 Raoul Tibes
Raoul Tibes James M. Bogenberger
James M. Bogenberger- 1NYU School of Medicine & Perlmutter Cancer Center, NYU Langone Health, New York, NY, United States
- 2Mayo Clinic, Scottsdale, AZ, United States
Acute myeloid leukemia (AML) is the most common adult acute leukemia. Survival remains poor, despite decades of scientific advances. Cytotoxic induction chemotherapy regimens are standard-of-care for most patients. Many investigations have highlighted the genomic heterogeneity of AML, and several new targeted therapeutic options have recently been approved. Additional novel therapies are showing promising clinical results and may rapidly transform the therapeutic landscape of AML. Despite the emerging clinical success of B-cell lymphoma (BCL)-2 targeting in AML and a large body of preclinical data supporting myeloid leukemia cell (MCL)-1 as an attractive therapeutic target for AML, MCL-1 targeting remains relatively unexplored, although novel MCL-1 inhibitors are under clinical investigation. Inhibitors of cyclin-dependent kinases (CDKs) involved in the regulation of transcription, CDK9 in particular, are being investigated in AML as a strategy to target MCL-1 indirectly. In this article, we review the basis for CDK inhibition in oncology with a focus on relevant preclinical mechanism-of-action studies of CDK9 inhibitors in the context of their therapeutic potential specifically in AML.
Introduction
In 2018, an estimated 21,380 people in the United States were diagnosed with acute myeloid leukemia (AML), making AML the most common adult acute leukemia (1). AML accounts for up to 32% of all adult leukemias and 1.3% of all new cancer cases in the United States (1). Survival outcomes for patients diagnosed with AML are poor. For patients <60 years of age treated with conventional chemotherapy, 35–45% achieve long-term survival (2). Older patients (≥65 years of age) have an even worse prognosis, with long-term survival rates of only 10–15% (2).
Cytotoxic induction therapy (7 days of standard-dose cytarabine and 3 days of an anthracycline or an anthracenedione, e.g., daunorubicin or idarubicin; also known as 7 + 3), which was developed in the 1970s, remains the standard of care for most patients with AML undergoing induction treatment (3, 4); however, recently a novel liposomal formulation of daunorubicin and cytarabine was approved by the US Food and Drug Administration (FDA) for high-risk and secondary AML (5, 6). Hypomethylating agents (HMAs), such as azacitidine and decitabine, while not formally approved by the FDA for the treatment of AML as single agents, have been used as de facto standard-of-care therapy for elderly patients with AML unfit for induction therapy. Response rates for azacitidine and decitabine monotherapy are low in elderly patients (10–50%), with a median overall survival of <1 year (7). Even for patients who achieve a complete remission with standard therapy, most will ultimately relapse and face a poor prognosis (8). Thus, there is a clear need for improved therapeutic options in AML. Since 2013, 65 drugs have been granted orphan designation specifically for the treatment of AML (9); however, there were only four US FDA approvals for new treatments in AML in 2017 (6, 10–16) and another four in 2018 (17–26).
Venetoclax combination therapy in particular has yielded promising results in elderly patients, with recent clinical trials showing a 65% complete remission rate in patients ≥75 years (7). As venetoclax is a selective inhibitor of B-cell lymphoma 2 (BCL-2), these outcomes highlight the importance of targeting BCL-2 family proteins for the treatment of AML. However, resistance to BCL-2 inhibition on venetoclax combination regimens is emerging, and alternative strategies to address resistance mechanisms are needed. Indeed, increased advances in the understanding of the role of BCL-2 family proteins and their interactors in apoptosis and AML pathogenesis have led to the discovery and clinical development of additional investigational treatments. Recent functional screens using CRISPR/Cas9 approaches highlight the central importance of mitochondrial function/architecture in resistance to BCL-2 inhibitor venetoclax (27, 28). Other BCL-2 family protein members may also play a role in AML patients refractory/resistant to BCL-2 inhibition, particularly MCL-1, which is an antiapoptotic multidomain protein regulated by distinct cyclin-dependent kinases (CDKs) in both apoptotic and cell-cycling pathways (8, 29–31).
In this review, we will address advances in the clinical development of CDK inhibitors as a strategy for indirectly targeting MCL-1 in the treatment of AML. We will briefly discuss the BCL-2 family of proteins that underlie AML pathogenesis and treatment resistance, as well as the therapeutic potential of targeting CDKs that regulate transcription, focusing on CDK9 inhibition.
BCL-2 Family Of Proteins, Including MCL-1, In AML Pathogenesis
Many studies have sought to identify critical, pathogenic mechanisms in AML. However, these efforts are complicated by the inherent heterogeneity of the disease (32) and its relatively low mutational load compared with some malignancies (33). Deregulated expression of one or more of the apoptosis-controlling BCL-2 family members, central regulators of cell survival and apoptosis, is common in AML (34). This family of proteins, which includes more than 20 members, has pro- or antiapoptotic functions converging on mitochondrial apoptosis, also commonly known as intrinsic apoptosis (35, 36), a critical cell-death regulatory mechanism (Figure 1). Impairment of apoptosis represents one of the postulated hallmarks of cancer and is highly relevant to AML, as antiapoptotic mechanisms are upregulated in AML (35, 37, 38).
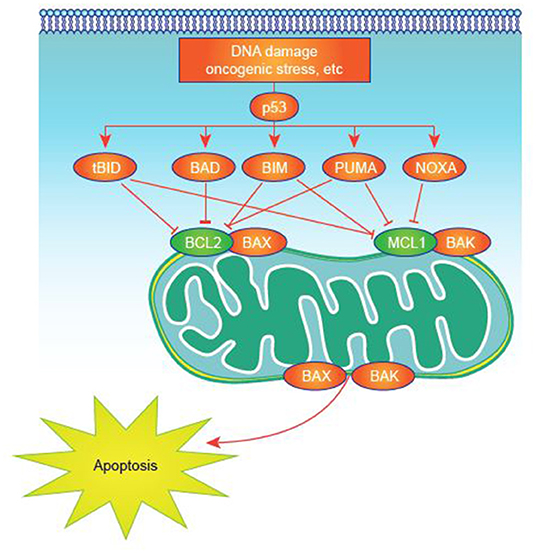
Figure 1. Apoptosis activation in normal and tumor cells. Apoptosis signaling is normally triggered by multiple death signals. There is a finely tuned balance between proapoptotic and antiapoptotic proteins that results in efficient apoptosis induction. MCL-1 and other antiapoptotic proteins block apoptotic effectors like BAK on the surface of the mitochondria. BH3-only proteins, such as NOXA, untether BAK from MCL-1, permitting BAK to cause events that result in cell death.
Based on sequence and structural homologies, BCL-2 family proteins can be classified into three groups, each containing at least one BCL-2 homology (BH) domain (BH1-4) (31):
• Proapoptotic multidomain effector proteins (including BAK, BAX, and BOK), which mediate the release of critical proapoptotic factors (e.g., cytochrome c, SMAC/Diablo) from mitochondria by inducing mitochondrial outer membrane permeabilization (MOMP).
• Proapoptotic, which contain only the BH3 domain (e.g., BID, BIM, PUMA, BAD, NOXA, HRK, BIK, BMF, BNIP3, and NIX) and are activated or induced by cell-death stimuli to promote cell death. This “BH3-only” group can be further subdivided into activators and/or sensitizers (39, 40). BH3-only activators directly and/or indirectly activate effector proteins to induce MOMP (31), and BH3-only sensitizers bind to antiapoptotic proteins to allow activator and effector proteins to drive MOMP (39). BIM has been reported to have the capacity to act as both sensitizer and activator, making it a powerful BH3-only protein actively involved in the response to drug therapy in blood-related malignancies (35, 41–45). BH3-only proteins and their role in apoptosis have been extensively reviewed elsewhere (36, 46).
• Antiapoptotic multidomain proteins (including BCL-2, BCL-XL, myeloid cell leukemia-1 [MCL-1], BCL-W, BCL-2-A1, and BCL-B) interact with both the multidomain effector proteins and BH3-only proteins to inhibit MOMP.
Overexpression of MCL-1 is recognized as having a critical role in several hematologic malignancies including diffuse large B-cell lymphoma (47), multiple myeloma (MM) (48), chronic lymphocytic lymphoma (49), and in AML cell survival and treatment resistance (50–52). Further, increased MCL-1 expression is associated with treatment resistance to myelodysplastic syndrome (MDS) (53, 54), which can evolve into AML. Preclinical studies support the potential for inhibition of MCL-1 to attenuate the underlying pathogenesis of AML (48, 50, 51, 55–59), including when used in combination with BCL-2 and/or BCL2L1 (also known as BCL-XL) inhibition (60–64). MCL-1 sequesters mitochondrial BAK, thereby preventing homo- or hetero-oligomerization, pore formation, and ultimately, apoptosis. MCL-1 is distinct from other antiapoptotic BCL-2 proteins as it has a very short half-life, ~30 min (56, 65), and therefore requires active transcription and translation to maintain functional levels in the cell (66).
Abundant evidence supports the concept that MCL-1 represents an attractive therapeutic target in AML (59, 67, 68). High MCL-1 expression appears to play a critical role in the survival of leukemic stem cells in patients with AML, and deletion of MCL-1 resulted in the cell death of murine AML cells in mice (50, 69). Further, some patients with AML have shown a ≥2-fold increase of MCL-1 expression at relapse (70). While initially intuitive to inhibit MCL-1 directly, several MCL-1 inhibitors failed in early preclinical development because of their toxicity [the development of small molecule and peptide MCL-1 inhibitors over the past decade has been reviewed in multiple articles (71–74)]. An alternative approach is to indirectly inhibit the antiapoptotic function of MCL-1. Indirect targeting of MCL-1 by inhibition of specific CDK isoforms that regulate transcription, particularly CDK9 or CDK7, has shown promising preclinical and clinical outcomes (8, 29, 30).
