 Valeria Maffeis
Valeria Maffeis Lorenzo Nicolè
Lorenzo Nicolè Rocco Cappellesso
Rocco Cappellesso
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 19 November 2019
Sec. Molecular and Cellular Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01255
This article is part of the Research Topic Oncogenic RAS-dependent Reprogramming of Cellular Plasticity View all 10 articles
The high morbidity and mortality of colorectal cancer (CRC) remain a worldwide challenge, despite the advances in prevention, diagnosis, and treatment. RAS alterations have a central role in the pathogenesis of CRC universally recognized both in the canonical mutation-based classification and in the recent transcriptome-based classification. About 40% of CRCs are KRAS mutated, 5% NRAS mutated, and only rare cases are HRAS mutated. Morphological and molecular correlations demonstrated the involvement of RAS in cellular plasticity, which is related to invasive and migration properties of neoplastic cells. RAS signaling has been involved in the initiation of epithelial to mesenchymal transition (EMT) in CRC leading to tumor spreading. Tumor budding is the morphological surrogate of EMT and features cellular plasticity. Tumor budding is clinically relevant for CRC patients in three different contexts: (i) in pT1 CRC the presence of tumor buds is associated with nodal metastasis, (ii) in stage II CRC identifies the cases with a prognosis similar to metastatic disease, and (iii) intratumoral budding could be useful in patient selection for neoadjuvant therapy. This review is focused on the current knowledge on RAS in CRC and its link with cellular plasticity and related clinicopathological features.
Colorectal cancer (CRC) is a malignant epithelial tumor originating in the large bowel and in almost all cases it features as an adenocarcinoma, a neoplasia with glandular characteristics (1). Despite the big efforts of the last decades resulting in the widespread implementation of screening programs, that have proved effective in reducing the burden of the disease in the population, and in the advances of the surgical and systemic treatments, that have improved the outcome of the patients, CRC is still the third cancer for incidence and the second for mortality in both sexes worldwide (2–4). This highlights the urgent need to identify novel diagnostic, prognostic, and predictive markers and to develop new strategies for CRC prevention, early detection, and therapy to drastically reduce CRC morbidity and mortality. Indeed, the identification of circulating markers would allow to anticipate the identification of CRC in the population, to early detect interval cancers, and to better select patients really needing colonoscopy. The current categorization based on tumor histology, grade, and stage provides limited understanding of CRC biology and often fails to recognize the true high-risk population after surgery. Consistent prognostic markers would allow to tailor the treatment according to the aggressiveness of the tumor. The development of reliable sentinel lymph node methods would modify the surgical management of the disease. The discovery of mechanisms impairing the response to current drugs and of novel targetable molecular alterations would allow a more appropriate therapy in specific subgroups of patients.
In the past, pioneering morphological, and molecular studies allowed to disclose the chain of events underlying the “adenoma to carcinoma cascade” theorized by Fearon and Vogelstein characterized by chromosomal instability (CIN) and sequential mutations of Adenomatous Polyposis Coli (APC), Kirsten rat sarcoma viral oncogene homolog (KRAS), and tumor protein p53 (TP53) genes (5). The importance of this model is such that it is the foundations on which CRC secondary prevention is based. However, it was soon clear that this model of carcinogenetic progression was not applicable to all cases of CRC since it is a heterogeneous disorder with a great variability in response to the therapies and presumed to arise from distinct precursor lesions (6). Subsequent molecular studies led to the identification of various subtypes of CRC, then grouped into a mutation-centered classification (6). However, even this approach partially failed to grasp the biological behavior of CRC and was inadequate in explaining the diversity in patient outcomes (7). More recently, research focused on gene expression profiling and characterization of tumor microenvironment pressures and stimuli to try to fill the gap in the understanding of the disease. Such strategies deepened the knowledge about cellular mechanisms of tumor progression, allowed to discover novel morphological clues of cancer aggressiveness, and provided a huge amount of data finally condensed in a new molecular classification (8).
In this article, we summarize the most meaningful molecular classifications of CRC highlighting the role of RAS in this tumor and its link with cellular plasticity, invasion, and migration at both molecular and morphological levels.
In the “adenoma to carcinoma” model, CRC carcinogenesis is presented as a stepwise process based on the accumulation of molecular alteration contributing to the malignant transformation of the mucosa. In this cascade, APC inactivation initiates the evolution of the mucosa into the adenoma and subsequent KRAS and TP53 mutations drive the emergence of increasingly aggressive subclones (5). However, the evidence that a consistent number of CRCs lacks APC and KRAS mutations has slowly eroded the foundations of this linear theory. Thus, a different categorization was needed because tumor classification is not just to give a name to the entities, but to differentiate them according to the clarification of the clinicopathological correlations, the determination of the etiologies, and the understanding of the evolution of the disease to achieve the best response to treatment.
The first attempt to organize CRC subgroups based on correlation of clinical, morphological, and molecular features used two main molecular alterations: genetic instability and DNA methylation (6, 9–11). Genetic instability can occur in two mutually exclusive forms, one affecting whole chromosomes or portions of chromosomes (namely CIN), the other affecting small repetitive sequences of DNA [namely DNA microsatellite instability (MSI)] (12). Thus, a CRC with CIN is DNA microsatellite stable (MSS). MSI was further stratified in MSI-high (MSI-H), and MSI-low (MSI-L) depending on the frequency of the mutations in the repetitive DNA sequences throughout the genome (13). These two conditions are also linked to different onset mechanisms. While MSI-H is related to the loss of expression of one or more members of the DNA mismatch repair machinery (namely MLH1, MSH2, MSH6, and PMS2), MSI-L is connected to extensive DNA methylation of the genome due to partial methylation and loss of expression of MLH1 or loss of expression of 0-6-Methylguanine DNA Methyltransferase (MGMT) (14–16). Epigenetic instability due to aberrant promoter CpG island hypermethylation is the second cornerstone on which CRC classification is based. According to the frequency of methylation of CpG loci, CRCs are separated into negative, low, and high CpG island methylator phenotype (CIMP) groups (17–20). The combination of these features results in a classification outlining five molecular subgroups of CRC whose alterations can be found also in definite precancerous lesions (Figure 1). The first subtype is the conventional CRC originating from adenoma. The tumor may be sporadic or associated with inherited conditions such as familial adenomatous polyposis (FAP) and mutY DNA glycosylase (MUTYH)-associated polyposis (MAP) (21). It is the most common type of CRC accounting for ~57% of cases and is molecularly characterized by CIN, CIMP negativity, and MSS. APC, KRAS, and TP53 genes are usually mutated, accordingly to the “adenoma to carcinoma” sequence (6). Another CRC subtype following this mutational cascade is represented by tumors developing from adenomas in the context of Lynch syndrome (accounting for about 3% of CRCs). Indeed, these tumors are chromosomal stable and CIMP-negative, but have a hypermutator phenotype due to MSI-H caused by the inherited mutation affecting one or more components of the DNA mismatch repair system (22). BRAF gene is typically wild type, as opposed to the so-called sporadic MSI-H CRC that is characterized by chromosomal stability, CIMP-H, MLH1 methylation, MSI-H, and BRAF mutation (23). This sort of CRC accounts for about 12% of cases and is thought to derive from sessile serrated adenoma (6, 23, 24). Another subgroup of CRC (about 8% of cases) originating from sessile serrated adenoma has chromosomal stability, CIMP-H, only partial methylation of MLH1, MSS or MSI-L, and harbors more commonly mutation of BRAF than of KRAS (6). The last subtype of CRC may develop from both conventional adenoma and sessile serrated adenoma, includes about 20% of tumors, and is characterized by CIN, CIMP-L, MSS, or MSI-L due to MGMT methylation, and always KRAS mutations (6). In general, CRCs with CIN are relatively more aggressive than those with MSI (25–27) and CIMP-H tumors has a less favorable prognosis than CIMP-L ones, but if CIMP-H is associated with MSI-H the outcome is slightly better (28, 29). Moreover, MSI CRCs are known to be not responsive to adjuvant fluorouracil-based therapy but may benefit of immune checkpoint blockade with anti-PD1 immunotherapy (30, 31). The major limit of this categorization is that tumors in each subgroup are considered to be a homogeneous entity from a therapeutic point of view, however they show profound differences in drug response and prognosis.
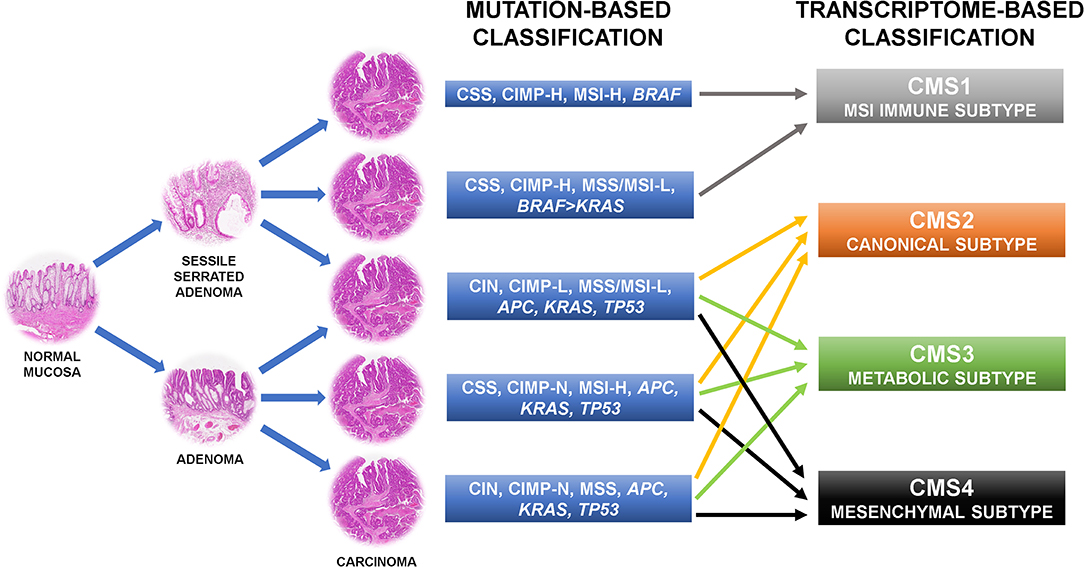
Figure 1. Colorectal cancer molecular classifications recently shifted from the mutation-based toward the transcriptome-based approach because this can better describe the behavior of the tumors. CIN, chromosomal instability; CSS, chromosomal stability; CIMP-N/L/H, CpG island methylator phenotype-negative/low/high; MSS, microsatellite stability; MSI-L/H, microsatellite instability-low/high.
