 Kendall L. Langsten1*
Kendall L. Langsten1* Jong Hyuk Kim2,3,4
Jong Hyuk Kim2,3,4 Aaron L. Sarver2,4,5
Aaron L. Sarver2,4,5 Mark Dewhirst6
Mark Dewhirst6 Jaime F. Modiano2,3,4,7,8,9,10*
Jaime F. Modiano2,3,4,7,8,9,10*- 1Department of Veterinary Population Medicine, University of Minnesota, St. Paul, MN, United States
- 2Animal Cancer Care and Research Program, University of Minnesota, St. Paul, MN, United States
- 3Department of Veterinary Clinical Sciences, College of Veterinary Medicine, University of Minnesota, St. Paul, MN, United States
- 4Masonic Cancer Center, University of Minnesota, Minneapolis, MN, United States
- 5Institute for Health Informatics, University of Minnesota, Minneapolis, MN, United States
- 6Radiation Oncology Department, Duke University Medical School, Durham, NC, United States
- 7Department of Laboratory Medicine and Pathology, University of Minnesota Medical School, Minneapolis, MN, United States
- 8Center for Immunology, University of Minnesota, Minneapolis, MN, United States
- 9Stem Cell Institute, University of Minnesota, Minneapolis, MN, United States
- 10Institute for Engineering in Medicine, University of Minnesota, Minneapolis, MN, United States
The complex ecosystem in which tumor cells reside and interact, termed the tumor microenvironment (TME), encompasses all cells and components associated with a neoplasm that are not transformed cells. Interactions between tumor cells and the TME are complex and fluid, with each facet coercing the other, largely, into promoting tumor progression. While the TME in humans is relatively well-described, a compilation and comparison of the TME in our canine counterparts has not yet been described. As is the case in humans, dog tumors exhibit greater heterogeneity than what is appreciated in laboratory animal models, although the current level of knowledge on similarities and differences in the TME between dogs and humans, and the practical implications of that information, require further investigation. This review summarizes some of the complexities of the human and mouse TME and interjects with what is known in the dog, relaying the information in the context of the temporo-spatial organization of the TME. To the authors' knowledge, the development of the TME over space and time has not been widely discussed, and a comprehensive review of the canine TME has not been done. The specific topics covered in this review include cellular invasion and interactions within the TME, metabolic derangements in the TME and vascular invasion, and the involvement of the TME in tumor spread and metastasis.
Introduction
Cancer, the uncontrolled proliferation of cells, is a significant cause of morbidity and mortality in humans and their canine companions worldwide (1, 2). The process of neoplastic transformation is similar amongst species and can most easily be conceptualized in the three steps of initiation, promotion, and progression toward malignancy (3), although it is now apparent that these steps are neither sequential nor obligate. In the seminal work by Hanahan and Weinberg (4), tumors were introduced as complex heterotypic tissues where a non-transformed milieu influences the progression of transformed cells with which it co-exists in the same space and time. This milieu, the tumor microenvironment (TME), may be thought of as the ecosystem or community within which neoplastic cells survive and reside. The genomic landscape of the malignant cells and the composition and behavior of the TME are shaped by intense selection that can be described as prototypical Darwinian evolution in a microscopic scale. All non-transformed cells that interact with tumor cells, including inflammatory cells, endothelium, adipocytes, and fibroblasts, among others, as well as the non-cellular components, including structural scaffold surrounding the cells and the soluble factors secreted by the tumor and non-tumor components, compose the TME [Figure 1; (5)]. In a non-neoplastic environment, these components have a vast range of functions, including forming the interstitium that creates a scaffold for parenchyma to organize, sequestering growth factors, supplying nutrients, draining waste from tissue, and creating a competent immune system to protect the body against invaders.
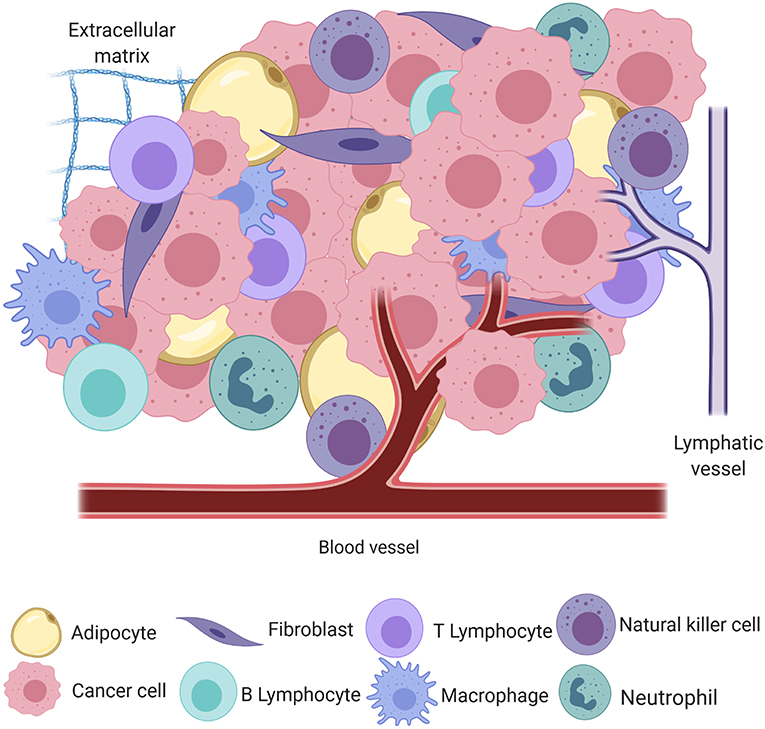
Figure 1. A simplified schematic of the cellular and structural component of the tumor microenvironment, including adipocytes, fibroblasts, B and T lymphocytes, macrophages, natural killer cells, neutrophils, blood and lymphatic vessels, and the extracellular matrix, all intermingled with transformed cancer cells (created with Biorender.com).
The interplay between the TME and tumor cells is paramount in the progression and response to neoplastic growth. While our understanding of the TME in dogs is rudimentary, there are the similarities in tumor heterogeneity between dogs and humans (Table 1), which are often not appreciated in laboratory animal models. Although the current level of knowledge on similarities and differences in the TME between dogs and humans, and the practical implications of that information require further investigation. This review provides an overview of the complexity observed in the human and mouse TME, interjects known similarities and differences in the dog, and relays them in the context of the temporo-spatial organization of the TME. In short, the proposed temporo-spatial organization of the TME involves neoplastic tells transforming, the transformation of adjacent TME into a cancer associated phenotype, and vascular invasion, potentially culminating in tumor cell spread and metastasis (Figure 2). To the authors' knowledge, this approach to the organization and conceptualization of the TME, as well as a review of the TME in the dog, have not been described before. Discussions include cellular invasion and interactions within the TME, metabolic derangements in the TME and vascular invasion, and the involvement of the TME in tumor spread and metastasis.
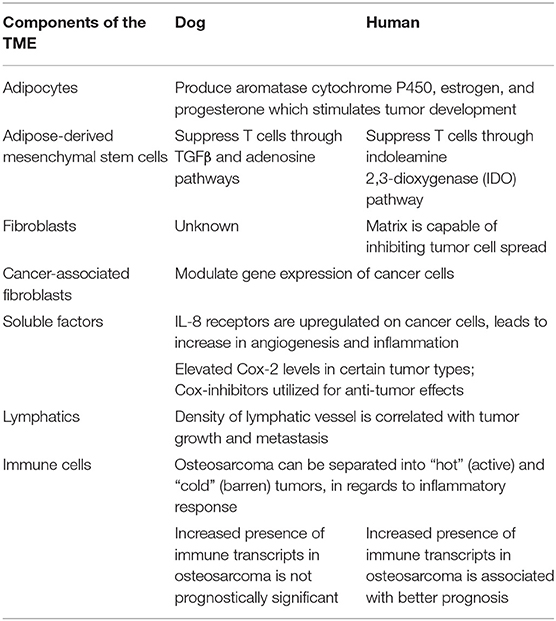
Table 1. Comparative features of the TME between dogs and humans.
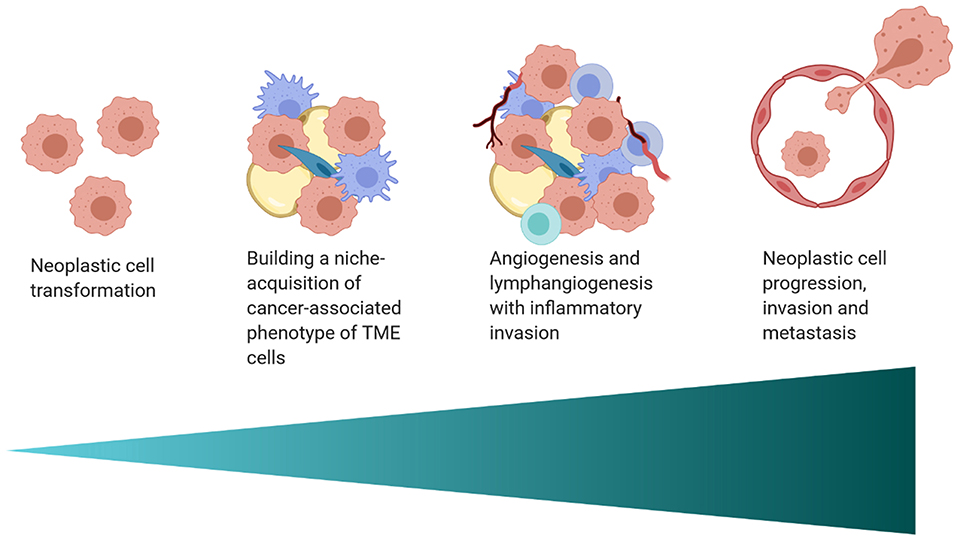
Figure 2. A schematic of the proposed temporo-spatial organization of the TME. Cells must first undergo neoplastic transformation, allowing for the creation of a permissive micro-ecological niche. Neighboring cells, including adipocytes, fibroblasts, and macrophages, among others, can adopt a cancer-associated phenotype, with complex, pro-tumorigenic effects. Hypoxia induced by cell proliferation and metabolic changes encourage lymph and blood vessel invasion, increasing infiltration by inflammatory cells. Furthermore, angiogenesis and lymphangiogenesis create increased opportunity for neoplastic cell spread and metastasis (created with Biorender.com).
Cellular Invasion and Interactions Within the TME
While the definition of “cancer stem cells” (CSCs; also called tumor-initiating cells or tumor-propagating cells) is mostly semantic, the importance of cells that retain or acquire stem-like features in the tumor cannot be underestimated. Whether they be few, as in traditional hierarchically-organized tumors, or many, as in stochastically organized tumors, these cells contribute to remodeling the TME (6).
Recent work characterized genome-wide gene expression signatures in canine tumor models (hemangiosarcoma, osteosarcoma, and glioblastoma) that were grouped according to their hierarchical organization (7). Cell lines derived from these three tumor types were cultured under non-adherent low serum conditions that promote sphere formation and enrich CSCs. The steady state gene expression associated with CSC maintenance in tumors with high sphere-forming efficiency (i.e., hierarchically organized with relatively few CSCs) showed metabolic skewing toward fatty acid synthesis and secretion of immunosuppressive cytokines. On the other hand, tumors with low sphere-forming efficiency (i.e., stochastically organized with many or most cells having CSC potential) showed metabolic skewing toward fatty acid oxidation and potential immunoevasion through upregulation of CD40. In the incipient tumor, CSCs create reactive microenvironments that support tumor growth (8), importantly, by producing cytokines that leverage the innate properties of resident macrophages to remodel the microenvironment. CSCs also interact bidirectionally with myeloid cells, which can remain in an incompletely differentiated state and become myeloid-derived suppressor cells (MDSCs, a highly heterogeneous population of cells that contributes to cancer stemness as well as to the functional immunosuppressive barrier (9, 10).
Intriguingly, the CSC condition appears to be at least partly under extrinsic control. Depletion of CSCs in cultured canine and human cell lines leads to reprogramming of differentiated cells to become CSCs, maintaining the population in a steady state (11). The signals that regulate this process are poorly understood, but they might involve dysregulated expression of Snail family transcription factors Snail (SNAI1), Slug (SNAI2), as well as TWIST1 and Zeb1 (12–14), perhaps through epigenetic modification of their respective promoters (15). These responses are tightly regulated by environmental cues. For example, expression of SNAI2 and its targets, CDH1, VIM, and JUP in hemangiosarcoma cells showed a biphasic response to interkeukin-8 (IL-8), with small amounts of IL-8 favoring self-renewal and abundant IL-8 favoring expansion of bulk (differentiated) tumor cells (8, 16).