CDK Inhibitors As Cancer Therapeutics
CDKs are a family of serine/threonine kinases discovered in yeast, classically shown to promote cell-cycle transitions via interaction with various fluctuating cyclins (75, 76). A plethora of evidence suggest that the classical cell cycle CDKs, such as CDK1 and CDK2, are targets for treating various malignancies driven by uncontrolled proliferation (77, 78). Interfering with CDK function by targeting CDK regulatory kinases upstream of CDK1/2, such as CHK1 (79) and WEE1 kinase (80), or their combination (81), has also shown proof-of-principle activity in AML (79), and other cancers (82).
The concept of targeting cell-cycle dysregulation as a therapeutic approach has been a major focus of cancer research for many decades. Research and development has been centered around multiple efforts with small molecules directed against CDKs, including for leukemia and MDS (83–85). Several CDK inhibitors have been investigated as cancer therapies (29), with approvals in recent years in solid tumors for palbociclib (86–89), ribociclib (90, 91), and abemaciclib (67, 92–94).
CDKs have other functions beyond direct cell-cycle regulation (Table 1) (66, 76, 114, 124–130). Additionally, CDKs that regulate gene transcription are of increasing interest as potential therapeutic targets in cancer (130–132).
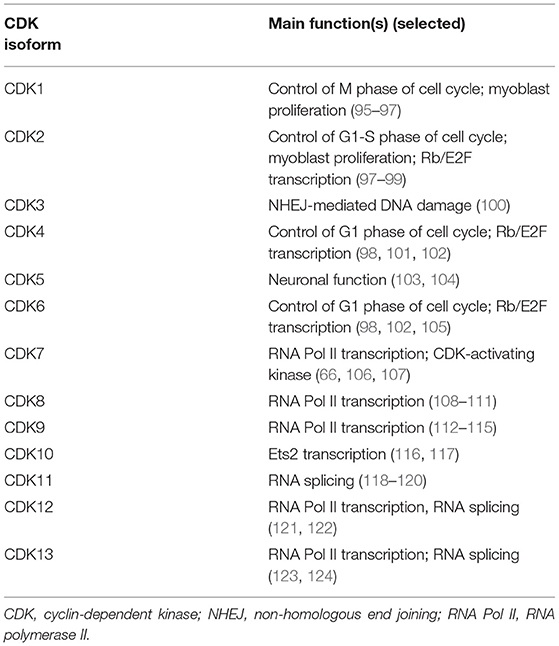
Table 1. Functions of CDK isoforms.
Transcriptional CDKs
As gene transcription has been found to be dysregulated in several cancers, including AML, transcription-associated CDKs are a natural target for cancer therapy. Inhibition of CDK7, CDK8, and CDK9 have been of particular interest in AML (59, 133, 134). These CDKs phosphorylate the carboxyl terminal domain (CTD) of RNA polymerase II (RNA Pol II), to facilitate the production of mature transcripts (128, 129, 135, 136). These transcriptional CDKs have been found to be dysregulated in AML (58, 137).
CDK8
CDK8 regulates the Mediator complex, a highly conserved multiprotein complex that functions as a coactivator of transcription (138, 139). Among its functions, the Mediator complex interacts with the pre-initiation complex consisting of RNA Pol II and general transcription factors, such as TFIIH to initiate the process of transcription (140). A CDK8 subcomplex, consisting of CDK8, cyclin C, and Mediator complex subunits Med12, and Med13, prevents re-initiation of transcription after RNA Pol II promoter clearance (i.e., transcription elongation) by binding to the Mediator complex in a manner mutually exclusive with RNA Pol II and independent of CDK8 kinase activity (140). CDK8 kinase activity is reported to both negatively and positively regulate transcription, which may be promoter-specific (141–143). For example, CDK8 phosphorylation of general transcription factors, such as TFIIH or transcription factor Notch, promotes disassembly of the pre-initiation complex to negatively regulate transcription (141, 142), while CDK8 phosphorylation of histone H3 facilitates cooperative histone H3 acetylation that activates transcription (143) by phosphorylating cyclin H (139, 140).
SEL120-34A is a novel, potent, and selective CDK8 inhibitor being evaluated preclinically (144). In AML cell lines, CDK8 inhibition with SEL120-34A was effective in cells expressing high levels of STAT5 and STAT1 (144). SEL120-34A also showed in vivo activity in mice with xenografted KG-1 and MV4-11 AML tumors with repression of oncogenic MCL-1 in MV4-11 tumors (144).
CDK7 and CDK9
RNA transcription is catalyzed by both CDK7 and CDK9 working in sequence (Figures 2A,B). CDK9 is the catalytic subunit of the positive transcription elongation factor b (P-TEFb) complex that, together with a regulatory subunit (cyclins T or K), phosphorylates the CTD of RNA Pol II (145). This process is essential to escape from abortive initiation and facilitate RNA Pol II elongation to generate mature mRNA/transcripts (113, 146–148). The CTD of RNA Pol II is first phosphorylated by CDK7 as part of the transcription factor IIH complex, allowing polymerase to initiate transcription (76, 125, 149). However, the CTD of RNA Pol II must be further phosphorylated by CDK9/P-TEFb for productive elongation of transcription to occur (113, 150). Application of CDK9 and CDK7 inhibitors modulate the activity of all RNA Pol II-regulated genes. There are a few preclinical studies evaluating CDK7 inhibition for AML; however, much more focus has been on inhibition of CDK9, as upregulation of CDK9 is associated with MCL-1 synthesis and treatment-resistance (58, 151).
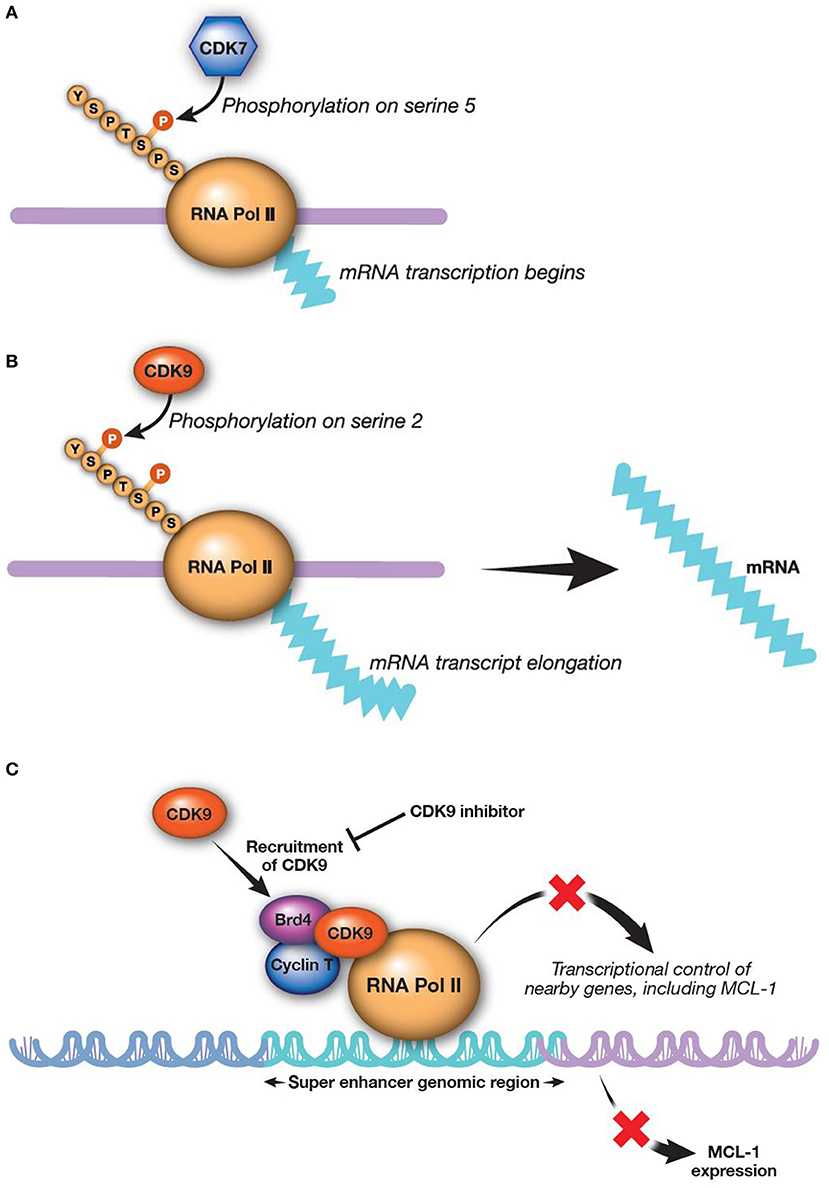
Figure 2. CDK9 and CDK7 work sequentially to produce mature transcripts. CDK9 is the catalytic subunit of the P-TEFb complex, which is essential in generating mature transcripts. (A) CDK7 phosphorylates the fifth serine on the carboxyl-terminal domain of RNA Pol II, thereby activating RNA Pol II to begin transcribing RNAa. (B) CDK9 then phosphorylates the second carboxyl-terminal serine to enable elongation of RNA transcripts (113)a. (C) Inhibition of CDK9 reduces MCL-1 expression (8). aReprinted from Morales and Giordano (113), with permission from Taylor & Francis. Brd4, bromodomain-containing protein 4; CDK, cyclin-dependent kinase; MCL-1, myeloid cell leukemia-1; P-TEFb, positive transcription elongation factor b; RNA Pol II, RNA polymerase II.
THZ1 is a CDK7 inhibitor currently in preclinical study for treatment of AML and peripheral T-cell lymphomas (152, 153). THZ1 prevents Pol II CTD phosphorylation, which disrupts pausing, capping, and elongation, thereby inhibiting transcription (154). In preclinical studies, THZ1 appeared to inhibit transcription factors, such as RUNX1, a key regulator of hematopoiesis that is involved in the common t(8, 21) translocation event in AML (152, 154). Further studies are needed to determine appropriate dosing for clinical trials.