For this reason, more recent approaches shifted from the mutation-based toward the transcriptome-based classification thinking that it can better describe the behavior of the tumors. Indeed, several of such categorizations found CRC gene expression profiles more adherent to the outcome of the patients than the previous system (7, 32–37). These patient stratifications could be useful for the therapeutic decision-making process and are attractive for a rapid translation into the clinic, thus there are many expectations in this regard (7). However, several inconsistencies have emerged by the comparison of the results of these new classification systems. Indeed, each study has attained its own taxonomy including a different number of CRC subtypes. These substantial discrepancies were mostly due to the different CRC populations investigated, the various analysis platforms used, the distinct methods of bioinformatic analysis applied, and the interpretation of data performed (7, 32–37). To clear these hurdles, the CRC Subtyping Consortium (CRCSC) was formed with the purpose of evaluating potential overlaps among the different transcriptome-based CRC classifications to identify core subtype patterns (Figure 1) (8). Four consensus molecular subtypes (CMSs) were delineated using a network-based meta-analysis method of six different taxonomies followed by comprehensive multi-omic and clinical characterization (8). The CMS1 sort of CRCs accounts for about 14% of cases and corresponds to the “MSI immune subtype” characterized by MSI, CIMP-H, BRAF mutations, and intense and widespread immune infiltrate (8). CSM2, the so-called “canonical subtype,” is the most common subtype of CRC accounting for ~37% of tumors. Epithelial characteristics, CIN, activation of WNT and MYC signaling pathways, and upregulation of the miR-17-92 cluster feature this CRC (8). About 13% of CRCs are included in the “metabolic subtype” or CMS3 group, characterized by loss of regulation of metabolic pathways, CIN, CIMP-L, heterogeneous MSI-status, KRAS mutations, and let-7 miR family downregulation (8). Overexpression of epithelial to mesenchymal transition (EMT) markers, miR-200 family downregulation, activation of TGF-β pathway, neoangiogenesis, and stromal infiltration feature the CRC subgroup related to the worst prognosis: the “mesenchymal subtype,” namely CMS4 (8). This subtype accounts for about 23% of CRC cases. Of note, ~13% of CRCs are not classifiable in any of these categories because of intratumoral heterogeneity or a phenotype mixing molecular features of several CMS subtypes (8). The frequency of KRAS mutation varies among the CRC subtypes (23% in CMS1, 38% in CMS2, 28% in CMS3, and 68% in CMS4) and this could explain the different behavior of mutated tumors (7).
The human RAS gene family includes three members, namely KRAS, neuroblastoma RAS viral oncogene homolog (NRAS), and Harvey rat sarcoma viral oncogene homolog (HRAS), encoding four proteins: KRAS4A and KRAS4B (secondary and prevalent isoforms, respectively, deriving from alternative splicing of the RNA), NRAS, and HRAS (38). By means of their GTPase enzymatic site, these small proteins play as molecular switches transducing extracellular signals, such as growth factors, differentiation factors, and mitogens, to transcription factors and cell cycle proteins in the nucleus thus triggering cell growth, differentiation, proliferation, and survival. This site cycles between the guanosine diphosphate (GDP)-bound inactive and the guanosine-5′-triphosphate (GTP)-bound active forms. In normal conditions, extracellular cues stimulate transmembrane tyrosine kinase receptors which recruit guanine nucleotide exchange factors (RASGEFs) promoting activation of the RAS GTPase through the hydrolysis of GDP to GTP (39). In turn, RAS recruits and activates several downstream effectors in different pathways, mainly the phosphoinositide 3-kinase (PI3K)-AKT pathway and the cascade comprising RAF kinase, which activate mitogen-activated protein kinase kinases 1 and 2 (MEK1 and MEK2), and subsequent activation of extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2), thus promoting cell survival, proliferation, invasion, and migration (39, 40). Missense gain-of-function mutations in members of the RAS family have been found in about 25% of all human cancers. Usually, these are single nucleotide point mutations involving few hotspot regions: the codons 12 and 13 in exon 2, the codons 59–61 in exon 3, and the codons 117 and 146 in exon 4. Such mutations result in a conformation of the RAS active site having intrinsic hydrolytic capability (39). Thus, in mutated cells occurs an accumulation of constitutively GTB-bound active RAS proteins able to trigger downstream signaling even in the absence of extracellular stimuli.
KRAS is the most frequently mutated isoform accounting for about 20% of all human cancers. NRAS and HRAS mutations, instead, are found in about 8 and 3% of cancers, respectively (39). Interestingly, different cancer types are related to mutation of a precise RAS isoform, suggesting that the carcinogenetic role of RAS is tissue-specific (39). Indeed, KRAS mutations are usually detected in colorectal, pancreatic, biliary tract, and lung carcinomas, NRAS mutation in malignant melanomas, and HRAS mutation in head and neck carcinomas (41, 42). This feature has been investigated in an adenomatous polyposis coli (APC)-deficient mouse model where mutations of KRAS were able to promote the development of colorectal cancers, while NRAS mutations were ineffective (43).
About 40% of colorectal cancers are KRAS mutated, 5% NRAS mutated, and rarely HRAS mutated. Of note, mutations in different RAS isoforms seems to be mutually exclusive. For this reason, from now on we focus mostly on KRAS. KRAS mutations are considered to play a pivotal role both in the early phases of malignant transformation of colorectal cells and in the advanced metastatic disease (44). In colorectal cancer, most KRAS mutations are in the codons 12 (about 80%) and 13 (about 15%) of exon 2 and in the codon 146 of exon 4 (about 4%); the remaining are in the codons 59-61 of exon 3 and in the codon 117 of exon 4 (45). Mutation frequency in each hotspot varies significantly among the diverse cancer types, exactly as it happens for the RAS-mutated isoforms (38). This could underlie that also the functional consequences of RAS mutation could be divergent in different cancer settings, up to assume paradoxical effects as the induction of cellular senescence as reported by Serrano et al. (46). Moreover, in the same cancer type the effects of a RAS mutation could vary depending on the codon involved. Indeed, a proteomic study found that in colorectal cancer cells a KRAS mutation in codon 12 leads to the overexpression of doublecortin like kinase 1 (DCLK1) and tyrosine-protein kinase MET, while in codon 13 brings to the overexpression of tight junction protein ZO-2 (47).
Metastatic dissemination results from tumor cell invasion and migration through the tissues and represents a major challenge in cancer management (48). The cornerstones of these cancer cell characteristics are deregulation of cell-cell adhesion, acquisition of cytoskeletal deformability, gaining of cellular motility, turnover of cell-matrix interactions, and extracellular matrix (ECM) breakdown (49). Cancer invasion and migration are heterogeneous and adaptive processes based on changes in the usual morphology of the cells, generation of new cell polarization, and cell body displacement that finally leads to the translocation of the entire cells. This may happen in different ways (48, 50). Indeed, tumor cell migration may be either individual, with loss of cell-cell junctions, or collective, with retention of intercellular bonds (Figure 2) (49). Two main types of individual cell motility have been recognized: elongated-mesenchymal and rounded-amoeboid modes. As for collective cell migration, it can happen as multicellular streaming or collective invasion. All these patterns of migration are closely linked to the ECM features, resulting from the coordinated actions of actin cytoskeleton, actomyosin contraction, cell polarity, and cell surface receptors interacting with the surrounding cells and ECM structures. Collective and individual invasion may also coexist, enhancing the efficiency of the metastatic process (51).
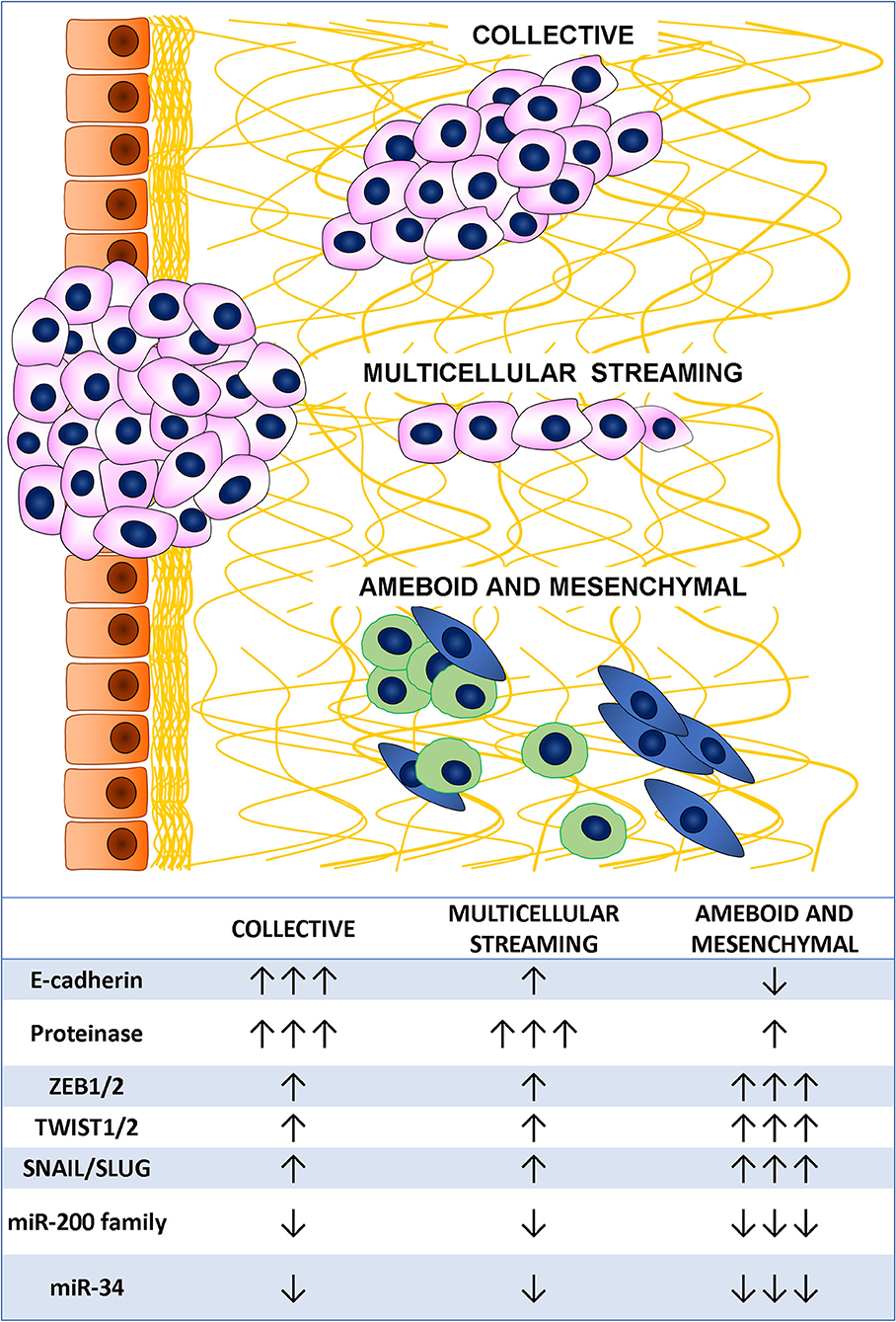
Figure 2. Tumor cell migration mode and main associated markers.
