs being developed as potential markers or therapeutic targets in the future.
Enhancers are often occupied by multiple signature TFs. The typical chromatin signature of enhancers includes a high H3K4me1 to H3K4me3 ratio, histone H3 lysine 27 acetylation, P300 acetyltransferase binding, CREB binding protein (CBP) binding, mediator complex subunit 12 binding, and a high sensitivity to nucleases (17–22). A typical enhancer activates its target gene transcription via its cis-acting function along with interactions with the promoter and multiple TFs, including Yin-Yang 1 (YY1) and myogenic differentiation 1 (MYOD) (9, 23). Enhancers can exert their function in an orientation and distance-independent manner, being capable of targeting both upstream and downstream genes (24). RNAP II occupation at some enhancers leads to the transcription of eRNAs, which is considered as another hallmark of an active enhancer (12). SEs are tissue specific regulatory regions of DNA consisting of clusters of enhancers. In various murine cell types (macrophages, Th cells, pro-B cells, embryonic stem cells, and myotubes), SEs and their target genes, which encode cell-type specific TFs, have been identified (20). By investigating the distribution of disease-associated DNA sequence variation in enhancers and SEs in human cells and tissues, Hnisz et al. found that trait-associated single-nucleotide polymorphisms (SNPs) were highly enriched in SEs, indicating their potential disease-associated role (2). In addition, SEs are characterized by specific histone modifications and they bind with a higher level of mediators, nipped-B-like protein, P300, chromodomain-helicase-DNA-binding protein 7 (CHD7), Bromodomain-containing protein 4 (BRD4), kruppel-like factor 4, estrogen-related receptor beta, and cohesin compared with typical enhancers (2, 20). The level of histone modifications H3K27ac and H3K4me1 at SEs exceeded those at typical enhancers significantly (20). Moreover, RNAP II is clustered at SEs at a greater density than at typical enhancers, resulting in a higher level of super-enhancer RNA (seRNA) production (25) (Figure 1).
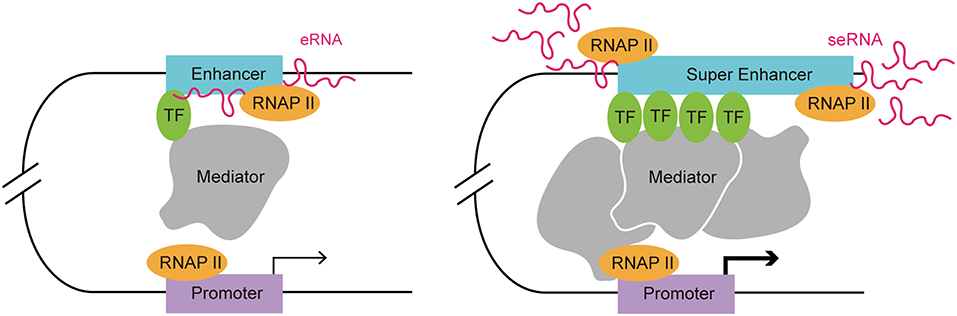
Figure 1. Comparison of a typical enhancer and super enhancer. Compared with typical enhancers, super enhancers are enriched with more transcription factors, mediators, and RNAP II. Hence, the transcription activity at a super enhancer is usually higher than at a typical enhancer. Functionally, super enhancers have a higher potential to promote target cell-identity-related gene transcription.
RNAs transcribed from enhancers can be classified as short, bi-directional, and non-polyadenylated eRNAs, and long, unidirectional, and polyadenylated eRNAs. The majority of seRNAs are capped and polyadenylated RNAs (25). This feature makes seRNAs more stable and capable of having a wider effect in physiological and pathological conditions. eRNAs are transcribed by the binding of RNAP II to enhancer DNA in various types of cells, such as macrophages, neurons, keratinocytes, and breast cancer cells (26, 27). It was proposed that TFs that are bound at enhancers interact directly or indirectly with the promoter via a cofactor to exert a stimulatory effect on RNAP II. Upon this dynamic interaction, RNAP II and its accessory effectors come close to enhancer DNA, resulting in initiation of eRNA transcription (28).
The exact function of eRNAs is incompletely understood; nevertheless, evidence suggests that at least some eRNAs play an active part in the regulation of enhancer activity and gene expression. Nicholas et al. found that the synthesis of eRNAs precedes the transcription of target gene transcription in lipopolysaccharide-activated macrophages (29). This indicated that eRNAs might be associated with transcription activation of target gene. The transcript level of the majority eRNAs correlates highly correlated with the mRNA expression level of the nearby target gene, suggesting an activating function in promoting mRNA synthesis (27, 30). Consistently, knockdown of eRNAs leads to decreased expression of nearby target genes (31, 32).
The actions of eRNAs have been widely studied in recent decades and several functional mechanisms have been proposed. The biological functions of eRNA are associated with TF recruitment, enhancer-promoter looping, chromatin conformation, and histone acetylation (Figure 2) (7–12, 33, 34). In addition to their functional contributions, eRNAs are also markers of enhancer activity. As an independent indicator of enhancer activity, the presence of eRNAs can distinguish whether the enhancer is active or silent. In macrophages, nearly all SEs express seRNAs (93.3%) within intergenic regions, which indicated that the presence of seRNAs could be used to mark SEs (35).
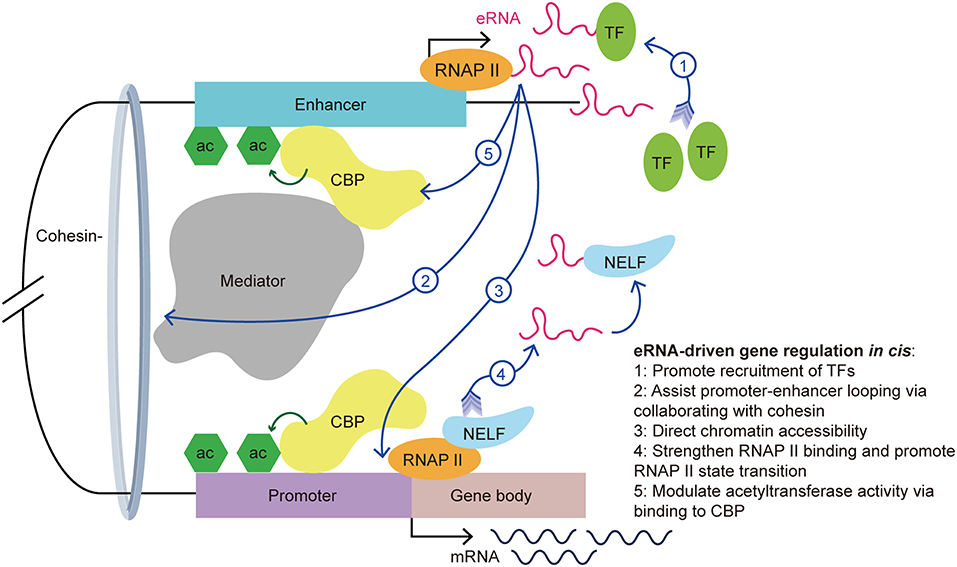
Figure 2. eRNA-driven gene regulation in cis. Cis-regulatory elements, distal enhancers, and proximal promoters interact with transcription-associated proteins by forming an enhancer-promoter loop. The looping conformation brings the eRNA near to the promoter of the target gene, allowing it exert its function in cis. eRNAs exert their gene regulation function via interaction with a variety of transcription-associated proteins, including transcription factors, cohesin, mediators, RNAP II, and CBP.
First, recent studies showed that eRNAs can promote the recruitment of TFs. YY1 is a TF that not only binds to active enhancers and promoters, but also binds to eRNA and RNA transcribed from promoters in murine embryonic stem cells. Further investigations revealed that YY1 binding to DNA is stabilized by an eRNA that is tethered by RNAP II (23). It is possible that the eRNA captures free YY1, which allows this TF to bind to a nearby DNA locus. This creates a positive feedback loop in the stimulation of local transcription and allows more TFs to bind the genomic locus. Recently, Weintraub et al. found that YY1 could form dimers and bind to enhancers and promoters to facilitate enhancer-promoter looping (36), which suggested an indirect facilitation effect of the eRNA on enhancer-promoter looping. Similarly, Charles et al. found a novel group of interferon gamma (IFNG) eRNAs that bind nuclear factor kappa B (NF-κB) to enhance its function. Treatment of chromatin with ribonuclease led to decreased NF-κB binding to the IFNG genomic sites. Using cell transfection techniques, the authors illustrated that knockout of IFNG-R-49, an IFNG eRNA, resulted in the reduction of NF-κB binding to the IFNG-D-49 genomic site, which demonstrated that IFNG eRNAs contribute to maintaining the binding between NF-κB and the IFNG locus (7).
Second, Amartya et al. identified a significant correlation between promoter-enhancer looping, the presence of eRNAs, and gene expression, which suggested that eRNAs are involved in the interaction between enhancers and promoters (37). As part of the gene regulatory mechanism, enhancer-promoter looping is necessary for gene activation (38). A previous study showed that enhancer-promoter looping was modulated in part by the mediator complex and cohesin (21). Following the binding of the mediator complex and cohesin to the enhancer and promoter, looping of the enhancer and promoter brings the eRNA close to the target gene promoter to allow coordination and activation. Knockdown of specific eRNAs reduced enhancer-promoter looping and limited the interplay between transcription effectors that are located within the loop, such as mediator 1, P300 and early growth response 1 (8, 31, 39, 40). Knockdown of the growth regulating estrogen receptor binding (GREB) eRNA led to suppression of enhancer-promoter looping and inhibition of GREB1 gene induction. Further investigations showed a reduction in cohesin recruitment after eRNA knockdown. This finding suggested that eRNAs promote enhancer-promoter looping via collaborating with cohesin (33).
Third, eRNAs also contribute to directing chromatin accessibility and thus promote specific gene expression. Mousavi et al. identified two seRNAs transcribed from CE and DRR enhancers in MYOD1, a recently labeled SE, in skeletal muscle satellite cells. Depletion of these eRNAs caused reduced chromatin accessibility and RNAP II occupancy at the MYOD1 and MYOG (Myogenin) loci, respectively. Normally, the MYOG locus remains inaccessible to nucleases, and chromatin remodeling is needed for the transcription activation of this locus. Using deoxyribonulcease I (DNase I) accessibility as an indicator for remodeling, the authors detected a reduction of DNase I accessibility at specific loci in eRNA-depleted cells. Additionally, they hypothesized that eRNAs are involved in regulating the assembly of transcriptional systems by observing that eRNAs affects RNAP II residency at target genes (9).