ICEC0942 is an orally bioavailable CDK7 inhibitor also being evaluated in preclinical studies (155). Whereas, THZ1 has a covalent mechanism of action and may interact with other kinases, ICE0942 has a non-covalent mechanism of action, which could possibly limit side effects (154, 155). Oral administration of ICE0942 in mice showed favorable safety with no adverse effects on liver or kidney function (155).
SNS-032 is an inhibitor of CDK2, CDK7, and CDK9. In vitro studies of SNS-032 (BMS-387032) alone or in combination with cytarabine, induced AML cell cytotoxicity through inhibition of CDK2, CDK7, and CDK9 transcription, resulting in the attenuation of RNA Pol II-mediated transcription (156).
Alvocidib, previously described as flavopiridol, is a pan-CDK inhibitor structurally related to a natural alkaloid derived from Dysoxylum binectariferum, a plant indigenous to India (157). The initial clinical use of alvocidib was based on evidence for the compound as a cell-cycle modulator (157–160). However, further investigation revealed that alvocidib is more potent for inhibiting CDKs directly associated with transcription machinery, most prominently CDK9, to regulate RNA Pol II as described above. Down-regulation of transcription by CDK9 inhibition broadly and preferentially affects proteins with a short half-life, such as MCL-1 (161–163). Consistent with this evidence, alvocidib treatment has been shown to decrease MCL-1 levels in AML and chronic lymphocytic leukemia cells (Figure 2C) (30, 162, 164). Alvocidib induces apoptosis in many tumor cell lines, including those derived from lymphoma, MM, and AML (8). Despite exhibiting its own unique toxicities, alvocidib could speculatively have a more tractable therapeutic index owing to its transient and reversible reduction of MCL-1 via CDK9 inhibition, in contrast to other direct inhibitors of MCL-1. Synergistic therapeutic combinations harnessing lower doses of alvocidib than what may otherwise not be effective as monotherapy, are thus also speculatively attractive, as discussed further in the section Combination Therapy Utilizing CDK9 Inhibition.
Atuveciclib (BAY 1143572) is a potent, selective P-TEFb/CDK9 inhibitor (165). Early discovery efforts identified in vivo activity in both MOLM-13 and MV4-11 xenograft models of AML with a 14-day continuous oral course of atuveciclib at the maximum tolerated dose. Oral bioavailability was improved over the existing lead compound, and atuveciclib was ultimately selected for phase 1 clinical evaluation on the basis of the totality of its in vitro and in vivo activity (165). A phase 1 dose-escalation trial of atuveciclib in subjects with advanced leukemia has completed, but results have not yet been reported (NCT02345382).
Dinaciclib is a pan-CDK inhibitor that targets CDK9, but for which potency is stronger for other CDKs, namely CDK2 and CDK5 (166). Based on preclinical observations involving mostly AML cell lines, dinaciclib treatments resulted in MCL-1 downregulation (167). In vitro and in vivo antitumor activity of dinaciclib in mixed lineage leukemia fusion protein AML, regarded as a chemotherapy resistant and poor-prognosis subtype, lends further support to the functionality and therapeutic effects of the anti-CDK9 properties of dinaciclib (168).
LY2857785 is another pan-CDK inhibitor (including CDK7, CDK8, and CDK9) and also inhibits other kinases (130). As part of efforts to characterize the relative contributions of CDK9 vs. CDK7 inhibition, this study suggests that inhibition of cell proliferation was primarily mediated by CDK9. LY2857785 was shown to confer significant inhibition among all 24 tested hematologic cancer lines, with AML cell lines found to be most sensitive. Similarly, an in vivo model (MV4-11 xenograft) showed that tumor growth was significantly inhibited by LY2857785 (130).
TG02 (169–171) has exhibited an IC50 < 10 nM for CDK9 and CDKs 1, 2, 3, and 5 along with potent inhibition, albeit less pronounced relative to CDKs, against other targets that include JAK1, JAK2, and FLT3 (169). Using ex vivo expanded AML blast cells (N = 16), more potent inhibition of cell proliferation was demonstrated for TG02 relative to SNS-032 and the non-CDK-targeted FLT3 inhibitor sunitinib. CDK9 was identified as the most sensitive target of TG02, effecting RNA Pol II-mediated transcription at lower concentrations than those for SNS-032 while downregulation of MCL-1 was confirmed. Findings from MV4-11 and HL-60 AML models collectively support tumor growth inhibition for TG02-treated animals.
Voruciclib is an oral, selective inhibitor of CDK9. Preclinical data has shown that voruciclib in combination with BCL-2 inhibitor venetoclax enhances cell death in AML cell lines (172).
These preclinical studies have shown that targeted CDK9 inhibition results in down-regulation of MCL-1 and subsequent cell death in AML cell lines. Clinical trials have been initiated and further support CDK9 inhibition as a potentially effective treatment for patients with AML.
Clinical Development of CDK9 Inhibitors in AML
Alvocidib was the first CDK inhibitor to be tested in a clinical trial (159, 160). As of 2019, ~65 clinical trials had been conducted investigating alvocidib alone or in combination with other antineoplastic drugs. Of these trials, 31 assessed alvocidib in hematologic malignancies, including AML (173). While an overview of clinical trial data is beyond the scope of this review, it is important to note that several clinical trials have been conducted or are in progress with the purpose of determining the optimal use of alvocidib in AML. For example, a randomized phase 2 multicenter study, which compared ACM/FLAM to 7 + 3 in newly diagnosed adult AML patients with intermediate- and adverse-risk cytogenetics, reported the activity of alvocidib when it was used as timed sequential therapy in combination with cytarabine and mitoxantrone (3). The ACM/FLAM regimen led to higher complete response rates compared with patients who received either 7 + 3 alone (46%; P = 0.003) or 7 + 3 followed by an additional dose of cytarabine on days 1–5 and daunorubicin on days 1–2 (5 + 2; 57%; P = 0.08), and without an increase in toxicity; however, relapse and overall survival rates remained similar (3). Final results of the study data (median follow-up, 1,644 days) confirmed the similar overall survival and higher CR rates in the ACM/FLAM treatment arm compared with the 7 + 3 arm (174). Phase 1 trials of atuveciclib in AML and other acute leukemias (NCT02345382) and TG02 in AML and other advanced hematologic malignancies (NCT01204164) have been completed; however, results have not yet been published. A phase 1 trial of AZD4573 [a CDK9 inhibitor for which preclinical data have been presented (175, 176) but have not yet been published] in relapsed or refractory AML and other hematologic malignancies (NCT03263637) was initiated in October 2017. A phase 1 trial of voruciclib is currently recruiting patients with B-cell malignancies or AML (NCT03547115).
Combination Therapy Utilizing CDK9 Inhibition
Experimental evidence suggests that malignant cells, including leukemias, may be dependent on one or more of the multiple antiapoptotic proteins for survival (64, 177, 178). For example, while some cancer cells may depend on MCL-1 for survival, others may depend on BCL-2 or BCL-XL (177). Analysis of BCL-2, BCL-XL, and MCL-1 protein levels in primary AML samples (N = 577) showed that MCL-1, BCL-2, and BCL-XL exhibited variable protein expression levels, within and across differentiation stages (French-American-British [FAB] classification subgroups), as well as across different cytogenetic and molecular defined subgroups (178). This indicates heterogeneity of antiapoptotic protein expression in AML (64) and supports the concept of dual or triple targeting of BCL-2 family proteins in AML and myeloid malignancies. Conceivably, clonal heterogeneity with regard to antiapoptotic protein expression/dependence may be also observed within a single patient and could promote resistance to therapy. However, for all BCL-2 family members, there is also substantial interpatient variability. Agents that target MCL-1 may have no effect on cells dependent on BCL-2 for survival and vice versa. Thus, combination therapies based on dual-targeting of BCL-2 family members may be attractive strategies for treating AML.
With regard to BCL-2 family inhibition, combination strategies including CDK9 inhibitors aim to overcome MCL-1–dependent drug resistance via transcriptional silencing of MCL-1. CDK9 inhibitors can also overcome intrinsic apoptotic resistance via induction of pro-apoptotic BH3-only proteins, such as BIM, possibly occurring through transcriptional down-regulation of miRNAs that negatively regulate these pro-apoptotic BH3-only proteins (30, 161, 162, 179). Considering these premises, potential synergistic partners for CDK9 inhibitors include small molecules known to inhibit BCL-2 family proteins, such as navitoclax (ABT-263; AbbVie, Inc., North Chicago, IL, USA) and venetoclax (30, 62, 172). Pharmaceutical targeting of BCL-2 alone with venetoclax or BCL-XL/BCL-2 combined with navitoclax is active in pre clinical models of AML (177) but only modestly active in the clinic (180). Activity of BCL-2 and/or BCL-2/BCL-XL inhibition can be further enhanced by knocking down MCL-1 (45, 177, 181–183). Combination treatment of voruciclib and venetoclax showed synergistic activity in inducing apoptosis in both venetoclax-sensitive and venetoclax-resistant AML cell lines (172). Concurrent treatment with alvocidib plus venetoclax results in reductions of the half-maximal activity (EC50) values for venetoclax in both venetoclax-sensitive and -resistant AML cell lines, and also results in synergy in primary AML samples treated with the combination ex vivo, as well as in a mouse xenograft model of AML. The combination of alvocidib and venetoclax was largely dependent on BIM, and thus the combination is mechanistically founded in the intrinsic apoptotic pathway (30). Based on this work, a phase 1b clinical trial to evaluate combined alvocidib and venetoclax in AML was initiated in May 2018 and is in progress (NCT03441555). The combination of dinaciclib and venetoclax is also being actively evaluated in a phase 1b trial in patients with relapsed or refractory AML (NCT03484520), which was initiated in July 2018.