Individual migration patterns are featured by the absence of tumor cell-cell interactions and are strongly linked to the ECM structure. In the elongated-mesenchymal mode, the high ESM stiffness stimulates the cell to produce actin-rich protrusion, thus the cell assumes a spindle morphology with strong focal adhesion, matrix proteolysis, and actomyosin contractility localized at the rear (52). If the ECM surrounding the tumor is loose, the preferential individual invasion mode is the rounded-amoeboid pattern. The cell in this case forms small, unstable cellular protrusions (blebs or spikes) throughout its surface (53). These result from increased intracellular pressure, low degree of integrin-mediated adhesion, and reduced cell-cell interactions (54). Cyclic expansion and retraction of the cellular protrusion at the leading front of the cell are responsible for the cell progression (55).
In multicellular streaming migration mode, the cells move forming single cell files following the same path and are attracted by chemokines gradients or constrained by the ECM structure (56). Streaming cells can display rounded-amoeboid or spindle-mesenchymal phenotypes and advance by generating traction force on the surrounding ECM with weak and short-lived cell-cell interactions (57).
In collective migration pattern, the tumor advances through the neighboring tissues in compact clusters, strands, or cords of connected cells (49). These patterns are determined by a combination of parameters, such as cellular morphology, cell-cell adhesion, and ECM features. Unlike multicellular streaming migration, the collective migration mode is featured by cohesive cells forming solid strands or cords lined up for two or more cells, even to create broad clusters (58). This pattern is supported by long-lived cell-cell interactions, while the morphology varies according to cell nature, ECM features, and host tissue types (56). Main feature of an invasive multicellular mass is the specialization of the leading edge cells that express a mesenchymal phenotype, generate an integrin-mediated forward traction and ECM rearrangement by enzyme-mediated proteolysis of the surrounding structures (59). Interestingly, this invasion pattern has been described as the slowest migration mode (60), conferring some advantages to the tumor, such as secretion of higher amount of pro-invasive factors and immune escape (61).
The cellular plasticity needed to allow migration of cancer cells is achieved through complex mechanisms finely governed by several genes, most of them encoding for transcription factors. In CRC, the best delineated of these molecular programs driving cellular migration is EMT, that is characterized by the acquisition of a mesenchymal phenotype through tight junction dissolution, disruption of apical-basal polarity, and reorganization of the cytoskeletal architecture (62). A huge amount of studies has shown that EMT plays a pivotal role in cancer progression and metastasis in several tumor types, including CRC (63). EMT requires a precisely regulated cooperation of a complex molecular network, which comprises factors categorized into three groups: the extracellular cues activating EMT (EMT inducers), the transcription factors orchestrating the EMT program (EMT core regulators), and the effector molecules executing the EMT-related cellular transformation (EMT effectors) (64). The best characterized external inducers are the transforming growth factor- β (TGF-β) signaling and the WNT/β-catenin pathway. Both these pathways may induce the expression of the three main family of EMT regulators: (i) the SNAIL family of zinc-finger transcription factors comprising SNAIL and SLUG; (ii) the zinc finger E-box binding homeobox (ZEB) family of transcription factors including ZEB1 and ZEB2; (iii) the TWIST family of basic helix-loop-helix (bHLH) transcription factors encompassing TWIST1 and TWIST2. The roles of these transcription factors in EMT have been well-established in a variety of cancers including CRC, and most of them showed correlation with the prognosis (65, 66). Final effects of EMT regulators are the overexpression of genes encoding for proteins linked to mesenchymal phenotype, such as vimentin, fibronectin, α-smooth muscle actin, and N-cadherin, and the down-regulation of epithelial markers, such as E-cadherin, claudins, and occludins (64). Post-transcriptional regulation of gene expression by EMT-related miRNAs showed a great impact in promoting epithelial or mesenchymal phenotype targeting specific mRNA (67). Members of the miR-200 family (miR-200a, miR-200b, miR-200c, miR-141, and miR-429) promote epithelial phenotype preventing the translation of ZEB1 and ZEB2 mRNA (68–70) that, in turn, act in a negative feedback loop down-regulating the miR-200 family expression (71). Moreover, ZEB2 is also identified as a direct target of miR-132, miR-192, and miR-335. Downregulation of these miRNAs is usually associated with the acquisition of an aggressive mesenchymal phenotype leading to distant metastasis and dismal prognosis (72, 73). MiR-34a/b/c is another caretaker of the epithelial phenotype through the down-regulation of SNAIL, SLUG, and ZEB1 (74). Suppression of miR-34a/b/c causes up-regulation of SNAIL resulting in the enhanced expression of EMT markers, mesenchymal features, and improved cell invasion and motility.
As above mentioned, KRAS mutation is common in CRC and activates several effector pathways involved in cell proliferation, invasion, and migration. In particular, RAS signaling has been reported to play a crucial role in EMT initiation (75, 76). It has been shown that in CRC cell lines mutated KRAS can activate downstream effectors of the PI3K pathway, such as Ras homolog gene family member A (RhoA), Ras-related C3 botulinum toxin substrate 1 (Rac1), and cell division cycle 42 (Cdc42), and in synergy with TGF-β signaling can promote EMT inducing a decrease of E-cadherin expression and an increase of vimentin expression (Figure 3) (40, 77, 78). Thus, it seems that KRAS mutation alone is not able to modify the epithelial morphology of CRC cells but requires the cooperation of growth factor cues to accomplish the cell transformation.
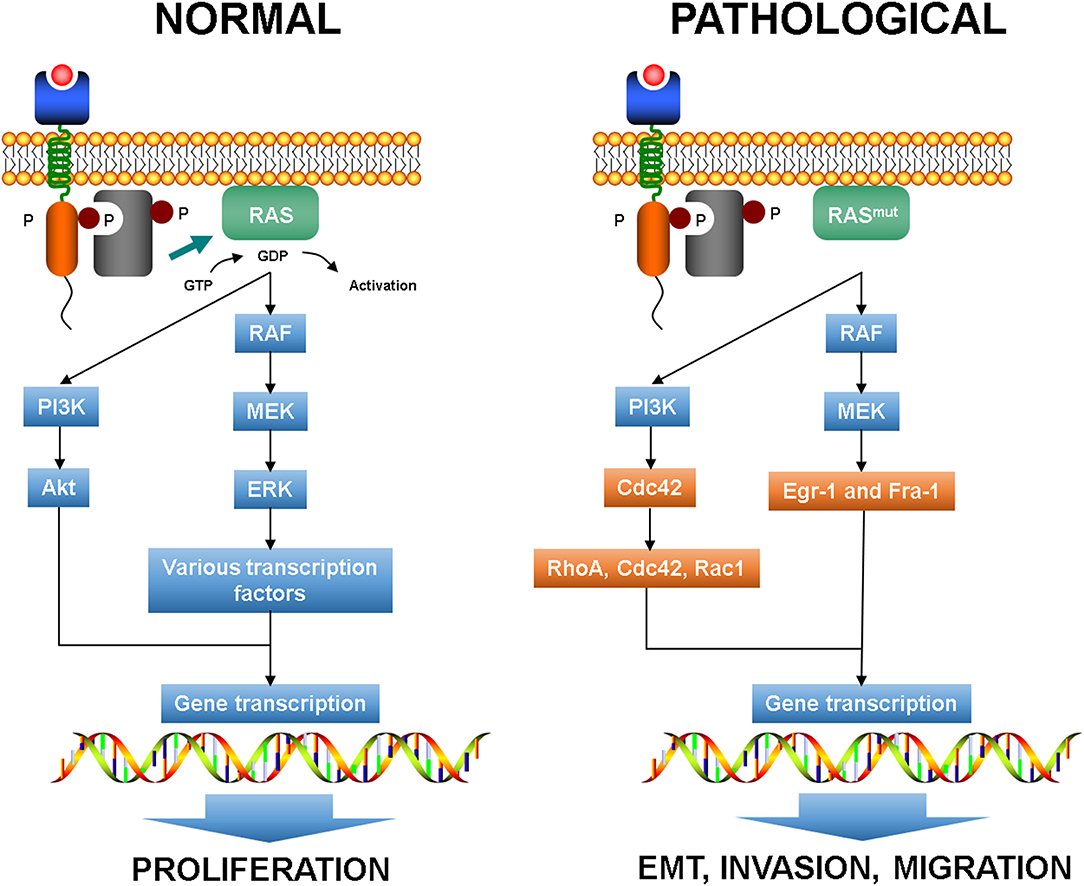
Figure 3. Normal RAS pathways and plasticity-related aberrant pathways. EMT, epithelial to mesenchymal transition.
RAS activation is a crucial connector between receptor and cytoskeleton during chemotaxis in normal conditions (79). Indeed, PI3K-triggered RAS acts on F-actin forming a coupled excitable system that leads to short-lived RAS-F-actin patches that anticipates the extension of cellular protrusions (80).
Moreover, the activation of MEK1 in the RAS-RAF-MEK cascade allows the enrollment of the downstream effectors Egr-1 and Fra-1 that can promote the expression of SNAIL and SLUG, which in turn downregulate E-cadherin expression (81). In EMT, the pathways that regulate actomyosin and cytoskeleton dynamics drive plasticity and KRAS mutation can determine the mode and effectiveness of migration by means RhoA and Rac1 signaling (82, 83).
Several miRNAs were linked to K-RAS-driven tumorigenesis. In experimental models down-regulation of miR-1, Let-7a, miR-16, miR-18a, miR-30a, miR-217, miR-622 results in increased K-RAS expression (84). In particular, miR-30a directly targets KRAS and PI3K inhibiting anchorage-independent growth, cell migration and invasion, and in vivo tumorigenesis by KRAS-mutant CRC cells (85, 86). Moreover, low expression of miR-30a has been found in highly metastatic CRC cell lines and liver metastases (86). Clinically, down-regulation of Let-7a was correlated with increased risk of nodal metastasis and with shortened overall and disease-free survival (87).