Fourth, studies have suggested that eRNAs exert various roles in the interaction with RNAP II. For example, eRNAs strengthen the binding of RNAP II to enhancer regions and promoters (9). Maruyama et al. disclosed the attenuation of diethyl maleate-induced RNAP II binding to promoters in eRNA knockdown cells (41). Moreover, eRNA promotes the paused RNAP II transition into the gene body by acting as a decoy. Arc eRNA depletion resulted in a decrease in the elongating form of RNAP II, which indicated that eRNAs promote the state transition of RNAP II. This hypothesis was further supported by the finding that knockdown of Arc eRNA led to maintenance of the negative elongation factor complex (NELF) on the promoter. NELF induces RNAP II pausing by binding to RNAP II, the promoter, and the newly generated RNA. Katie et al. suggested that eRNAs bind to NELF via competing with the nascent RNA, leading to the detachment of NELF from RNAP II, thereby enabling RNAP II elongation and mRNA synthesis (11). The state transition of RNAP II from paused to productive elongation is extremely important for target transcript production.
Last but not the least, eRNAs can bind to CBP and modulate the acetyltransferase activity at the enhancer, thus increasing the transcription of target genes. CBP binding to P300 and the resulting high levels of H3K27ac, are hallmarks of enhancers. Having noticed that there was more active transcription from loci with CBP bound to eRNAs than from the no-RNA binding control CBP binding sites, Daniel et al. found that this effect is stimulated by eRNAs binding to the histone acetyltransferase (HAT) domain of CBP. This domain determines the HAT enzymatic activity and this process promotes CBP acetyltransferase activity. The authors demonstrated that eRNA binding exposes the activation loop in CBP/P300. Thus, there is an increase of H3K27ac and H3K18ac at the enhancer and promoter, which increases transcription of the target genes (12). Taken together, eRNAs stimulate target gene transcription in part by stimulating histone acetylation.
The functions of seRNAs can be classified as cis-regulation and trans-regulation. The cis regulation by eRNAs has been widely accepted, in which the enhancer-promoter looping structure brings the eRNA close to the target gene. Using chromosome conformation capture (3C) technology, this looping model has been supported by a wide range of studies. As previously discussed, the cis-acting function of eRNAs is accomplished via their dynamic interactions with TFs, modifiers, and cohesin subunits within or near the enhancer-promoter loop. Depletion of seRNAs from distal super enhancers at the NANOG locus led to significantly decreased expression of DPPA3 (encoding developmental pluripotency associated 3). DPPA3 is the nearest gene to the NANOG super enhancer apart from NANOG itself. Using 3C, the authors demonstrated that the looping of the distal super-enhancer at the DPPA3 promoter decreased by ~50%, suggesting that the distal seRNA stabilizes the looping and chromatin interactions in cis, thereby regulating the expression of DPPA3 (31). Similarly, the transcription of another seRNA, named CARMEN (Cardiac mesoderm enhancer-associated non-coding RNA), was found to cause activation the expression of direct downstream genes (42). Taken together, these results demonstrated that seRNA functions as a scaffold that guides TFs and looping-associated protein complexes in cis.
Intriguingly, some recent studies suggested seRNAs might also function interchromosomally (trans activity) to direct target gene expression (Figure 3) (8, 25, 32, 43). For instance, the MYOD Upstream Non-coding RNA (MUNC), an eRNA originating from the distal regulatory region enhancer of MYOD, was observed to induce the transcription of specific myogenic genes [e.g., MYOD, MYOG, and MYH3 (myosin heavy chain 3)] in trans. Overexpression of MUNC in MYOD−/− cells caused MYOG and MYH3 transcription and expression. Notably, these two genes are located on different chromosomes, validating MUNC's trans activity (32). Alcarez-Dominguez et al. reported that a polyadenylated-eRNA-producing Band 3 SE transcribes an seRNA called Bloodlinc that can facilitate gene expression and stimulate red cell production in trans (25). Strikingly, they found that Bloodlinc diffused beyond its domain of transcription and 81 direct gene targets located across multiple chromosomes were identified as regulated reciprocally upon Bloodlinc depletion or overexpression. This is quite different from typical eRNAs, which usually remain in proximity to their parent enhancers. Many of the regulated genes were located outside the super-enhancer domain. Further investigations showed that Bloodlinc binds to trans-chromosomal loci that encode key erythroid modulators and TFs. Using mass spectrometry [comprehensive identification of RNA-binding proteins by mass spectrometry (ChiIRP-MS)] techniques, the authors found that Bloodlinc interacted with multiple protein complexes that function as RNA helicases (e.g., DExD-box helicase 21), RNA transporters (e.g., heterogeneous nuclear ribonucleoprotein A1), RNA splicers (e.g., KH-type splicing regulatory protein), chromatin organization regulators (e.g., marker of proliferation Ki-67 (MKI67), and Lamin A/C), and transcription coactivators or co-repressors [e.g., MYB binding protein 1a (MYBBP1A) and heat shock protein family A member 8 (HSPA8)]. Moreover, immunoprecipitates of endogenous heterogeneous nuclear ribonucleoprotein U (HNRNPU), a nuclear matrix protein that stabilizes RNA-chromatin associations, were specifically enriched for Bloodlinc, which confirmed the interaction between Bloodlinc and HNRNPU (25). Thus, these findings suggested a model for how Bloodlinc acts in trans. Specifically, Bloodlinc accesses its trans target genes via chromatin interactions stabilized by HNRNPU. Bloodlinc stimulates or represses target genes expression via interacting with transcription coactivators (e.g., MYBBP1A) or transcriptional repressors (e.g., HSPA8). This process is stabilized by chromatin organization regulators that also interact with Bloodlinc.
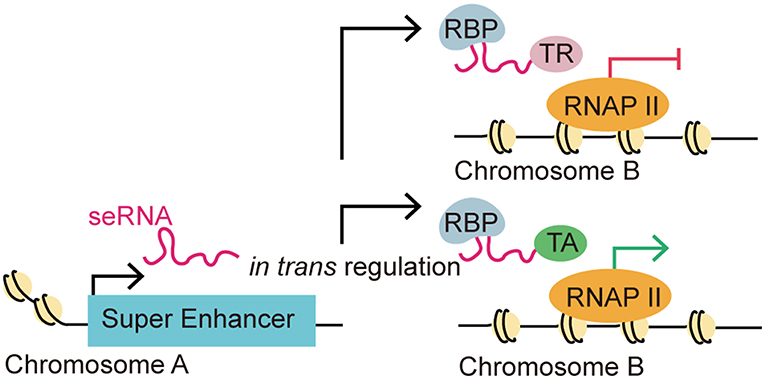
Figure 3. eRNA-driven gene regulation in trans. Recent studies identified that eRNAs could regulate gene transcription in trans, which means that an eRNA could affect gene regulation in a distal target gene in a different chromosome. This distal regulation function is accomplished via interactions with RNA-binding proteins (RBPs) and other transcription-associated proteins. The trans regulation by eRNAs can be classified as repressive or activating depending on their interaction with transcription repressors (TRs) or transcription activators (TAs).
To determine whether enhancers or eRNAs correlate with disease-associated DNA sequence variations, Hnisz et al. investigated the distribution of SNPs within enhancers and super-enhancers. They found that SNPs were enriched in enhancers and SEs, with trait-associated SNPs occurring in SEs at a strikingly higher rate than in enhancers. Analysis in a colorectal cancer cell line demonstrated that more than one-third of SE genes have functions that are closely related with cancer hallmarks, such as evading growth suppressors (e.g., Cyclin D1, Epiregulin), avoiding immune destruction (e.g., F2R like trypsin receptor 1) and sustaining proliferative signaling (e.g., insulin receptor substrate 1, KIT ligand) (2, 16). Consistently, other studies found that SEs are associated with critical tumor oncogenes in various types of cancers. Lovén et al. found that in multiple myeloma, disruption of BRD4 (bromodomain-containing Protein-4) and mediator occupancy in an SE led to inhibition of tumor oncogenes, including MYC (44). Recently, eRNAs were found to participate in regulating gene transcription and cell-cycle progression with TP53 (p53 tumor suppressor). Melo et al. found some of the TP53 binding regions encompass enhancer activity and produce eRNAs in a p53-dependent manner. Knockdown of these eRNAs significantly inhibited downstream target gene transcription upon TP53 activation, suggesting the eRNAs produced from TP53 bound enhancer regions that are required for efficient TP53 transcription enhancement and p53-dependent cell-cycle arrest (45). Moreover, Jiao et al. found a heparanase eRNA that is elevated in cancer cell lines, and enhances tumorgenesis and aggressiveness of cancer cells by facilitating chromatin looping between a super enhancer and the HPSE (heparanase) promoter (39). The results from these studies indicated that seRNAs might have significant roles in tumorgenesis and could serve as potential targets for cancer clinical therapy.
Immune checkpoints, including stimulatory and inhibitory pathways, are regulatory signals that play vital roles in the maintenance of the delicate balance between activation of adaptive immunity and retaining self-tolerance from autoimmunopathy. Stimulatory checkpoint molecules encompass CD28 and the inducible T cell costimulator (both from the B7-CD28 superfamily) as well as CD27, CD40, OX40 (TNF receptor superfamily member 4), and CD137 (TNF receptor superfamily member 9), which are all from the tumor necrosis factor (TNF) receptor superfamily. Inhibitory checkpoint molecules include members of the B7-family [such as CTLA-4 (CD 152), PD-1 (CD 279)], Lymphocyte Activation gene-3, and nicotinamide adenine dinucleotide phosphate. Through unique and non-redundant pathways, these molecules work as secondary signals to determine the activation or inhibition of immune cells upon antigen recognition, which modulates the duration and amplitude of physiological immune responses.