The role of adipocytes in the TME has received more attention as evidence mounts for a link between obesity and cancer risk in dogs and humans (17, 18). Adipocytes adjacent to tumor cells, known as cancer-associated adipocytes (CAAs), are recruited to be actively involved in tumor initiation, promotion, and progression. The mechanism of CAA development is unclear, but likely involves a bidirectional communication stream that includes adipokines and extracellular vesicles, among other factors. Adipokines, metabolically active substances secreted by adipocytes to create a permissive TME, include substances such as leptin, tumor necrosis factor-α (TNFα), C-C Motif Chemokine Ligand 2 (CCL2), and adiponectin. A concise review of adipokines in domestic animals was recently published (19).
Adipocytes promote neoplastic development through a variety of mechanisms, including supporting angiogenesis (described later in this review), manipulating tumor cell metabolism, and encouraging a pro-inflammatory state, leading to the recruitment of macrophages. Adipocytes play an important role in reprogramming tumor cell metabolism. For example, ovarian cancer cells co-cultured with abdominal adipose cells were shown to coerce neighboring adipocytes into supplying free fatty acids, thereby providing substrates for sustained tumor cell replication (20). The role of adipocytes in promoting chronic inflammation has been the subject of numerous studies. Adipocytes produce pro-inflammatory adipokines and cytokines [including CCL2, interleukin-6 (IL-6), and TNFα], which increase inflammation and metastatic risk, supporting tumor survival (20, 21). In the case of mammary and breast carcinomas in dogs and humans, respectively, adipocyte-derived aromatase cytochrome P450, estrogen, and progesterone have been reported to stimulate tumor development and enhance invasive potential [Table 1; (22, 23)]. Finally, extracellular remodeling in tumors, including increased collagen deposition by adipocytes, can lead to adipocyte apoptosis and necrosis. Macrophages are then recruited into the tumor due to the release of pro-inflammatory damage-associated molecular patterns (DAMPs) from the dying adipocytes (17).
Recently, attention has been given to the influence of adipose-derived mesenchymal stem cells (ad-MSCs) in tumor progression. The secretome of ad-MSCs is incompletely understood but is thought to have overarching immunomodulatory and pro-angiogenic properties (24). The immunomodulatory properties of these cells are dependent on the inflammatory milieu in which the cells reside. Some of the anti-inflammatory properties of human and dog MSCs seem to differ mechanistically. Ad-MSC dependent T cell suppression in humans is through the indoleamine 2,3-dioxygenase (IDO) pathway, resulting in decreased T cell function through tryptophan depletion (25). Alternatively, in dogs ad-MSCs most likely decrease T cell activity through TGFβ and adenosine pathways [Table 1; (26)]. While a solid body of knowledge about the influence of adipocytes and ad-MSCs in human tumor growth and progression has been developed in recent years, the influence of these cells on the TME in dogs remains to be elucidated.
In a non-cancer associated microenvironment, fibroblasts play a major role in producing components of the extracellular matrix (ECM) including fibrillar collagen, elastin, laminin, fibronectin, and glycosaminoglycans (27). Fibroblasts are critical in wound healing, inflammatory reactions, fibrosis, promoting angiogenesis, and cancer progression. Tumors are often conceptualized as a “wound that will not heal” with abundant collagen deposition (28). In vitro studies using cell lines from various species, although to the authors' knowledge not from dogs, have demonstrated that normal, non-cancer associated fibroblasts and the matrix they produce are capable of inhibiting the spread of tumor cells, a phenomenon termed neighbor suppression (29–31). Since neighbor suppression was first recognized by Stoker et al. (29), many theories have developed around the molecular mechanisms influencing this finding, including heterologous communication between transformed and non-transformed cells through junctional complexes and through soluble factors within the ECM (32, 33). Neighbor suppression has not yet been recognized in canine tumors (Table 1).
Cancer-associated fibroblasts (CAFs) are corrupted by the neoplastic cells in their proximity and have drastically different functions than their non-transformed counterparts. The origin of CAFs is not entirely clear; many theories on their origin claim CAFs originate from resident mesodermal precursors (34–38). An influential paper by Erez et al. (39) demonstrated that the transcription factor NFκB induces the CAF phenotype through upregulation of pro-inflammatory genes. These findings suggest a necessity for innate immune involvement in the education of CAFs. Furthermore, epigenetic changes also play a role in the development of CAFs. Albrengues et al. (36) demonstrated that CAFs have constitutively activated JAK1/STAT3 signaling pathways secondary to epigenetic changes. Histone acetylation of STAT3 in CAFs by leukemia inhibitory factor (LIF) caused subsequent activation of DNMT3b (a DNA methyltransferase). This in turn led to decreased SHP-1 expression with subsequent sustained activation of JAK1. Interestingly, inhibition of DNMTs caused CAFs to convert to a non-cancer associated fibroblast phenotype (36). CAFs have diverse phenotypes without unique markers, although phenotypic similarities to myofibroblasts, including reduced caveolin-1 (CAV-1) expression and increased expression of α-SMA, vimentin, fibroblast-activating protein, and MCT-4 (40, 41) have been described. Additionally, CAFs have been shown to increase tumor cell growth, motility, and local invasion through ECM remodeling and cytokine release (37, 42, 43). In both humans and dogs, CAFs modulate gene expression of cancer cells (44, 45). However, it is difficult to compare their transcriptional programs across species, as experimental protocols and genes of interest differ between published studies. Functionally, CAFs differ from normal fibroblasts in the products and quantities of enzymes that they produce. For example, in both canine mammary carcinoma and human breast carcinoma CAFs exhibit increased aromatase activity, which is associated with hormone-driven tumor progression (46, 47).
Mesenchymal stem cells (MSCs), also known as undifferentiated fibroblasts or mesenchymal stromal cells, are another important component of the TME. These cells are phenotypically plastic cells that originate from the mesoderm (48). MSCs home from bone marrow, spleen and other locations to sites of injury and inflammation, including tumors (49). The role of MSCs in the TME are numerous; one of the better-studied functions is their influence in changing the immune landscape (for more information, see the section on metabolism, vascular invasion, and immune cells within the TME).
Tumor-associated ECM is markedly different from ECM in a non-pathologic milieu. As an active driver of tumor progression, tumor-associated ECM is reorganized, directing tumor cell migration and promoting local invasion along collagen fibers (50, 51). Furthermore, tumor-associated ECM is associated with increased pro-inflammatory cytokines, promotes angiogenesis, and factors that increase fibroblast proliferation (52). As all components of the TME are simultaneously interacting with one another and tumor cells, it stands to reason that by encouraging inflammation, tumor-associated ECM likely contributes to the production of CAFs. Collagen is one of the most abundant components of the ECM and is known to exhibit tumor-associated collagen signatures. Differences in collagen density, width, length, and straightness, as well as reorganization of the boundary between tumor and stroma, are some of the collagen signatures appreciated (53, 54). In dogs and humans, collagen signatures are important prognostic indicators in mammary and breast carcinoma (53, 54). For example, in a study analyzing characteristics of mammary carcinoma in dogs, tumor-associated ECM had upregulated collagen1α1, α-SMA, fibroblast activation protein (FAP), platelet-derived growth factor (PDGF)-β, and paradoxically, CAV-1 (55).
Interactions between tumor cells, stromal cells, and the ECM are heterogeneous and tumor-specific. However, fragmentation of hyaluronic acid, which is pro-inflammatory, and deposition of tenascin-C seem to occur in most tumor types (56–58). The hyaluronic acid receptor, CD44, is expressed by most tumor and stromal cells, but the highest levels are seen in CSCs (6). In addition to HA, the ECM is composed of collagens, elastins, laminins, fibrinogen, and tissue-specific proteoglycans. The stoichiometry and topology of these components regulates adhesion (for example, by interaction with cell surface integrins) and stiffness of the ECM. Tumor cells and inflammatory cells secrete proteases that degrade the ECM, and proteins and proteoglycans to remodel it. The interactions of the ECM with integrins, mechanoreceptors, and signaling proteins that activate contractility, such as focal adhesion kinase, modulate cellular motility, proliferation and survival (59–61). The interactions are bidirectional, as the cytoskeleton “pushes back” into the ECM, maintaining integrins and focal adhesions in a state of isometric tension. Increased tension also activates the small G protein Rho and its target Rho-associated kinase (ROCK), which controls myosin light chain phosphorylation. The ECM in most tumors is several orders of magnitude stiffer than their normal tissue counterparts, making it permissive for cell migration and ultimately, metastasis (59). There are myriad studies documenting the importance of ROCKs in tumor progression, but a noteworthy study showed that ROCK inhibitors were able to push chemoresistant mouse osteosarcoma cells away from a malignant phenotype and into terminal adipocyte differentiation (62). Perhaps more interestingly, cells that escaped terminal differentiation in the presence of ROCK inhibitors regained sensitivity to chemotherapy and could be eliminated by treatment with doxorubicin (62).
The extensive heterogeneity and adaptation of the tumor niche is partly dependent on intercellular communication. Malignant cells co-opt developmental programs of intercellular communication to create and maintain a niche with unique properties that promote growth and survival (6). Intercellular communication involves a multitude of interactions mediated by cell-to-cell contacts and soluble mediators. Cell-to-cell contacts include adhesion molecules, stable ligand-receptor interactions, and promiscuous, transient to stable interactions between cell surface proteins, glycans, and lipids (63, 64). Emerging evidence also suggests that cells can communicate in the local environment by exchanging genetic and biochemical mediators through tunneling nanotubes (65, 66). Soluble mediators of communication include hormones, cytokines, chemokines, lipids, and microvesicles (67–70). Cells also interact with their external environment through pressure receptors and by molding the ECM (59, 71–73).
Soluble mediators of communication have been relatively well-described in humans, although there is little information available as to the impact of the stroma and soluble factors in dogs. Kim et al. (8) showed that IL-8, a cytokine produced by fibroblasts, neoplastic cells, and other cell types, supports tumor progression by modulating the TME in canine hemangiosarcoma into a more “reactive” state; increasing the propensity toward inflammation, fibrosis, and coagulation. Intriguingly, IL-8 blockade reduced tumor cell survival and engraftment in a xenograft model of canine hemangiosarcoma, indicating this cytokine may be necessary to establish the initial niche for this disease (8). Similar findings have been reported in humans, with tumor cells of various tumor types upregulating IL-8 production and IL-8 receptors on cancer cells as well as other cells types with increases in angiogenesis and inflammation within those tissues [Table 1; (74, 75)]. The implications and utilization of soluble factors in cancer treatment is a topic that in recent years has begun to gain traction as an important area for investigation.
Intercellular interactions are also critical to establish and maintain the tumor immunosuppressive barrier, by excluding or incapacitating host immune cells. For example, expression of pro-apoptotic molecules, such as FasL can target infiltrating effector T cells in the tumor environment, while sparing apoptosis resistant tumor cells, CAFs, and cancer-associated endothelial cells. For more information on immune cells within the TME, please see the next section on metabolism, vascular invasion, and immune cells within the TME.
Metabolism, Vascular Invasion, and Immune Cells Within the TME
Formation of blood vessels is an absolute requirement for tumor growth, survival, and progression. Without access to oxygen and nutrients supplied by the blood, tumor growth is restricted to an ~1–3 mm diameter mass ex vivo and ~100–500 microns in vivo (76–78). It stands to reason that the aspects of the TME reviewed might precede angiogenesis, lymphangiogenesis, and immune invasion due to the size of the tumor where these processes occur. Tumor neovascularization is a complex and multifaceted process driven by tissue hypoxia, defined as tissue with oxygen concentrations below 10 mmHg, which is a common feature of solid tumors (79, 80). Below this threshold, cells upregulate a host of adaptive proteins; a response mostly driven by the heterodimeric transcription factor, hypoxia-inducible factor (HIF-1) (81). In normoxic conditions, prolyl hydroxylases (which have oxygen dependent enzymatic activity), hydroxylate proline residues in the oxygen degradation domain of HIF-1α. The von Hippel-Lindau (VHL) complex is then able to recognize HIF-1α for subsequent proteasomal degradation (81). Under hypoxic conditions, VHL itself undergoes proteasomal degradation, leading to stabilization of HIF-1α and subsequent binding to its constitutively regulated partner, HIF-1β (82). Once this occurs, the HIF-1α/HIF-1β heterodimer enters the nucleus and binds to hypoxia-regulated-elements (HREs) of hundreds of genes (83). Binding targets of HIF-1 are in part controlled by epigenetic changes that promote active chromatin states at HIF binding sites (84). The impacts of HIF-1 binding are numerous, from reducing oxygen consumption to increasing angiogenesis through regulation of vascular endothelial growth factor (VEGF), the angiopoietin-1 regulated tyrosine kinase receptor TIE2 (also known as TEK), and angiopoietin, among others (83, 85).