According to the definition of the International Tumor Budding Consensus Conference (ITBCC) proposed in 2016 (88) and then validated in 2018 (89, 90), CRC tumor budding (TB) consists of single neoplastic cells or cell clusters of up to four neoplastic cells at the invasive front of the tumor (peritumoral TB) (Figure 4) or within the tumor mass (intratumoral TB) (88). In Western countries, these recommendations were incorporated into the College of American Pathologists (CAP) cancer protocol for patients with primary CRC (91), in the 8th edition of the American Joint Committee on Cancer (AJCC) staging manual (92) and in the European Society for Medical Oncology consensus guidelines (93). This acknowledgment derives from the increasing and established evidences of TB as reliable and independent prognostic factor in CRC, regardless of the scoring method applied for the evaluation (3, 90, 94–96). However, the inclusion of TB in the pathologist report is not yet mandatory, but merely recommended. This is due to its apparent poor reproducibility along with the lack of a standardized scoring system before the ITBCC (97–99). Indeed, TB definition and evaluation method have been controversial throughout its development and different diagnostic criteria are present in the literature (3, 100). The recent agreement reached upon the definition and scoring system method (89) is an essential step to implement TB in the routine CRC assessment.
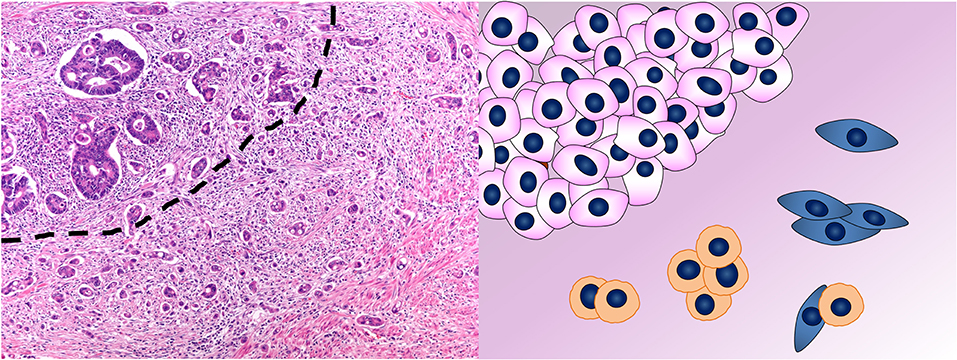
Figure 4. Tumor budding in colorectal cancer. In the photomicrograph, the dashed line separates the tumor mass on the top left from the tumor buds on the bottom right. This phenomenon is depicted in the cartoon where single cells or aggregates of up to four cells detach from the mass of neoplastic cells in the top left and infiltrate. Hematoxylin & eosin stain. Original magnification 200x.
The morphological feature now called TB was originally described in Japan by Imai in 1949 (101) and firstly reported in the English language literature by Gabbert in 1985 (102). Histologically, TB cells show a more marked atypia than their counterparts in the tumor bulk, thus TB was initially termed “tumor dedifferentiation” (102). Imai, instead, proposed the term “sprouting” to describe the tumor cells detaching from the tumor mass along its invasive edge. Moreover, he suggested to use this feature, peritumoral stromal reaction, and lymphovascular invasion in a prognostic system for gastric cancer (101). Some Japanese researches observed the same phenomenon in CRC (103–105) and it was called TB by Morodomi in 1989 (106). In the last decades, a growing number of data reinforced the value of TB as CRC prognostic marker (107–114). Besides CRC, TB has been found in a variety of other solid tumors, such as oral squamous cell carcinoma (115, 116), invasive ductal breast cancer (117), pancreatic (118), and esophageal cancer (119).
Invasion and metastasis are some of the hallmarks of cancer (120), which requires the ability of tumor cells to detach from the primary tumor, move through the ECM, invade lymphovascular vessels, and finally reach and colonize lymph nodes and distant organs (121, 122). TB is the histological demonstration of this ability, which is intrinsically dynamic. Thus, it is conceivable that tumor buds possess cellular plasticity properties, such as cytoskeletal deformability, motility, and full or partial EMT characteristics (122).
Tumor buds often show typical features of EMT (Table 1): loss of E-cadherin expression, β-catenin translocation in the nucleus (sign of WNT pathway activation), and acquisition of vimentin expression (122). The motile and invasive phenotype of TB cells is depicted by the loss of cell adhesion molecules (such as E-cadherin), overexpression of proteins involved in ECM degradation and cell invasion (such as MMP2, MMP9, and cathepsin B), and cell migration (such as laminin, fascin, and α-smooth muscle actin) (121, 138, 142, 143). However, some studies failed in confirming the expression of the classic EMT-related transcription factors ZEB1, TWIST, SNAIL, and SLUG in tumor buds (131).
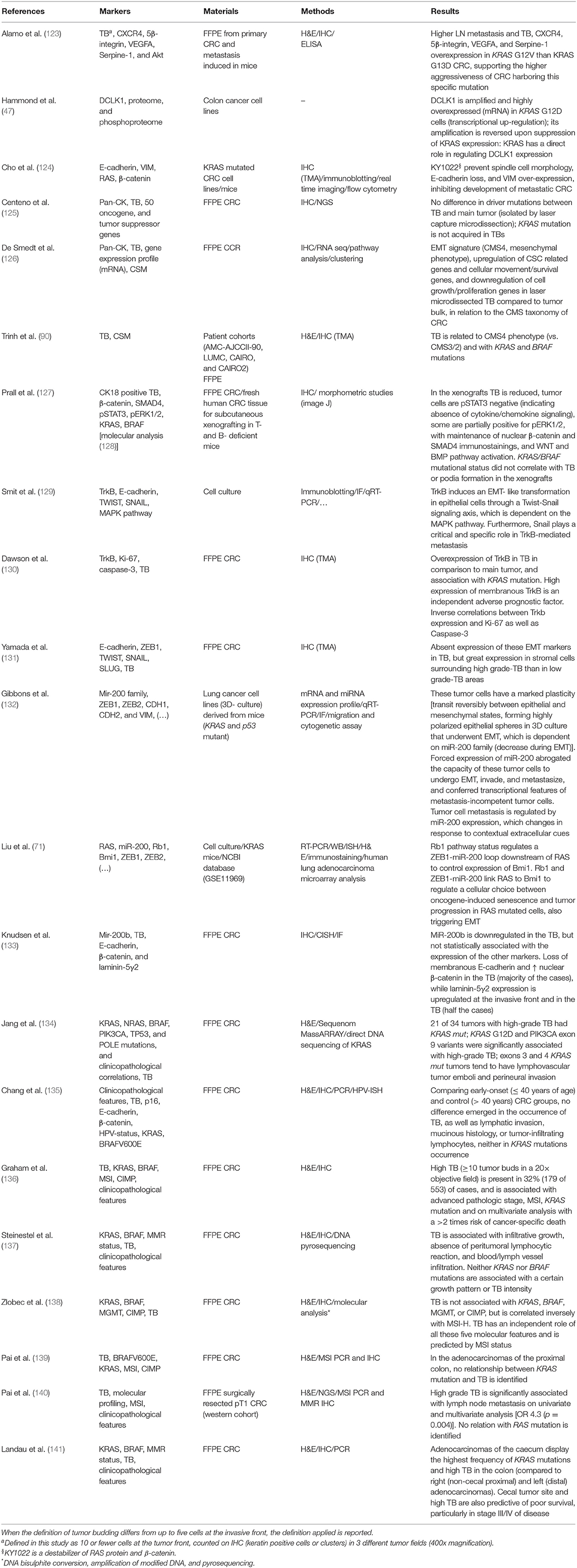
Table 1. Studies which investigated the KRAS status and/or TB in relation to cell morphology and/or cellular plasticity, also considered as EMT, or partial-EMT phenotype.
Tumor buds and their corresponding tumor bulk share the same driver mutations (125). De Smedt et al. found 296 differentially expressed genes by the comparison of neoplastic cells in the tumor mass and those microdissected from the tumor buds (126). TB cells undergo phenotype switching while detaching from the main tumor, with upregulation of genes related to cellular motility and downregulation of genes involved in cell growth and proliferation (126). This is consistent with the hypothesis that migration and proliferation are spatially and temporally exclusive (122). Regarding the CRCSC categories, TB cells showed a gene expression profile consistent with the “mesenchymal phenotype” (CMS4), while the cells in the main tumor had a molecular signature similar to the “canonical subtype” (CMS2) (126). This finding is supported by the results of another study in a large series of CRCs highlighting the association of TB with CMS4 phenotype—a greater number of tumor buds was found in CMS4 than in CMS2 and CMS3 tumors—and KRAS mutations (90). A significant association between KRAS mutations and the presence of high-grade TB has been reported in CRC (Table 1) (122, 131, 138, 142, 143). In vitro, KRAS mutations can induce expression of ZEB1, which promotes EMT, invasion, and metastasis (71, 132). Moreover, TB cells in CRC patients show increased expression of ZEB1 and a concomitant reduction of miR-200b and miR-200c, supporting the association between miR-200 family members and EMT (133). Resistance to anoikis, the cell death mechanism that occurs to non-neoplastic cells when detach from ECM, is a prerequisite for TB cells to survive during invasion. Neurothropic tyrosine receptor kinase B (TrkB) is a potent anoikis suppressor, which is overexpressed in tumor buds and in CRC with high-grade tumor budding and KRAS mutations (130). Indeed, RAS signaling promotes TrkB-induced EMT, anoikis resistance, and metastasis through TWIST and SNAIL (129). Morphologically, treatment of KRAS mutated cell lines with a destabilizer of β-catenin and RAS proteins can prevent spindle cell morphology as well as E-cadherin loss and vimentin over-expression (124). A xenograft model of CRC was also studied, but KRAS mutational status did not correlate with TB or podia formation (127).
TB can be considered as a snapshot of the dynamic process of invasion and a surrogate morphological marker of EMT. The translation into the clinics of TB, for a long time believed as a sign of biological aggressiveness, fits with its demonstration as an adverse and independent prognostic marker in all stages of CRC (Table 2) (3, 90, 94–96, 122, 136, 141, 146, 148). Regardless of the assessing method, evidences suggest that TB has a prognostic effect independent of age, sex, and stage of disease (3, 90, 94–96, 122, 123, 135, 146, 148). TB is usually associated with high tumor grade, advanced stage, lymphovascular invasion, nodal and distant metastasis, locoregional and distant recurrence, and worse overall, disease free, and recurrence free survival (122). The clinical implications are not only prognostic but also therapeutic. In metastatic patients, the presence of high tumor budding can predict resistance to anti-EGFR therapies (149). Moreover, KRAS status assessment seems to be useful to identify possible non-responder patients in the metastatic setting (149). Recently, it has been demonstrated that intratumoral TB is related to nodal and distant metastasis in CRC (90, 150–152). Apparently, intratumoral TB has a prognostic effect assessed on a continuous scale (90), and similarly to peritumoral TB has been associated with higher stages, vascular invasion, infiltrative margin, poor survival, and to peritumoral TB itself (122, 147). To date, the prognostic impact of TB has been associated with three major clinical scenarios.