Cancer cells are capable of evading immune recognition and immune-mediated destruction by downregulating the expression of tumor antigens, seizing inhibitory immune checkpoints, and inducing immune exhaustion, which leads to the increased expression of inhibitory receptors on T cells, such as CTLA-4 and PD-1 (16). Exhausted T cells often feature CTLA-4 expression. CTLA-4, as a B7/CD28 family member, is involved in tumor immune evasion via down-regulation of CD4+ effector cells (Teff) and promotion of Treg cell activity (13). PD-1, another marker of T cell exhaustion, is expressed at a characteristically high level in tumor infiltrating T cells, which is in consistent with a reduction in interleukin (IL)-2 and IFNγ production and cell cycle arrest in T cells (46). In addition, tumor cells and tumor associated antigen presenting cells (APCs) often express higher levels of co-inhibitory molecules than co-stimulatory molecules, which enhances the activation threshold of T cell and leads to T cell anergy (47).
Rheumatic diseases are characterized by abnormal activation of the immune system, which leads to chronic inflammation and tissue damage. Immune checkpoints are actively involved in the manifestation of rheumatic disease. Genetic polymorphisms in the PD-1 gene (PDCD1) in humans correlate with a variety of autoimmune diseases, including type 1 diabetes (T1D), rheumatoid arthritis (RA), multiple sclerosis, and systemic lupus erythematosus (SLE) (48). Mice deficient for a single inhibitory receptor (such as CTLA-4 or PD1) often display enhanced susceptibility to experimentally-induced autoimmune diseases or may spontaneously develop a lupus-like disease (49). CTLA-4 has a fundamental role in establishing immune tolerance. Ctla4 knockout mice showed premature death caused by the development of lymphoproliferative disease with multiple organ involvement (50), while human patients with mutations that caused loss of function of CTLA-4 also manifested widespread immune dysregulation (51). Jury et al. identified CTLA-4 dysfunction as a possible cause of abnormal T-cell activation in patients with SLE (52). In addition, autoimmunity activation and inflammatory toxicities, such as colitis, hepatitis, pneumonitis, dermatitis, and myasthenia gravis, are major adverse events caused by the use of immune-checkpoint blockers in tumor immunotherapy (53–56). The use of Ipilimumab, a CTLA-4-blocking antibody, was reported to cause inflammatory exacerbation in 25% of patients who had preexisting autoimmune diseases (57). This led to the hypothesis that enhancing inhibitory pathways would be beneficial to treat autoimmune disease. Abatacept, a CTLA4–Fc fusion protein, is the first checkpoint-targeting drug to be approved to treat rheumatic diseases. CTLA-4 Fc prevents costimulatory signaling, thus reducing T cell activation in RA, SLE, and psoriatic arthritis (58–60).
PD-1 and CTLA-4 are the most widely studied inhibitory checkpoint molecules because of their superior performance in the treatment of tumors (Table 1). PD-1 is expressed on T cells, B cells, dendritic cells (DCs), monocytes, natural killer (NK) T cells, exhausted cells, and Treg cells. When engaged to its receptor PD-L1, which is widely expressed on antigen-presenting cells, CD4+ T cells, and non-lymphoid tissues, PD-1 delivery an inhibitory signal via direct and indirect pathways. In the direct pathway, PD-L1-engaged PD1 potently counteracts CD28-co-stimulation and T cell receptor (TCR) signal transduction via terminating zeta chain of T cell receptor associated protein kinase 70 and phosphoinositide-3-kinase (PI3K) phosphorylation, leading to recruitment of protein tyrosine phosphatase non-receptor type 11 (PTPN11), which in turn inhibits IL2 production and reduces the transcription of pro-survival factor BCL-2-like protein 1 (73). In the indirect inhibitory mechanism, engaged PD1 decreases casein kinase 2 alpha 1 expression and activity, which results in the maintenance of phosphatase and tensin homolog activity, shutting off both the protein kinase B (AKT) pathway and subsequent T cell growth (73). In addition, the inhibitory function of PD-1 is exemplified by the promotion of TCR endocytosis and shifting the metabolic status of T cells toward fatty acid beta-oxidation, which leads to metabolic restriction (73). While CTLA-4 plays a pivotal role in attenuating the activation of naïve and memory T cells via competing with CD28-mediated signaling (74). Downstream of both CTLA-4 and PD-1 abrogates AKT activity, which is related to limiting cellular metabolism (70).
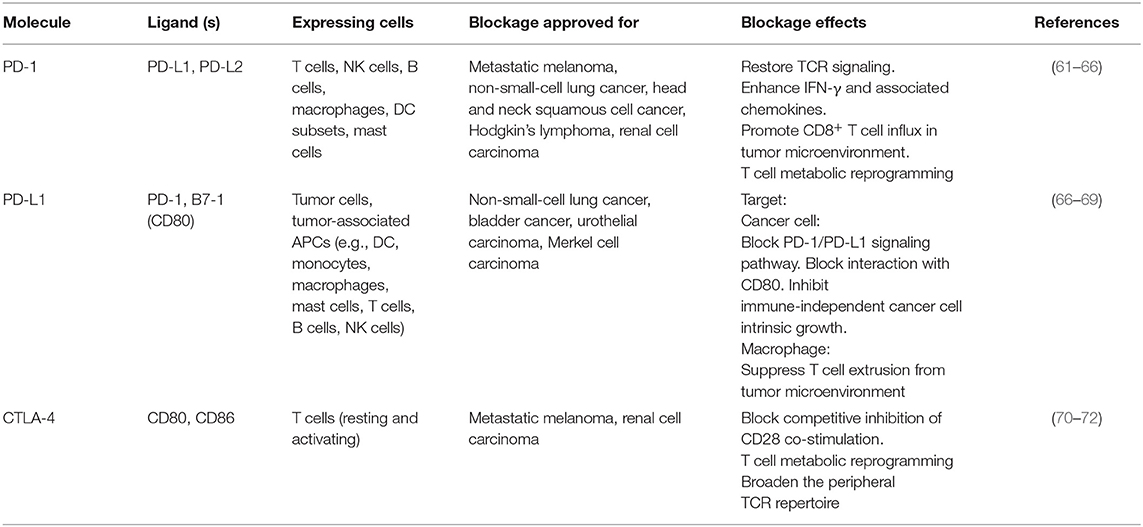
Table 1. Overview of PD-1/PD-L1 and CTLA-4 blockage.
Immunotherapy, especially PD-1/PD-L1 blockage and CTLA-4 blockage, has revolutionized the landscape of cancer treatment in recent years. The FDA has approved immune checkpoint inhibitors for the treatment of a range of tumor types, such as melanoma, non-small cell lung cancer, renal cell carcinoma, Hodgkin lymphoma, and head and neck squamous cell carcinoma.
By counteracting the pathological function of PD-1, antibodies that block PD-1 (e.g., Pembrolizumab and Nivolumab) and its ligand PD-L1 (e.g., Atezolizumab, Avelumab, and Duralumab) inhibit adaptive immune resistance and reinvigorate the immune response against cancer cells. PD-1 mediates immune suppression via a variety of mechanisms in cancer. In T cells, PD-L1-bound PD-1 inhibits TCR signaling by recruiting PTPN11 to the immunoreceptor tyrosine-based switch motif domain, which results in dephosphorylation of downstream signaling molecules, decreased IL-2 production, reduction in cell cycle progression, and reduced expression of TFs involved in effector function (T-bet and eomesodermin) (74). An elevated level of circulating IFNγ and its associated chemokines [C-X-C motif chemokine ligand (CXCL)-9 and CXCL-10] and T cell activation markers (MKI67 and major histocompatibility complex, class II, DR) were detected in the serum of patients undergoing anti-PD-1 and anti-PD-L1 treatment (61). PD-1 blockage restores T cell activation and an influx of CD8+ T cells was detected in the tumor microenvironment (62). In addition, PD-1 signaling interferes with CD28-mediated activation of PI3K and AKT, which in turn limits glucose metabolism (70). The resulting bioenergetic insufficiencies inhibit mammalian target of rapamycin (mTOR) activity and IFNγ production, impair EZH2 (enhancer of zeste 2 polycomb repressive complex 2 subunit) expression in T cells, and reduce the level of phosphoenolpyruvate, which is linked with a lack of activation of CD4+ tumor-infiltrating lymphocytes (61). The process of metabolic restriction is a driver of T cell exhaustion. Antagonists of PD-1 cause T cell metabolic reprogramming and restore their glycolytic capacity, as well as the subsequent effector function (63). PD-1 and PD-L1 blockage also decrease E3 ubiquitin ligase CBL-B expression thus inhibiting the downregulation of TCR (64).
PD-L1 expression is especially high in tumor cells and tumor-associated APCs (e.g., tumor environment DCs, macrophages, and fibroblasts) (75). As a result of adaptive immune resistance, PD-L1 overexpression on tumor cells is induced by IFNγ that is produced by activated T cells. High levels of PD-L1 expression have been associated with poor prognosis in many types of cancer. PD-L1 antibodies exert their antitumor effect partly by blocking the PD-1–PD-L1 interaction. Manish et al. found that PD-L1 also interacts with B7-1 (CD80) to inhibit T cell activation and proliferation (67). Therefore, PD-L1 blockage also may restore T cell activation by inhibiting the CD80–PD-L1 interaction. A recent study showed that tumor-expressed PD-L1 has tumor-intrinsic effects in addition to delivering an inhibitory signal to PD-1 on T cells. PD-L1 promotes cell-intrinsic growth in an immune-independent manner in both melanoma and ovarian cancer. PD-L1 represses tumor autophagy and enhances the mTOR pathway in both ovarian cancer and melanoma (68). Thus, PD-L1 blockage may exert its effect by mediating PD-L1-related intrinsic tumor signaling. PD-L1 expression on macrophages may result in active extrusion of T cells from the tumor microenvironment, indicating another possible pathway for PD-L1 blockage (69).