Bromodomain and extra-terminal (BET) family proteins, such as BRD4 interact with cyclin T1 and CDK9 to positively regulate P-TEFb [(183–186); Figure 2C]. Thus, BET inhibitors are emerging as a new, additional therapeutic class for alternatively targeting P-TEFb. Several BET inhibitors, such as CPI-0610 (Constellation Pharmaceuticals, Cambridge, MA, USA) (NCT02158858; AML and MDS), MK-8628 (Merck and Co., Inc., Whitehouse Station, NJ, USA) (NCT02698189; AML, MDS, and diffuse large B-cell lymphoma), and R06870810 (Roche, Basel, Switzerland) (NCT02308761; AML and MDS) are being tested in early clinical trials in AML and other hematologic malignancies, including myelodysplastic syndromes. While BET inhibitors are being investigated as monotherapy, their use in combination with CDK9 inhibitors may show even more promising outcomes for treatment of AML. For example, BET inhibitor BI 894999 has shown activity in AML cell lines, primary patient samples, and xenograft models as a monotherapy and in combination with CDK9 inhibitors LDC000067 and alvocidib (186). Combined BET and CDK9 inhibition results in rapid induction of apoptosis in vivo and in cultured cells, perhaps due to a global arrest of transcriptional elongation (186). A phase 1 dose-finding clinical study of BI 894999 monotherapy in solid tumors has been initiated (NCT02516553). It will be interesting to compare the therapeutic window and efficacy between CDK9 and BET inhibitors in AML, as well as the potential for combined BET and CDK9 inhibition.
The combination of a CDK9 inhibitor with an immune checkpoint inhibitor is another intriguing approach for the treatment of AML. Programmed cell death protein 1 receptor (PD-1) is expressed in T-cell subpopulations, and increased PD-1 expression may lead to inhibition of the immune response as AML progresses (187). In murine syngeneic tumor models, use of dinaciclib increased immunogeneic properties of tumor cells, including early expression of type I interferon response genes, and increased expression of PD-1. When dinaciclib was combined with an anti-PD-1 antibody, T-cell and APC activation within the tumor was increased, and antitumor efficacy was improved as compared with either monotherapy (188). The combination of anti-PD-1 (pembrolizumab) and dinaciclib is currently being evaluated in a phase 1 trial in patients with hematologic malignancies (KEYNOTE-155, NCT02684617).
Proteomic And BH3 Profiling As Putative Biomarker Approaches
In addition to an expanded portfolio of potential treatment options, patients may benefit from proteomic profiling, which may predict response to certain treatments. For example, proteomic profiling of several proteins involved in apoptosis, differentiation, and signal transduction identified clusters that correlated with AML FAB subtypes and cytogenetic defined subgroups. In this large proteomic dataset, differences in BH3-mimetic and BCL-2 protein expression were found in FAB and cytogenetic subgroups, supporting a preferential usage of different molecules within the intrinsic apoptotic pathway in subsets of AML (178). Proteomic profiling may also reveal prognostic subgroups that have not been uncovered through transcriptomic methods. For instance, a study utilizing a reverse-phase protein array (RPPA) found that increased chromosomal region maintenance 1 (CRM1) expression in AML is associated with poor survival. Further, inhibition of CRM1 induces apoptosis in AML cells in a p53-dependent manner (189) (see Figure 1). Protein signatures could therefore predict survival and may be useful in directing therapy, including for BCL-2 targeting drugs and combinations. RPPA methods may also provide greater accuracy into protein expression levels and post-translational modifications in cancer cells, which can lead to novel therapeutics (190). In conjunction with genomic and transcriptomic analysis, proteomics can identify commonalities as well as novel networks in cancer biology (190). However, there are certain challenges in implementing RPPA into clinical practice including a centralized database, high-quality patient samples, and establishment of best practices (191).
BH3 profiling is another approach in predicting response of novel therapeutics in AML cells. BH3 profiling is a functional assay for assessing the apoptotic capacity of a cell. This assay specifically determines how readily cells will undergo apoptosis, i.e., whether the cells are primed for apoptosis, as well as which antiapoptotic BCL-2 family proteins may be most important for survival (192–194). Additionally, BH3 profiling of AML cell lines can indicate possible cooperativity of agents targeting different molecules in the apoptotic process, suggesting that BH3 profiling may function as a potential tool to assess combination therapies in AML (171). Using BH3 profiling, Bhola et al. found that AML and other cancer types are highly heterogeneous in regard to apoptotic priming (192). In primary AML cells and AML xenograft models, BH3 profiling showed a strong correlation between the level of apoptotic priming and the relative sensitivity of the cells to the BCL-2 inhibitor venetoclax (39).
BH3 profiling also has the potential to distinguish clinical responses to azacitidine in AML (64). Vo et al. used BH3 profiling to predict responses to targeted BCL-2 inhibition, as well as to conventional chemotherapy in AML (40). In this study, a subset of patients with low-primed samples displayed a higher risk of relapse. In addition, retrospective studies profiling the BCL-2 family proteins (BCL-2, BCL-XL, and MCL-1) identified potential biomarkers of clinical response to venetoclax (180). As the HRK peptide has the highest affinity for BCL-XL and denotes BCL-XL dependency and the BAD peptide denotes BCL-2 and/or BCL-XL dependency, a BAD-HRK mathematical derivation is considered a rough metric for BCL-2 dependency. It was shown that dependence on BCL-XL and MCL-1 correlated with a decrease in response after short-term therapy. Addition of the HRK peptide to BH3 profiling as a post-hoc metric predicted that longer-term therapy could be used without development of resistance (180).
NOXA is a pro-apoptotic BH3-only protein that exhibits the greatest affinity for MCL-1 and is thought to specifically bind to and disrupt MCL-1 function in vivo (Figure 1) (36, 59). Thus, due to NOXA selectivity for MCL-1, NOXA peptides are thought to be a specific read-out of MCL-1 dependency. Thus, NOXA as a BH3 profiling metric may be an ideal biomarker for CDK9 inhibitors given strong mechanistic links to MCL-1. Mitochondrial profiling in patients with AML treated with alvocidib followed by cytarabine and mitoxantrone (ACM, previously FLAM [ACM/FLAM]) revealed that patients who experienced complete response had higher NOXA priming in bone marrow samples than non-responding patients (164, 195). This rationale has also been used in a study of patients receiving combined vorinostat and gemtuzumab ozogamicin therapy. Responders displayed significantly higher MCL-1 dependence (observed as higher NOXA priming) than non-responders (P = 0.027), with a high correlation also seen between MCL-1 dependence and overall survival (P = 0.026 per logistic regression) (196). It is unknown which BH3 profiling metrics may be best for predicting response to combination therapies, such as BCL-2 combined with CDK9 inhibition.
It is important to note that, while BH3 profiling has demonstrated clinical utility, a lack of standardized protocols may limit broader clinical applicability. The use of different platforms, as well as the use of either whole-cell or mitochondrial preparations, may lead to different conclusions (197). Difficulty in determining the optimal peptide concentration for profiling highlights further need for standardization (198). Finally, existing BH3 profiling techniques are labor-intensive and require a large volume of starting tissue, which increases error and decreases clinical applicability (199). Despite these limitations, further refinement of BH3 profiling is promising for precision medicine-based treatment.
Conclusion
Inhibitors of transcriptional CDKs are potential novel therapies for AML. While preclinical studies are currently investigating CDK7 and CDK8 inhibitors, there is abundant preclinical and clinical evidence to support targeting CDK9 whose inhibition downregulates, amongst others, the important anti-apoptotic protein MCL-1. CDK9 inhibitors represent a novel therapeutic class of small molecules for indirectly inhibiting MCL-1. Thus, over-expression of MCL-1 could be a predictive biomarker for treatment response to CDK9 inhibition. BH3 profiling, measuring specific priming for MCL-1 and other BCL-2 family proteins, also has the potential to serve as a biomarker for identifying/selecting patients who could favorably respond to CDK9 inhibition. CDK9 inhibitors have the potential to be combined with other agents in AML, and results are awaited from several completed and ongoing clinical trials.
Author Contributions
RT and JMB participated in designing the concept of this manuscript, reviewed the literature, and drafted the article.
Funding
Medical writing and editorial support were provided by Adelphi Communications and Ashfield Healthcare Communications, part of UDG Healthcare plc, and were funded by Boston Biomedical, Inc. Neither of the named authors received any compensation for their contributions to this work.
Conflict of Interest
RT and JMB report medical writing support for this publication. RT and JMB have published work on alvocidib that is being used by pharmaceutical companies designing clinical trials and did not receive any personal or research compensation for their work.
References
1. American Cancer Society. Cancer Facts & Figures 2018. Atlanta, GA: American Cancer Society (2018). Available online at: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2018.html (accessed June 6, 2019).
2. Short NJ, Rytting ME, Cortes JE. Acute myeloid leukaemia. Lancet. (2018) 392:593–606. doi: 10.1016/S0140-6736(18)31041-9
3. Zeidner JF, Foster MC, Blackford AL, Litzow MR, Morris LE, Strickland SA, et al. Randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine, and mitoxantrone (FLAM) versus cytarabine/daunorubicin (7 + 3) in newly diagnosed acute myeloid leukemia. Haematologica. (2015) 100:1172–9. doi: 10.3324/haematol.2015.125849
4. Kadia TM, Ravandi F, Cortes J, Kantarjian H. New drugs in acute myeloid leukemia. Ann Oncol. (2016) 27:770–8. doi: 10.1093/annonc/mdw015
5. U.S. Food and Drug Administration. FDA Approves Liposome-Encapsulated Combination of Daunorubicin-Cytarabine for Adults With Some Types of Poor Prognosis AML. (2017). Available online at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-liposome-encapsulated-combination-daunorubicin-cytarabine-adults-some-types-poor (accessed June 5, 2019).
6. Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. (2018) 36:2684–92. doi: 10.1200/JCO.2017.77.6112
7. DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. (2019) 133:7–17. doi: 10.1182/blood-2018-08-868752
8. Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res. (2015) 39:1312–8. doi: 10.1016/j.leukres.2015.10.010
9. U.S. Food and Drug Administration. Orphan Drug Designations and Approvals. Available online at: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm (accessed June 6, 2019).
10. U.S. Food and Drug Administration. FDA Granted Regular Approval to Enasidenib for the Treatment of Relapsed or Refractory AML. (2017). Available online at: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm569482.htm (accessed June 6, 2019).
11. U.S. Food and Drug Administration. FDA Approves Liposome-Encapsulated Combination of Daunorubicin-Cytarabine for Adults With Some Types of Poor Prognosis AML. (2017). Available online at: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm569950.htm (accessed June 6, 2019).
12. U.S. Food and Drug Administration. Midostaurin. (2017). Available online at: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555756.htm (accessed June 6, 2019).
13. U.S. Food and Drug Administration. FDA Approves Mylotarg for Treatment of Acute Myeloid Leukemia. (2017). Available online at: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm574507.htm (accessed June 6, 2019).
14. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. (2017) 130:722–31. doi: 10.1182/blood-2017-04-779405
15. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. (2017) 377:454–64. doi: 10.1056/NEJMoa1614359
16. Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie JN, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. (2012) 379:1508–16. doi: 10.1016/S0140-6736(12)60485-1
17. U.S. Food and Drug Administration. FDA Approves Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia. (2018). Available online at: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm614128.htm (accessed June 6, 2019).
18. U.S. Food and Drug Administration. FDA Approves Venetoclax in Combination for AML in Adults. (2018). Available online at: https://www.fda.gov/drugs/fda-approves-venetoclax-combination-aml-adults (accessed June 6, 2019).
19. U.S. Food and Drug Administration. FDA Approves Glasdegib for AML in Adults Age 75 or Older or Who Have Comorbidities. (2018). Available online at: https://www.fda.gov/drugs/fda-approves-glasdegib-aml-adults-age-75-or-older-or-who-have-comorbidities (accessed June 6, 2019).
20. U.S. Food and Drug Administration. FDA Approves Gilteritinib for Relapsed or Refractory Acute Myeloid Leukemia (AML) With a FLT3 Mutatation. (2018). Available online at: https://www.fda.gov/drugs/fda-approves-gilteritinib-relapsed-or-refractory-acute-myeloid-leukemia-aml-flt3-mutatation (accessed June 6, 2019).
21. Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. (2017) 18:1061–75. doi: 10.1016/S1470-2045(17)30416-3
22. Cortes JE, Douglas Smith B, Wang ES, Merchant A, Oehler VG, Arellano M, et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: phase 2 study results. Am J Hematol. (2018) 93:1301–10. doi: 10.1002/ajh.25238
23. Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. (2019) 33:379. doi: 10.1038/s41375-018-0312-9
24. DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. (2018) 19:216–28. doi: 10.1016/S1470-2045(18)30010-X
25. Wei A, Strickland SA, Roboz GJ, Hou JZ, Fiedler W, Lin TL, et al. Safety and efficacy of venetoclax plus low-dose cytarabine in treatment-naive patients aged ≥65 years with acute myeloid leukemia. Blood. (2016) 128:102. doi: 10.1182/blood.V128.22.102.102
26. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. (2018) 378:2386–98. doi: 10.1056/NEJMoa1716984
27. Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to Venetoclax treatment. Cancer Discov. (2019) 9:890–909. doi: 10.1158/2159-8290.CD-19-0117
28. Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D'Alessandro A, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML Cells. Cancer Discov. (2019) 9:910–25. doi: 10.1158/2159-8290.CD-19-0125
29. Aleem E, Arceci RJ. Targeting cell cycle regulators in hematologic malignancies. Front Cell Dev Biol. (2015) 3:16. doi: 10.3389/fcell.2015.00016
30. Bogenberger J, Whatcott C, Hansen N, Delman D, Shi CX, Kim W, et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget. (2017) 8:107206–22. doi: 10.18632/oncotarget.22284
31. Phillips DC, Xiao Y, Lam LT, Litvinovich E, Roberts-Rapp L, Souers AJ, et al. Loss in MCL-1 function sensitizes non-Hodgkin's lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J. (2016) 6:e403. doi: 10.1038/bcj.2016.12
32. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. (2015) 373:1136–52. doi: 10.1056/NEJMra1406184
33. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
34. Shafer D, Grant S. Update on rational targeted therapy in AML. Blood Rev. (2016) 30:275–83. doi: 10.1016/j.blre.2016.02.001
35. Pierceall WE, Kornblau SM, Carlson NE, Huang X, Blake N, Lena R, et al. BH3 profiling discriminates response to cytarabine-based treatment of acute myelogenous leukemia. Mol Cancer Ther. (2013) 12:2940–9. doi: 10.1158/1535-7163.MCT-13-0692
36. Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: orchestrators of apoptosis. Biochim Biophys Acta. (2011) 1813:508–20. doi: 10.1016/j.bbamcr.2010.11.024
37. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
38. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
39. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. (2014) 4:362–75. doi: 10.1158/2159-8290.CD-13-0609
40. Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. (2012) 151:344–55. doi: 10.1016/j.cell.2012.08.038
41. Fiskus W, Sharma S, Qi J, Valenta JA, Schaub LJ, Shah B, et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther. (2014) 13:1142–54. doi: 10.1158/1535-7163.MCT-13-0770
42. LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. (2012) 122:2018–31. doi: 10.1172/JCI46231
43. Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, et al. Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. (2016) 22:4440–51. doi: 10.1158/1078-0432.CCR-15-3057
44. Rahmani M, Aust MM, Attkisson E, Williams DC Jr., Ferreira-Gonzalez A, Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. (2013) 73:1340–51. doi: 10.1158/0008-5472.CAN-12-1365
45. Zhang W, Ruvolo VR, Gao C, Zhou L, Bornmann W, Tsao T, et al. Evaluation of apoptosis induction by concomitant inhibition of MEK, mTOR, and Bcl-2 in human acute myelogenous leukemia cells. Mol Cancer Ther. (2014) 13:1848–59. doi: 10.1158/1535-7163.MCT-13-0576
46. Shukla S, Saxena S, Singh BK, Kakkar P. BH3-only protein BIM: an emerging target in chemotherapy. Eur J Cell Biol. (2017) 96:728–38. doi: 10.1016/j.ejcb.2017.09.002
47. Wenzel SS, Grau M, Mavis C, Hailfinger S, Wolf A, Madle H, et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia. (2013) 27:1381. doi: 10.1038/leu.2012.367
48. Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. (2005) 19:1248–52. doi: 10.1038/sj.leu.2403784
49. Awan FT, Kay NE, Davis ME, Wu W, Geyer SM, Leung N, et al. Mcl-1 expression predicts progression-free survival in chronic lymphocytic leukemia patients treated with pentostatin, cyclophosphamide, and rituximab. Blood. (2009) 113:535–7. doi: 10.1182/blood-2008-08-173450
50. Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. (2012) 26:120–5. doi: 10.1101/gad.182980.111
51. Xiang Z, Luo H, Payton JE, Cain J, Ley TJ, Opferman JT, et al. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest. (2010) 120:2109–18. doi: 10.1172/JCI39964
52. Cassier PA, Castets M, Belhabri A, Vey N. Targeting apoptosis in acute myeloid leukaemia. Br J Cancer. (2017) 117:1089–98. doi: 10.1038/bjc.2017.281
53. Jilg S, Reidel V, Müller-Thomas C, Konig J, Schauwecker J, Hockndorf U, et al. Blockade of BCL-2 proteins efficiently induces apoptosis in progenitor cells of high-risk myelodysplastic syndromes patients. Leukemia. (2016) 30:112–23. doi: 10.1038/leu.2015.179
54. Reidel V, Kauschinger J, Hauch RT, Muller-Thomas C, Nadarajah N, Burgkart R, et al. Selective inhibition of BCL-2 is a promising target in patients with high-risk myelodysplastic syndromes and adverse mutational profile. Oncotarget. (2018) 9:17270–81. doi: 10.18632/oncotarget.24775
55. Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nat Rev Cancer. (2013) 13:455–65. doi: 10.1038/nrc3538
56. Wei G, Margolin AA, Haery L, Brown E, Cucolo L, Julian B, et al. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell. (2012) 21:547–62. doi: 10.1016/j.ccr.2012.02.028
57. Wei SH, Dong K, Lin F, Wang X, Li B, Shen JJ, et al. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. (2008) 62:1055–64. doi: 10.1007/s00280-008-0697-7
58. Yeh YY, Chen R, Hessler J, Mahoney E, Lehman AM, Heerema NA, et al. Up-regulation of CDK9 kinase activity and Mcl-1 stability contributes to the acquired resistance to cyclin-dependent kinase inhibitors in leukemia. Oncotarget. (2015) 6:2667–79. doi: 10.18632/oncotarget.2096
59. Bose P, Grant S. Mcl-1 as a therapeutic target in acute myelogenous leukemia (AML). Leuk Res Rep. (2013) 2:12–4. doi: 10.1016/j.lrr.2012.11.006
60. Luedtke DA, Niu X, Pan Y, Zhao J, Liu S, Edwards H, et al. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct Target Ther. (2017) 2:17012. doi: 10.1038/sigtrans.2017.12
61. Teh T-C, Nguyen N-Y, Glaser S, Moujalled D, Pomilio G, Huang DCS, et al. Eradication of acute myeloid leukemia is enhanced by combined Bcl-2 and Mcl-1 targeting. Blood. (2014) 124:988. doi: 10.1182/blood.V124.21.988.988
62. Teh TC, Nguyen NY, Moujalled DM, Segal D, Pomilio G, Rijal S, et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia. (2018) 32:303–12. doi: 10.1038/leu.2017.243
63. Zhang Q, Han L, Pan R, Shi C, Ma H, Leverson J, et al. Synergistic induction of apoptotic cell death in AML by targeting Bcl-2 with ABT-199 in combination with Bcl-Xl/Mcl-1 inhibition or selective kinase inhibitors. Clin Lymphoma Myeloma Leuk. (2015) 15:S16. doi: 10.1016/j.clml.2015.07.037
64. Bogenberger JM, Kornblau SM, Pierceall WE, Lena R, Chow D, Shi CX, et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia. (2014) 28:1657–65. doi: 10.1038/leu.2014.44
65. Adams KW, Cooper GM. Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J Biol Chem. (2007) 282:6192–200. doi: 10.1074/jbc.M610643200
66. Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. (2015) 163:174–86. doi: 10.1016/j.cell.2015.08.063
67. Bose P, Grant S. Orphan drug designation for pracinostat, volasertib and alvocidib in AML. Leuk Res. (2014) 38:862–5. doi: 10.1016/j.leukres.2014.06.007
68. Bose P, Simmons GL, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Invest Drugs. (2013) 22:723–38. doi: 10.1517/13543784.2013.789859
69. Yoshimoto G, Miyamoto T, Jabbarzadeh-Tabrizi S, Iino T, Rocnik JL, Kikushige Y, et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood. (2009) 114:5034–43. doi: 10.1182/blood-2008-12-196055
70. Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, et al. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. (1998) 91:991–1000. doi: 10.1182/blood.V91.3.991
71. Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. (2013) 12:1691–700. doi: 10.1158/1535-7163.MCT-13-0058
72. Chen L, Fletcher S. Mcl-1 inhibitors: a patent review. Expert Opin Ther Pat. (2017) 27:163–78. doi: 10.1080/13543776.2017.1249848
73. Wan Y, Dai N, Tang Z, Fang H. Small-molecule Mcl-1 inhibitors: emerging anti-tumor agents. Eur J Med Chem. (2018) 146:471–82. doi: 10.1016/j.ejmech.2018.01.076
74. Zou J, Ardecky R, Pinkerton AB, Sergienko E, Su Y, Stonich D, et al. Selective Bcl-2 inhibitor probes. In: Probe Reports From the NIH Molecular Libraries Program. Bethesda, MD: National Center for Biotechnology Information (US) (2010). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK133437/ (accessed October 31, 2019).
