 Lourdes M. Mendez
Lourdes M. Mendez Ryan R. Posey
Ryan R. Posey Pier Paolo Pandolfi
Pier Paolo Pandolfi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 07 November 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01162
This article is part of the Research TopicThe Biological Landscape of Immunotherapy in AMLView all 7 articles
AML holds a unique place in the history of immunotherapy by virtue of being among the first malignancies in which durable remissions were achieved with “adoptive immunotherapy,” now known as allogeneic stem cell transplantation. The successful deployment of unselected adoptive cell therapy established AML as a disease responsive to immunomodulation. Classification systems for AML have been refined and expanded over the years in an effort to capture the variability of this heterogeneous disease and risk-stratify patients. Current systems increasingly incorporate information about cytogenetic alterations and genetic mutations. The advent of next generation sequencing technology has enabled the comprehensive identification of recurrent genetic mutations, many with predictive power. Recurrent genetic mutations found in AML have been intensely studied from a cell intrinsic perspective leading to the genesis of multiple, recently approved targeted therapies including IDH1/2-mutant inhibitors and FLT3-ITD/-TKD inhibitors. However, there is a paucity of data on the effects of these targeted agents on the leukemia microenvironment, including the immune system. Recently, the phenomenal success of checkpoint inhibitors and CAR-T cells has re-ignited interest in understanding the mechanisms leading to immune dysregulation and suppression in leukemia, with the objective of harnessing the power of the immune system via novel immunotherapeutics. A paradigm has emerged that places crosstalk with the immune system at the crux of any effective therapy. Ongoing research will reveal how AML genetics inform the composition of the immune microenvironment paving the way for personalized immunotherapy.
Acute myeloid leukemia (AML) is a disease of the myeloid lineage in which rapidly proliferating progenitors are transformed, leading to a block in terminal differentiation and the ability to self-renew, the latter normally being the privilege of only hematopoietic stem cells. Two decades of research have demonstrated that these expanded clones are the result of a differentiation hierarchy which mirrors normal hematopoiesis, with “stem-like” progenitors giving rise to partially committed myeloid progenitors, themselves giving rise to committed but undifferentiated myeloid precursors (1). This dysfunctional hierarchy, referred to as malignant hematopoiesis, occurs at the expense of normal hematopoiesis and is characterized by progressive cytopenias and immunosuppression with concomitant increased risk of infection and bleeding.
AML is an aggressive disease; in the U.S. from 2009 to 2015, the 5-years survival rate was 28.3% (2). For decades the backbone of therapy for fit patients has been intensive induction chemotherapy, such as the 7 + 3 regimen with the combination of an anthracycline and infusional cytarabine. Despite complete remission rates of 60–80% in younger patients and 40–60% in older patients with such regimens (3–5), rates of overall survival are far more sobering highlighting the critical problem of relapsed leukemia, following an initial response. Thus far, the only exception to this somber picture is acute promyelocytic leukemia (APL), which as a result of the high rates of cure achieved with ATRA- and ATO- containing therapy, is considered separately from non-APL AML for the purpose of risk classification/stratification (6). For patients with intermediate or high risk disease who are fit, allogeneic stem cell transplantation offers the chance of durable remission and long term survival weighed against treatment related morbidity and mortality (7, 8). Elderly patients and/or patients with multiple comorbidities, have faced more limited options mainly hypomethylating agents (HMA) and low dose cytarabine (6) until recently. The last few years have seen a flurry of FDA approvals for AML including a liposomal formulation of daunarubicin and cytarabine, the anti-CD33 antibody drug conjugate gemtuzumab ozogamicin, the IDH1/2 mutant inhibitors ivosidenib and enasidenib, the FLT3-ITD inhibitors midostaurin (in combination with chemotherapy) and gilternitib and the BCL2 inhibitor venetoclax (in combination with HMA or low dose cytarabine).