VEGF is a potent growth factor influencing vascular permeability and angiogenesis (86). VEGF-A is one of the best-characterized forces in the development of new vessels and binds to VEGF receptors-1 and -2 (VEGFR-1 and VEGFR-2). VEGF-A can be secreted along with other pro-angiogenic factors by numerous cell types, including adipocytes, within the TME (87). VEGFR-1 and VEGFR-2 are both receptor tyrosine kinases that contain a split tyrosine-kinase domain, although they function differently within the TME (88). VEGFR-2 is upregulated in endothelial cells of newly forming blood vessels within tumors and is commonly implicated in neovascularization. A recent study demonstrated that the α4β1-integrin is capable of VEGFR2 binding and activation, presenting a novel potential target for therapy (89). Alternatively, VEGFR-1 has relatively weak pro-angiogenic properties and can recruit and activate tumor-associated macrophages (TAMs) and myeloid cells, promoting tumor cell metastasis and proliferation [Figure 3; (90)]. Little is known about the dynamic balance between VEGFR-2 and VEGFR-1 in tumors of dogs, but there is one report suggesting that heritable traits or the breed background might influence the expression and function of these receptors in vascular sarcomas (91). A second major regulator of angiogenesis is Tie-2. In the presence of active Tie-2 signaling, the vasculature remains in a mature state surrounded by pericytes (92). Angiopoietin 2 (ANGPT2), an angiopoietin 1 competitive antagonist and HIF-1 target gene, binds to endothelial cells, preventing Tie-2 signaling. This causes the vasculature to become less mature with fewer pericytes (93). This microvasculature is then primed for maximum response to VEGF.
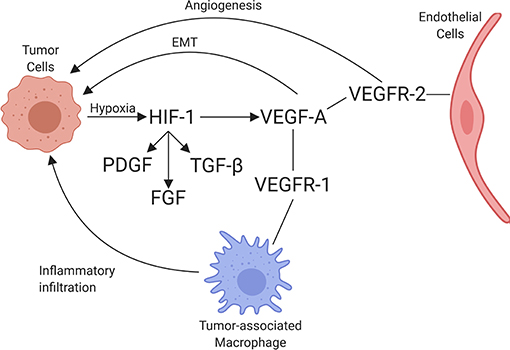
Figure 3. Schematic of the interaction between tumor cells and endothelial cells, including the associated recruitment of inflammatory cells by endothelium. Through hypoxia, HIF-1 is upregulated, increasing PDGF, FGF, TGFβ, and VEGF-A expression. VEGF-A binds to its receptors, VEGFR-1 and VEGFR-2, leading to angiogenesis (through VEGFR-2 signaling), immune cell invasion (through VEGFR-1 signaling), and promotion of epithelial-mesenchymal transition (EMT) in tumor cells (created with Biorender.com).
Cyclo-oxygenase 2 (Cox-2), which is involved in the formation of some types of prostaglandin production, has been shown to increase expression of VEGF-A mRNA in tumors, thus having pro-angiogenic properties (94). The mechanism by which Cox-2 increases VEGF-A expression in tumors likely involves p38 mitogen activated protein kinase (MAPK) and Janus kinase (JNK) pathways. These pathways are integral in the transcriptional and post-transcriptional regulation of VEGF-A (95, 96). Elevated Cox-2 levels have been reported in canine and human prostatic carcinoma, transitional cell carcinoma, and squamous cell carcinoma, among others [Table 1; (97–102)]. As such, anti-Cox-2 therapies, including non-steroidal anti-inflammatory drugs (NSAIDs) which inhibit the production of Cox-2, have been the subject of anti-cancer initiatives. In dogs with urothelial cell carcinoma, treatment with the NSAIDs piroxicam and deracoxib have shown promising clinical results, including decreasing tumor volume and increasing apoptosis of neoplastic cells (103, 104). Similarly, NSAIDs have been used for their anti-tumor effects in humans, such as a chemopreventative agent for colorectal cancer and certain subtypes of breast cancer (105, 106). Another promising anti-tumor therapy that leverages increased Cox-2 expression in tumors utilizes conditionally replicative oncolytic adenoviruses. To overcome the traditionally poor infectivity of these viruses, an oncolytic adenovirus with Cox-2 promoter-based targeting control mechanisms was designed. This viral therapy is specific to Cox-2 positive cells with the potential to specifically target a variety of Cox-2 positive tumors, thereby increasing efficacy and safety of this potential therapy (107, 108).
The clinical implications of HIF-1 and VEGF expression and regulation have been the subjects of recent investigation. Moeller et al. (109) were the first to demonstrate that radiotherapy upregulates HIF-1 protein levels, even at a time when the tumor is re-oxygenated. The mechanism for this effect was shown to be related to two factors: (1) release of HIF-1 mediated transcripts of HIF-1 stored in stress granules during hypoxia, and (2) an increase in oxidative stress after radiotherapy, preventing the activity of prolyl hydroxylases to prime HIF-1α for degradation. As a follow on, Li et al. (110) demonstrated that infiltration of macrophages into irradiated tumors stabilized HIF-1 via a nitric oxide mediated mechanism. Other cytotoxic treatments have been shown to increase HIF-1α levels via mechanisms involving oxidative stress. Doxorubicin can upregulate HIF-1α levels in aerobic tumor cells by stimulating inducible nitric oxide synthase (iNOS) activity (111). Hyperthermia increases the activity of NADPH-oxidase in tumor cells, thereby stabilizing HIF-1α. There are multiple potential consequences of chronic HIF-1 transcriptional upregulation, but of central importance is the upregulation of VEGF. As part of a clinical trial conducted in dogs with soft tissue sarcomas, treated with fractionated hyperthermia and radiotherapy, Chi et al. (112) examined the hypothesis that there would be an increase in HIF-1 mediated transcripts and associated physiologic modification early in the course of treatment. Tumor tissues were removed prior to, and 24 h after, radiotherapy and the first hyperthermia treatment. Tissues were examined for changes in gene expression and, concomitantly, the apparent diffusion constant of water of these tumors was measured using magnetic resonance imaging (ADC-MRI; a biomarker of hyperpermeability). Unsupervised gene expression analysis showed two main groupings, distinguished by whether ADC-MRI increased (indicating increased water content) or remained unchanged. Among several HIF-1 regulated genes observed in the subgroup that showed increased ADC, VEGF upregulation was one of the most predominant (113). Thus, this canine clinical study supported pre-clinical results; that the combination of hyperthermia and radiotherapy increases HIF-1 transcriptional activity. The fact that ADC only increased in a fraction of tumors, suggests that ADC may be a viable biomarker for understanding how the physiologic microenvironment responds to cytotoxic therapy. An important future direction of these observations includes ascertaining whether changes in ADC are associated with treatment outcome.
Hypoxia affects innate and adaptive immune function in multiple and complex ways (114). Macrophage response to hypoxia is multifaceted and relies on the presence and concentration of cytokines and other immune cells. TAMs, which are believed to arise from the resident macrophage pool, have been categorized as “M0” (uncommitted), “M1” (pro-immune), and “M2” (pro-angiogenic and immunosuppressive) (115–117). However, both resident and recruited macrophages are remarkably plastic and can revert among these phenotypes, with all three co-existing in different stages of tumor development and progression. M2 TAMs tend to accumulate in hypoxic regions due to hypoxia-mediated chemokine expression by both tumor and stromal cells (116, 118). The presence of macrophages in hypoxic regions promotes immunosuppressive functions, including release of immunosuppressive cytokines such as TGFβ, recruitment of regulatory T cells (Tregs), and binding of programmed death-1 (PD-1) receptor on cytotoxic T-cells by the HIF-1 target, programmed death ligand-1 (PD-L1) (118, 119). Additionally, hypoxia inhibits the adaptive immune system by downregulating T-cell motility and upregulating the HIF-1 targets SDF-1 and its ligand CXCR4, thereby stimulating intratumoral recruitment of immunosuppressive MDSCs (120–122). Hypoxia disturbs the balance between effector T cells and Tregs, tipping the balance toward the latter (123).
In the absence of oxygen, cells are obligated to use glycolysis to produce ATP. Reprogramming energy metabolism is regarded as a hallmark of cancer, as described by Hananhan and Weinberg (124). The “Warburg Effect,” the unique process of tumor cells utilizing aerobic glycolysis, was first described by Warburg (125). Lactate is the product of both aerobic and anaerobic glycolysis (126). The relative predominance of hypoxia in tumors, therefore, is a major contributor to the production of lactate. In addition, the relative inefficiency in solute transport by tumor vasculature leads to accumulation of lactate to substantially elevated levels. Concentrations of lactate can range from normal levels of 1–2 mM to as high as 15–40 mM in both pre-clinical and human clinical samples (127, 128).
Lactic acid is a major component that fuels metabolic symbiosis between the aerobic and hypoxic tumor compartments (129). Lactate produced by hypoxic tumor cells is transported by passive monocarboxylic acid transporters (MCTs), which enable lactate to be excreted by cells that produce it and to be taken up by aerobic cells where in high concentrations, is back converted to pyruvate, where it enters the tricarboxylic acid (TCA) cycle to produce alanine and glutamate (126, 130). The affinity of aerobic tumor cells for lactate is 10 times higher than glucose, indicating that aerobic tumor cells preferentially use lactate (130). If the ability of aerobic tumor cells to use lactate is blocked, the cells will switch to utilizing glucose, thereby depleting local glucose concentrations. Excess glucose present in aerobic tumor regions can diffuse to hypoxic regions, where the glucose is catabolized to lactate (129). Since hypoxic tumor cells are reliant on glucose, even though some can use glutamine in its stead, this can lead to death of the cell (129, 131).
Utilization of lactate has also been described in tumor-associated fibroblasts, which have low expression of CAV-1, an inhibitor of myofibroblast differentiation (132). An informative study showed that tumor-associated myofibroblasts could use aerobic glycolysis to produce lactate. Lactate was then used by aerobic tumor cells to fuel the TCA cycle through its conversion to pyruvate. The authors termed this symbiosis the “Reverse Warburg Effect” because the myofibroblasts were responsible for aerobic glycolysis instead of the tumor cells (132). Lactate can also stimulate the stabilization of HIF-1α in aerobic tumor and endothelial cells (133). Like aerobic tumor cells, endothelial cells uptake lactate (133). The conversion of lactate to pyruvate interferes with the activity of the prolyl hydroxylases responsible for HIF-1α degradation. In the presence of elevated pyruvate, HIF-1α levels, and consequently VEGF levels increase, which promotes angiogenesis. The negative influence of lactate on cancer prognosis in humans is most likely attributed to downstream stabilization of HIF-1α in tumor and stromal cells (134). Lora-Michiels et al. (135) demonstrated that in 39 dogs with soft tissue sarcoma, those with relatively low pH tumors were associated with shorter progression free interval and overall survival than dogs with higher tumor pH. Extracellular pH, which is simpler to measure, can be used as a surrogate of lactate, since transport of lactate across a cell membrane via the MCT transporters includes a hydrogen ion (135). High lactate levels and associated extracellular acidosis also contribute to immune suppression (136).
Once tumor-associated blood vessels are formed, they are structurally and epigenetically abnormal, which facilitates metastatic spread. These vessels tend to be irregularly dilated and tortuous with increased permeability, decreased pericyte numbers, and abnormal deposition of collagen type IV in the basement membrane. Endothelial cell adhesion molecules, such as selectins and integrins are required for leukocyte transmigration into tissues (137, 138). It has been reported that these adhesion molecules are often absent in tumor microvessels, thereby reducing the ability of immune cells to gain access into tumors (137, 139). The downregulation of adhesion molecules is regulated by VEGF (139) and can be reversed by blocking VEGF or by altering IL-6 trans-signaling (138). Thus, the first line of defense that tumors use to inhibit immune surveillance is the blockade of transmigration of immune cells. Furthermore, there is substantial signaling between endothelial cells and tumor cells, especially CSCs, which have a tendency to seek out or create vascular niches (140). Several signaling pathways, including Sonic Hedgehog, and Notch, to name a few, emanate from endothelial cells and promote acquisition of CSC properties and proliferation within vascular niches (141). In the tumor microenvironment, it is likely that the balance between these pro and anti-inflammatory mediators dictates the extent of leukocyte-endothelial cell interactions that occur naturally and in response to therapy. Modulation of these interactions is likely essential for optimization of immunotherapy.