Super-enhancers play a critical role in the regulation of genes that define cell identity, and increasing evidence suggests that SEs and eRNAs have functions in tumorgenesis. However, the exact function and mechanism of seRNAs in the regulation of tumorgenesis and tumor immunotolerance is not fully understood. Lovén et al. found that cancer cells acquire SEs at oncogenes and at genes that are important for tumor pathogenesis. They found that SEs assist the high level transcription of genes [e.g., MYC, IRF4 (interferon regulatory factor 4), XBP1 (X-box binding protein 1), CCND2 (Cyclin D2)] that are deregulated in multiple myeloma cells (44). Ding et al. revealed the oncogenic role of eGREB1, an eRNA of an estrogen-responsive gene enhancer, growth regulating estrogen receptor binding 1 (GREB1), in bladder cancer. Upregulated eGREB1 is associated with higher level TNM stages of bladder cancer. Consistently, proliferation, migration, and invasion were inhibited upon eGREB1 knockdown, while apoptosis was promoted (76). In addition, for a variety of cancer cells (e.g., pancreatic cancer and T cell leukemia), SEs were identified around the MYC gene in cancer cells. However, these SEs were not identified in their healthy counterparts (2). Besides, Hnisz et al. provided a list of tumor-specific SEs that fall into different categories of hallmark cancer genes in colorectal cancer. For instance, the identified SEs of PCDH7 (protocadherin 7), CCND1 (Cyclin D1), and F2RL1 are associated with activating invasion and metastasis, evading growth suppressors, and avoiding immune destruction, respectively (2). Taken together, these findings indicated that SEs might act as keys for amplified oncogene expression. This hypothesis was supported by the results of Wong et al., who found that multiple oncogenic TFs are regulated by SEs in acute T cell lymphoblastic leukemia. Disruption of SE-related gene expression and cancer cell death were identified after treating cancer cells with RNAP II activation blocker (77). SeRNAs play an active role in promoting SE function; therefore, we suspected seRNAs could also function in this inhibitory process.
MYC is an oncogene that has been studied in depth. The activation and overexpression of MYC is a characteristic feature of tumorgenesis and cancer maintenance. As a TF, MYC activates the expression of many pro-proliferative genes by binding at enhancers and recruiting HATs (78). One of the mechanisms by which the MYC gene is believed to maintain cancer cell survival is to exempt itself from immune surveillance and the anti-tumor immune response (79). This hypothesis was supported by the finding that MYC expression correlated highly with PDCD1L1 (PD-L1) gene expression in non-small cell lung cancer cell (79). Kim et al. also identified poorer clinical outcome for patients with both MYC and PD-L1 dysregulation and overexpression (79). Consistently, Casey et al. demonstrated that MYC upregulates the expression of immune checkpoints, CD47 and PDCD1L1, on cancer cells by direct interaction with the promoters of these two genes. In multiple types of cancer, silencing of MYC leads to a significant reduction in the transcription and expression of CD47 and PDCD1L1, both in vitro and in vivo (80). Therefore, MYC may be a key regulator for immune checkpoint expression in cancer. For a variety of cancer cells, SEs are found specifically clustered at genes surrounding the MYC gene (2). SEs tend to express seRNAs at higher levels than typical enhancers; therefore, we wondered whether seRNAs participate in the regulation of immune checkpoint expression. Human colorectal cancer-specific nucleus retained Colorectal Cancer Associated Transcript 1-long isoform (CCAT1-L) is a 2,600 nucleotide long lncRNA that is transcribed from an SE and therefore is considered as an seRNA. A recent study suggested that the seRNA CCAT1-L contributes to the regulation of MYC expression in cis in colorectal cancer. CCAT1-L is transcribed from a locus 515 kb upstream of the MYC gene (MYC-515) in the human 8q24 region. This SE forms an enhancer-promoter loop with the MYC promoter, thus bringing CCAT1-L in to close proximity with the promoter. Such chromatin interactions are present specifically in colorectal, breast, and prostate cancer (81). Xiang et al. reported that CCAT1-L assists in the maintenance of chromatin looping between the SE and the MYC promoter (82). Previous studies revealed that eRNAs participate in gene regulation by stabilizing enhancer-promoter looping and by dynamic interactions with TFs and mediators in the surrounding area. The accumulation of CCAT1-L surrounding its SE indicates its possible function in the regulation of its target gene. By examining the expression of mRNA after CCAT1-L knockdown, reduced MYC transcription and expression were detected. However, overexpression of CCAT1-L from a plasmid showed no apparent activation of MYC expression. This could be explained by the possibility that extrinsic CCAT1-L localized to many nuclear sites but not its in cis site. To confirm this hypothesis, Xiang et al. (82) used transcription activator-like engineered nucleases to achieve in cis overexpression of CCAT1-L in a low CCAT1-L expression cell line. CCAT1-L overexpression resulted in higher MYC expression and faster cell growth than that in the control cancer cell group. Thus, CCAT1-L enhances MYC expression in cis. The most common form of seRNA function is in cis regulation; however, how is CCAT1-L brought close to its target gene from 515 kb away? To answer this question, Xiang et al. applied the 3C technique to investigate the interaction frequencies between possible enhancers and MYC promoter segments. Intriguingly, they found an interaction between a locus 335 kb upstream of the MYC promoter (MYC-335), and the MYC promoter showed the highest interaction frequency with this site, while the interaction between MYC-515 and MYC-355 ranked as second. Earlier Pomerantz et al. found an enhancer located at MYC-355, which forms a loop between the MYC promoters to promote its transcription (83). The result of 3C analysis suggested that CCAT1-L locates to MYC-335, bringing it closer to MYC. Knockdown of CCAT1-L resulted in a prominent decrease in the chromatin interactions between MYC-335 and the MYC promoter and between MYC-515 and MYC-335. Taken together, the results suggested that a looping conformation is formed between MYC and MYC-335 and between MYC-335 and MYC-515. In addition, CCAT1-L is required to maintain the specific loops between the MYC enhancers and the MYC promoter. Further investigation into the exact mechanism by which CCAT1-L functions to promote enhancer-promoter looping revealed that TFs transcription factor 4 and CCCTC-binding factor (CTCF) are enriched at the loops of the MYC promoter, and the MYC-335 and MYC-515 segments. Moreover, knockdown of CTCF is associated with decreased transcription of MYC and CCAT1-L, suggesting that the enhancer-promoter looping at MYC is CTCF-mediated. In addition, RNA immunoprecipitation showed a specific interaction between CTCF and CCAT1-L. Reduced CTCF occupation of the loop region at MYC was detected after depletion of CCAT1-L, indicating that CCAT1-L assists the binding of CTCF to chromatin and contributes to the looping formation at the MYC locus (82). In summary, Xiang et al. demonstrated the involvement of seRNA CCAT1-L in the regulation of key oncogene MYC in colon rectal cancer. CCAT1-L regulates the expression of MYC by interacting with CTCF and assisting its binding with chromatin to sustain the enhancer-promoter looping conformation between the MYC promoter and MYC-335 and between MYC-335 and MYC-515. MYC expression has been linked to the regulation of a variety of cancer hallmark-related genes in tumors. As mentioned above, MYC upregulates the expression of immune checkpoints CD47 and PDCD1L1 on cancer cells by direct interaction with promoters of these two genes. Therefore, it is possible that seRNA CCAT1-L participates in the regulation of CD47 and PDCD1L1 expression indirectly by promoting MYC gene expression in cis.
Additionally, Jiang et al. recently demonstrated that CCAT1 interacts with the TFs tumor protein p63 (TP63) and SRY-box 2 (SOX2) to regulate the expression of the epidermal growth factor receptor (EGFR) in esophageal squamous cell carcinoma (ESCC) (84). With identification of TP63 and SOX2 co-occupied at an SE, the authors wondered whether there is interplay between the seRNA and TP63 and SOX2. Further investigation showed that transcription of seRNA CCAT1 is activated or inhibited by TP63 and SOX2 co-binding or depletion at the promoter and SE of CCAT1. Thus, seRNA CCAT1 was validated as the downstream interaction target for TP63 and SOX2. Moreover, seRNA CCAT1 forms a complex with TP63 and SOX2 and then binds to SEs of EGFR to enhance its transcription and expression. The transcription of EGFR activates the downstream PD-L1 expression related pathways, including RAS/mitogen activated protein kinase (MAPK) and PI3K/AKT signaling pathways (84). Consistently, the expression of PD-L1 is elevated significantly in response to EGFR signaling activation in esophageal squamous cancer cells. By contrast, inhibition of the EGFR pathway led to a sharp decline in PD-L1 expression (85, 86). Further investigation showed that EGFR-dependent expression of PD-L1 in ESCC is affected by EGFR/PI3K/AKT, EGFR/RAS/MAPK and EGR-phospholipase C gamma 1 (PLC-γ) signaling pathways (84). Many studies have revealed the close connection between PD-L1 expression and EGFR mutation (87–89). Higher PD-L1 expression usually indicates poor prognosis (88, 89). For instance, in non-small-cell lung cancer, mutation in EGFR lead to upregulation of PD-L1 by activating PI3K/AKT and RAS/extracellular signal-regulated kinase (ERK) signaling (90). Notably, the EGFR gene is located in the 7p11 region in the human genome, indicating that CCAT1 promotes EGFR transcription in trans. Taken together, we propose that seRNA CCAT1 could be involved in the regulation of PDCD1L1 transcription in trans by forming a seRNA-TF complex to promote EGFR expression and activate the downstream signaling pathways (Figure 4).