Efforts over the last years to understand the impact of recurrent genetic alterations in AML on prognosis have resulted in genetic classification systems that summarize the risk of relapse following intensive induction chemotherapy (3, 6). A similar system is not available to guide treatment with HMAs. Identifying the predictors, genetic and others, of response and resistance to the recently approved targeted agents, such as the IDH1/2 mutant inhibitors and the BCL2 inhibitor, venetoclax, is an active field of research. Aberrant mitochondrial metabolism and the regulation of mitochondrial apoptosis have emerged as central themes in the discussion about response and resistance to existing and emerging therapies; how these processes tie into the genetics of AML remains largely unknown. Durable remissions following allogeneic transplantation for the treatment of leukemia was an early success story in the history of immunotherapy, demonstrating that leukemia could be eradicated by an effective immune response. The successes of checkpoint inhibitors in solid tumor oncology has resulted in a renaissance for immunotherapies against AML with promising modalities including cellular therapies, such as dendritic cell fusion vaccines to address minimal residual disease (9, 10). However, there is a considerable gap between the overwhelming progress in basic research in AML and advances in AML immunotherapy. While the genetics of AML have been extensively studied to classify and predict responses to chemotherapy, the relationship between genetic aberrations and the immune system has yet to be comprehensively described. This review aims to cover this topic, as well as how existing therapies affect the immune milieu. While the immune modulatory properties of a number of genetic mutations present in AML would seem to be an obvious connection, this remains an emerging area of investigation in AML. The experience to date suggests that co-occurring mutations together with additional key modifiers, such as stage of differentiation and metabolic profile, will converge on the immune microenvironment dictating biology and prognosis.
It has been appreciated for some time that outcomes with intensive induction chemotherapy are quite variable, and that a minority of patients attain durable remissions. Older age (>60 years old) and certain genetic alterations predict for markedly inferior outcomes. Multiple studies have demonstrated the prognostic utility of classifying patients by genetic mutations and cytogenetic abnormalities (11–14). The ability of the ELN genetic classification system, to discriminate outcomes, was reported by Mrozek et al. in 2012 in 1,550 adult patients treated with intensive chemotherapy. The overall survival at 3 years varied in younger patients (<60 years), from 66% in the favorable risk group to 10% in the high risk group compared to the older patients (>60 years), 33% in the favorable risk group and 3% in the high risk group. Such variability in outcomes has been understood to reflect underlying heterogeneity in disease biology and propelled efforts to further define this heterogeneity (11). The advent of next-generation sequencing technology and ensuing insight into the genetic and clonal architecture of AML, opened a new chapter in AML pathogenesis, classification and risk stratification.
Large-scale sequencing efforts have provided a catalog of recurrently mutated genes in AML. Whole exome and whole genome sequencing performed on 200 de novo AML samples by The Cancer Genome Atlas Network revealed that AML is characterized by few mutations in coding genes, on average 13 per patient, 5 of these recurrent mutations (15). Recurrently mutated genes can be grouped into functional categories revealing mutual exclusivity between different combinations of mutations. This suggests that alterations of different genes may converge on common pathways to give rise to AML. Mutual exclusivity was observed between, but is not limited to, mutations in NPM1, CEBPA, RUNX1, TP53, and transcription factor-fusion genes. Co-occurrence of mutations was also noted, such as among FLT3-ITD, DNMT3A, and NPM1 (15). Interestingly, RNAseq expression data revealed clustering that correlated with FAB subtypes and thus stage of differentiation, in accordance with other publications (15–17).