Like neoangiogenesis, lymphangiogenesis can act as an important gateway to tumor metastasis. The density of lymphatic vessels within a tumor has been correlated with tumor growth and metastasis in both dogs and humans [Table 1; (142–145)]. Mechanistic control of lymphangiogenesis is complex, involving a multitude of factors including many of the same factors described in tumor-associated neovascularization. Two of the major mechanisms controlling lymphangiogenesis are well-described. One is dependent on VEGF-C and VEGF-D produced by both tumor cells and TAMs, which bind VEGFR-3 on lymphatic endothelial cells (LECs) (146). The other is the SRY-related HMG-box (SOX18) pathway through prospero homeobox-1 activation (147, 148). Lymphangiogenic factors not only increase the number of lymphatic vessels within solid tumors, but also are capable of enlarging the diameter of the lymphatic vessels, increasing tumor cell metastasis to local lymph nodes (149). Furthermore, VEGF-C secreted by tumor cells can promote lymphangiogenesis within draining lymph nodes, increasing the number and diameter of lymphatic vessels thereby increasing the overall metastatic potential of the tumor (150). Although little is known about tumor cell entry into lymphatic vessels, multiple studies have demonstrated that cancer cells can express CC-chemokine receptor 7 (CCR7), which lymphocytes use to home to lymph nodes via CCL21 binding, in a sense, hijacking the lymphatic system to gain entry to lymphatic vessels and lymph nodes (151). LECs have additionally been implicated in immunomodulation within the TME, including multifaceted mechanisms to promote immune evasion. These include local deletional tolerance of CD8+ T cells, inhibition of dendritic cell maturation leading to decreased effector T cell activity, and tumor antigen trapping and retainment to archive for antigen-presenting cells (152–154).
In their landmark update of the Hallmarks of Cancer in 2011 (124), Hanahan and Weinberg called tumor-promoting inflammation an enabling characteristic of cancer. Inflammation is critical for the formation and maintenance of the tumor niche. It persists throughout the existence of the tumor, through therapy, remission, stabilization of disease, and relapse, and it is foundational in creating the metastatic niche. In its steady state, inflammation in the TME can promote or inhibit the capacity of innate and adaptive immune cells to infiltrate the tumor stroma and eliminate the tumor cells. However, the inflammatory TME is highly dynamic (155), characterized by a recurring cycle that established an evolutionary arms race at microscopic scale. Whether the balance tips toward immunosuppression or toward productive anti-tumor immunity is a critical determinant in the ultimate rate of tumor progression and patient outcomes.
Thousands of studies have examined the composition of the TME in humans and animals. Most studies focused on one or a few features in isolation, such as infiltration by immunosuppressive elements like Tregs or by tumoricidal NK cells or cytolytic T cells. For example, increased CD4+:CD8+ T cell ratios were correlated with decreased survival in dogs with mammary carcinomas (156), and enrichment of Foxp3+ regulatory T cells within tumors was associated with tumor progression in mammary and testicular cancers (157, 158). As another example, the immunomodulatory properties of MSCs follow licensing by inflammatory cytokines such as interferon-γ (IFNγ) and TNFα (159, 160). Licensed MSCs are resistant to apoptosis, and thus impervious to immune attack. In both syngeneic and xenograft models, MSCs reorganize the TME, excluding T cells, macrophages, and other host effector cells, tilting the balance away from tumor host control and toward tumor progression (161, 162). MSCs are also able to inhibit T cell proliferation and inhibit natural killer (NK) cell function through soluble factors, and cell-cell communication (163–165). Paradoxically, these cells can inhibit TNFα and IFNγ which are initially necessary for licensing or “tumor-mediated education,” while also increasing IL-10, an immunosuppressive cytokine (164). In dogs, MSCs induced from skin fibroblasts have shown similar immunomodulatory effects to naturally sourced MSCs (166).
Recent advances in next generation sequencing and bioinformatics, as well as the availability of high-quality samples that comprise The Cancer Genome Atlas (TCGA), made it possible to divide the immune landscape of human tumors into six distinct steady states or subtypes (167). Thorsson et al. based these subtypes on their respective transcriptional programs (Table 2), which in turn allowed them to establish the predicted cellular composition for each tumor (167). While these subtypes are probably not static, their dominance at any given type in any given tumor is prognostically significant. It should be noted that there was extensive overlap among genes, and therefore among predicted cell types comprising these subtypes, underscoring the futility of trying to understand the relationship between cancer and the immune system without the benefit of comprehensive information. For this reason, the extensive literature describing unique components of the immune TME will not be further summarized in this review. Emerging technologies such as single cell sequencing (168, 169) and highly multiplexed 3-dimensional optical imaging (170, 171), individually and combined, will bring about the next transformation in the understanding of the immune landscape of cancer.
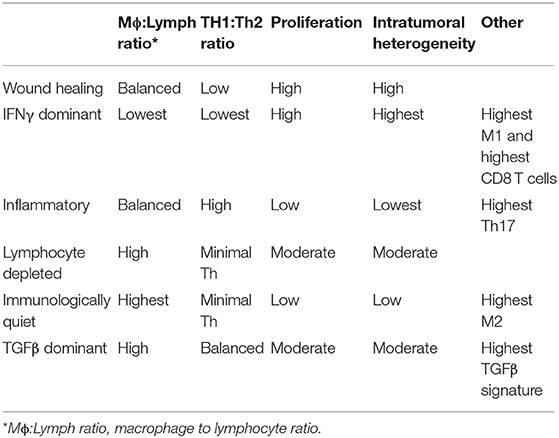
Table 2. Immune subtypes of cancer.
Recent advances in sequencing technology and bioinformatics (172) are being applied to studies of canine immuno-oncology. Specifically, genome-wide gene expression profiles using RNA-Seq transcriptomic data can be utilized to estimate the abundance of distinct subsets of immune infiltrate in the tumor tissues and to examine the features of the inflammatory response (167, 173, 174). Scott et al. (175) showed that, even though osteosarcomas are immunologically “cold” (barren) tumors, RNA sequencing was sufficiently sensitive to detect transcripts (Table 1). This points to the presence of immune cellular infiltrates that allow stratification of spontaneous osteosarcomas of humans and dogs into immunologically “hot” and “cold” tumors. The transcriptional programs associated with immune cells were remarkably well-conserved across tumors from both species and did not show specificity regarding cell type or upregulation of specific molecules, such as those associated with immune checkpoints. While the increased presence of immune transcripts in tumors was associated with significantly better prognosis in human patients, such relationship was absent in dogs (Table 1). This observation is especially paradoxical when considering the reproducible success of experimental immunotherapies in canine osteosarcoma models (176), and even though the basis for it is not yet understood, it raises an important cautionary note in the application of canine osteosarcoma as a model for immunotherapy of human osteosarcoma.
Gorden et al. (177) showed that spontaneous canine hemangiosarcomas can be classified into three distinct molecular subtypes. Preliminary data suggest that these tumors in virtually all dogs that achieved durable remissions after conventional therapy show enrichment of immune gene signatures. Other groups have reported the immune characteristics of canine gliomas (178) and canine malignant melanoma (179), both showing similar patterns of immune infiltration to those reported for bone and vascular sarcomas.
It is widely accepted that macrophages play a major role in molding the TME, making them attractive targets for tumor therapy. Myeloid antigen presenting cells (APCs), and especially dendritic cells, control the initial steps in the cancer-immunity cycle, engulfing tumor cells and tumor debris and presenting it to T cells in the draining lymph nodes (155). Since tumors are derived from “self,” immune recognition can be compromised, and this process can lead to tolerance (155, 180). Immune recognition and activation, however, is aided by genomic instability. Tumors show a direct relationship between mutational burden and immune infiltration, and this relationship extends to the observed response to immunotherapies (167, 181, 182). After immune recognition, T cells must traffic to the tumor, extravasate and infiltrate the tumor stroma, recognize the cancer cells, and selectively kill them (155). Each of these steps creates opportunities for the tumor to inhibit or evade the immune response—and concomitantly, a potential step for therapeutic intervention.
Immune recognition of the tumor intensifies the selective pressures that drive tumor evolution through the process of immunoediting (183). T cells can potentially eliminate the majority of cells in a tumor that display particular mutations. The T cell receptor repertoire in tumors is becoming increasingly well-understood, following conventional roles of clonal evolution. In virally induced tumors, such as those associated with Epstein Barr virus, narrowing of the repertoire through strong selection for foreign viral epitopes is associated with worse prognosis (184). This observation extends to tumors without viral etiologies, where tumor epitopes promote selection of a narrow diversity of clones. Greater clonal heterogeneity is associated with a more favorable prognosis in multiple tumor types (185–187). Lymphocyte clonal diversity and the potential to modulate it therapeutically in canine osteosarcoma has been examined. Preliminary results document feasibility and show variation in the diversity of the T cell repertoire across different tumors (188). However, the influence of clonal diversity on outcomes for dogs with cancer remains to be determined.
The elimination phase of immunoediting gives way to an equilibrium phase where the immune system appears to control the tumor. However, editing is not static, and the process eventually favors outgrowth of resistant tumor cells that “edit” the epitopes recognized by the immune system. Editing can occur through downregulation of major histocompatibility complex (MHC) proteins, upregulation of proteins that resist T cell activation and killing, acquisition of the ability to kill activated T cells or resist T-cell induced apoptosis, or alteration of target epitopes by further mutation or epigenetic regulation (183). The host is able to respond to these immune evasive mechanisms, for example by deploying NK cells that recognize tumor (and other) cells that downregulate MHC (180, 189). However, most cases progress to the third phase of escape (183). The immune system has evolved over millions of years to defend hosts against lethal pathogens, with the function of cancer immunosurveillance probably arising as a secondary benefit (190). Cancer is rare before reproductive age and even into young adulthood, and so robust anti-tumor immunity is unlikely to create sufficiently large selective pressure to favor individual survival.
Some common steps in the evolution of the immune microenvironment of cancer has led to the development of highly effective immunotherapies. Immune checkpoint blockade using antibodies that interfere with binding of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) to CD80 and CD86, or with binding of PD-1 to PD-L1 and PD-L2, are the first therapies directed against the TME that have been effective at achieving meaningful cancer control. Durable remissions in as many as 50% of patients with advanced cutaneous melanoma, various types of tobacco-related malignancies, gastrointestinal tumors, and certain blood cancers have been achieved via these therapies (182, 191–193). The best responses cluster in tumors (or tumor types) with high mutational burden and robust immune infiltration (182). Multitudes of other immune-enhancing therapies that can modulate the TME, and especially that can shift the inflammatory response toward T helper-1 (Th1) programs are under development, alone or in combination with immune checkpoint blockade. These include Toll-like receptor (TLR) agonists, oncolytic viruses, VEGF inhibitors that promote blood vessel normalization and improve T cell trafficking to tumors, among others (194). Indeed, the first chimeric antigen receptor redirected T cells (CAR T cells) directed against mesothelin, a protein expressed exclusively in the TME, have completed early phase clinical trials (ClinicalTrials.gov Identifier: NCT01583686).
Spontaneous canine cancers provide a rich resource to understand both conserved and species-specific mechanisms that create and maintain the tumor immune landscape. Dogs can teach us much about therapeutic immune system manipulation in the context of cancer, including the potential to alter the TME to enhance immune responses. There are numerous published studies on the subject, including using pharmacologic intratumoral delivery of vectors encoding FasL (195), a variety of tumor vaccine approaches that activate molecular pattern receptors (196–198), applications of CAR-T cell immunotherapies (199), and others (200, 201). That being said, these data must be interpreted with due caution. It should be recognized that the canine and human immune systems are separated by millions of years of evolution and were adapted to distinct pathogens in distinct environments until both species collided into shared social structures about 20,000 years ago (202), that became more intimate over the past three to five decades (203). The timeline of the canine-human relationship is rather short, and the strong influence of artificial selection will inevitably diminish the role of immunosurveillance in adaptive evolution for both species.
Involvement of the TME in Tumor Invasion and Metastasis
The components of the TME work in concert through epigenetic and functional means to promote tumor cell invasion and metastasis. Although metastasis can occur at any point in space and time during the course of tumor evolution, the cellular, structural, and molecular components of the TME are able to enhance numerous pro-tumorigenic activities, which in turn facilitate invasion and enhance the metastatic potential of tumor cells. The organization of the primary tumor niche requires significant alterations to the normal extracellular environment. Molecular interactions are highly specific to different tumors and can vary substantially even within tumor types; but the general features include “loosening” of stable cell-cell adhesion, loss of cell polarity, and reorganization of the cytoskeleton, as well as stiffening of the extracellular matrix, which enhances motility, facilitates invasion, and enables the metastatic phenotype (59, 71, 73). Collectively, these alterations are linked to the “epithelial to mesenchymal transition” (EMT). EMT is characterized by genetic and epigenetic changes that alter expression of genes encoding cadherins, occludins, claudins, integrins, and a multitude of other adhesion and cell surface proteins, as well as cytokines, extracellular proteases, and many others. Reduced expression of epithelial (E) cadherin (CDH1) with a concomitant increase in neuronal (N) cadherin (CDH2) is among the most well-studied features of EMT (12, 15, 64). Loss of E-cadherin, which is widely conserved in epithelial tumors across species, is an indicator of more aggressive behavior and poor prognosis for a multitude of human as well as canine (204, 205) tumors. Tumor cells themselves can enhance EMT potential; for example VEGF-A produced by tumor cells, in contrast to VEGF-A produced by the TME, has been shown to promote EMT (206). Regardless, the influence of the TME on EMT should not be overlooked. For example, collagen type I in the adjacent ECM has been implicated in promoting EMT through numerous mechanisms, including upregulation of NFκB, Snail, and lymphoid enhancer-binding factor-1 (LEF-1) in tumor cells. These factors promote a mesenchymal phenotype with subsequent cell migration [Figure 4; (207)]. Other pro-EMT transcripts, such as TWIST1, are expressed in higher concentration in tumor cells adjacent to collagen dense stroma (208). Additionally, intercellular interactions establish a pro-inflammatory environment where autocrine and paracrine loops, such as those mediated by interactions between colony-stimulating factor-1 (CSF-1) and its receptor in CSCs and TAMs, support the EMT transcriptional programs (209).