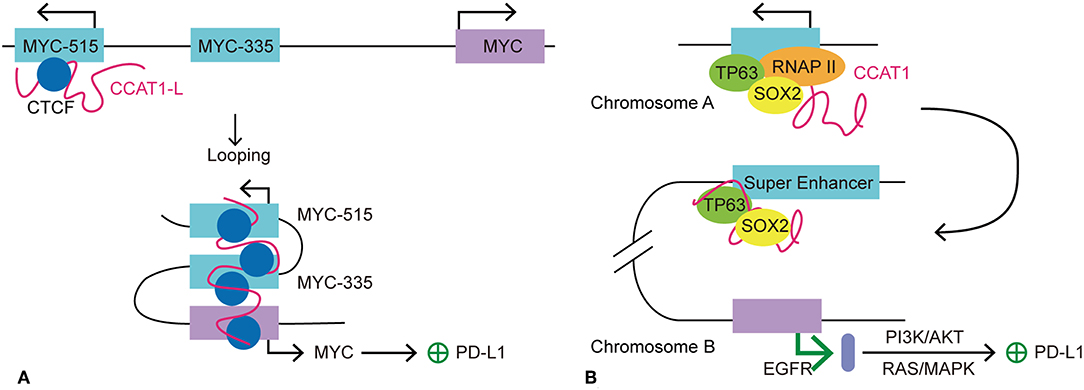
Figure 4. Models of seRNA CCAT1 in the regulation of PD-L1 expression. CCAT1 is an oncogenic seRNA that can regulate the expression of PD-L1 via cis and trans actions. (A) SeRNA CCAT1-L induces the expression of PD-L1 via enhancing the expression of MYC in cis. With the help of CTCF, CCAT1-L is brought close to the MYC promoter via MYC-515-MYC-335-MYC looping. This looping conformation promotes MYC transcription, which allows its further promotion of PD-L1 expression. (B) TP63 and SOX2 are two transcription factors that promote seRNA CCAT1 transcription. CCAT1 interacts with TP63 and SOX2 to further act in trans on another chromosome to promote the transcription of EGFR. EGFR promotes PD-L1 expression via its downstream pathways, PI3K/AKT and RAS/MAPK.
CDK7 affects transcription initiation and elongation by blocking SE normal function. As for its potential clinical application, Chipumuro et al. found cyclin-dependent kinase 7 (CDK7) inhibitor, THZ1, selectively downregulates SE-regulated MYCN overexpression and MYCN-driven transcription amplification in neuroblastoma (91). Similarly, other researchers revealed THZ1 suppressed SE-driven oncogenic transcriptional amplification in other cancers (92, 93).
BRD4 is a member of BET family, which includes BRD2, BRD3, and bromodomain testis-specific proteins (BRDT). As a transcription co-activator, BRD4 is often required for the expression of oncogenes, including MYC (94–96). The functions of BET proteins include initiation and elongation of transcription, and cell cycle control. BRD4 recruits a variety of transcription complexes, including mediator and positive transcriptional elongation factor b (P-TEFb), to acetylated chromatin, leading to the activation of gene expression via phosphorylation of RNAP II (97, 98). Studies showed accumulation of BRD4 at SEs, which facilitated eRNA transcription via interacting with acetylated histones to assist RNAP II progression (99). Recently, BET inhibitors (i.e., JQ1) have been developed for anticancer treatment. JQ1 displaces BRD4 from chromatin, breaks the cell cycle, and induces apoptosis in tumor cells. JQ1 also inhibits BRD4-associated seRNA synthesis by targeting SEs preferentially (44, 99). When treated with JQ1, SEs displayed a higher level of loss of BRD4 accumulation than typical enhancers (44). eRNAs have a role in enhancer function; therefore, it is possible that BRD4 might regulate target gene expression indirectly via its effect on eRNA synthesis. A recent study demonstrated co-occupancy of BRD4 and mediator at MYC SEs, and the use of BET inhibitor JQ1 resulted in transcription elongation defects in MYC (44). JQ1 displacement of BRD4 from the MYC promoter/enhancer also led to suppression of MYC-driven malignancies, such as multiple myeloma and acute myeloid leukemia (95, 96). Zhu et al. found that treatment with a BET inhibitor suppressed PD-L1 expression in ovarian cancer (100). In a mouse model, the authors observed a dose- and time-dependent reduction in the level of PDCD1L1 transcription and expression during treatment with JQ1 in tumor cells, macrophages and tumor-associated DCs. Knockdown of BRD4 decreased PD-L1 expression, indicating that PD-L1 expression is at least partly regulated by BRD4. Further investigation demonstrated that the PD-L1 encoding gene, PDCD1L1 (also known as CD274), is a direct target of BRD4. In addition, the application of JQ1 increased CD8+ cytotoxic T cell activity; thus promoting anti-tumor immunity, limiting ovarian cancer progression, and improving mouse survival (101). The mechanism by which BRD4 regulates PDCD1L1 expression in cancer cell has been determined. Hogg et al. demonstrated that the inhibition of PDCD1L1 transcription by the BET inhibitor is independent of MYC expression, which is usually involved in PD-L1 expression regulation and is also as a target for BET inhibitors in hematologic malignancies (102, 103). The authors found that downregulation of PDCD1L1 under JQ1 treatment was not associated with changes in MYC regulation. Ectopic overexpression of MYC did not affect JQ1's suppression of PD-L1 expression. In addition, depletion of BRD4 led to PDCD1L1 transcription inhibition in the absence of putative changes in MYC expression. Using chromatin immunoprecipitation sequencing and analysis of RNAP II occupancy, the authors further identified a distal SE proximal to the PDCD1L1 gene. Moreover, this distal SE bridged to the PDCD1L1 transcription start site (TSS), forming a chromatin loop. The accumulation of BRD4 at the PDCD1L1 TSS decreased substantially upon JQ1 treatment, indicating that disruption of the TSS and SE loop might contribute to JQ1-mediated PD-L1 expression inhibition (103). As mentioned previously, BRD4 promotes eRNA transcription by interacting with acetylated histones. A recent study demonstrated that the BRD4–eRNA interaction promotes the binding of BRD4 to acetylated histone. This interaction further potentiates BRD4 recruitment to enhancers and promotes subsequent transcription events (104). Therefore, we hypothesized that seRNAs might be involved in BRD4-mediated PD-L1 regulation by contributing to stability of the chromatin loop formed by the distal SE and the PDCD1L1 TSS, which promotes RNAP II progression. BRD4 regulates the expression of PDCD1L1 indirectly by promoting the transcription of an SE, and the resulting seRNA contributes to promoting enhancer activity, thus affecting the transcription of PDCD1L1 (Figure 5).
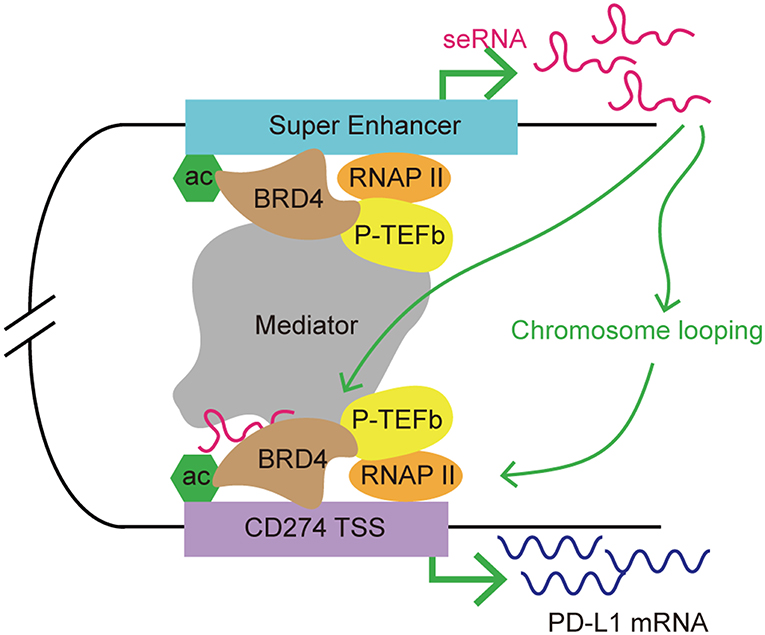
Figure 5. A model of seRNA in the regulation of PD-L1 expression via BRD4. BRD4 is enriched at super enhancers and contributes to seRNA transcription. PDCD1L1 (also known as CD274; encoding PD-L1) is a direct target of BRD4. BRD4 recruits mediator and P-TEFb to acetylated chromatin, leading to the activation of gene expression via phosphorylating RNAP II. An seRNA transcribed from a distal super enhancer proximal to CD274 is found to loop with the CD274 transcription start site (TSS). This seRNA enhances the chromosome looping stability and interacts with BRD4 to promote CD274 expression.