A large data set of 1,540 patients, treated on one of three German-Austrian AML Study Group trials, integrated clinical data with genetic profiling (cytogenetics and sequencing of 111 driver mutations) enabling a more detailed view of the mutational landscape of AML, prompting a new proposed genomic classification scheme for diagnosis beyond the current WHO subgroups, and allowing the authors to tackle the problem of the prognostic implications of co-occurring mutations. Analysis of allele frequencies allowed for establishment of clonal relationships identifying mutations in the epigenetic modifiers ASXL1, TET2, IDH1/2, and DNMT3A as the earliest event occurring in the founding clone whereas mutations in receptor tyrosine kinase-RAS pathway genes occurred late as previously described (18–20) with more than one such mutation in a given patient (12). The proposed, new classification system is composed of 11 genomic subgroups of AML including AML with NPM1 mutation; AML with mutated chromatin, RNA-splicing genes or both; AML with TP53 mutations, chromosomal aneuploidy or both; AML with inv (16) or t (16, 16); AML with biallelic CEBPA mutations; AML with t (8, 21); AML with MLL fusion genes, AML with inv (3); t(3, 3), GATA2, MECOM; AML with IDH2-R172 and no other class-defining lesions; AML with t (6, 9). The chromatin-spliceosome and TP53-aneuploidy groups in particular represent new genomic subgroups with their respective class defining lesions imparting a deleterious effect on survival. Interestingly, the initial, recently reported findings of the Beat AML programme found that TP53 and ASXL1 (one of the recurrent mutated chromatin genes) were associated with a general pattern of drug resistance in an ex vivo 122 small molecule inhibitor screen (21).
The results of efforts over the last years to understand the impact of recurrent genetic alterations on outcomes following intensive chemotherapy are summarized in the updated ELN 2017 genetic classification system for AML (6). The ELN 2017 system starts to incorporate knowledge about the impact of co-occurring mutations on outcome; specifically, the favorable prognosis of NPM1-mutated AML is noted to be contingent on a low FLT3-ITD mutational burden. At the present time, the ELN 2017 system does not include other interactions between/among genes in its risk stratification algorithm and this remains a frontier in AML that is actively being explored (12, 21). The complexity of mutation co-occurrence is such that the negative prognostic impact of FLT3-ITD may be most relevant to AML with the most frequent three-gene co-occurrence of mutations in NPM1, DNMT3A, and FLT-3 whereas, the negative impact on survival in AML with FLT3-ITD and NPM1 mutation or FLT3-ITD and DNMT3A mutation is less pronounced irrespective of the FLT3-ITD allelic frequency (12).
More recently, single cell sequencing-based assays have been performed to finely resolve clonal and subclonal architectures in AML, offering insights into clonal evolution during both leukemogenesis and disease progression following treatment (22, 23). Van Galen et al. recently applied single cell RNAseq and genotyping to profile 40 bone marrow aspirates (from 16 AML samples, five healthy controls). By transcriptomics analysis, they identified six malignant cell types that resembled normal bone marrow cell types and correlated with leukemia cell differentiation state: HSC-like, progenitor-like, GMP-like, promonocyte-like, monocyte-like, conventional dendritic cells-like. The composition of a patient's leukemia with respect to the malignant cell types varied considerably and could be predominantly composed of one cell type or contain the spectrum of cell types. The abundance of malignant cell types correlated with morphologic and cell surface marker metrics in clinical use. Moreover, gene signatures for each malignant cell type allowed interrogation of the composition of the TCGA AML sample collection revealing close correlations with the genetics of AML. Intriguingly, they further found that monocyte-like AML cells express various immunomodulatory genes and inhibit T cell activation in vitro, suggesting that this malignant cell type participates in the immunosuppressive leukemia microenvironment (23). This study represents a pioneering effort to capture and integrate multiple facets of AML biology—differentiation state, intratumoral heterogeneity, genetics and the immune milieu in AML. In spite of a growing wealth of genetic data, at the present time, comparatively little is published on the immunologic consequences of AML genetic perturbations, representing a significant gap in our knowledge of AML biology.
AML was among the first malignancies successfully treated with “adoptive immunotherapy,” a term coined by G. Mathe in 1965 referring to allogeneic stem cell transplantation (24). This provided proof-of-concept that leukemia could be eradicated by an effective immune response, the graft-vs.-leukemia effect, and also implicated a suppressed endogenous anti-leukemia immune response as part of the pathogenesis of AML. The successes of immune check point inhibitors in solid tumors has invigorated research into immunotherapies, that like allogeneic stem cell transplantation, could result in durable remissions, with the hope that novel, selected adoptive cell therapies and biologics would additionally be better tolerated (9, 25).