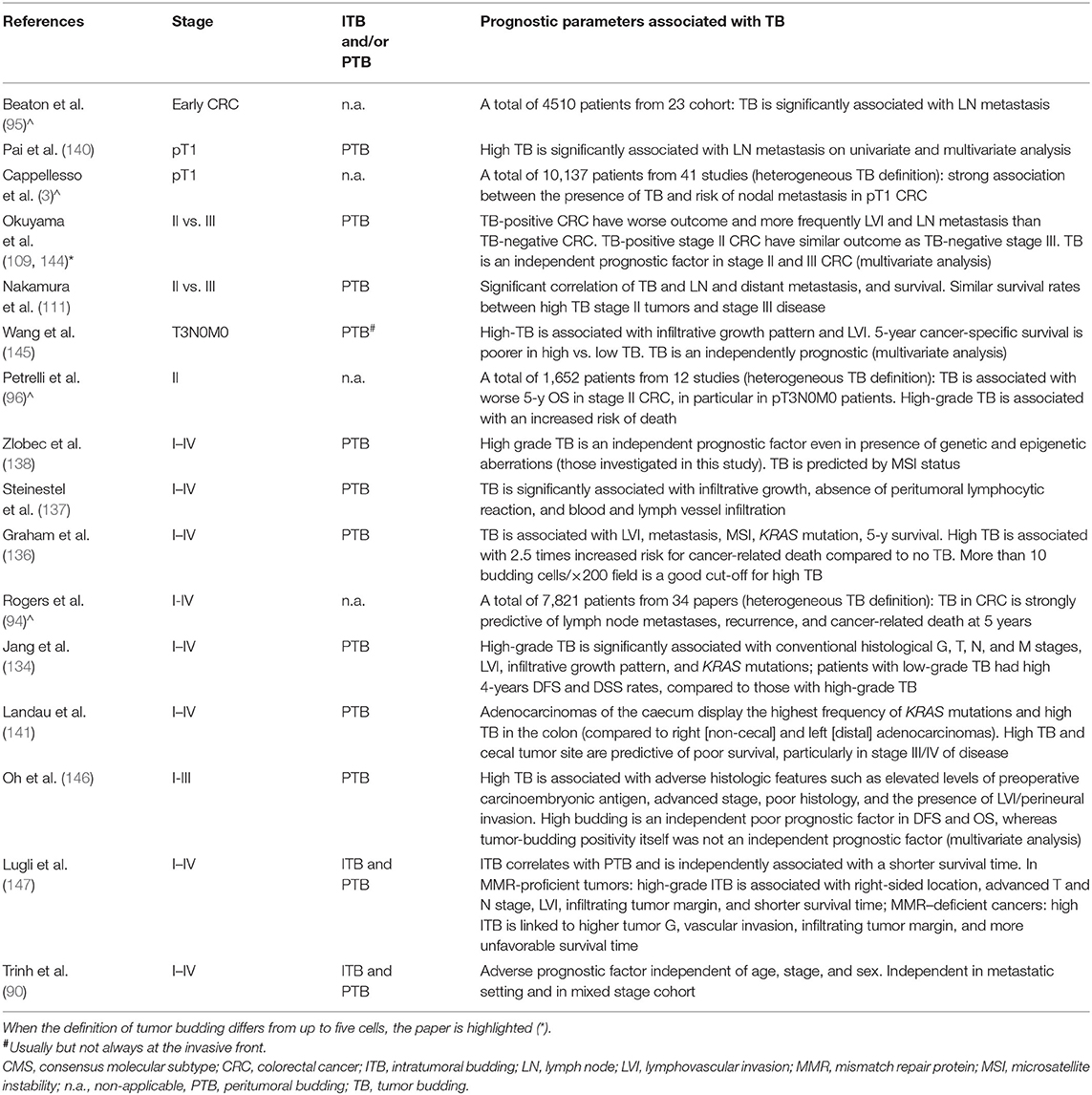
Table 2. Selected studies and reviews (∧) which investigated tumor budding as a prognostic marker in colorectal cancer.
First, in CRC infiltrating the submucosa (categorized as pT1 according to the current staging system), TB is an accurate predictor of nodal metastasis (3, 88, 95, 153–155). A recent meta-analysis including over a thousand of patients with endoscopically removed pT1 CRCs has shown that tumors with TB are strongly associated with lymph node involvement (3). In a western cohort of 116 surgically resected pT1 CRCs, high grade TB has been significantly associated with lymph node metastasis on univariate and multivariate analysis (140). While the Japanese Society for Cancer of the Colo-Rectum has already incorporated TB among the mandatory prognostic variables for pT1 CRC reports, in Western countries this has not yet happened. However, the available evidences strongly support its incorporation also in Western guidelines to improve lymphadenectomy planning (3, 88, 89, 140).
Second, in stage II CRC (namely a tumor without nodal and distant metastasis) the presence of high-grade TB confers a more aggressive behavior similarly to stage III CRC (namely a tumor with nodal metastasis but without distant metastasis) (96, 109, 111, 113, 126, 144, 145, 156). A metanalysis including over a thousand and a half stage II CRC patients highlighted that tumors with high grade TB are associated with worse overall survival, with a difference of survival of about 25%, mostly in pT3N0M0 patients (96). The survival rate of stage II CRC patients stratified as low or high grade TB vs. stage III CRC ones has been directly studied showing significantly differences depending on TB level (111). In particular, the survival rates of stage II CRC patients with high grade TB resulted comparable to those of patients with metastatic disease. These findings raise the opportunity of offering adjuvant chemotherapy to these patients, since they are expected to have a more aggressive disease.
Third, pre-operative biopsies could benefit of intratumoral TB assessment. Indeed, in CRC surgical samples intratumoral and peritumoral TB are strongly related and associated with a shorter survival (147). Moreover, high-grade intratumoral TB correlates with higher tumor grade, more advanced primary tumor, lymphatic and vascular invasion, and nodal metastasis (147). Intratumoral TB could be used as predictive parameter in the selection of candidates for neo-adjuvant therapy (88, 90).
The deepening of the knowledge on the molecular mechanisms linking common gene mutations, such as those affecting RAS, to specific gene-expression profiles, tumor cell characteristics, and biological behavior will disclose novel opportunities for the prevention, detection, and tailored treatment of CRC.
VM and LN reviewed the literature, wrote the first draft, and edited the manuscript. RC planned the article, coordinated and supervised the work, reviewed the literature, wrote the first draft, and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CIN, chromosomal instability; CSS, chromosomal stability; CIMP-N/L/H, CpG island methylator phenotype-negative/low/high; MSS, microsatellite stability; MSI-L/H, microsatellite instability-low/high; EMT, epithelial to mesenchymal transition; ECM, extra-cellular matrix; TB, tumor budding; APC, adenomatous polyposis coli; KRAS, Kirsten rat sarcoma viral oncogene homolog; NRAS, neuroblastoma RAS viral oncogene homolog; HRAS, Harvey rat sarcoma viral oncogene homolog; TP53, tumor protein p53; TGF-β, transforming growth factor β; FAP, familial adenomatous polyposis; MUTYH, mutY DNA glycosylase; MAP, MUTYH-associated polyposis; CRCSC, CRC Subtyping Consortium; CMS, consensus molecular subtypes; GDP, guanosine diphosphate; GTP, guanosine-5′-triphosphate; RASGEF, recruit guanine nucleotide exchange factor; PI3K, phosphoinositide 3-kinase; MEK, mitogen-activated protein kinase kinase; ERK, extracellular signal-regulated kinase; DCLK1, doublecortin like kinase 1; ZEB, zinc finger E-box binding homeobox; TrkB, tyrosine receptor kinase B; EGFR, epidermal growth factor receptor.
1. Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon: International Agency for Research on Cancer (IARC). (2010).
2. Cremolini C, Schirripa M, Antoniotti C, Moretto R, Salvatore L, Masi G, et al. First-line chemotherapy for mCRC—a review and evidence-based algorithm. Nat Rev Clin Oncol. (2015) 12:607–19. doi: 10.1038/nrclinonc.2015.129
3. Cappellesso R, Luchini C, Veronese N, Lo Mele M, Rosa-Rizzotto E, Guido E, et al. Tumor budding as a risk factor for nodal metastasis in pT1 colorectal cancers: a meta-analysis. Hum Pathol. (2017) 65:62–70. doi: 10.1016/j.humpath.2017.04.013
4. GLOBOCAN 2018 Graph production: IARC. World Health Organization. http://gco.iarc.fr/today (accessed May 15, 2019).
5. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. (1990) 61:759–67. doi: 10.1016/0092-8674(90)90186-I
6. Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. (2007) 50:113–30. doi: 10.1111/j.1365-2559.2006.02549.x
7. Wang W, Kandimalla R, Huang H, Zhu L, Li Y, Gao F, et al. Molecular subtyping of colorectal cancer: recent progress, new challenges and emerging opportunities. Semin Cancer Biol. (2019) 55:37–52. doi: 10.1016/j.semcancer.2018.05.002
8. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. (2015) 21:1350–6. doi: 10.1038/nm.3967
9. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. (1993) 363:558–61. doi: 10.1038/363558a0
10. Okugawa Y, Grady WM, Goel A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. (2015) 149:1204–25.e12. doi: 10.1053/j.gastro.2015.07.011
11. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. (2010) 138:2059–72. doi: 10.1053/j.gastro.2009.12.065
12. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. (1996) 87:159–70. doi: 10.1016/S0092-8674(00)81333-1
13. Mori Y, Selaru FM, Sato F, Yin J, Simms LA, Xu Y, et al. The impact of microsatellite instability on the molecular phenotype of colorectal tumors. Cancer Res. (2003) 63:4577–82.
14. Whitehall VL, Walsh MD, Young J, Leggett BA, Jass JR. Methylation of O-6-methylguanine DNA methyltransferase characterizes a subset of colorectal cancer with low-level DNA microsatellite instability. Cancer Res. (2001) 61:827–830.
15. Tuppurainen K, Mäkinen JM, Junttila O, Liakka A, Kyllönen AP, Tuominen H, et al. Morphology and microsatellite instability in sporadic serrated and non-serrated colorectal cancer. J Pathol. (2005) 207:285–94. doi: 10.1002/path.1850
16. Mahooti S, Hampel H, LaJeunesse J, Sotamaa K, De La Chapelle A, Frankel WL. MLH1 and PMS2 protein expression in 103 colorectal carcinomas with MLH1 promoter methylation and without MLH1 or PMS2 germline mutation. Gastrointestinal Lab Invest. (2006) 86:113A. doi: 10.1038/sj.labinvest.3700611
17. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. (1999) 96:8681–86. doi: 10.1073/pnas.96.15.8681
18. Ang PW, Loh M, Liem N, Lim PL, Grieu F, Vaithilingam A, et al. Comprehensive profiling of DNA methylation in colorectal cancer reveals subgroups with distinct clinicopathological and molecular features. BMC Cancer. (2010) 10:227. doi: 10.1186/1471-2407-10-227
19. Bae JM, Kim JH, Kang GH. Epigenetic alterations in colorectal cancer: the CpG island methylator phenotype. Histol Histopathol. (2013) 28:585–95. doi: 10.14670/HH-28.585
20. Sideris M, Papagrigoriadis S. Molecular biomarkers and classification models in the evaluation of the prognosis of colorectal cancer. Anticancer Res. (2014) 34:2061–8.
21. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, et al. Inherited variants of MYH associated with somatic G:C–>T:A mutations in colorectal tumors. Nat Genet. (2002) 30:227–32. doi: 10.1038/ng828
22. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. (2010) 138:2073–87.e3. doi: 10.1053/j.gastro.2009.12.064
23. Wang L, Cunningham JM, Winters JL, Guenther JC, French AJ, Boardman LA, et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. (2003) 63:5209–12.
24. Cappellesso R, Lo Mele M, Munari G, Rosa-Rizzotto E, Guido E, De Lazzari F, et al. Molecular characterization of “sessile serrated” adenoma to carcinoma transition in six early colorectal cancers. Pathol Res Pract. (2019) 215:957–62. doi: 10.1016/j.prp.2019.02.001
25. Matsui A, Ihara T, Suda H, Mikami H, Semba K. Gene amplification: mechanisms and involvement in cancer. Biomol Concepts. (2013) 4:567–82. doi: 10.1515/bmc-2013-0026
26. Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, et al. Chromosomal instability. (CIN) phenotype, CIN high or CIN low, predicts survival for colorectal cancer. J Clin Oncol. (2012) 30:2256–64. doi: 10.1200/JCO.2011.38.6490
27. Malesci A, Laghi L, Bianchi P, Delconte G, Randolph A, Torri V, et al. Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clin Cancer Res. (2007) 13:3831–9. doi: 10.1158/1078-0432.CCR-07-0366
28. Dahlin AM, Palmqvist R, Henriksson ML, Jacobsson M, Eklöf V, Rutegård J, et al. The role of the CpG island methylator phenotype in colorectal cancer prognosis depends on microsatellite instability screening status. Clin Cancer Res. (2010) 16:1845–55. doi: 10.1158/1078-0432.CCR-09-2594
29. Ward RL, Cheong K, Ku SL, Meagher A, O'Connor T, Hawkins NJ. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. J Clin Oncol. (2003) 21:3729–36. doi: 10.1200/JCO.2003.03.123
30. Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. (2010) 28:3219–26. doi: 10.1200/JCO.2009.27.1825
31. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1200/jco.2015.33.18_suppl.lba100
32. De Sousa E, Melo F, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LPMH, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. (2013) 19:614–8. doi: 10.1038/nm.3174
33. Schlicker A, Beran G, Chresta CM, McWalter G, Pritchard A, Weston S, et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med Genom. (2012) 5:66. doi: 10.1186/1755-8794-5-66
34. Budinska E, Popovici V, Tejpar S, D'Ario G, Lapique N, Sikora KO, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. (2013) 231:63–76. doi: 10.1002/path.4212
35. Roepman P, Schlicker A, Tabernero J, Majewski I, Tian S, Moreno V, et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer. (2014) 134:552–62. doi: 10.1002/ijc.28387
36. Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. (2013) 10:e1001453. doi: 10.1371/journal.pmed.1001453
37. Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. (2013) 19:619–25. doi: 10.1038/nm.3175
38. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. (2016) 129:1287–92. doi: 10.1242/jcs.182873
39. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. (2012) 72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612
40. Makrodouli E, Oikonomou E, Koc M, Andera L, Sasazuki T, Shirasawa S, et al. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: a comparative study. Mol Cancer. (2011) 10:118. doi: 10.1186/1476-4598-10-118
41. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. (2014) 13:828–51. doi: 10.1038/nrd4389
42. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. (2011) 39:D945–50. doi: 10.1093/nar/gkq929
43. Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. (2008) 40:600–8. doi: 10.1038/ng.115
44. Han C-B, Li F, Ma J-T, Zou H-W. Concordant KRAS mutations in primary and metastatic colorectal cancer tissue specimens: a meta-analysis and systematic review. Cancer Invest. (2012) 30:741–7. doi: 10.3109/07357907.2012.732159
45. Edkins S, O'Meara S, Parker A, Stevens C, Reis M, Jones S, et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol Ther. (2006) 5:928–32. doi: 10.4161/cbt.5.8.3251
46. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. (1997) 88:593–602. doi: 10.1016/S0092-8674(00)81902-9
47. Hammond DE, Mageean CJ, Rusilowicz EV, Wickenden JA, Clague MJ, Prior IA. Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J Proteome Res. (2015) 14:1535–46. doi: 10.1021/pr501191a
48. Pandya P, Orgaz JL, Sanz-Moreno V. Modes of invasion during tumour dissemination. Mol Oncol. (2017) 11:5–27. doi: 10.1002/1878-0261.12019
49. Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. (2011) 147:992–1009. doi: 10.1016/j.cell.2011.11.016
50. Clark AG, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol. (2015) 36:13–22. doi: 10.1016/j.ceb.2015.06.004
51. Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. (2012) 14:777–83. doi: 10.1038/ncb2548
52. Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. (2003) 160:267–77. doi: 10.1083/jcb.200209006
53. Lorentzen A, Bamber J, Sadok A, Elson-Schwab I, Marshall CJ. An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J Cell Sci. (2011) 124:1256–67. doi: 10.1242/jcs.074849
54. Bergert M, Erzberger A, Desai RA, Aspalter IM, Oates AC, Charras G, et al. Force transmission during adhesion-independent migration. Nat Cell Biol. (2015) 17:524–9. doi: 10.1038/ncb3134
55. Charras G, Paluch E. Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol. (2008) 9:730–6. doi: 10.1038/nrm2453
56. Haeger A, Wolf K, Zegers MM, Friedl P. Collective cell migration: guidance principles and hierarchies. Trends Cell Biol. (2015) 25:556–66. doi: 10.1016/j.tcb.2015.06.003
57. Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. (2009) 10:445–57. doi: 10.1038/nrm2720
58. Alexander S, Koehl GE, Hirschberg M, Geissler EK, Friedl P. Dynamic imaging of cancer growth and invasion: a modified skin-fold chamber model. Histochem Cell Biol. (2008) 130:1147–54. doi: 10.1007/s00418-008-0529-1
59. Friedl P, Hegerfeldt Y, Tusch M. Collective cell migration in morphogenesis and cancer. Int J Dev Biol. (2004) 48:441–9. doi: 10.1387/ijdb.041821pf
60. Weigelin B, Bakker G-J, Friedl P. Third harmonic generation microscopy of cells and tissue organization. J Cell Sci. (2016) 129:245–55. doi: 10.1242/jcs.152272
61. Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. (2003) 3:362–74. doi: 10.1038/nrc1075
62. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. (2009) 139:871–90. doi: 10.1016/j.cell.2009.11.007
63. Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, et al. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology. (2006) 131:830–40. doi: 10.1053/j.gastro.2006.06.016
64. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. (2013) 27:2192–206. doi: 10.1101/gad.225334.113
65. Vu T, Datta PK. Regulation of EMT in colorectal cancer: a culprit in metastasis. Cancers. (2017) 9:E171. doi: 10.3390/cancers9120171
66. Cappellesso R, Marioni G, Crescenzi M, Giacomelli L, Guzzardo V, Mussato A, et al. The prognostic role of the epithelial-mesenchymal transition markers E-cadherin and Slug in laryngeal squamous cell carcinoma. Histopathology. (2015) 67:491–500. doi: 10.1111/his.12668
67. Chi Y, Zhou D. MicroRNAs in colorectal carcinoma–from pathogenesis to therapy. J Exp Clin Cancer Res. (2016) 35:43. doi: 10.1186/s13046-016-0320-4
68. Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. (2008) 283:14910–14. doi: 10.1074/jbc.C800074200
69. Park S-M, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. (2008) 22:894–907. doi: 10.1101/gad.1640608
70. Hur K, Toiyama Y, Takahashi M, Balaguer F, Nagasaka T, Koike J, et al. MicroRNA-200c modulates epithelial-to-mesenchymal transition. (EMT) in human colorectal cancer metastasis. Gut. (2013) 62:1315–26. doi: 10.1136/gutjnl-2011-301846
71. Liu Y, Sánchez-Tilló E, Lu X, Huang L, Clem B, Telang S, et al. The ZEB1 transcription factor acts in a negative feedback loop with miR200 downstream of Ras and Rb1 to regulate Bmi1 expression. J Biol Chem. (2014) 289:4116–25. doi: 10.1074/jbc.M113.533505
72. Sun Z, Zhang Z, Liu Z, Qiu B, Liu K, Dong G. MicroRNA-335 inhibits invasion and metastasis of colorectal cancer by targeting ZEB2. Med Oncol. (2014) 31:982. doi: 10.1007/s12032-014-0982-8
73. Geng L, Chaudhuri A, Talmon G, Wisecarver JL, Are C, Brattain M, et al. MicroRNA-192 suppresses liver metastasis of colon cancer. Oncogene. (2014) 33:5332–40. doi: 10.1038/onc.2013.478
74. Hahn S, Jackstadt R, Siemens H, Hünten S, Hermeking H. SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J. (2013) 32:3079–95. doi: 10.1038/emboj.2013.236
75. Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. (2002) 156:299–313. doi: 10.1083/jcb.200109037
76. Gotzmann J, Mikula M, Eger A, Schulte-Hermann R, Foisner R, Beug H, et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. (2004) 566:9–20. doi: 10.1016/S1383-5742(03)00033-4
77. Guarino M. Epithelial-mesenchymal transition and tumour invasion. Int J Biochem Cell Biol. (2007) 39:2153–60. doi: 10.1016/j.biocel.2007.07.011
78. Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. (2015) 36:103–12. doi: 10.1016/j.ceb.2015.08.005
79. Sasaki AT, Janetopoulos C, Lee S, Charest PG, Takeda K, Sundheimer LW, Meili R, et al. G protein-independent Ras/PI3K/F-actin circuit regulates basic cell motility. J Cell Biol. (2007) 178:185–91. doi: 10.1083/jcb.200611138
80. van Haastert PJM, Keizer-Gunnink I, Kortholt A. Coupled excitable Ras and F-actin activation mediates spontaneous pseudopod formation and directed cell movement. Mol Biol Cell. (2017) 28:922–34. doi: 10.1091/mbc.e16-10-0733
81. Lemieux E, Bergeron S, Durand V, Asselin C, Saucier C, Rivard N. Constitutively active MEK1 is sufficient to induce epithelial-to-mesenchymal transition in intestinal epithelial cells and to promote tumor invasion and metastasis. Int J Cancer. (2009) 125:1575–86. doi: 10.1002/ijc.24485
82. Yamazaki D, Kurisu S, Takenawa T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene. (2009) 28:1570–83. doi: 10.1038/onc.2009.2
83. Sanz-Moreno V, Marshall CJ. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr Opin Cell Biol. (2010) 22:690–6. doi: 10.1016/j.ceb.2010.08.020
84. Jinesh GG, Sambandam V, Vijayaraghavan S, Balaji K, Mukherjee S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene. (2018) 37:839–46. doi: 10.1038/onc.2017.377
85. Shen H, Xing C, Cui K, Li Y, Zhang J, Du R, et al. MicroRNA-30a attenuates mutant KRAS-driven colorectal tumorigenesis via direct suppression of ME1. Cell Death Differ. (2017) 24:1253–62. doi: 10.1038/cdd.2017.63
86. Zhong M, Bian Z, Wu Z. miR-30a suppresses cell migration and invasion through downregulation of PIK3CD in colorectal carcinoma. Cell Physiol Biochem. (2013) 31:209–18. doi: 10.1159/000343362
87. Liu T-P, Huang C-C, Yeh K-T, Ke T-W, Wei P-L, Yang J-R, et al. Down-regulation of let-7a-5p predicts lymph node metastasis and prognosis in colorectal cancer: implications for chemotherapy. Surg Oncol. (2016) 25:429–34. doi: 10.1016/j.suronc.2016.05.016
88. Lugli A, Kirsch R, Ajioka Y, Bosman F, Cathomas G, Dawson H, et al. Recommendations for reporting tumor budding in colorectal cancer based on the International Tumor Budding Consensus Conference. (ITBCC) 2016. Modern Pathol. (2017) 30:1299–311. doi: 10.1038/modpathol.2017.46
89. Dawson H, Galuppini F, Träger P, Berger MD, Studer P, Brügger L, et al. Validation of the International Tumor Budding Consensus Conference 2016 recommendations on tumor budding in stage I-IV colorectal cancer. Hum Pathol. (2019) 85:145–51. doi: 10.1016/j.humpath.2018.10.023
90. Trinh A, Lädrach C, Dawson HE, ten Hoorn S, Kuppen PJK, Reimers MS, et al. Tumour budding is associated with the mesenchymal colon cancer subtype and RAS/RAF mutations: a study of 1320 colorectal cancers with Consensus Molecular Subgroup. (CMS) data. Br J Cancer. (2018) 119:1244–51. doi: 10.1038/s41416-018-0230-7
91. Kakar S, Shi C, Berho ME, Driman DK, Fitzgibbons P, Frankel WL, et al. Protocol for the Examination of Specimens From Patients With Primary Carcinoma of the Colon and Rectum. (V4.0.0.1). College of American Pathologists. (CAP) website. http://www.cap.org/cancerprotocols (accessed December 20, 2017).