The application of immune checkpoints in oncology sometimes triggers auto-inflammatory adverse effects, which has prompted further investigations of the contribution of immune checkpoints to autoimmunity. The expression and functions of inhibitory immune checkpoints are often dysregulated in autoimmune diseases. Promoting the inhibitory function of immune checkpoints should be beneficial to restore the immune balance in rheumatic disease. However, Farh et al. found that distinct enhancers and eRNAs are involved in autoimmune disease (Table 2). Genome-wide association studies revealed that risk loci for autoimmunity are enriched in immune cell-specific SEs and enhancers (111). Peeters et al. revealed a disease-specific super-enhancer signature in CD4+ memory/effector cells in the synovial fluid of patients with juvenile idiopathic arthritis (105). These SEs are associated with inflammatory arthritis SNPs, indicating the contribution of SEs and, possibly, seRNAs in the control of disease pathogenesis. Moreover, the application of BET inhibitors suppressed SE-associated gene expression subsequent to a reduction in proinflammatory markers (105). A previous study found that in T cells, one-third of the non-coding RNAs are transcribed from SEs, indicating their potential role in regulating the T cell immune response (112). In T cells, cytokine receptors and cytokines are the predominant type of genes that have an SE architecture (112). ADAM like decysin 1 (ADAMDEC1) is a member of the ADAMs (A Disintegration And Metalloproteinase) protein family. ADAMDEC1 and ADAM28, which is located upstream of ADAMDEC1, are overexpressed in SLE and are upregulated in inflammatory states. Further investigation showed that the interaction between eRNAs and P300 is involved in ADAMDEC1 expression regulation (106). One of the functions of eRNAs is to bind CBP and modulate the acetyltransferase activity at enhancers. NF-κB is recruited to the enhancer upon activation of the inflammatory signal cascade. The accumulation of NF-κB leads to assembly of P300 on the enhancer, which can be activated by eRNAs. Activation of P300 leads to increased histone acetylation and transcription elongation (106). That study revealed the participation of eRNAs in the regulation of autoimmune-associated gene expression. Therefore, it was suggested that there is crosstalk between immune checkpoints and seRNAs in the context of autoimmunity. BTB domain and CNC homolog 2 (BACH2), a TF that functions to suppress effector programs to maintain the Treg-mediated immune homeostasis, has the most prominent super-enhancer in its gene locus in T cells (113). BACH2 regulates the expression of a variety of cytokines in T cells. Genetic variation in the BACH2 locus is associated with autoimmune-related diseases, such as Crohn's disease, RA, and T1D (114–116). Vahedi et al. found that the BACH2 locus is SE-regulated, with high P300 occupancy. Knockdown of BACH2 led to a significant increase in the expression of genes with an SE architecture in T cells, including those encoding cytokines and cytokine receptors (112). In addition, seRNA-related transcription is inhibited by BACH2 (112). Therefore, the authors identified a network in which the expression levels of genes and eRNAs are negatively regulated by BACH2, which itself is also SE-regulated (112). Recent research found that BACH2 promotes tumor immunosuppression via IFNγ and Treg-mediated intratumoral CD8+ T cell inhibition. Elevated levels of IFNγ were observed in tumors of mice with BACH2 deficiency. Further analysis revealed that suppression of IFNγ is caused by BACH2-mediated Treg-dependent tumor immunosuppression (117). Thus, the results demonstrated a pathway for BACH2 to regulate the expression of IFNγ immunosuppression. There are two interferon regulatory factor-1 (IRF-1) binding sites on the promoter of PDCD1L1. Diaz et al. revealed that IFNγ signaling is the primarily regulating signal for PDCD1L1 expression in melanoma cells. Diaz et al. identified the IFNγ-Janus kinase (JAK)-signal transducer and activator of transcription (STAT)-IRF1 axis that regulates PD-L1 expression (118). Such findings link BACH2 with PD-L1 expression, indicating its potential influence on immune checkpoint expression. Similarly, in the context of autoimmune disease, Osum et al. demonstrated that IFNγ drives PD-L1 expression on islet beta cells in T1D (119). T cell-directed beta cell destruction is the main cause of T1D. In vivo and in vitro experiments showed that T cell infiltration-dependent islet beta cell PD-L1 upregulation is mediated by IFNγ (119). In addition, the increased PD-L1 expression correlated with the level of T cell infiltration and insulitis, indicating that elevated PD-L1 expression is a salvage response to islet destruction (119). These findings led us to hypothesize that SE-regulated TF BACH2 might play a role in regulating the expression of PD-L1 indirectly by mediating the expression of IFNγ in autoimmune disease. Moreover, seRNAs might also contribute to this regulation by promoting SE function. However, little research has been performed on the contribution of seRNAs to the regulation of immune checkpoint expression in autoimmune diseases, and the exact contribution of seRNAs to autoimmune diseases remains poorly understood. Further investigation and more direct evidence are required to reveal the details of the crosstalk between seRNAs and immune checkpoints in autoimmune diseases.
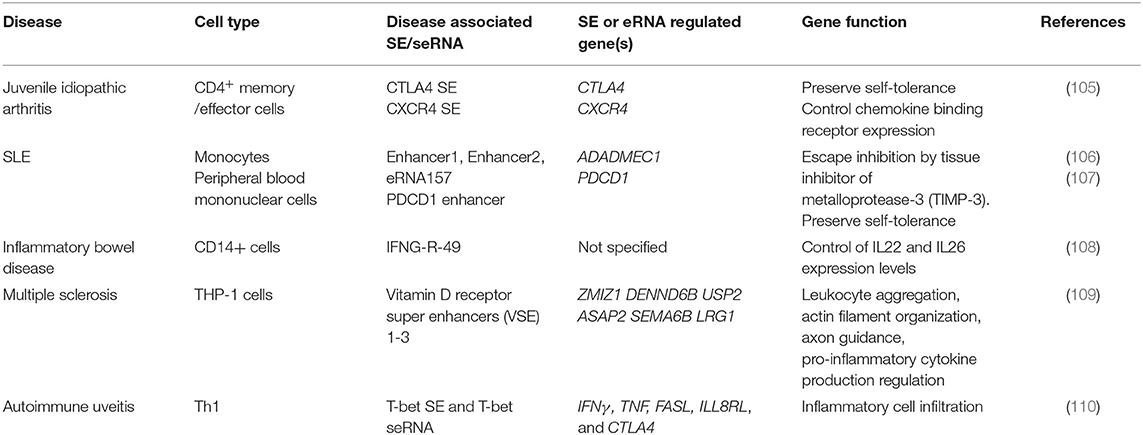
Table 2. Involvement of SE in autoimmune diseases.
Current knowledge of immune checkpoints and SEs has increased our understanding of immune checkpoint expression in oncology and autoimmunity. Evading immune destruction is a major hallmark of cancer, suggesting that the expression of inhibitory immune checkpoints could be a critical identity character of tumor cells (16). Super enhancers are clusters of enhancers that facilitate gene expression that is important for cell identity. eRNAs contribute to enhancer function via multiple approaches, including enhancing promoter-enhancer looping, facilitating TF assembly, and promoting RNAP II activation. In the present review, we proposed several ways by which seRNAs contribute to immune checkpoint regulation indirectly by mediating the expression of key genes that regulate immune checkpoint expression, and play critical roles in determining cell identity.
As a potential therapeutic target, many researchers had focus on exploring the application of SE blockers in cancers and autoimmune diseases (Figure 6). He et al. recently summarized the role of SE as therapeutic target in cancer treatment. By using BET inhibitor, CDK7 inhibitor, AKT inhibitors, demethylases, and acetyltransferase, researchers target carcinogenic SEs to inhibit cancer growth, invasion, immune escape and progression (121).
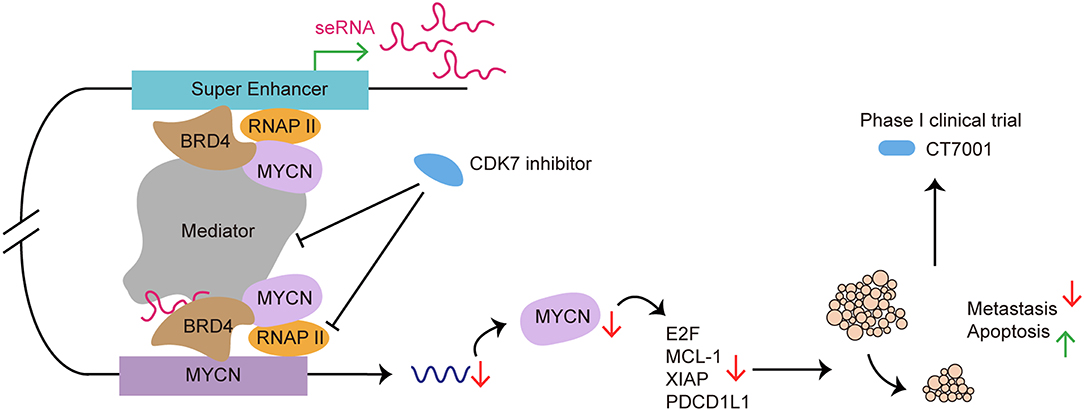
Figure 6. SE as a therapeutic target in cancer treatment: from preclinical to clinical. CDK7 inhibitor targets SE to suppress SE-regulated MYCN driven oncogenic transcription amplification, including E2F, MCL-1, XIAP (93). CDK7 inhibitor blocks SE functioning by affecting RNAP II and Mediator complex. Use of CDK7 inhibitor induces tumor regression, cancer cell apoptosis and reduces metastasis both in vivo and in vitro (120). CT7001, a CDK7 inhibitor, has been approved for phase I clinical trial in patients with advanced malignancies.
Although remarkable process has been made in revealing the regulation of immune checkpoint expression and eRNA function, many questions and challenges remain. For instance, the exact function and mechanism of seRNAs have not been clearly demonstrated. Additional and direct evidence of seRNA function in immune checkpoint expression is required to further support the indirect gene regulation effect by seRNAs. Recent findings also revealed that the exact boundaries between eRNAs and lncRNAs are not absolute, suggesting that some eRNAs might have been mistakenly identified as lncRNAs (28). Despite having a similar frequency of transcription to protein-coding genes, eRNAs have a shorter half-life compared with that of mRNAs and lncRNAs, which represents an obstacle for their thorough study (122). Such characteristics and uncertainty of seRNA make research into the interaction between seRNAs and TFs or chromatin a challenge. Joint efforts should be made by biologists and immunologists to further identify the correlations between SEs, seRNAs, and immune checkpoints. We are only starting to comprehend the full panoply of eRNAs' functions. In the future, a thorough understanding of the mechanism by which seRNAs regulate gene expression and their contribution to disease pathogenesis might help to identify new therapeutic targets and disease biomarkers.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by grants from National Natural Science Foundation of China (No. 81770545), and by MDT Project of Clinical Research Innovation Foundation, Renji Hospital, School of Medicine, Shanghai Jiao Tong University (PYI-17-003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge support from National Natural Science Foundation of China and Renji Hospital, School of Medicine, Shanghai Jiao Tong University.