Components of a successful anti-tumor immune response include engagement of the innate immune system via danger signals, presentation of tumor-associated antigens to T cell receptors (TCR) together with activating co-stimulatory signals and immunologic memory (26). In AML, remodeling of the leukemia microenvironment and barriers to an effective immune response include but are not limited to (1) low neoantigen burden and defective antigen presentation, (2) imbalance between T effector (Teff) and regulatory populations (Treg) in favor of regulatory/suppressive cells, (3) T cell exhaustion, such as through upregulation of immune checkpoint ligands and receptors, chronic inflammation, (3) increased myeloid derived suppressor cell populations (MDSC), (4) increased suppressive macrophage populations, and (5) production of immunosuppressive soluble factors including metabolites (9, 25). Multiple agents including biologics, selected adoptive cell therapies and targeted agents are currently the subject of clinical investigation in an effort to address the ineffective anti-leukemia immune response. A non-exhaustive list is provided as an example, along with the proposed immunologic effect in Table S1; a subset of these agents is briefly discussed below.
Eliciting specific antitumor immunity requires the presentation of polypeptide fragments of tumor-associated antigens bound to major histocompatibility complex (MHC) class I or II, in conjunction with co-stimulatory signals to a cognate CD8+ (MHC I bound antigen) or CD4+ (MHC II bound antigen) TCR. MHC class I is expressed by most cells and presents self-antigens, whereas MHC class II expression is normally restricted to professional antigen presenting cells, such as dendritic cells, B cells and macrophages and binds extracellular antigens. The critical role of antigen presentation in AML is highlighted by a recent publication reporting that relapse following allogeneic stem cell presentation is associated with down-regulation of MHC class II genes (27). Neoantigens are thought to be the ideal tumor antigens in part because the corresponding TCR repertoire may have escaped negative selection during central tolerance. As discussed later in this review, neoantigens encoded by recurrent AML mutations have been identified, including those derived from NPM1c, FLT3-ITD, and mutant p53. High mutational/neoantigen burden correlates with response to checkpoint blockade (28, 29). With an average of 13 mutations/patient (15), AML is considered to be a malignancy with low mutational burden and thus, at least in this sense, low immunogenicity. On the other hand, genetic subsets, such as TP53-mutated AML or AML with loss of TP53 may be associated with an increased mutational burden and complex karytotype and thus, increased epitopes, which could predict for a higher likelihood of response to checkpoint blockade (30). In addition, the efficacy of allogeneic stem cell transplantation in a subset of patients indicates, that at least within certain contexts, AML is sufficiently immunogenic.
In addition to the challenges imposed by low mutational/neoantigen burden, multiples studies have reported on defective antigen presentation by AML cells and APC in the leukemia microenvironment (25). AML cells are aberrant myeloid precursors with their normal counterparts maturing into dendritic cells, monocytes, macrophages and neutrophils. Dendritic cells and macrophages are professional APC while monocytes and neutrophils though not professional APC, have been described to participate in antigen presentation and stimulation of the adaptive immune response (31–35). As discussed in the next section, multiple reports substantiate the ability of HMA to enhance antigen presentation pathways in AML and recalibrate the immune microenvironment to elicit anti-tumor immunity (Table S1). AML vaccines are an emerging, promising modality of immunotherapy that may address the problem of antigenicity/immunogenicity (Table S1). Vaccine strategies include neoantigen vaccines (36), leukemia-derived dendritic cell vaccines (37), GM-CSF transduced/secreting leukemia cell vaccines (38) and modified, autologous dendritic cell-based vaccines. AML-dendritic cell fusion vaccines have the benefit of overcoming defective antigen presenting functions of leukemic cells and of not requiring a priori selection of antigens. Moreover, by presenting multiple antigens with the potential to engage multiple TCRs, a diverse T cell response could be elicited (39, 40).