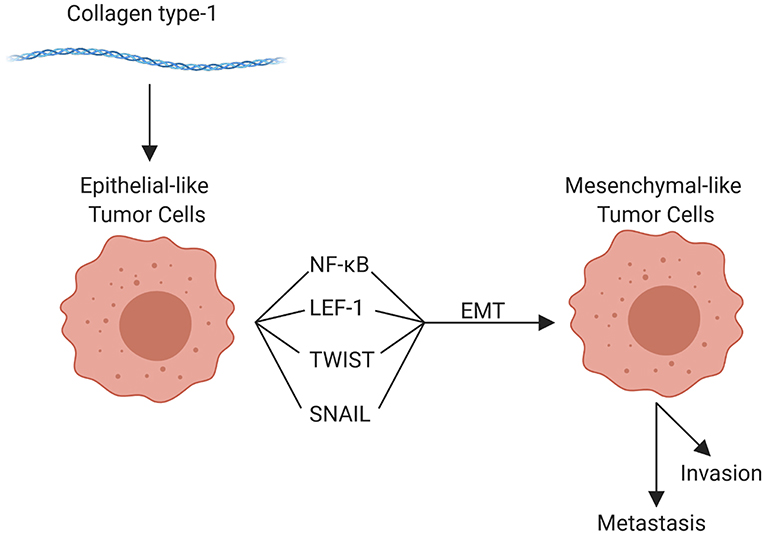
Figure 4. Role of collagen type 1 in the tumor microenvironment and in epithelial-mesenchymal transition (EMT). Collagen type 1 leads to the upregulation of numerous factors that promote EMT, promoting invasion, and metastasis (created with Biorender.com).
Changes in cell-to-cell contacts in sarcomas have been less well-studied, almost certainly due to the rarity of these tumors in humans. The concept of EMT in sarcomas presents a paradox: some, but not all sarcomas exhibit aggressive, rapidly metastatic phenotypes, and cells in these tumors undergo phenotypic changes that resemble EMT. Yet, all sarcoma cells possess a mesenchymal phenotype. This has given rise to a more nuanced vision of EMT, and the reverse process called mesenchymal to epithelial transition (MET), where the transcriptional and epigenetic mechanisms that regulate these transitions give rise to metastable phenotypes that are adaptive (12). In other words, cells acquire these phenotypes in response to environmental cues, as well as to natural selection on a microscopic scale.
Emerging evidence suggests that exosomes are critically important mediators that mold the distant or metastatic tumor niche in blood-derived and solid tumors. Exosomes are formed by inward budding of early endosome membranes by the endosomal sorting complex required for the transport (ESCRT) complex. Mature endosomes, also known as multi-vesicular bodies (MVBs) fuse with plasma membranes releasing exosomes vesicles to the extracellular space. Exosomes circulate systemically and can bind to and merge with other cells, creating a mechanism for horizontal transfer of activated oncoproteins, oncogenic DNA, and oncogenic and regulatory microRNAs (210). For example, CAAs and ad-MSCs, a developmentally plastic cell type that can be derived from, or differentiate to adipocytes within the TME, can produce extracellular vesicles (211, 212). To the authors' knowledge, there are no reports characterizing the role of CAAs in dogs; however, extracellular vesicles from human adipocytes have been shown to enhance tumor cell invasiveness by providing substrates for increased fatty acid oxidation in the tumor cells (211). Tumor exosomes carry biologically active molecules; thus, they can reprogram the activity of cells locally, as well as at distant sites, in essence “conditioning” these sites for metastatic tumor growth. Exosomes contribute to the formation of each component of the primary tumor niche, including the metabolic, immune, hypoxic, and infiltrating regions (68, 213). However, their role in metastatic conditioning makes them attractive targets for diagnosis and therapy. Early data in the field of exosome biology and metastasis showed that secreted exosomes could condition regional lymph nodes to create a favorable metastatic niche for melanoma cells (214). These results have been extensively reproduced in multiple experimental tumor systems, extending to other niches such as liver and lungs (213). Strategies to leverage the capacity and specificity of exosomes to home to the metastatic niche are under development as means to improve delivery and activity of drugs that can delay or arrest metastasis. Perhaps most promising is the use of exosomes in early detection and targeted chemoprevention. Canine osteosarcoma was instrumental in the development of an innovative discovery platform to distinguish RNA signatures in serum exosomes originating, respectively from tumor and host cells (215, 216). While this work is still in the early stages, there is potential that these signatures can be used in rationally designed screening programs aimed at detecting changes in the TME in the earliest stages of tumor formation. Novel therapies may be developed that are able to arrest the development of tumors before they form, creating a completely new outlook on cancer prevention.
Summary
Cancers are complex and dynamic multicellular tissues; multiple distinct events contribute to initiation, promotion, and progression. Ultimately, these events converge into more rigid molecular programs and create recognizable histological tumor phenotypes that are widely conserved across species. Tumor formation is a tightly orchestrated process, molded by selection to support growth and survival of a clonal population of immortalized cells. This review has demonstrated the complexity and intricacies of the TME in the human and mouse, and established, to the best of the author's abilities, the same complexities within the dog. While there is much room for growth in the understanding of the TME in the dog, the current knowledge base in conjunction with the information known about the TME in humans and mice, provides a solid foothold for the development of further basic and clinical endeavors.
Author Contributions
KL, JK, MD, and JM contributed to the writing of this manuscript. Figures were rendered by KL and AS. Editing was performed by all authors of this paper.
Funding
The authors wish to acknowledge support from grants R21CA208529 (JM) and R50CA211249 (AS) from the National Cancer Institute of the National Institutes of Health, grant CA170218 (JM) from the United States Department of Defense, grant T2018-018 (JM) from the V Foundation for Cancer Research, and by the Zach Sobiech Osteosarcoma Fund of the Children's Cancer Research Fund. JK receives support from the Animal Cancer Care and Research Program of the University of Minnesota. AS receives support from the Masonic Cancer Center Comprehensive Cancer Center Support Grant (P30CA077598; bioinformatics core) from the National Cancer Institute of the National Institutes of Health. JM was supported by the Alvin and June Perlman Endowed Chair in Animal Oncology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, U.S. Department of Defense, the V Foundation for Cancer Research, or the Children's Cancer Research Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge Erin Dickerson, Doug Fearon, Derek Korpela, Ben Langsten, and Ashleigh McKinnon for their invaluable editorial suggestions and advice on this manuscript. We extend our deepest appreciation to Jim McCarthy for his insightful and constructive editing during the writing process.
References
1. Albuquerque TAF, Drummond do Val L, Doherty A, de Magalhães JP. From humans to hydra: patterns of cancer across the tree of life. Biol Rev Camb Philos Soc. (2018) 93:1715–34. doi: 10.1111/brv.12415
2. Schiffman JD, Breen M. Comparative oncology: what dogs and other species can teach us about humans with cancer. Philos Trans R Soc Lond B Biol Sci. (2015) 370:20140231. doi: 10.1098/rstb.2014.0231
3. Pitot HC. The molecular biology of carcinogenesis. Cancer. (1993) 72:962–70. doi: 10.1002/1097-0142(19930801)72:3+<962::aid-cncr2820721303>3.0.co;2-h
4. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
5. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. (2012) 125(Pt 23):5591–6. doi: 10.1242/jcs.116392
6. Prager BC, Xie Q, Bao S, Rich JN. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell. (2019) 24:41–53. doi: 10.1016/j.stem.2018.12.009
7. Kim JH, Frantz AM, Sarver AL, Gorden Klukas BH, Lewellen M, O'Brien TD, et al. Modulation of fatty acid metabolism and immune suppression are features of in vitro tumour sphere formation in ontogenetically distinct dog cancers. Vet Comp Oncol. (2018) 16:E176–84. doi: 10.1111/vco.12368
8. Kim JH, Frantz AM, Anderson KL, Graef AJ, Scott MC, Robinson S, et al. Interleukin-8 promotes canine hemangiosarcoma growth by regulating the tumor microenvironment. Exp Cell Res. (2014) 323:155–64. doi: 10.1016/j.yexcr.2014.02.020
9. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. (2013) 39:611–21. doi: 10.1016/j.immuni.2013.08.025
10. Maccalli C, Parmiani G, Ferrone S. Immunomodulating and immunoresistance properties of cancer-initiating cells: implications for the clinical success of immunotherapy. Immunol Invest. (2017) 46:221–38. doi: 10.1080/08820139.2017.1280051
11. Lin W, Modiano JF, Ito D. Stage-specific embryonic antigen: determining expression in canine glioblastoma, melanoma, and mammary cancer cells. J Vet Sci. (2017) 18:101–4. doi: 10.4142/jvs.2017.18.1.101
12. Sannino G, Marchetto A, Kirchner T, Grunewald TGP. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transition in mesenchymal tumors: a paradox in sarcomas? Cancer Res. (2017) 77:4556–61. doi: 10.1158/0008-5472.CAN-17-0032
13. Li Y, Laterra J. Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res. (2012) 72:576–80. doi: 10.1158/0008-5472.CAN-11-3070
14. Zhou H, Neelakantan D, Ford HL. Clonal cooperativity in heterogenous cancers. Semin Cell Dev Biol. (2017) 64:79–89. doi: 10.1016/j.semcdb.2016.08.028
15. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. (2013) 19:1438–49. doi: 10.1038/nm.3336
16. Kim JH, Anderson KL, Frantz AM, Graef AJ, Dickerson EB, Modiano JF. IL-8 and Slug regulate cancer cell self-renewal and microenvironment interactions in hemangiosarcoma. In: Proceedings of the Annual Meeting of the Veterinary Cancer Society VCS. Minneapolis, MI (2013).
17. Cozzo AJ, Fuller AM, Makowski L. Contribution of adipose tissue to development of cancer. Compr Physiol. (2017) 8:237–82. doi: 10.1002/cphy.c170008
18. Perez Alenza MD, Peña L, del Castillo N, Nieto AI. Factors influencing the incidence and prognosis of canine mammary tumours. J Small Anim Pract. (2000) 41:287–91. doi: 10.1111/j.1748-5827.2000.tb03203.x
19. Radin MJ, Sharkey LC, Holycross BJ. Adipokines: a review of biological and analytical principles and an update in dogs, cats, and horses. Vet Clin Pathol. (2009) 38:136–56. doi: 10.1111/j.1939-165X.2009.00133.x
20. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. (2011) 17:1498–503. doi: 10.1038/nm.2492
21. Himbert C, Delphan M, Scherer D, Bowers LW, Hursting S, Ulrich CM. Signals from the adipose microenvironment and the obesity-cancer link-a systematic review. Cancer Prev Res. (2017) 10:494–506. doi: 10.1158/1940-6207.CAPR-16-0322
22. Lim HY, Im KS, Kim NH, Kim HW, Shin JI, Yhee JY, et al. Effects of obesity and obesity-related molecules on canine mammary gland tumors. Vet Pathol. (2015) 52:1045–51. doi: 10.1177/0300985815579994
23. Bulun SE, Price TM, Aitken J, Mahendroo MS, Simpson ER. A link between breast cancer and local estrogen biosynthesis suggested by quantification of breast adipose tissue aromatase cytochrome P450 transcripts using competitive polymerase chain reaction after reverse transcription. J Clin Endocrinol Metab. (1993) 77:1622–8. doi: 10.1210/jcem.77.6.8117355
24. Kapur SK, Katz AJ. Review of the adipose derived stem cell secretome. Biochimie. (2013) 95:2222–8. doi: 10.1016/j.biochi.2013.06.001
25. Rivera-Cruz CM, Shearer JJ, Figueiredo Neto M, Figueiredo ML. The immunomodulatory effects of mesenchymal stem cell polarization within the tumor microenvironment niche. Stem Cells Int. (2017) 2017:4015039. doi: 10.1155/2017/4015039
26. Chow L, Johnson V, Coy J, Regan D, Dow S. Mechanisms of immune suppression utilized by canine adipose and bone marrow-derived mesenchymal stem cells. Stem Cells Dev. (2017) 26:374–89. doi: 10.1089/scd.2016.0207
28. Dvorak HF. Tumors: wounds that do not heal-A historical perspective with a focus on the fundamental roles of increased vascular permeability and clotting. Semin Thromb Hemost. (2019) 45:576–92. doi: 10.1055/s-0039-1687908