1. International Human Genome Sequencing C. Finishing the euchromatic sequence of the human genome. Nature. (2004) 431:931–45. doi: 10.1038/nature03001
2. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. (2013) 155:934–47. doi: 10.1016/j.cell.2013.09.053
3. Chen L, Toke NH, Luo S, Vasoya RP, Fullem RL, Parthasarathy A, et al. A reinforcing HNF4-SMAD4 feed-forward module stabilizes enterocyte identity. Nat Genet. (2019) 51:777–85. doi: 10.1038/s41588-019-0384-0
4. Li X, Zheng Y. Regulatory T cell identity: formation and maintenance. Trends Immunol. (2015) 36:344–53. doi: 10.1016/j.it.2015.04.006
5. Di Micco R, Fontanals-Cirera B, Low V, Ntziachristos P, Yuen Stephanie K, Lovell Claudia D, et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. (2014) 9:234–47. doi: 10.1016/j.celrep.2014.08.055
6. Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. (2007) 14:103–5. doi: 10.1038/nsmb0207-103
7. Spurlock CF III, Shaginurova G, Tossberg JT, Hester JD, Chapman N, Guo Y, et al. Profiles of long noncoding RNAs in human naive and memory T cells. J Immunol. (2017) 199:547–58. doi: 10.4049/jimmunol.1700232
8. Hsieh CL, Fei T, Chen Y, Li T, Gao Y, Wang X, et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci USA. (2014) 111:7319–24. doi: 10.1073/pnas.1324151111
9. Mousavi K, Zare H, Dell'orso S, Grontved L, Gutierrez-Cruz G, Derfoul A, et al. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell. (2013) 51:606–17. doi: 10.1016/j.molcel.2013.07.022
10. Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, et al. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. (2013) 494:497–501. doi: 10.1038/nature11884
11. Schaukowitch K, Joo JY, Liu X, Watts JK, Martinez C, Kim TK. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. (2014) 56:29–42. doi: 10.1016/j.molcel.2014.08.023
12. Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, Berger SL. RNA binding to CBP stimulates histone acetylation and transcription. Cell 168:135–49.e122. doi: 10.1016/j.cell.2016.12.020
13. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. (2017) 8:561. doi: 10.3389/fphar.2017.00561
14. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
15. Rhie SK, Guo Y, Tak YG, Yao L, Shen H, Coetzee GA, et al. Identification of activated enhancers and linked transcription factors in breast, prostate, and kidney tumors by tracing enhancer networks using epigenetic traits. Epigenet Chrom. (2016) 9:50. doi: 10.1186/s13072-016-0102-4
16. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
17. Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. (2011) 470:279–83. doi: 10.1038/nature09692
18. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. (2010) 107:21931–6. doi: 10.1073/pnas.1016071107
19. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. (1996) 87:953–9 doi: 10.1016/S0092-8674(00)82001-2
20. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. (2013) 153:307–19. doi: 10.1016/j.cell.2013.03.035
21. Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. (2010) 467:430–5. doi: 10.1038/nature09380
22. May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, et al. Large-scale discovery of enhancers from human heart tissue. Nat Genet. (2011) 44:89–93. doi: 10.1038/ng.1006
23. Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, et al. Transcription factor trapping by RNA in gene regulatory elements. Science. (2015) 350:978–81. doi: 10.1126/science.aad3346
24. Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. (2007) 39:311–8. doi: 10.1038/ng1966
25. Alvarez-Dominguez JR, Knoll M, Gromatzky AA, Lodish HF. The super-enhancer-derived alncRNA-EC7/bloodlinc potentiates red blood cell development in trans. Cell Rep. (2017) 19:2503–14. doi: 10.1016/j.celrep.2017.05.082
26. Lam MT, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. (2014) 39:170–82. doi: 10.1016/j.tibs.2014.02.007
27. Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. (2010) 465:182–7. doi: 10.1038/nature09033
28. Espinosa JM. Revisiting lncRNAs: how do you know yours is not an eRNA? Mol Cell. (2016) 62:1–2. doi: 10.1016/j.molcel.2016.03.022
29. Nicholas NE II, Heward JA, Roux B, Tsitsiou E, Fenwick PS, Lenzi L, et al. Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat Comm. (2014) 5:3979. doi: 10.1038/ncomms4979
30. Ren C, Liu F, Ouyang Z, An G, Zhao C, Shuai J, et al. Functional annotation of structural ncRNAs within enhancer RNAs in the human genome: implications for human disease. Sci Rep. (2017) 7:15518. doi: 10.1038/s41598-017-15822-7
31. Blinka S, Reimer MH Jr, Pulakanti K, Rao S. Super-enhancers at the nanog locus differentially regulate neighboring pluripotency-associated genes. Cell Rep. (2016) 17:19–28. doi: 10.1016/j.celrep.2016.09.002
32. Cichewicz MA, Kiran M, Przanowska RK, Sobierajska E, Shibata Y, Dutta A. MUNC, an eRNA upstream from the MYOD gene, induces a subgroup of myogenic transcripts in trans, independently of MyoD. Mol Cell Biol. (2018) 38:e00655–17. doi: 10.1128/MCB.00655-17.
33. Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. (2013) 498:516–20. doi: 10.1038/nature12210
34. Bose DA, Berger SL. eRNA binding produces tailored CBP activity profiles to regulate gene expression. RNA Biol. (2017) 14:1655–9. doi: 10.1080/15476286.2017.1353862
35. Hah N, Benner C, Chong LW, Yu RT, Downes M, Evans RM. Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proc Natl Acad Sci USA. (2015) 112:E297–302. doi: 10.1073/pnas.1424028112
36. Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, et al. YY1 is a structural regulator of enhancer-promoter loops. Cell. (2017) 171:1573–88.e1528. doi: 10.1016/j.cell.2017.11.008
37. Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. (2012) 489:109–13. doi: 10.1038/nature11279
38. Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. (2012) 149:1233–44. doi: 10.1016/j.cell.2012.03.051
39. Jiao W, Chen Y, Song H, Li D, Mei H, Yang F, et al. HPSE enhancer RNA promotes cancer progression through driving chromatin looping and regulating hnRNPU/p300/EGR1/HPSE axis. Oncogene. (2018) 37:2728–45. doi: 10.1038/s41388-018-0128-0
40. Pnueli L, Rudnizky S, Yosefzon Y, Melamed P. RNA transcribed from a distal enhancer is required for activating the chromatin at the promoter of the gonadotropin alpha-subunit gene. Proc Natl Acad Sci USA. (2015) 112:4369–74. doi: 10.1073/pnas.1414841112
41. Maruyama A, Mimura J, Itoh K. Non-coding RNA derived from the region adjacent to the human HO-1 E2 enhancer selectively regulates HO-1 gene induction by modulating Pol II binding. Nucleic Acids Res. (2014) 42:13599–614. doi: 10.1093/nar/gku1169
42. Ounzain S, Micheletti R, Arnan C, Plaisance I, Cecchi D, Schroen B, et al. CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J Mol Cell Cardiol. (2015) 89(Pt A):98–112. doi: 10.1016/j.yjmcc.2015.09.016
43. Feng J, Bi C, Clark BS, Mady R, Shah P, Kohtz JD. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. (2006) 20:1470–84. doi: 10.1101/gad.1416106
44. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. (2013) 153:320–34. doi: 10.1016/j.cell.2013.03.036
45. Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell. (2013) 49:524–35. doi: 10.1016/j.molcel.2012.11.021
46. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. (2013) 25:214–21. doi: 10.1016/j.coi.2012.12.003
47. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. (2008) 8:467–77. doi: 10.1038/nri2326
48. Zamani MR, Aslani S, Salmaninejad A, Javan MR, Rezaei N. PD-1/PD-L and autoimmunity: a growing relationship. Cell Immunol. (2016) 310:27–41. doi: 10.1016/j.cellimm.2016.09.009
49. Olde Nordkamp MJ, Koeleman BP, Meyaard L. Do inhibitory immune receptors play a role in the etiology of autoimmune disease? Clin Immunol. (2014) 150:31–42. doi: 10.1016/j.clim.2013.11.007
50. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. (1995) 270:985–8. doi: 10.1126/science.270.5238.985
51. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. (2014) 345:1623–7. doi: 10.1126/science.1255904
52. Jury EC, Flores-Borja F, Kalsi HS, Lazarus M, Isenberg DA, Mauri C, et al. Abnormal CTLA-4 function in T cells from patients with systemic lupus erythematosus. Eur J Immunol. (2010) 40:569–78. doi: 10.1002/eji.200939781
53. Siakavellas SI, Bamias G. Checkpoint inhibitor colitis: a new model of inflammatory bowel disease? Curr Opin Gastroenterol. (2018) 34:377–83. doi: 10.1097/MOG.0000000000000482
54. Dougan M. Checkpoint blockade toxicity and immune homeostasis in the gastrointestinal tract. Front Immunol. (2017) 8:1547. doi: 10.3389/fimmu.2017.01547
55. Makarious D, Horwood K, Coward JIG. Myasthenia gravis: an emerging toxicity of immune checkpoint inhibitors. Eur J Cancer. (2017) 82:128–36. doi: 10.1016/j.ejca.2017.05.041
56. Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events associated with immune checkpoint blockade in patients with cancer: a systematic review of case reports. PLoS ONE. (2016) 11:e0160221. doi: 10.1371/journal.pone.0160221
57. Johnson DB, Sullivan RJ, Ott PA, Carlino MS, Khushalani NI, Ye F, et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol. (2016) 2:234–40. doi: 10.1001/jamaoncol.2015.4368
58. Blair HA, Deeks ED. Abatacept: a review in rheumatoid arthritis. Drugs. (2017) 77:1221–33. doi: 10.1007/s40265-017-0775-4
59. Pimentel-Quiroz VR, Ugarte-Gil MF, Alarcon GS. Abatacept for the treatment of systemic lupus erythematosus. Expert Opin Invest Drugs. (2016) 25:493–9. doi: 10.1517/13543784.2016.1154943
60. Mease PJ, Gottlieb AB, van der Heijde D, FitzGerald O, Johnsen A, Nys M, et al. Efficacy and safety of abatacept, a T-cell modulator, in a randomised, double-blind, placebo-controlled, phase III study in psoriatic arthritis. Ann Rheum Dis. (2017) 76:1550–8. doi: 10.1136/annrheumdis-2016-210724
61. Granier C, De Guillebon E, Blanc C, Roussel H, Badoual C, Colin E, et al. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open. (2017) 2:e000213. doi: 10.1136/esmoopen-2017-000213
62. Choueiri TK, Fishman MN, Escudier B, McDermott DF, Drake CG, Kluger H, et al. Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma. Clin Cancer Res. (2016) 22:5461–71. doi: 10.1158/1078-0432.CCR-15-2839
63. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