92. Amin MB, Edge SB, Greene FL, Byrd DR, Brookland RK, Washington MK, et al., editors. AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer (2017).
93. Glynne-Jones R, Wyrwicz L, Tiret E, Brown G, Rödel C, Cervantes A, et al. Rectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2017) 28:iv22–40. doi: 10.1093/annonc/mdx224
94. Rogers AC, Winter DC, Heeney A, Gibbons D, Lugli A, Puppa G, et al. Systematic review and meta-analysis of the impact of tumour budding in colorectal cancer. Br J Cancer. (2016) 115:831–40. doi: 10.1038/bjc.2016.274
95. Beaton C, Twine CP, Williams GL, Radcliffe AG. Systematic review and meta-analysis of histopathological factors influencing the risk of lymph node metastasis in early colorectal cancer. Colorectal Dis. (2013) 15:788–97. doi: 10.1111/codi.12129
96. Petrelli F, Pezzica E, Cabiddu M, Coinu A, Borgonovo K, Ghilardi M, et al. Tumour budding and survival in stage II colorectal cancer: a systematic review and pooled analysis. J Gastrointest Cancer. (2015) 46:212–8. doi: 10.1007/s12029-015-9716-1
97. Quirke P, Risio M, Lambert R, von Karsa L, Vieth M. International Agency for Research on Cancer. European guidelines for quality assurance in colorectal cancer screening and diagnosis. First Edition–Quality assurance in pathology in colorectal cancer screening and diagnosis. Endoscopy. (2012) 44 Suppl 3:SE116–30. doi: 10.1055/s-0032-1309797
98. Lino-Silva LS, Gamboa-Domínguez A, Zúñiga-Tamayo D, López-Correa P. Interobserver variability in colorectal cancer and the 2016 ITBCC concensus. Mod Pathol. (2019) 32:159–60. doi: 10.1038/s41379-018-0027-5
99. Dawson H, Kirsch R, Lugli A, participants of the International Tumor Budding Consensus Conference 2016. (1). Reply to: interobserver variability in colorectal cancer and the 2016 ITBCC consensus. Mod Pathol. (2019) 32:161–2. doi: 10.1038/s41379-018-0104-9
100. Lino-Silva LS, Salcedo-Hernández RA, Gamboa-Domínguez A. Tumour budding in rectal cancer. A comprehensive review. Współczesna Onkol. (2018) 22:61–74. doi: 10.5114/wo.2018.77043
101. Imai T. Histological comparison of cancer of the stomach in autopsy and operation cases. Jpn J Cancer Res. (1949) 40:199–201.
102. Gabbert H, Wagner R, Moll R, Gerharz C-D. Tumor dedifferentiation: an important step in tumor invasion. Clin Exp Metast. (1985) 3:257–79. doi: 10.1007/BF01585081
103. Hayashida K, Isomoto I, Shirouzu K, Morodomi T, Kurohiji T, Sou M, et al. A study of invasive colorectal carcinoma with reference mainly to vessel invasion and budding. Nippon Daicho-komonbyo Gakkai Zasshi. (1987) 40:119–126. doi: 10.3862/jcoloproctology.40.119
104. Morodomi T. Clinicopathological studies of advanced rectal cancers–predicting the degree of lymph node metastases from histopathological findings in pre-operative biopsy specimens. Nippon Geka Gakkai Zasshi. (1988) 89:352–63.
105. Hase K, Mochizuki H, Kurihara H, et al. Clinicopathological studies on local recurrence of rectal cancer. Nippon Shokakigeka Gakkai Zasshi. (1989) 22:1401.
106. Morodomi T, Isomoto H, Shirouzu K, Kakegawa K, Irie K, Morimatsu M. An index for estimating the probability of lymph node metastasis in rectal cancers. Lymph node metastasis and the histopathology of actively invasive regions of cancer. Cancer. (1989) 63:539–543. doi: 10.1002/1097-0142(19890201)63:3<539::AID-CNCR2820630323>3.0.CO;2-S
107. Hase K, Shatney C, Johnson D, Trollope M, Vierra M. Prognostic value of tumor “budding” in patients with colorectal cancer. Dis. Colon Rectum. (1993) 36:627–35. doi: 10.1007/BF02238588
108. Ueno H, Murphy J, Jass JR, Mochizuki H, Talbot IC. Tumour ‘budding’ as an index to estimate the potential of aggressiveness in rectal cancer. Histopathology. (2002) 40:127–32. doi: 10.1046/j.1365-2559.2002.01324.x
109. Okuyama T, Nakamura T, Yamaguchi M. Budding is useful to select high-risk patients in stage II well-differentiated or moderately differentiated colon adenocarcinoma. Dis Colon Rectum. (2003) 46:1400–6. doi: 10.1007/s10350-004-6757-0
110. Ha SS, Choi HJ, Park KJ, Kim JM, Kim SH, Roh YH, et al. Intensity of tumor budding as an index for the malignant potential in invasive rectal carcinoma. Cancer Res Treat. (2005) 37:177–82. doi: 10.4143/crt.2005.37.3.177
111. Nakamura T, Mitomi H, Kanazawa H, Ohkura Y, Watanabe M. Tumor budding as an index to identify high-risk patients with stage II colon cancer. Dis Colon Rectum. (2008) 51:568–72. doi: 10.1007/s10350-008-9192-9
112. Ohtsuki K, Koyama F, Tamura T, Enomoto Y, Fujii H, Mukogawa T, et al. Prognostic value of immunohistochemical analysis of tumor budding in colorectal carcinoma. Anticancer Res. (2008) 28:1831–6.