29. Stoker MG, Shearer M, O'Neill C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J Cell Sci. (1966) 1:297–310.
30. Kirk D, Kaighn ME. Non-reciprocal interactions in normal-neoplastic human cells. A quantitative, kinetic approach to cell interactions in vitro. Cell Biol Int Rep. (1980) 4:599–608. doi: 10.1016/0309-1651(80)90027-2
31. Flaberg E, Markasz L, Petranyi G, Stuber G, Dicso F, Alchihabi N, et al. High-throughput live-cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts. Int J Cancer. (2011) 128:2793–802. doi: 10.1002/ijc.25612
32. Mehta PP, Bertram JS, Loewenstein WR. Growth inhibition of transformed cells correlates with their junctional communication with normal cells. Cell. (1986) 44:187–96. doi: 10.1016/0092-8674(86)90497-6
33. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
34. Madar S, Goldstein I, Rotter V. 'Cancer associated fibroblasts'–more than meets the eye. Trends Mol Med. (2013) 19:447–53. doi: 10.1016/j.molmed.2013.05.004
35. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. (2016) 16:582–98. doi: 10.1038/nrc.2016.73
36. Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. (2015) 6:10204. doi: 10.1038/ncomms10204
37. Klymenko Y, Nephew KP. Epigenetic crosstalk between the tumor microenvironment and ovarian cancer cells: a therapeutic road less traveled. Cancers. (2018) 10:E295. doi: 10.3390/cancers10090295
38. Bu L, Baba H, Yoshida N, Miyake K, Yasuda T, Uchihara T, et al. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene. (2019) 38:4887–901. doi: 10.1038/s41388-019-0765-y
39. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. (2010) 17:135–47. doi: 10.1016/j.ccr.2009.12.041
40. Sappino AP, Skalli O, Jackson B, Schürch W, Gabbiani G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int J Cancer. (1988) 41:707–12. doi: 10.1002/ijc.2910410512
41. Yoshimoto S, Hoshino Y, Izumi Y, Takagi S. α-smooth muscle actin expression in cancerassociated fibroblasts in canine epithelial tumors. Japanese J Vet Res. (2017) 65:135–44. doi: 10.14943/jjvr.65.3.135
42. Buganim Y, Madar S, Rais Y, Pomeraniec L, Harel E, Solomon H, et al. Transcriptional activity of ATF3 in the stromal compartment of tumors promotes cancer progression. Carcinogenesis. (2011) 32:1749–57. doi: 10.1093/carcin/bgr203
43. Goetz JG, Minguet S, Navarro-Lérida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. (2011) 146:148–63. doi: 10.1016/j.cell.2011.05.040
44. Król M, Pawłowski KM, Szyszko K, Maciejewski H, Dolka I, Manuali E, et al. The gene expression profiles of canine mammary cancer cells grown with carcinoma-associated fibroblasts (CAFs) as a co-culture in vitro. BMC Vet Res. (2012) 8:35. doi: 10.1186/1746-6148-8-35
45. Rozenchan PB, Carraro DM, Brentani HLD, de Carvalho Mota LD, Bastos EP, e Ferreira EN, et al. Reciprocal changes in gene expression profiles of cocultured breast epithelial cells and primary fibroblasts. Int J Cancer. (2009) 125:2767–77. doi: 10.1002/ijc.24646
46. van Duursen MB, Nijmeijer SM, de Morree ES, de Jong PC, van den Berg M. Genistein induces breast cancer-associated aromatase and stimulates estrogen-dependent tumor cell growth in in vitro breast cancer model. Toxicology. (2011) 289:67–73. doi: 10.1016/j.tox.2011.07.005
47. Purohit A, Ghilchik MW, Duncan L, Wang DY, Singh A, Walker MM, et al. Aromatase activity and interleukin-6 production by normal and malignant breast tissues. J Clin Endocrinol Metab. (1995) 80:3052–8. doi: 10.1210/jcem.80.10.7559896
48. Salem HK, Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. (2010) 28:585–96. doi: 10.1002/stem.269
49. Lin W, Huang L, Li Y, Fang B, Li G, Chen L, et al. Mesenchymal stem cells and cancer: clinical challenges and opportunities. Biomed Res Int. (2019) 2019:2820853. doi: 10.1155/2019/2820853
50. Fang M, Yuan J, Peng C, Li Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. (2014) 35:2871–82. doi: 10.1007/s13277-013-1511-7
51. Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. (2006) 4:38. doi: 10.1186/1741-7015-4-38
52. Yamauchi M, Barker TH, Gibbons DL, Kurie JM. The fibrotic tumor stroma. J Clin Invest. (2018) 128:16–25. doi: 10.1172/JCI93554
53. Case A, Brisson BK, Durham AC, Rosen S, Monslow J, Buza E, et al. Identification of prognostic collagen signatures and potential therapeutic stromal targets in canine mammary gland carcinoma. PLoS ONE. (2017) 12:e0180448. doi: 10.1371/journal.pone.0180448
54. Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. (2011) 178:1221–32. doi: 10.1016/j.ajpath.2010.11.076
55. Ettlin J, Clementi E, Amini P, Malbon A, Markkanen E. Analysis of gene expression signatures in cancer-associated stroma from canine mammary tumours reveals molecular homology to human breast carcinomas. Int J Mol Sci. (2017) 18:E1101. doi: 10.3390/ijms18051101
56. Jia D, Liu Z, Deng N, Tan TZ, Huang RY, Taylor-Harding B, et al. A COL11A1-correlated pan-cancer gene signature of activated fibroblasts for the prioritization of therapeutic targets. Cancer Lett. (2016) 382:203–14. doi: 10.1016/j.canlet.2016.09.001
57. Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. (2004) 4:839–49. doi: 10.1038/nrc1477
58. Trembley JH, Unger GM, Korman VL, Abedin MJ, Nacusi LP, Vogel RI, et al. Tenfibgen ligand nanoencapsulation delivers bi-functional anti-CK2 RNAi oligomer to key sites for prostate cancer targeting using human xenograft tumors in mice. PLoS ONE. (2014) 9:e109970. doi: 10.1371/journal.pone.0109970
59. Poltavets V, Kochetkova M, Pitson SM, Samuel MS. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front Oncol. (2018) 8:431. doi: 10.3389/fonc.2018.00431
60. Jang I, Beningo KA. Integrins, CAFs and mechanical forces in the progression of cancer. Cancers. (2019) 11:E721. doi: 10.3390/cancers11050721
61. Stupack DG. Integrins as a distinct subtype of dependence receptors. Cell Death Differ. (2005) 12:1021–30. doi: 10.1038/sj.cdd.4401658
62. Takahashi N, Nobusue H, Shimizu T, Sugihara E, Yamaguchi-Iwai S, Onishi N, et al. ROCK inhibition induces terminal adipocyte differentiation and suppresses tumorigenesis in chemoresistant osteosarcoma cells. Cancer Res. (2019) 79:3088–99. doi: 10.1158/0008-5472.CAN-18-2693
63. Cohen DJ, Nelson WJ. Secret handshakes: cell-cell interactions and cellular mimics. Curr Opin Cell Biol. (2018) 50:14–9. doi: 10.1016/j.ceb.2018.01.001
64. Gloushankova NA, Zhitnyak IY, Rubtsova SN. Role of epithelial-mesenchymal transition in tumor progression. Biochemistry. (2018) 83:1469–76. doi: 10.1134/S0006297918120052
65. Lou E, Zhai E, Sarkari A, Desir S, Wong P, Iizuka Y, et al. Cellular and molecular networking within the ecosystem of cancer cell communication via tunneling nanotubes. Front Cell Dev Biol. (2018) 6:95. doi: 10.3389/fcell.2018.00095
66. Yamashita YM, Inaba M, Buszczak M. Specialized intercellular communications via cytonemes and nanotubes. Annu Rev Cell Dev Biol. (2018) 34:59–84. doi: 10.1146/annurev-cellbio-100617-062932
67. Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. (2016) 30:836–48. doi: 10.1016/j.ccell.2016.10.009
68. Maia J, Caja S, Strano Moraes MC, Couto N, Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. (2018) 6:18. doi: 10.3389/fcell.2018.00018
69. Robinson SC, Coussens LM. Soluble mediators of inflammation during tumor development. Adv Cancer Res. (2005) 93:159–87. doi: 10.1016/S0065-230X(05)93005-4
70. Rodriguez AM, Graef AJ, LeVine DN, Cohen IR, Modiano JF, Kim JH. Association of sphingosine-1-phosphate (S1P)/S1P receptor-1 pathway with cell proliferation and survival in canine hemangiosarcoma. J Vet Intern Med. (2015) 29:1088–97. doi: 10.1111/jvim.13570
71. Huang S, Ingber DE. Cell tension, matrix mechanics, and cancer development. Cancer Cell. (2005) 8:175–6. doi: 10.1016/j.ccr.2005.08.009
72. Wei L, Surma M, Shi S, Lambert-Cheatham N, Shi J. Novel insights into the roles of rho kinase in cancer. Arch Immunol Ther Exp. (2016) 64:259–78. doi: 10.1007/s00005-015-0382-6
73. Holle AW, Young JL, Spatz JP. In vitro cancer cell-ECM interactions inform In vivo cancer treatment. Adv Drug Deliv Rev. (2016) 97:270–9. doi: 10.1016/j.addr.2015.10.007
74. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. (2008) 14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843
75. De Larco JE, Wuertz BR, Furcht LT. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin Cancer Res. (2004) 10:4895–900. doi: 10.1158/1078-0432.CCR-03-0760
76. Gimbrone MA, Leapman SB, Cotran RS, Folkman J. Tumor dormancy In vivo by prevention of neovascularization. J Exp Med. (1972) 136:261–76. doi: 10.1084/jem.136.2.261
77. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. (1971) 285:1182–6. doi: 10.1056/NEJM197111182852108
78. Bielenberg DR, Zetter BR. The contribution of angiogenesis to the process of metastasis. Cancer J. (2015) 21:267–73. doi: 10.1097/PPO.0000000000000138
79. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. (2007) 26:225–39. doi: 10.1007/s10555-007-9055-1
80. Snyder SA, Dewhirst MW, Hauck ML. The role of hypoxia in canine cancer. Vet Comp Oncol. (2008) 6:213–23. doi: 10.1111/j.1476-5829.2008.00163.x
81. Samanta D, Semenza GL. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim Biophys Acta Rev Cancer. (2018) 1870:15–22. doi: 10.1016/j.bbcan.2018.07.002
82. Liu W, Xin H, Eckert DT, Brown JA, Gnarra JR. Hypoxia and cell cycle regulation of the von Hippel-Lindau tumor suppressor. Oncogene. (2011) 30:21–31. doi: 10.1038/onc.2010.395
83. Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. (2003) 9:677–84. doi: 10.1038/nm0603-677
84. Watson JA, Watson CJ, McCann A, Baugh J. Epigenetics, the epicenter of the hypoxic response. Epigenetics. (2010) 5:293–96. doi: 10.4161/epi.5.4.11684
85. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. (2010) 40:294–309. doi: 10.1016/j.molcel.2010.09.022
86. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. (2000) 6:389–95. doi: 10.1038/74651
87. Corvera S, Gealekman O. Adipose tissue angiogenesis: impact on obesity and type-2 diabetes. Biochim Biophys Acta. (2014) 1842:463–72. doi: 10.1016/j.bbadis.2013.06.003
88. Rahimi N. Vascular endothelial growth factor receptors: molecular mechanisms of activation and therapeutic potentials. Exp Eye Res. (2006) 83:1005–16. doi: 10.1016/j.exer.2006.03.019
89. Gutiérrez-González A, Aguilera-Montilla N, Ugarte-Berzal E, Bailón E, Cerro-Pardo I, Sánchez-Maroto C, et al. α4β1 integrin associates with VEGFR2 in CLL cells and contributes to VEGF binding and intracellular signaling. Blood Adv. (2019) 3:2144–8. doi: 10.1182/bloodadvances.2019000019
90. Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. (2006) 312:549–60. doi: 10.1016/j.yexcr.2005.11.012
91. Tamburini BA, Trapp S, Phang TL, Schappa JT, Hunter LE, Modiano JF. Gene expression profiles of sporadic canine hemangiosarcoma are uniquely associated with breed. PLoS ONE. (2009) 4:e5549. doi: 10.1371/journal.pone.0005549
92. Peters KG, Kontos CD, Lin PC, Wong AL, Rao P, Huang L, et al. Functional significance of Tie2 signaling in the adult vasculature. Recent Prog Horm Res. (2004) 59:51–71. doi: 10.1210/rp.59.1.51
93. Pichiule P, Chavez JC, LaManna JC. Hypoxic regulation of angiopoietin-2 expression in endothelial cells. J Biol Chem. (2004) 279:12171–80. doi: 10.1074/jbc.M305146200
94. Harada S, Nagy JA, Sullivan KA, Thomas KA, Endo N, Rodan GA, et al. Induction of vascular endothelial growth factor expression by prostaglandin E2 and E1 in osteoblasts. J Clin Invest. (1994) 93:2490–6. doi: 10.1172/JCI117258
95. Wu G., Luo J., Rana J. S., Laham R., Sellke F. W., Li J. (2006). Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells. Cardiovasc Res 69, 512–519. doi: 10.1016/j.cardiores.2005.09.019
96. Yoshino Y, Aoyagi M, Tamaki M, Duan L, Morimoto T, Ohno K. Activation of p38 MAPK and/or JNK contributes to increased levels of VEGF secretion in human malignant glioma cells. Int J Oncol. (2006) 29:981–7. doi: 10.3892/ijo.29.4.981
97. Tremblay C, Doré M, Bochsler PN, Sirois J. Induction of prostaglandin G/H synthase-2 in a canine model of spontaneous prostatic adenocarcinoma. J Natl Cancer Inst. (1999) 91:1398–403. doi: 10.1093/jnci/91.16.1398
98. Madaan S, Abel PD, Chaudhary KS, Hewitt R, Stott MA, Stamp GW, et al. Cytoplasmic induction and over-expression of cyclooxygenase-2 in human prostate cancer: implications for prevention and treatment. BJU Int. (2000) 86:736–41. doi: 10.1046/j.1464-410x.2000.00867.x
99. Khan KN, Knapp DW, Denicola DB, Harris RK. Expression of cyclooxygenase-2 in transitional cell carcinoma of the urinary bladder in dogs. Am J Vet Res. (2000) 61:478–81. doi: 10.2460/ajvr.2000.61.478
100. Mohammed SI, Knapp DW, Bostwick DG, Foster RS, Khan KN, Masferrer JL, et al. Expression of cyclooxygenase-2 (COX-2) in human invasive transitional cell carcinoma (TCC) of the urinary bladder. Cancer Res. (1999) 59:5647–50. doi: 10.1097/00005392-199904020-00268
101. Zhi H, Wang L, Zhang J, Zhou C, Ding F, Luo A, et al. Significance of COX-2 expression in human esophageal squamous cell carcinoma. Carcinogenesis. (2006) 27:1214–21. doi: 10.1093/carcin/bgi304
102. Pestili de Almeida EM, Piché C, Sirois J, Doré M. Expression of cyclo-oxygenase-, in naturally occurring squamous cell carcinomas in dogs. J Histochem Cytochem. (2001) 49:867–75. doi: 10.1177/002215540104900707
103. Mohammed SI, Bennett PF, Craig BA, Glickman NW, Mutsaers AJ, et al. Effects of the cyclooxygenase inhibitor, piroxicam, on tumor response, apoptosis, and angiogenesis in a canine model of human invasive urinary bladder cancer. Cancer Res. (2002) 62:356–8.