75. Nurse P, Thuriaux P. Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. Genetics. (1980) 96:627–37.
76. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. (2013) 140:3079–93. doi: 10.1242/dev.091744
77. Blachly JS, Byrd JC. Emerging drug profile: cyclin-dependent kinase inhibitors. Leuk Lymphoma. (2013) 54:2133–43. doi: 10.3109/10428194.2013.783911
78. Bai J, Li Y, Zhang G. Cell cycle regulation and anticancer drug discovery. Cancer Biol Med. (2017) 14:348–62. doi: 10.20892/j.issn.2095-3941.2017.0033
79. Webster JA, Tibes R, Morris L, Blackford AL, Litzow M, Patnaik M, et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk Res. (2017) 61:108–16. doi: 10.1016/j.leukres.2017.09.005
80. Tibes R, Bogenberger JM, Chaudhuri L, Hagelstrom RT, Chow D, Buechel ME, et al. RNAi screening of the kinome with cytarabine in leukemias. Blood. (2012) 119:2863–72. doi: 10.1182/blood-2011-07-367557
81. Chaudhuri L, Vincelette ND, Koh BD, Naylor RM, Flatten KS, Peterson KL, et al. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica. (2014) 99:688–96. doi: 10.3324/haematol.2013.093187
82. Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. (2016) 34:4371–80. doi: 10.1200/JCO.2016.67.5991
83. Tibes R, McDonagh KT, Lekakis L, Bogenberger JM, Kim S, Frazer N, et al. Phase I study of the novel Cdc2/CDK1 and AKT inhibitor terameprocol in patients with advanced leukemias. Invest New Drugs. (2015) 33:389–96. doi: 10.1007/s10637-014-0198-y
84. Xie S, Jiang H, Zhai XW, Wei F, Wang SD, Ding J, et al. Antitumor action of CDK inhibitor LS-007 as a single agent and in combination with ABT-199 against human acute leukemia cells. Acta Pharmacol Sinica. (2016) 37:1481–9. doi: 10.1038/aps.2016.49
85. Yang C, Boyson CA, Di Liberto M, Huang X, Hannah J, Dorn CD, et al. CDK4/6 inhibitor PD 0332991 sensitizes acute myeloid leukemia to cytarabine-mediated cytotoxicity. Cancer Res. (2015) 75:1838–45. doi: 10.1158/0008-5472.CAN-14-2486
86. U.S. Food and Drug Administration. Palbociclib (IBRANCE Capsules). (2016). Available online at: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm487080.htm (accessed June 6, 2019).
87. U.S. Food and Drug Administration. Palbociclib (IBRANCE). (2017). Available online at: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm549978.htm (accessed June 6, 2018).
88. Loibl S, Turner NC, Ro J, Cristofanilli M, Iwata H, Im SA, et al. Palbociclib combined with fulvestrant in premenopausal women with advanced breast cancer and prior progression on endocrine therapy: PALOMA-3 results. Oncologist. (2017) 22:1028–38. doi: 10.1634/theoncologist.2017-0072
89. Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. (2016) 375:1925–36. doi: 10.1056/NEJMoa1607303
90. U.S. Food and Drug Administration. Ribociclib (Kisqali). (2017). Available online at: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm (accessed June 6, 2019).
91. Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol. (2018) 29:1541–7. doi: 10.1093/annonc/mdy155
92. U.S. Food and Drug Administration. FDA Approves Abemaciclib for HR-Positive, HER2-Negative Breast Cancer. (2017). Available online at: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm578081.htm (accessed June 6, 2019).
93. Dickler MN, Tolaney SM, Rugo HS, Cortés J, Diéras V, Patt D, Wildiers H, Hudis CA, O'Shaughnessy J, Zamora E, Yardley DA. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR+/HER2− metastatic breast cancer. Clin Cancer Res. (2017) 23:5218–24. doi: 10.1158/1078-0432.CCR-17-0754
94. Sledge GW Jr., Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2– advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. (2017) 35:2875–84. doi: 10.1200/JCO.2017.73.7585
95. Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. (2002) 9:1287–93. doi: 10.1038/sj.cdd.4401130
96. Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div. (2010) 5:11. doi: 10.1186/1747-1028-5-11
97. Kitzmann M, Vandromme M, Schaeffer V, Carnac G, Labbe JC, Lamb N, et al. Cdk1- and cdk2-mediated phosphorylation of MyoD Ser200 in growing C2 myoblasts: role in modulating MyoD half-life and myogenic activity. Mol Cell Biol. (1999) 19:3167–76. doi: 10.1128/MCB.19.4.3167
98. Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. (2013) 14:518–28. doi: 10.1038/nrm3629
99. Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem. (2000) 275:40235–43. doi: 10.1074/jbc.M004356200
100. Tomashevski A, Webster DR, Grammas P, Gorospe M, Kruman II. Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ. (2010) 17:1189–98. doi: 10.1038/cdd.2009.221
101. Chen H, Xu X, Wang G, Zhang B, Wang G, Xin G, et al. CDK4 protein is degraded by anaphase-promoting complex/cyclosome in mitosis and reaccumulates in early G1 phase to initiate a new cell cycle in HeLa cells. J Biol Chem. (2017) 292:10131–41. doi: 10.1074/jbc.M116.773226
102. Donjerkovic D, Scott DW. Regulation of the G1 phase of the mammalian cell cycle. Cell Res. (2000) 10:1–16. doi: 10.1038/sj.cr.7290031
103. Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. (2006) 50:13–8. doi: 10.1016/j.neuron.2006.02.024
104. Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. (2012) 22:169–75. doi: 10.1016/j.tcb.2011.11.003
105. Foster DA, Yellen P, Xu L, Saqcena M. Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer. (2010) 1:1124–31. doi: 10.1177/1947601910392989
106. Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. (2005) 118:5171–80. doi: 10.1242/jcs.02718
107. Coin F, Egly JM. Revisiting the function of CDK7 in transcription by virtue of a recently described TFIIH kinase inhibitor. Mol Cell. (2015) 59:513–4. doi: 10.1016/j.molcel.2015.08.006
108. Elmlund H, Baraznenok V, Lindahl M, Samuelsen CO, Koeck PJ, Holmberg S, et al. The cyclin-dependent kinase 8 module sterically blocks Mediator interactions with RNA polymerase II. Proc Natl Acad Sci USA. (2006) 103:15788–93. doi: 10.1073/pnas.0607483103
109. Jeronimo C, Robert F. The mediator complex: at the nexus of RNA polymerase II transcription. Trends Cell Biol. (2017) 27:765–83. doi: 10.1016/j.tcb.2017.07.001
110. Rzymski T, Mikula M, Wiklik K, Brzozka K. CDK8 kinase-An emerging target in targeted cancer therapy. Biochim Biophys Acta. (2015) 1854:1617–29. doi: 10.1016/j.bbapap.2015.05.011
111. Tsai KL, Sato S, Tomomori-Sato C, Conaway RC, Conaway JW, Asturias FJ. A conserved mediator-CDK8 kinase module association regulates mediator-RNA polymerase II interaction. Nat Struct Mol Biol. (2013) 20:611–9. doi: 10.1038/nsmb.2549
112. Napolitano G, Majello B, Lania L. Role of cyclinT/Cdk9 complex in basal and regulated transcription. Int J Oncol. (2002) 21:171–7. doi: 10.3892/ijo.21.1.171
113. Morales F, Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. (2016) 15:519–27. doi: 10.1080/15384101.2016.1138186
114. Franco LC, Morales F, Boffo S, Giordano A. CDK9: a key player in cancer and other diseases. J Cell Biochem. (2018) 119:1273–84. doi: 10.1002/jcb.26293
115. Cress WD. A stitch in time and CDK9. Cell Cycle. (2017) 16:823–4. doi: 10.1080/15384101.2017.1308150
116. Bagella L, Giacinti C, Simone C, Giordano A. Identification of murine cdk10: association with Ets2 transcription factor and effects on the cell cycle. J Cell Biochem. (2006) 99:978–85. doi: 10.1002/jcb.20981
117. Kasten M, Giordano A. Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulates its transactivation activity. Oncogene. (2001) 20:1832–8. doi: 10.1038/sj.onc.1204295