113. Betge J, Kornprat P, Pollheimer MJ, Lindtner RA, Schlemmer A, Rehak P, et al. Tumor budding is an independent predictor of outcome in AJCC/UICC stage II colorectal cancer. Ann Surg Oncol. (2012) 19:3706–12. doi: 10.1245/s10434-012-2426-z
114. Horcic M, Koelzer VH, Karamitopoulou E, Terracciano L, Puppa G, Zlobec I, et al. Tumor budding score based on 10 high-power fields is a promising basis for a standardized prognostic scoring system in stage II colorectal cancer. Hum Pathol. (2013) 44:697–705. doi: 10.1016/j.humpath.2012.07.026
115. Jensen DH, Dabelsteen E, Specht L, Fiehn AMK, Therkildsen MH, Jønson L, et al. Molecular profiling of tumour budding implicates TGFβ-mediated epithelial-mesenchymal transition as a therapeutic target in oral squamous cell carcinoma. J Pathol. (2015) 236:505–16. doi: 10.1002/path.4550
116. Almangush A, Pirinen M, Heikkinen I, Mäkitie AA, Salo T, Leivo I. Tumour budding in oral squamous cell carcinoma: a meta-analysis. Br J Cancer. (2018) 118:577–86. doi: 10.1038/bjc.2017.425
117. Liang F, Cao W, Wang Y, Li L, Zhang G, Wang Z. The prognostic value of tumor budding in invasive breast cancer. Pathol Res Pract. (2013) 209:269–75. doi: 10.1016/j.prp.2013.01.009
118. Karamitopoulou E, Zlobec I, Born D, Kondi-Pafiti A, Lykoudis P, Mellou A, et al. Tumour budding is a strong and independent prognostic factor in pancreatic cancer. Eur J Cancer. (2013) 49:1032–9. doi: 10.1016/j.ejca.2012.10.022
119. Niwa Y, Yamada S, Koike M, Kanda M, Fujii T, Nakayama G, et al. Epithelial to mesenchymal transition correlates with tumor budding and predicts prognosis in esophageal squamous cell carcinoma. J Surg Oncol. (2014) 110:764–9. doi: 10.1002/jso.23694
120. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
121. Zlobec I, Lugli A. Epithelial mesenchymal transition and tumor budding in aggressive colorectal cancer: tumor budding as oncotarget. Oncotarget. (2010) 1:651–61. doi: 10.18632/oncotarget.199
122. Grigore A, Jolly M, Jia D, Farach-Carson M, Levine H. Tumor budding: the name is EMT. partial EMT. J Clin Med. (2016) 5:51. doi: 10.3390/jcm5050051
123. Alamo P, Gallardo A, Di Nicolantonio F, Pavón MA, Casanova I, Trias M, et al. Higher metastatic efficiency of KRas G12V than KRas G13D in a colorectal cancer model. FASEB J. (2015) 29:464–76. doi: 10.1096/fj.14-262303
124. Cho YH, Cha PH, Kaduwal S, Park JC, Lee SK, Yoon JS, et al. KY1022, a small molecule destabilizing Ras via targeting the Wnt/β-catenin pathway, inhibits development of metastatic colorectal cancer. Oncotarget. (2016) 7:81727–40. doi: 10.18632/oncotarget.13172
125. Centeno I, Paasinen Sohns A, Flury M, Galván JA, Zahnd S, Koelzer VH, et al. DNA profiling of tumor buds in colorectal cancer indicates that they have the same mutation profile as the tumor from which they derive. Virchows Archiv. (2017) 470:341–6. doi: 10.1007/s00428-017-2071-9
126. De Smedt L, Palmans S, Andel D, Govaere O, Boeckx B, Smeets D, et al. Expression profiling of budding cells in colorectal cancer reveals an EMT-like phenotype and molecular subtype switching. Br J Cancer. (2017) 116:58–65. doi: 10.1038/bjc.2016.382
127. Prall F, Maletzki C, Hühns M, Krohn M, Linnebacher M. Colorectal carcinoma tumour budding and podia formation in the xenograft microenvironment. PLoS ONE. (2017) 12:e0186271. doi: 10.1371/journal.pone.0186271
128. Ostwald C, Linnebacher M, Weirich V, Prall F. Chromosomally and microsatellite stable colorectal carcinomas without the CpG island methylator phenotype in a molecular classification. Int J Oncol. (2009) 35:321–7. doi: 10.3892/ijo_00000343
129. Smit MA, Geiger TR, Song JY, Gitelman I, Peeper DS. A twist-snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol Cell Biol. (2009) 29:3722–37. doi: 10.1128/MCB.01164-08
130. Dawson H, Grundmann S, Koelzer VH, Galván JA, Kirsch R, Karamitopoulou E, et al. Tyrosine kinase receptor B. (TrkB) expression in colorectal cancers highlights anoikis resistance as a survival mechanism of tumour budding cells. Histopathology. (2015) 66:715–25. doi: 10.1111/his.12603
131. Yamada N, Sugai T, Eizuka M, Tsuchida K, Sugimoto R, Mue Y, et al. Tumor budding at the invasive front of colorectal cancer may not be associated with the epithelial-mesenchymal transition. Hum Pathol. (2017) 60:151–9. doi: 10.1016/j.humpath.2016.10.007
132. Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. (2009) 23:2140–51. doi: 10.1101/gad.1820209
133. Knudsen KN, Lindebjerg J, Nielsen BS, Hansen TF, Sørensen FB. MicroRNA-200b is downregulated in colon cancer budding cells. PLoS ONE. (2017) 12:e0178564. doi: 10.1371/journal.pone.0178564
134. Jang S, Hong M, Shin MK, Kim BC, Shin H-S, Yu E, et al. KRAS and PIK3CA mutations in colorectal adenocarcinomas correlate with aggressive histological features and behavior. Hum Pathol. (2017) 65:21–30. doi: 10.1016/j.humpath.2017.01.010
135. Chang DT, Pai RK, Rybicki LA, Dimaio MA, Limaye M, Jayachandran P, et al. Clinicopathologic and molecular features of sporadic early-onset colorectal adenocarcinoma: an adenocarcinoma with frequent signet ring cell differentiation, rectal and sigmoid involvement and adverse morphologic features. Modern Pathol. (2012) 25:1128–39. doi: 10.1038/modpathol.2012.61
136. Graham RP, Vierkant RA, Tillmans LS, Wang AH, Laird PW, Weisenberger DJ, et al. Tumor budding in colorectal carcinoma: confirmation of prognostic significance and histologic cutoff in a population-based cohort. Am J Surg Pathol. (2015) 39:1340–6. doi: 10.1097/PAS.0000000000000504
137. Steinestel K, Lennerz JK, Eder S, Kraft K, Arndt A. Invasion pattern and histologic features of tumor aggressiveness correlate with MMR protein expression, but are independent of activating KRAS and BRAF mutations in CRC. Virchows Arch. (2014) 465:155–63. doi: 10.1007/s00428-014-1604-8
138. Zlobec I, Bihl MP, Foerster A, Rufle A, Lugli A. The impact of CpG island methylator phenotype and microsatellite instability on tumour budding in colorectal cancer. Histopathology. (2012) 61:777–87. doi: 10.1111/j.1365-2559.2012.04273.x
139. Pai RK, Jayachandran P, Koong AC, Chang DT, Kwok S, Ma L, et al. BRAF-mutated, microsatellite-stable adenocarcinoma of the proximal colon: an aggressive adenocarcinoma with poor survival, mucinous differentiation, and adverse morphologic features. Am J Surg Pathol. (2012) 36:744–52. doi: 10.1097/PAS.0b013e31824430d7
140. Pai RK, Cheng Y-W, Jakubowski MA, Shadrach BL, Plesec TP, Pai RK. Colorectal carcinomas with submucosal invasion. (pT1): analysis of histopathological and molecular factors predicting lymph node metastasis. Mod Pathol. (2017) 30:113–22. doi: 10.1038/modpathol.2016.166
141. Landau MA, Zhu B, Akwuole FN, Pai RK. Site-specific differences in colonic adenocarcinoma: KRAS mutations and high tumor budding are more frequent in cecal adenocarcinoma. Am J Surg Pathol. (2018) 42:351–8. doi: 10.1097/PAS.0000000000001004
142. Jass JR, Barker M, Fraser L, Walsh MD, Whitehall VLJ, Gabrielli B, et al. APC mutation and tumour budding in colorectal cancer. J Clin Pathol. (2003) 56:69–73. doi: 10.1136/jcp.56.1.69
143. Dawson H, Lugli A. Molecular and pathogenetic aspects of tumor budding in colorectal cancer. Front Med. (2015) 2:11. doi: 10.3389/fmed.2015.00011
144. Okuyama T, Oya M, Ishikawa H. Budding as a useful prognostic marker in pT3 well- or moderately-differentiated rectal adenocarcinoma. J Surg Oncol. (2003) 83:42–7. doi: 10.1002/jso.10230
145. Wang LM, Kevans D, Mulcahy H, O'Sullivan J, Fennelly D, Hyland J, et al. Tumor budding is a strong and reproducible prognostic marker in T3N0 colorectal cancer. Am J Surg Pathol. (2009) 33:134–41. doi: 10.1097/PAS.0b013e318184cd55
146. Oh BY, Park YA, Huh JW, Yun SH, Kim HC, Chun H-K, et al. Prognostic impact of tumor-budding grade in stages 1-3 colon cancer: a retrospective cohort study. Ann Surg Oncol. (2018) 25:204–11. doi: 10.1245/s10434-017-6135-5
147. Lugli A, Vlajnic T, Giger O, Karamitopoulou E, Patsouris ES, Peros G, et al. Intratumoral budding as a potential parameter of tumor progression in mismatch repair-proficient and mismatch repair-deficient colorectal cancer patients. Hum Pathol. (2011) 42:1833–40. doi: 10.1016/j.humpath.2011.02.010
148. van Wyk HC, Park JH, Edwards J, Horgan PG, McMillan DC, Going JJ. The relationship between tumour budding, the tumour microenvironment and survival in patients with primary operable colorectal cancer. Br J Cancer. (2016) 115:156–63. doi: 10.1038/bjc.2016.173
149. Zlobec I, Molinari F, Martin V, Mazzucchelli L, Saletti P, Trezzi R, et al. Tumor budding predicts response to anti-EGFR therapies in metastatic colorectal cancer patients. World J Gastroenterol. (2010) 16:4823–31. doi: 10.3748/wjg.v16.i38.4823
150. Giger OT, Comtesse SCM, Lugli A, Zlobec I, Kurrer MO. Intra-tumoral budding in preoperative biopsy specimens predicts lymph node and distant metastasis in patients with colorectal cancer. Mod Pathol. (2012) 25:1048–53. doi: 10.1038/modpathol.2012.56
151. Zlobec I, Hädrich M, Dawson H, Koelzer VH, Borner M, Mallaev M, et al. Intratumoural budding. (ITB) in preoperative biopsies predicts the presence of lymph node and distant metastases in colon and rectal cancer patients. Br J Cancer. (2014) 110:1008–13. doi: 10.1038/bjc.2013.797
152. Rogers AC, Gibbons D, Hanly AM, Hyland JMP, O'Connell PR, Winter DC, et al. Prognostic significance of tumor budding in rectal cancer biopsies before neoadjuvant therapy. Mod Pathol. (2014) 27:156–162. doi: 10.1038/modpathol.2013.124
153. Bosch SL, Teerenstra S, de Wilt JHW, Cunningham C, Nagtegaal ID. Predicting lymph node metastasis in pT1 colorectal cancer: a systematic review of risk factors providing rationale for therapy decisions. Endoscopy. (2013) 45:827–34. doi: 10.1055/s-0033-1344238
154. Ueno H, Mochizuki H, Hashiguchi Y, Shimazaki H, Aida S, Hase K, et al. Risk factors for an adverse outcome in early invasive colorectal carcinoma. Gastroenterology. (2004) 127:385–94. doi: 10.1053/j.gastro.2004.04.022
155. Kawachi H, Eishi Y, Ueno H, Nemoto T, Fujimori T, Iwashita A, et al. A three-tier classification system based on the depth of submucosal invasion and budding/sprouting can improve the treatment strategy for T1 colorectal cancer: a retrospective multicenter study. Mod Pathol. (2015) 28:872–9. doi: 10.1038/modpathol.2015.36
Keywords: RAS, colorectal cancer, plasticity, epithelial to mesenchymal transition, tumor budding
Citation: Maffeis V, Nicolè L and Cappellesso R (2019) RAS, Cellular Plasticity, and Tumor Budding in Colorectal Cancer. Front. Oncol. 9:1255. doi: 10.3389/fonc.2019.01255
Received: 19 July 2019; Accepted: 30 October 2019;
Published: 19 November 2019.
Edited by:
Georgia Konstantinidou, University of Bern, SwitzerlandReviewed by:
Saverio Marchi, Marche Polytechnic University, ItalyCopyright © 2019 Maffeis, Nicolè and Cappellesso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rocco Cappellesso, cm9jY28uY2FwcGVsbGVzc29AZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.