104. McMillan SK, Boria P, Moore GE, Widmer WR, Bonney PL, Knapp DW. Antitumor effects of deracoxib treatment in 26 dogs with transitional cell carcinoma of the urinary bladder. J Am Vet Med Assoc. (2011) 239:1084–9. doi: 10.2460/javma.239.8.1084
105. Jolly K, Cheng KK, Langman MJ. NSAIDs and gastrointestinal cancer prevention. Drugs. (2002) 62:945–56. doi: 10.2165/00003495-200262060-00006
106. Brasky TM, Bonner MR, Moysich KB, Ambrosone CB, Nie J, Tao MH, et al. Non-steroidal anti-inflammatory drugs (NSAIDs) and breast cancer risk: differences by molecular subtype. Cancer Causes Control. (2011) 22:965–75. doi: 10.1007/s10552-011-9769-9
107. Sato-Dahlman M, Yamamoto M. The development of oncoltyic adenovirus therapy in the past and future - for the case of pancreatic cancer. Curr Cancer Drug Targets. (2018) 18:153–61. doi: 10.2174/1568009617666170222123925
108. Yamamoto M, Davydova J, Wang M, Siegal GP, Krasnykh V, Vickers SM, et al. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology. (2003) 125:1203–18. doi: 10.1016/S0016-5085(03)01196-X
109. Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. (2004) 5:429–41. doi: 10.1016/S1535-6108(04)00115-1
110. Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, et al. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. (2007) 26:63–74. doi: 10.1016/j.molcel.2007.02.024
111. Cao Y, Eble JM, Moon E, Yuan H, Weitzel DH, Landon CD, et al. Tumor cells upregulate normoxic HIF-1α in response to doxorubicin. Cancer Res. (2013) 73:6230–42. doi: 10.1158/0008-5472.CAN-12-1345
112. Chi JT, Thrall DE, Jiang C, Snyder S, Fels D, Landon C, et al. Comparison of genomics and functional imaging from canine sarcomas treated with thermoradiotherapy predicts therapeutic response and identifies combination therapeutics. Clin Cancer Res. (2011) 17:2549–60. doi: 10.1158/1078-0432.CCR-10-2583
113. Thrall DE, Maccarini P, Stauffer P, Macfall J, Hauck M, Snyder S, et al. Thermal dose fractionation affects tumour physiological response. Int J Hyperthermia. (2012) 28:431–40. doi: 10.3109/02656736.2012.689087
114. Zhang X, Ashcraft KA, Betof Warner A, Nair SK, Dewhirst MW. Can exercise-induced modulation of the tumor physiologic microenvironment improve antitumor immunity? Cancer Res. (2019) 79:2447–56. doi: 10.1158/0008-5472.CAN-18-2468
115. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/fimmu.2014.00514
116. Leblond MM, Gerault AN, Corroyer-Dulmont A, MacKenzie ET, Petit E, Bernaudin M, et al. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology. (2016) 5:e1056442. doi: 10.1080/2162402X.2015.1056442
117. Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol. (2015) 53:676–88. doi: 10.1165/rcmb.2015-0012OC
118. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
119. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
120. Huang JH, Cárdenas-Navia LI, Caldwell CC, Plumb TJ, Radu CG, Rocha PN, et al. Requirements for T lymphocyte migration in explanted lymph nodes. J Immunol. (2007) 178:7747–55. doi: 10.4049/jimmunol.178.12.7747
121. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. (2004) 10:858–64. doi: 10.1038/nm1075
122. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. (2003) 198:1391–402. doi: 10.1084/jem.20030267
123. Westendorf AM, Skibbe K, Adamczyk A, Buer J, Geffers R, Hansen W, et al. Hypoxia enhances immunosuppression by inhibiting CD4+ effector T cell function and promoting treg activity. Cell Physiol Biochem. (2017) 41:1271–84. doi: 10.1159/000464429
124. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
125. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
126. Kennedy KM, Dewhirst MW. Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. (2010) 6:127–48. doi: 10.2217/fon.09.145
127. Schroeder T, Yuan H, Viglianti BL, Peltz C, Asopa S, Vujaskovic Z, et al. Spatial heterogeneity and oxygen dependence of glucose consumption in R3230Ac and fibrosarcomas of the Fischer 344 rat. Cancer Res. (2005) 65:5163–71. doi: 10.1158/0008-5472.CAN-04-3900
128. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. (2000) 60:916–21.
129. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. (2008) 118:3930–42. doi: 10.1172/JCI36843
130. Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS ONE. (2013) 8:e75154. doi: 10.1371/journal.pone.0075154
131. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. (2017) 3:169–80. doi: 10.1016/j.trecan.2017.01.005
132. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
133. Sonveaux P, Copetti T, De Saedeleer CJ, Végran F, Verrax J, Kennedy KM, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE. (2012) 7:e33418. doi: 10.1371/journal.pone.0033418
134. Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, et al. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys. (2001) 51:349–53. doi: 10.1016/S0360-3016(01)01630-3
135. Lora-Michiels M, Yu D, Sanders L, Poulson JM, Azuma C, Case B, et al. Extracellular pH and P-31 magnetic resonance spectroscopic variables are related to outcome in canine soft tissue sarcomas treated with thermoradiotherapy. Clin Cancer Res. (2006) 12:5733–40. doi: 10.1158/1078-0432.CCR-05-2669
136. Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol. (2001) 69:522–30.
137. Wu NZ, Klitzman B, Dodge R, Dewhirst M W. (1992). Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Res 52, 4265–4268.
138. Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest. (2011) 121:3846–59. doi: 10.1172/JCI44952
139. Bessa X, Elizalde JI, Mitjans F, Piñol V, Miquel R, Panés J, et al. Leukocyte recruitment in colon cancer: role of cell adhesion molecules, nitric oxide, and transforming growth factor beta1. Gastroenterology. (2002) 122:1122–32. doi: 10.1053/gast.2002.32369
140. Ping YF, Zhang X, Bian XW. Cancer stem cells and their vascular niche: do they benefit from each other? Cancer Lett. (2016) 380:561–7. doi: 10.1016/j.canlet.2015.05.010
141. Cao X, Geradts J, Dewhirst MW, Lo HW. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene. (2012) 31:104–15. doi: 10.1038/onc.2011.219
142. Al-Rawi MA, Mansel RE, Jiang WG. Lymphangiogenesis and its role in cancer. Histol Histopathol. (2005) 20:283–98. doi: 10.1177/1947601911423028
143. Sleeckx N, Van Brantegem L, Fransen EG, Van den Eynden Casteleyn C, Veldhuis Kroeze E, Van Ginneken C. Evaluation of immunohistochemical markers of lymphatic and blood vessels in canine mammary tumours. J Comp Pathol. (2013) 148:307–17. doi: 10.1016/j.jcpa.2012.09.007
144. Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. (2010) 140:460–76. doi: 10.1016/j.cell.2010.01.045
145. Stacker SA, Baldwin ME, Achen MG. The role of tumor lymphangiogenesis in metastatic spread. FASEB J. (2002) 16:922–34. doi: 10.1096/fj.01-0945rev
146. Schoppmann SF, Fenzl A, Nagy K, Unger S, Bayer G, Geleff S, et al. VEGF-C expressing tumor-associated macrophages in lymph node positive breast cancer: impact on lymphangiogenesis and survival. Surgery. (2006) 139:839–46. doi: 10.1016/j.surg.2005.12.008
147. Karpanen T, Alitalo K. Molecular biology and pathology of lymphangiogenesis. Annu Rev Pathol. (2008) 3:367–97. doi: 10.1146/annurev.pathmechdis.3.121806.151515
148. François M, Caprini A, Hosking B, Orsenigo F, Wilhelm D, Browne C, et al. Sox18 induces development of the lymphatic vasculature in mice. Nature. (2008) 456:643–7. doi: 10.1038/nature07391
149. Kim HS, Park YW. Metastasis via peritumoral lymphatic dilation in oral squamous cell carcinoma. Maxillofac Plast Reconstr Surg. (2014) 36:85–93. doi: 10.14402/jkamprs.2014.36.3.85
150. Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood. (2007) 109:1010–7. doi: 10.1182/blood-2006-05-021758
151. Legler DF, Uetz-von Allmen E, Hauser MA. CCR7: roles in cancer cell dissemination, migration and metastasis formation. Int J Biochem Cell Biol. (2014) 54:78–82. doi: 10.1016/j.biocel.2014.07.002
152. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. (2012) 1:191–9. doi: 10.1016/j.celrep.2012.01.005
153. Kedl RM, Tamburini BA. Antigen archiving by lymph node stroma: a novel function for the lymphatic endothelium. Eur J Immunol. (2015) 45:2721–9. doi: 10.1002/eji.201545739
154. Swartz MA. Immunomodulatory roles of lymphatic vessels in cancer progression. Cancer Immunol Res. (2014) 2:701–7. doi: 10.1158/2326-6066.CIR-14-0115
155. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
156. Estrela-Lima A, Araujo MS, Costa-Neto JM, Teixeira-Carvalho A, Barrouin-Melo SM, Cardoso SV, et al. Immunophenotypic features of tumor infiltrating lymphocytes from mammary carcinomas in female dogs associated with prognostic factors and survival rates. BMC Cancer. (2010) 10:256. doi: 10.1186/1471-2407-10-256
157. Kim JH, Hur JH, Lee SM, Im KS, Kim NH, Sur JH. Correlation of Foxp3 positive regulatory T cells with prognostic factors in canine mammary carcinomas. Vet J. (2012) 193:222–7. doi: 10.1016/j.tvjl.2011.10.022
158. Kim JH, Chon SK, Im KS, Kim NH, Cho KW, Sur JH. Infiltrating Foxp3+ regulatory T cells and histopathological features in canine classical and spermatocytic seminomas. Reprod Domest Anim. (2013) 48:218–22. doi: 10.1111/j.1439-0531.2012.02135.x
159. Chinnadurai R, Copland IB, Patel SR, Galipeau J. IDO-independent suppression of T cell effector function by IFN-γ-licensed human mesenchymal stromal cells. J Immunol. (2014) 192:1491–501. doi: 10.4049/jimmunol.1301828
160. Dorronsoro A, Ferrin I, Salcedo JM, Jakobsson E, Fernández-Rueda J, Lang V, et al. Human mesenchymal stromal cells modulate T-cell responses through TNF-α-mediated activation of NF-κB. Eur J Immunol. (2014) 44:480–8. doi: 10.1002/eji.201343668
161. Modiano JF, Lindborg BA, McElmurry RT, Lewellen M, Forster CL, Zamora EA, et al. Mesenchymal stromal cells inhibit murine syngeneic anti-tumor immune responses by attenuating inflammation and reorganizing the tumor microenvironment. Cancer Immunol Immunother. (2015) 64:1449–60. doi: 10.1007/s00262-015-1749-6
162. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. (2015) 348:74–80. doi: 10.1126/science.aaa6204
163. Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells. (2006) 24:386–98. doi: 10.1634/stemcells.2005-0008
164. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. (2005) 105:1815–22. doi: 10.1182/blood-2004-04-1559
165. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. (2002) 99:3838–43. doi: 10.1182/blood.V99.10.3838