118. Choi HH, Choi HK, Jung SY, Hyle J, Kim BJ, Yoon K, et al. CHK2 kinase promotes pre-mRNA splicing via phosphorylating CDK11(p110). Oncogene. (2014) 33:108–15. doi: 10.1038/onc.2012.535
119. Hu D, Mayeda A, Trembley JH, Lahti JM, Kidd VJ. CDK11 complexes promote pre-mRNA splicing. J Biol Chem. (2003) 278:8623–9. doi: 10.1074/jbc.M210057200
120. Loyer P, Trembley JH, Grenet JA, Busson A, Corlu A, Zhao W, et al. Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: influence of cyclin L isoforms on splice site selection. J Biol Chem. (2008) 283:7721–32. doi: 10.1074/jbc.M708188200
121. Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. (2010) 24:2303–16. doi: 10.1101/gad.1968210
122. Paculova H, Kohoutek J. The emerging roles of CDK12 in tumorigenesis. Cell Div. (2017) 12:7. doi: 10.1186/s13008-017-0033-x
123. Even Y, Escande ML, Fayet C, Genevière AM. CDK13, a kinase involved in pre-mRNA splicing, is a component of the perinucleolar compartment. PLoS ONE. 11:e0149184. doi: 10.1371/journal.pone.0149184
124. Greifenberg AK, Hönig D, Pilarova K, Duster R, Bartholomeeusen K, Bosken CA, et al. Structural and functional analysis of the Cdk13/cyclin K complex. Cell Rep. (2016) 14:320–31. doi: 10.1016/j.celrep.2015.12.025
125. Paparidis NF, Durvale MC, Canduri F. The emerging picture of CDK9/P-TEFb: more than 20 years of advances since PITALRE. Mol Biosyst. (2017) 13:246–76. doi: 10.1039/C6MB00387G
126. Garriga J, Xie H, Obradovic Z, Grana X. Selective control of gene expression by CDK9 in human cells. J Cell Physiol. (2010) 222:200–8. doi: 10.1002/jcp.21938
127. Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, et al. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep. (2010) 11:876–82. doi: 10.1038/embor.2010.153
128. Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. (2012) 19:1108–15. doi: 10.1038/nsmb.2399
129. Donner AJ, Ebmeier CC, Taatjes DJ, Espinosa JM. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol. (2010) 17:194–201. doi: 10.1038/nsmb.1752
130. Yin T, Lallena MJ, Kreklau EL, Fales KR, Carballares S, Torrres R, et al. A novel CDK9 inhibitor shows potent antitumor efficacy in preclinical hematologic tumor models. Mol Cancer Ther. (2014) 13:1442–56. doi: 10.1158/1535-7163.MCT-13-0849
131. Lui GY, Grandori C, Kemp CJ. CDK12: An emerging therapeutic target for cancer. J Clin Pathol. (2018) 71:957–62. doi: 10.1136/jclinpath-2018-205356
132. Fisher RP. Cdk7: a kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription. (2019) 10:47–56. doi: 10.1080/21541264.2018.1553483
133. Wong R, Ishida T, Sanda T. Targeting general transcriptional machinery as a therapeutic strategy for adult T-cell leukemia. Molecules. (2018) 23:1057. doi: 10.3390/molecules23051057
134. Clark K, Ainscow E, Peall A, Thomson S, Leishman A, Elaine S, et al. CT7001, a novel orally bio-available CDK7 inhibitor, is highly active in in-vitro and in-vivo models of AML. Blood. (2017) 130:2645.
135. Bacon CW, D'Orso I. CDK9: a signaling hub for transcriptional control. Transcription. (2019) 10:57–75. doi: 10.1080/21541264.2018.1523668
136. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. (2017) 173:83–105. doi: 10.1016/j.pharmthera.2017.02.008
137. Powell JA, Thomas D, Barry EF, Kok CH, McClure BJ, Tsykin A, et al. Expression profiling of a hemopoietic cell survival transcriptome implicates osteopontin as a functional prognostic factor in AML. Blood. (2009) 114:4859–70. doi: 10.1182/blood-2009-02-204818
138. Kelleher III RJ, Flanagan PM, Kornberg RD. A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell. (1990) 61:1209–15. doi: 10.1016/0092-8674(90)90685-8
139. Yin JW, Wang G. The Mediator complex: a master coordinator of transcription and cell lineage development. Development. (2014) 141:977–87. doi: 10.1242/dev.098392
140. Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. (2009) 23:439–51. doi: 10.1101/gad.1767009
141. Chi Y, Huddleston MJ, Zhang X, Young RA, Annan RS, Carr SA, et al. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev. (2001) 15:1078–92. doi: 10.1101/gad.867501
142. Fryer CJ, White JB, Jones KA. Mastermind recruits CycC: CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. (2004) 16:509–20. doi: 10.1016/j.molcel.2004.10.014
143. Meyer KD, Donner AJ, Knuesel MT, York AG, Espinosa JM, Taatjes DJ. Cooperative activity of cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. EMBO J. (2008) 27:1447–57. doi: 10.1038/emboj.2008.78
144. Rzymski T, Mikula M, Zyłkiewicz E, Dreas A, Wiklik K, Golas A, et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget. (2017) 8:33779–95. doi: 10.18632/oncotarget.16810
145. Napolitano G, Majello B, Licciardo P, Giordano A, Lania L. Transcriptional activity of positive transcription elongation factor b kinase in vivo requires the C-terminal domain of RNA polymerase II. Gene. (2000) 254:139–45. doi: 10.1016/S0378-1119(00)00278-X
146. Ni Z, Saunders A, Fuda NJ, Yao J, Suarez JR, Webb WW, et al. P-TEFb is critical for the maturation of RNA polymerase II into productive elongation in vivo. Mol Cell Biol. (2008) 28:1161–70. doi: 10.1128/MCB.01859-07
148. Payne JM, Laybourn PJ, Dahmus ME. The transition of RNA polymerase II from initiation to elongation is associated with phosphorylation of the carboxyl-terminal domain of subunit IIa. J Biol Chem. (1989) 264:19621–9.
149. Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, et al. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature. (1995) 374:283–87. doi: 10.1038/374283a0
150. Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. (1995) 270:12335–8. doi: 10.1074/jbc.270.21.12335
151. Lyle L, Daver N. Current and emerging therapies for patients with acute myeloid leukemia: a focus on MCL-1 and the CDK9 pathway. Am J Manag Care. (2018) 24:S356–65. Available online at: https://ajmc.s3.amazonaws.com/_media/_pdf/EBO_A841_08_2018_AML_Article02.pdf
152. Sampathi S, Acharya P, Zhao Y, Wang J, Stengel K, Liu Q, et al. The CDK7 inhibitor THZ1 alters RNA polymerase dynamics at the 5′ and 3′ ends of genes. Blood. (2017) 130:1195. doi: 10.1093/nar/gkz127
153. Cayrol F, Praditsuktavorn P, Fernando TM, Kwiatkowski N, Marullo R, Calvo-Vidal MN, et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat Commun. (2017) 8:14290. doi: 10.1038/ncomms14747
154. Nilson KA, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS, et al. THZ1 reveals roles for Cdk7 in co-transcriptional capping and pausing. Mol Cell. (2015) 59:576–87. doi: 10.1016/j.molcel.2015.06.032
155. Patel H, Periyasamy M, Sava GP, Bondke A, Slafer BW, Kroll SHB, et al. ICEC0942, an orally bioavailable selective inhibitor of CDK7 for cancer treatment. Mol Cancer Ther. (2018) 17:1156–66. doi: 10.1158/1535-7163.MCT-16-0847
156. Walsby E, Lazenby M, Pepper C, Burnett AK. The cyclin-dependent kinase inhibitor SNS-032 has single agent activity in AML cells and is highly synergistic with cytarabine. Leukemia. (2011) 25:411–9. doi: 10.1038/leu.2010.290
157. Sedlacek H, Czech J, Naik R, Kaur G, Worland P, Losiewicz M, et al. Flavopiridol (L86 (8275) NSC 649890), a new kinase inhibitor for tumor therapy. Int J Oncol. (1996) 9:1143–68. doi: 10.3892/ijo.9.6.1143
158. Kaur G, Stetler-Stevenson M, Sebers S, Worland P, Sedlacek H, Myers C, et al. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86-8275. J Natl Cancer Inst. (1992) 84:1736–40. doi: 10.1093/jnci/84.22.1736
159. Senderowicz AM, Headlee D, Stinson SF, Lush RM, Kalil NVillalba L, et al. Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J Clin Oncol. (1998) 16:2986–99. doi: 10.1200/JCO.1998.16.9.2986