64. Ostrand-Rosenberg S, Horn LA, Haile ST. The programmed death-1 immune-suppressive pathway: barrier to antitumor immunity. J Immunol. (2014) 193:3835–41. doi: 10.4049/jimmunol.1401572
65. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Disc. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
66. Abril-Rodriguez G, Ribas A. SnapShot: immune checkpoint inhibitors. Cancer Cell. (2017) 31:848–e841. doi: 10.1016/j.ccell.2017.05.010
67. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. (2007) 27:111–22. doi: 10.1016/j.immuni.2007.05.016
68. Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, et al. Tumor-Intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. (2016) 76:6964–74. doi: 10.1158/0008-5472.CAN-16-0258
69. Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc cooperates with ras by programming inflammation and immune suppression. Cell. (2017) 171:1301–15.e1314. doi: 10.1016/j.cell.2017.11.013
70. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
71. Kim ES, Kim JE, Patel MA, Mangraviti A, Ruzevick J, Lim M. Immune checkpoint modulators: an emerging antiglioma armamentarium. J Immunol Res. (2016) 2016:4683607. doi: 10.1155/2016/4683607
72. Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. (2014) 20:2424–32. doi: 10.1158/1078-0432.CCR-13-2648
73. Arasanz H, Gato-Canas M, Zuazo M, Ibanez-Vea M, Breckpot K, Kochan G, et al. PD1 signal transduction pathways in T cells. Oncotarget. (2017) 8:51936–45. doi: 10.18632/oncotarget.17232
74. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. (2018) 8:86. doi: 10.3389/fonc.2018.00086
75. Zou W, Wolchok JD, Chen L. PD-L1. (B7-H1). and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. (2016) 8:328rv324. doi: 10.1126/scitranslmed.aad7118
76. Ding M, Liu Y, Li J, Yao L, Liao X, Xie H, et al. Oestrogen promotes tumorigenesis of bladder cancer by inducing the enhancer RNA-eGREB1. J Cell Mol Med. (2018) 22:5919–27. doi: 10.1111/jcmm.13861
77. Wong RWJ, Ngoc PCT, Leong WZ, Yam AWY, Zhang T, Asamitsu K, et al. Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood. (2017) 130:2326–38. doi: 10.1182/blood-2017-06-792184
78. Cotterman R, Jin VX, Krig SR, Lemen JM, Wey A, Farnham PJ, et al. N-Myc regulates a widespread euchromatic program in the human genome partially independent of its role as a classical transcription factor. Cancer Res. (2008) 68:9654–62. doi: 10.1158/0008-5472.CAN-08-1961
79. Kim EY, Kim A, Kim SK, Chang YS. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer. (2017) 110:63–7. doi: 10.1016/j.lungcan.2017.06.006
80. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. (2016) 352:227–31. doi: 10.1126/science.aac9935
81. Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci USA. (2010) 107:9742–6. doi: 10.1073/pnas.0910668107
82. Xiang JF, Yin QF, Chen T, Zhang Y, Zhang XO, Wu Z, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. (2014) 24:513–31. doi: 10.1038/cr.2014.35
83. Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. (2009) 41:882–4. doi: 10.1038/ng.403
84. Jiang Y, Jiang YY, Xie JJ, Mayakonda A, Hazawa M, Chen L, et al. Co-activation of super-enhancer-driven CCAT1 by TP63 and SOX2 promotes squamous cancer progression. Nat Commun. (2018) 9:3619. doi: 10.1038/s41467-018-06081-9
85. Zhang W, Pang Q, Yan C, Wang Q, Yang J, Yu S, et al. Induction of PD-L1 expression by epidermal growth factor receptor-mediated signaling in esophageal squamous cell carcinoma. OncoTargets Ther. (2017) 10:763–71. doi: 10.2147/OTT.S118982
86. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Disc. (2013) 3:1355–63. doi: 10.1158/2159-8290.CD-13-0310
87. Tang Y, Fang W, Zhang Y, Hong S, Kang S, Yan Y, et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget. (2015) 6:14209–19. doi: 10.18632/oncotarget.3694
88. Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol. (2014) 25:1935–40. doi: 10.1093/annonc/mdu242
89. Okita R, Maeda A, Shimizu K, Nojima Y, Saisho S, Nakata M. PD-L1 overexpression is partially regulated by EGFR/HER2 signaling and associated with poor prognosis in patients with non-small-cell lung cancer. Cancer Immunol Immunother. (2017) 66:865–76. doi: 10.1007/s00262-017-1986-y
90. Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. (2015) 21:4014–21. doi: 10.1158/1078-0432.CCR-15-0016
91. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. (2014) 159:1126–39. doi: 10.1016/j.cell.2014.10.024
92. Chen CH, Yang N, Zhang Y, Ding J, Zhang W, Liu R, et al. Inhibition of super enhancer downregulates the expression of KLF5 in basal-like breast cancers. Int J Biol Sci. (2019) 15:1733–42. doi: 10.7150/ijbs.35138
93. Rasool RU, Natesan R, Deng Q, Aras S, Lal P, Sander Effron S, et al. CDK7 inhibition suppresses castration-resistant prostate cancer through MED1 inactivation. Cancer Disc. (2019) 9:1538–55. doi: 10.1158/2159-8290.CD-19-0189
94. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. (2011) 478:529–33. doi: 10.1038/nature10509
95. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. (2011) 146:904–17. doi: 10.1016/j.cell.2011.08.017
96. Da Costa D, Agathanggelou A, Perry T, Weston V, Petermann E, Zlatanou A, et al. BET inhibition as a single or combined therapeutic approach in primary paediatric B-precursor acute lymphoblastic leukaemia. Blood Cancer J. (2013) 3:e126. doi: 10.1038/bcj.2013.24
97. Chiang CM. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol Rep. (2009) 1:98. doi: 10.3410/B1-98
98. Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. (2007) 282:13141–5. doi: 10.1074/jbc.R700001200
99. Kanno T, Kanno Y, LeRoy G, Campos E, Sun HW, Brooks SR, et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol. (2014) 21:1047–57. doi: 10.1038/nsmb.2912
100. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. (2016) 16:2829–37. doi: 10.1016/j.celrep.2016.08.032
101. Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM, et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. (2016) 76:6320–30. doi: 10.1158/0008-5472.CAN-16-0854
102. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. (2017) 28:1776–87. doi: 10.1093/annonc/mdx157
103. Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. (2017) 18:2162–74. doi: 10.1016/j.celrep.2017.02.011
104. Rahnamoun H, Lee J, Sun Z, Lu H, Ramsey KM, Komives EA, et al. RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat Struct Mol Biol. (2018) 25:687–97. doi: 10.1038/s41594-018-0102-0
105. Peeters JG, Vervoort SJ, Tan SC, Mijnheer G, de Roock S, Vastert SJ, et al. Inhibition of super-enhancer activity in autoinflammatory site-derived T cells reduces disease-associated gene expression. Cell Rep. (2015) 12:1986–96. doi: 10.1016/j.celrep.2015.08.046
106. Shi L, Li S, Maurer K, Zhang Z, Petri M, Sullivan KE. Enhancer RNA and NFkappaB-dependent P300 regulation of ADAMDEC1. Mol Immunol. (2018) 103:312–21. doi: 10.1016/j.molimm.2018.09.019
107. Prokunina L, Castillejo-López C, Oberg F, Gunnarsson I, Berg L, Magnusson V, et al. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet. (2002) 32:666–9. doi: 10.1038/ng1020
108. Aune TM, Crooke PS III, Patrick AE, Tossberg JT, Olsen NJ, Spurlock CF III. Expression of long non-coding RNAs in autoimmunity and linkage to enhancer function and autoimmune disease risk genetic variants. J Autoimm. (2017). 81:99–109. doi: 10.1016/j.jaut.2017.03.014
109. Lu M, McComish BJ, Burdon KP, Taylor BV, Korner H. The association between vitamin d and multiple sclerosis risk: 1,25. (OH)2D3 induces super-enhancers bound by VDR. Front Immunol. (2019) 10:488. doi: 10.3389/fimmu.2019.00488
110. Hertweck A, Evans CM, Eskandarpour M, Lau JC, Oleinika K, Jackson I, et al. T-bet activates Th1 genes through mediator and the super elongation complex. Cell Rep. (2016) 15:2756–70. doi: 10.1016/j.celrep.2016.05.054
111. Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. (2015) 518:337–43. doi: 10.1038/nature13835
112. Vahedi G, Kanno Y, Furumoto Y, Jiang K, Parker SC, Erdos MR, et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature. (2015) 520:558–62. doi: 10.1038/nature14154
113. Afzali B, Gronholm J, Vandrovcova J, O'Brien C, Sun HW, Vanderleyden I, et al. BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat Immunol. (2017) 18:813–23. doi: 10.1038/ni.3753
114. McAllister K, Yarwood A, Bowes J, Orozco G, Viatte S, Diogo D, et al. Identification of BACH2 and RAD51B as rheumatoid arthritis susceptibility loci in a meta-analysis of genome-wide data. Arthr Rheum. (2013) 65:3058–62. doi: 10.1002/art.38183
115. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. (2010) 42:1118–25. doi: 10.1038/ng.717
116. Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet. (2008) 40:1399–401. doi: 10.1038/ng.249
117. Roychoudhuri R, Eil RL, Clever D, Klebanoff CA, Sukumar M, Grant FM, et al. The transcription factor BACH2 promotes tumor immunosuppression. J Clin Invest. (2016) 126:599–604. doi: 10.1172/JCI82884
118. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
119. Osum KC, Burrack AL, Martinov T, Sahli NL, Mitchell JS, Tucker CG, et al. Interferon-gamma drives programmed death-ligand 1 expression on islet beta cells to limit T cell function during autoimmune diabetes. Sci Rep. (2018) 8:8295. doi: 10.1038/s41598-018-26471-9
120. Zhang J, Liu S, Ye Q, Pan J. Transcriptional inhibition by CDK7/9 inhibitor SNS-032 abrogates oncogene addiction and reduces liver metastasis in uveal melanoma. Mol Cancer. (2019) 18:140. doi: 10.1186/s12943-019-1070-7
121. Zhang Z, Lee JH, Ruan H, Ye Y, Krakowiak J, Hu Q, et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat Commun. (2019) 10:4562. doi: 10.1038/s41467-019-12543-5
Keywords: super-enhancer, non-coding RNA, cell identity, immune checkpoint, cancer, autoimmune disease
Citation: Wu M and Shen J (2019) From Super-Enhancer Non-coding RNA to Immune Checkpoint: Frameworks to Functions. Front. Oncol. 9:1307. doi: 10.3389/fonc.2019.01307
Received: 12 August 2019; Accepted: 11 November 2019;
Published: 22 November 2019.
Edited by:
Patricia P. Reis, São Paulo State University, BrazilReviewed by:
Giuseppe Palma, National Cancer Institute G. Pascale Foundation (IRCCS), ItalyCopyright © 2019 Wu and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Shen, c2hlbmp1bnJqQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.