166. Chow L, Johnson V, Regan D, Wheat W, Webb S, Koch P, et al. Safety and immune regulatory properties of canine induced pluripotent stem cell-derived mesenchymal stem cells. Stem Cell Res. (2017) 25:221–32. doi: 10.1016/j.scr.2017.11.010
167. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. (2018) 48:812–30.e14. doi: 10.1016/j.immuni.2018.03.023
168. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. (2017) 8:15081. doi: 10.1038/ncomms15081
169. Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D II, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. (2016) 352:189–96. doi: 10.1126/science.aad0501
170. Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. (2014) 20:436–42. doi: 10.1038/nm.3488
171. Li W, Germain RN, Gerner MY. High-dimensional cell-level analysis of tissues with Ce3D multiplex volume imaging. Nat Protoc. (2019) 14:1708–33. doi: 10.1038/s41596-019-0156-4
172. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
173. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. (2015) 12:453–7. doi: 10.1038/nmeth.3337
174. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. (2017) 18:220. doi: 10.1186/s13059-017-1349-1
175. Scott MC, Temiz NA, Sarver AE, LaRue RS, Rathe SK, Varshney J, et al. Comparative transcriptome analysis quantifies immune cell transcript levels, metastatic progression, and survival in osteosarcoma. Cancer Res. (2018) 78:326–37. doi: 10.1158/0008-5472.CAN-17-0576
176. Wycislo KL, Fan TM. The immunotherapy of canine osteosarcoma: a historical and systematic review. J Vet Intern Med. (2015) 29:759–69. doi: 10.1111/jvim.12603
177. Gorden BH, Kim JH, Sarver AL, Frantz AM, Breen M, Lindblad-Toh K, et al. Identification of three molecular and functional subtypes in canine hemangiosarcoma through gene expression profiling and progenitor cell characterization. Am J Pathol. (2014) 184:985–95. doi: 10.1016/j.ajpath.2013.12.025
178. Filley A, Henriquez M, Bhowmik T, Tewari BN, Rao X, Wan J, et al. Immunologic and gene expression profiles of spontaneous canine oligodendrogliomas. J Neurooncol. (2018) 137:469–79. doi: 10.1007/s11060-018-2753-4
179. Giannuzzi D, Marconato L, Elgendy R, Ferraresso S, Scarselli E, Fariselli P, et al. Longitudinal transcriptomic and genetic landscape of radiotherapy response in canine melanoma. Vet Comp Oncol. (2019) 17:308–16. doi: 10.1111/vco.12473
180. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. (2016) 39:44–51. doi: 10.1016/j.coi.2015.12.007
181. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
182. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
183. O'Donnell JS, Teng MW, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
184. Varn FS, Schaafsma E, Wang Y, Cheng C. Genomic characterization of six virus-associated cancers identifies changes in the tumor immune microenvironment and altered genetic programs. Cancer Res. (2018) 78:6413–23. doi: 10.1158/0008-5472.CAN-18-1342
185. Iglesia MD, Parker JS, Hoadley KA, Serody JS, Perou CM, Vincent BG. Genomic analysis of immune cell infiltrates across 11 tumor types. J Natl Cancer Inst. (2016) 108:djw144. doi: 10.1093/jnci/djw144
186. Keane C, Gould C, Jones K, Hamm D, Talaulikar D, Ellis J, et al. The T-cell receptor repertoire influences the tumor microenvironment and is associated with survival in aggressive B-cell lymphoma. Clin Cancer Res. (2017) 23:1820–8. doi: 10.1158/1078-0432.CCR-16-1576
187. Kikutake C, Yoshihara M, Sato T, Saito D, Suyama M. Pan-cancer analysis of intratumor heterogeneity associated with patient prognosis using multidimensional measures. Oncotarget. (2018) 9:37689–99. doi: 10.18632/oncotarget.26485
188. Naik S, Makielsi KM, Henson MS, Stuebner KM, Tabaran AF, Cornax IOSMG, et al. Characterization of anti-tumor immune responses and effects on survival of neoadjuvant oncolytic virotherapy in spontaneous osteosarcoma. Abstract O43. In: Society for Immunotherapy of Cancer 2018. Washington, DC: SITC (2018).
189. Miller JS, Lewis LL. Natural killer cells in cancer immunotherapy. Ann Rev Cancer Biol. (2019) 3:77–103. doi: 10.1146/annurev-cancerbio-030518-055653
190. Cooper EL. Evolution of immune systems from self/not self to danger to artificial immune systems (AIS). Phys Life Rev. (2010) 7:55–78. doi: 10.1016/j.plrev.2009.12.001
191. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. (2015) 33:1974–82. doi: 10.1200/JCO.2014.59.4358
192. Helmink BA, Gaudreau PO, Wargo JA. Immune checkpoint blockade across the cancer care continuum. Immunity. (2018) 48:1077–80. doi: 10.1016/j.immuni.2018.06.003
193. Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. (2017) 14:203–20. doi: 10.1038/nrclinonc.2016.168
194. Gajewski TF. The next hurdle in cancer immunotherapy: overcoming the non-T-cell-inflamed tumor microenvironment. Semin Oncol. (2015) 42:663–71. doi: 10.1053/j.seminoncol.2015.05.011
195. Modiano JF, Bellgrau D, Cutter GR, Lana SE, Ehrhart NP, Ehrhart E, et al. Inflammation, apoptosis, and necrosis induced by neoadjuvant fas ligand gene therapy improves survival of dogs with spontaneous bone cancer. Mol Ther. (2012) 20:2234–43. doi: 10.1038/mt.2012.149
196. Sorenmo KU, Krick E, Coughlin CM, Overley B, Gregor TP, Vonderheide RH, et al. CD40-activated B cell cancer vaccine improves second clinical remission and survival in privately owned dogs with non-Hodgkin's lymphoma. PLoS ONE. (2011) 6:e24167. doi: 10.1371/journal.pone.0024167
197. Mason NJ, Gnanandarajah JS, Engiles JB, Gray F, Laughlin D, Gaurnier-Hausser A, et al. Immunotherapy with a HER2-targeting listeria induces HER2-specific immunity and demonstrates potential therapeutic effects in a phase I trial in canine osteosarcoma. Clin Cancer Res. (2016) 22:4380–90. doi: 10.1158/1078-0432.CCR-16-0088
198. Andersen BM, Pluhar GE, Seiler CE, Goulart MR, SantaCruz KS, Schutten MM, et al. Vaccination for invasive canine meningioma induces in situ production of antibodies capable of antibody-dependent cell-mediated cytotoxicity. Cancer Res. (2013) 73:2987–97. doi: 10.1158/0008-5472.CAN-12-3366
199. Panjwani MK, Smith JB, Schutsky K, Gnanandarajah J, O'Connor CM, Powell DJ, et al. Feasibility and safety of RNA-transfected CD20-specific chimeric antigen receptor T cells in dogs with spontaneous B cell lymphoma. Mol Ther. (2016) 24:1602–14. doi: 10.1038/mt.2016.146
200. Regan D, Dow S. Manipulation of innate immunity for cancer therapy in dogs. Vet Sci. (2015) 2:423–39. doi: 10.3390/vetsci2040423
201. Anderson KL, Modiano JF. Progress in adaptive immunotherapy for cancer in companion animals: success on the path to a cure. Vet Sci. (2015) 2:363–87. doi: 10.3390/vetsci2040363
202. Larson G, Karlsson EK, Perri A, Webster MT, Ho SY, Peters J, et al. Rethinking dog domestication by integrating genetics, archeology, and biogeography. Proc Natl Acad Sci USA. (2012) 109:8878–83. doi: 10.1073/pnas.1203005109
203. Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D, et al. Cohabiting family members share microbiota with one another and with their dogs. Elife. (2013) 2:e00458. doi: 10.7554/eLife.00458
204. Raposo-Ferreira TMM, Brisson BK, Durham AC, Laufer-Amorim R, Kristiansen V, Pure E, et al. Characteristics of the epithelial-mesenchymal transition in primary and paired metastatic canine mammary carcinomas. Vet Pathol. (2018) 55:622–33. doi: 10.1177/0300985818776054
205. Im KS, Kim JH, Kim NH, Yu CH, Hur TY, Sur JH. Possible role of Snail expression as a prognostic factor in canine mammary neoplasia. J Comp Pathol. (2012) 147:121–8. doi: 10.1016/j.jcpa.2011.12.002
206. Fantozzi A, Gruber DC, Pisarsky L, Heck C, Kunita A, Yilmaz M, et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. (2014) 74:1566–75. doi: 10.1158/0008-5472.CAN-13-1641
207. Medici D, Nawshad A. Type I collagen promotes epithelial-mesenchymal transition through ILK-dependent activation of NF-kappaB and LEF-1. Matrix Biol. (2010) 29:161–5. doi: 10.1016/j.matbio.2009.12.003
208. Vellinga TT, den Uil S, Rinkes IH, Marvin D, Ponsioen B, Alvarez-Varela A, et al. Collagen-rich stroma in aggressive colon tumors induces mesenchymal gene expression and tumor cell invasion. Oncogene. (2016) 35:5263–71. doi: 10.1038/onc.2016.60
209. Sainz B Jr, Carron E, Vallespinos M, Machado HL. Cancer stem cells and macrophages: implications in tumor biology and therapeutic strategies. Mediators Inflamm. (2016) 2016:9012369. doi: 10.1155/2016/9012369
210. Rak J, Guha A. Extracellular vesicles–vehicles that spread cancer genes. Bioessays. (2012) 34:489–97. doi: 10.1002/bies.201100169
211. Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux-Petit M, LeGonidec S, et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: a novel mechanism linking obesity and cancer. Cancer Res. (2016) 76:4051–7. doi: 10.1158/0008-5472.CAN-16-0651
212. Robado de Lope L, Alcíbar OL, Amor López A, Hergueta-Redondo M, Peinado H. Tumour-adipose tissue crosstalk: fuelling tumour metastasis by extracellular vesicles. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20160485. doi: 10.1098/rstb.2016.0485
213. Ruivo CF, Adem B, Silva M, Melo SA. The biology of cancer exosomes: insights and new perspectives. Cancer Res. (2017) 77:6480–8. doi: 10.1158/0008-5472.CAN-17-0994
214. Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. (2011) 71:3792–801. doi: 10.1158/0008-5472.CAN-10-4455
215. Scott MC, Garbe JR, Tomiyasu H, Donnelly A, Bryan BA, Subramanian S, et al. Unbiased discovery of exosome-associated biomarkers using xenograft models. In: Proceedings of the American Association for Cancer Research Annual Meeting 2017. Washington, DC; Philadelphia, PA: AACR. doi: 10.1158/1538-7445.AM2017-817
Keywords: tumor microenvironment, temporo-spatial organization, dog, canine, human
Citation: Langsten KL, Kim JH, Sarver AL, Dewhirst M and Modiano JF (2019) Comparative Approach to the Temporo-Spatial Organization of the Tumor Microenvironment. Front. Oncol. 9:1185. doi: 10.3389/fonc.2019.01185
Received: 04 September 2019; Accepted: 21 October 2019;
Published: 07 November 2019.
Edited by:
Thomas Daubon, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2019 Langsten, Kim, Sarver, Dewhirst and Modiano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kendall L. Langsten, a2FubmV0dGlAdW1uLmVkdQ==; Jaime F. Modiano, bW9kaWFub0B1bW4uZWR1