160. Senderowicz AM. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest New Drugs. (1999) 17:313–20.
161. Gojo I, Zhang B, Fenton RG. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin Cancer Res. (2002) 8:3527–38. Available online at: https://clincancerres.aacrjournals.org/content/clincanres/8/11/3527.full.pdf
162. Kitada S, Zapata JM, Andreeff M, Reed JC. Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate antiapoptosis proteins in B-cell chronic lymphocytic leukemia. Blood. (2000) 96:393–7. doi: 10.1182/blood.V96.2.393
163. Nelson DM, Joseph B, Hillion J, Segal J, Karp JE, Resar LM. Flavopiridol induces BCL-2 expression and represses oncogenic transcription factors in leukemic blasts from adults with refractory acute myeloid leukemia. Leuk Lymphoma. (2011) 52:1999–2006. doi: 10.3109/10428194.2011.591012
164. Smith BD, Warner SL, Whatcott C, Siddiqui-Jain A, Bahr B, Dettman E, et al. An alvocidib-containing regimen is highly effective in AML patients through a mechanism dependent on MCL1 expression and function. J Clin Oncol. (2015) 33:7062. doi: 10.1200/jco.2015.33.15_suppl.7062
165. Lücking U, Scholz A, Lienau P, Siemeister G, Kosemund D, Bohlmann R, et al. Identification of atuveciclib (BAY 1143572), the first highly selective, clinical PTEFb/CDK9 inhibitor for the treatment of cancer. ChemMedChem. (2017) 12:1776–93. doi: 10.1002/cmdc.201700447
166. Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclindependent kinase inhibitor. Mol Cancer Ther. (2010) 9:2344–53. doi: 10.1158/1535-7163.MCT-10-0324
167. Gojo I, Sadowska M, Walker A, Iyer SP, Baer MR, Sausville EA, et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH727965) in acute leukemias. Cancer Chemother Pharmacol. (2013) 72:897–908. doi: 10.1007/s00280-013-2249-z
168. Baker A, Gregory GP, Verbrugge I, Kats L, Hilton JJ, Vidacs E, et al. The CDK9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of MLL-rearranged acute myeloid leukemia. Cancer Res. (2016) 76:1158–69. doi: 10.1158/0008-5472.CAN-15-1070
169. Goh KC, Novotny-Diermayr V, Hart S, Ong LC, Loh YK, Cheong A, et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia. (2012) 26:236–43. doi: 10.1038/leu.2011.218
170. Pallis M, Abdul-Aziz A, Burrows F, Seedhouse C, Grundy M, Russell N. The multi-kinase inhibitor TG02 overcomes signalling activation by survival factors to deplete MCL1 and XIAP and induce cell death in primary acute myeloid leukaemia cells. Br J Haematol. (2012) 159:191–203. doi: 10.1111/bjh.12018
171. Pallis M, Burrows F, Ryan J, Grundy M, Seedhouse C, Abdul-Aziz A, et al. Complementary dynamic BH3 profiles predict co-operativity between the multi-kinase inhibitor TG02 and the BH3 mimetic ABT-199 in acute myeloid leukaemia cells. Oncotarget. (2017) 8:16220–32. doi: 10.18632/oncotarget.8742
172. Luedtke DA, Su Y, Edwards H, Polin L, Kushner J, Dzinic SH, et al. Voruciclib, an oral, selective CDK9 inhibitor, enhances cell death induced by the Bcl-2 selective inhibitor venetoclax in acute myeloid leukemia. Blood. (2018) 132:1361. doi: 10.1182/blood-2018-99-118372
173. ClinicalTrials. gov. NCT02520011: Alvocidib Biomarker-Driven Phase 2 AML Study. Available online at: https://clinicaltrials.gov/ct2/show/NCT02520011 (accessed June 5, 2019).
174. Zeidner JF, Foster MC, Blackford AL, Litzow MR, Morris LE, Strickland SA, et al. Final results of a randomized multicenter phase II study of alvocidib, cytarabine, and mitoxantrone versus cytarabine and daunorubicin (7+ 3) in newly diagnosed high-risk acute myeloid leukemia (AML). Leuk Res. (2018) 72:92–5. doi: 10.1016/j.leukres.2018.08.005
175. Cidado J, Proia T, Boiko S, San Martin M, Criscione S, Ferguson D, et al. AZD4573, a novel CDK9 inhibitor, rapidly induces cell death in hematological tumor models through depletion of Mcl1. Cancer Res. (2018) 78:Abstract 310. doi: 10.1158/1538-7445.AM2018-310
176. Barlaam B, De Savi C, Drew L, Ferguson AD, Ferguson D, Gu C, et al. Discovery of AZD4573, a potent and selective inhibitor of CDK9 that enables transient target engagement for the treatment of hematologic malignancies. Cancer Res. (2018) 78:Abstract 1650. doi: 10.1158/1538-7445.AM2018-1650
177. Bogenberger JM, Delman D, Hansen N, Valdez R, Fauble V, Mesa RA, et al. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk Lymphoma. (2015) 56:226–9. doi: 10.3109/10428194.2014.910657
178. Kornblau SM, Tibes R, Qiu YH, Chen W, Kantarjian HM, Andreeff M, et al. Functional proteomic profiling of AML predicts response and survival. Blood. (2009) 113:154–64. doi: 10.1182/blood-2007-10-119438
179. Chen R, Keating MJ, Gandhi V, Plunkett W. Transcription inhibition by flavopiridol: mechanism of chronic lymphocytic leukemia cell death. Blood. (2005) 106:2513–9. doi: 10.1182/blood-2005-04-1678
180. Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. (2016) 6:1106–17. doi: 10.1158/2159-8290.CD-16-0313
181. Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell. (2017) 32:748–60. doi: 10.1016/j.ccell.2017.11.003
182. Pan R, Ruvolo VR, Wei J, Konopleva M, Reed JC, Pellecchia M, et al. Inhibition of Mcl-1 with the pan–Bcl-2 family inhibitor (–) BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood. (2015) 126:363–72. doi: 10.1182/blood-2014-10-604975
183. Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. (2005) 19:523–34. doi: 10.1016/j.molcel.2005.06.027
184. Winter GE, Mauer A, Buckley DL, Erb MA, Roderick JE, Vittori S, et al. BET bromeodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol Cell. (2017) 67:5–18.e19. doi: 10.1016/j.molcel.2017.06.004
185. Junwei S, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromeodomain inhibition. Mol Cell. (2014) 54:728–36. doi: 10.1016/j.molcel.2014.05.016
186. Gerlach D, Tontsch-Grunt U, Baum A, Popow J, Scharn D, Hofmann MH, et al. The novel BET bromeodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene. (2018) 37:2687–701. doi: 10.1038/s41388-018-0150-2
187. Giannopoulos K. Targeting immune signaling checkpoints in acute myeloid leukemia. J Clin Med. (2019) 8:1–11. doi: 10.3390/jcm8020236
188. Hossain DMS, Javaid S, Cai M, Zhang C, Sawant A, Hinton M, et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J Clin Invest. (2018) 128:644–54. doi: 10.1172/JCI94586
189. Kojima K, Kornblau SM, Ruvolo V, Dilip A, Duvvuri S, Davis RE, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. (2013) 121:4166–74. doi: 10.1182/blood-2012-08-447581
190. Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun. (2014) 5:3887. doi: 10.1038/ncomms4887
191. Akbani R, Becker KF, Carragher N, Goldstein T, de Koning L, Korf U, et al. Realizing the promise of reverse phase protein arrays for clinical, translational, and basic research: a workshop report: the RPPA (Reverse Phase Protein Array) society. Mol Cell Proteomics. (2014) 13:1625–43. doi: 10.1074/mcp.O113.034918
192. Bhola PD, Mar BG, Lindsley RC, Ryan JA, Hogdal LJ, Vo TT, et al. Functionally identifiable apoptosis-insensitive subpopulations determine chemoresistance in acute myeloid leukemia. J Clin Invest. (2016) 126:3827–36. doi: 10.1172/JCI82908
193. Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods. (2013) 61:156–64. doi: 10.1016/j.ymeth.2013.04.006
194. Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci USA. (2010) 107:12895–900. doi: 10.1073/pnas.0914878107
195. Dettman EJ, Warner SL, Doykan C, Arn M, Blake N, Bearss DJ, et al. Mitochondrial profiling in AML patients treated with an alvocidib containing regimen reveals MCL1 dependency in responder bone marrow. Cancer Res. (2015) 75:3400. doi: 10.1158/1538-7445.AM2015-3400
196. Pierceall WE, Lena RJ, Medeiros BC, Blake N, Doykan C, Elashoff M, et al. Mcl-1 dependence predicts response to vorinostat and gemtuzumab ozogamicin in acute myeloid leukemia. Leuk Res. (2014) 38:564–8. doi: 10.1016/j.leukres.2014.02.007
197. Del Gaizo Moore V, Letai A. BH3 profiling—measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett. (2013) 332:202–5. doi: 10.1016/j.canlet.2011.12.021
198. Butterworth M, Pettit A, Varadarajan S, Cohen GM. BH3 profiling and a toolkit of BH3-mimetic drugs predict anti-apoptotic dependence of cancer cells. Br J Cancer. (2016) 114:638–41. doi: 10.1038/bjc.2016.49
Keywords: acute myeloid leukemia, BCL-2, CDK, CDK9, MCL-1, transcriptional inhibition
Citation: Tibes R and Bogenberger JM (2019) Transcriptional Silencing of MCL-1 Through Cyclin-Dependent Kinase Inhibition in Acute Myeloid Leukemia. Front. Oncol. 9:1205. doi: 10.3389/fonc.2019.01205
Received: 03 July 2019; Accepted: 23 October 2019;
Published: 12 December 2019.
Edited by:
Naval Daver, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Alice Mims, The Ohio State University, United StatesAmol Padgaonkar, Prescient Healthcare Group, United States
Copyright © 2019 Tibes and Bogenberger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raoul Tibes, UmFvdWwuVGliZXNAbnl1bGFuZ29uZS5vcmc=