AML has been demonstrated to induce suppressive populations derived from the myeloid lineage, such as myeloid derived suppressor cells (MDSC) and M2 macrophages. MDSC are associated with a poor prognosis in solid and hematologic malignancies and have been associated with a blunted response to immunotherapy (41, 42). These immature myeloid cells suppress Teff through arginase-1, nitric oxygen synthase and ROS production (43). Pyzer et al. demonstrated that MDSC are increased in the peripheral blood of patients with AML compared to normal controls. Interestingly, MDSC appeared to be derived from both normal progenitors and leukemia cells in line with previous reports that AML cells employ arginase to suppress Teff (44–46). Given that HMA are known to possess differentiation activity, an open question is whether differentiation of blasts into leukemia-derived dendritic cells, macrophages or neutrophils down-regulates MDSC function originating from blasts and renders leukemia more immune competent. If this is indeed the case, it would have implications for approved and investigational targeted agents that induce differentiation of blasts, such as the recently approved selective inhibitors of IDH1/2-mutants.
The nomenclature M1/M2 macrophages, refers to the distinction between “classically activated” macrophages, M1, that engage in effective anti-microbial or tumor responses and “alternatively activated” macrophages, M2, that have anti-inflammatory activity and tumor supporting properties. AML promotes the polarization of leukemia-supporting macrophages (47). Yang and colleagues further found that, in patients, leukemia associated macrophages are heterogeneous, with both M1- and M2-like leukemia associated macrophages (48). M2-like leukemia associated macrophages were associated with a poor prognosis. In murine models of leukemia, treatment with polyIC (a synthetic analog of double stranded RNA) or inhibition of the SAPK/JNK pathway “repolarized” leukemia associated macrophages toward an M1 phenotype and resulted in prolonged survival.
Regulatory T cells (Treg) constitute another part of the immune suppressive leukemia microenvironment. Treg restrain immune competent helper T and cytotoxic T cells through inhibition of T cell proliferation and cytokine production and play an important role in preventing autoreactivity (49, 50). Treg also restrain the activity of B cells (51) and cells of the innate immune system (52, 53). Increased Treg compared to normal controls are present in AML patients at diagnosis, during chemotherapy-induced pancytopenia and following chemotherapy; the pretreatment Treg level has been observed to correlate with response to chemotherapy. Interestingly, the lowest levels of Treg were observed at the time of hematopoietic recovery following chemotherapy (54–57). Importantly, the available data indicates that interventions directed against Treg render chemotherapy and immunotherapy more effective in hematologic malignancies (49, 58). One strategy employed to address the suppressive leukemia microenvironment in which regulatory or suppressive cell types dominate, is to tip the scale through infusion of either manipulated autologous or allogeneic cells with cytolytic activity, such as chimeric antigen receptor T cells (CAR-T cells; see Table S1) (59).
Another means by which T cell effector function is restrained is T cell exhaustion, which was initially characterized in the context of chronic viral infection, but is by now a well-established mechanism cancer immune evasions (60). Cell surface expression of inhibitory co-receptors marks exhausted T cells. In contrast to the co-stimulatory signal required for naïve T cell activation in concert with TCR MHC-peptide engagement, signaling through inhibitory ligands/receptors, results in decreased T cell proliferation and cytokine production (Figure 1). The best studied inhibitory receptors are CTLA-4 and PD-1; others include Tim-3 and TIGIT (9). Inhibitory ligands/receptors are upregulated during acute infection/inflammation to limit the inflammatory response. Type I and II interferons induce the expression of PD-L1/2, as part of non-neoplastic immune circuits (61–63); interferons have also been reported to induce the expression of PD-L1/2 on AML blasts (64–66). Immune checkpoint inhibitors (ICI) that prevent signaling through inhibitory ligands and receptors, have been approved for the treatment of various solid tumors (67). Increased expression of inhibitory receptors/ligands including CTLA-4, PD-1, PD-L1/2 on T cells and myeloid cells has been described following treatment with HMA in patients with myeloid neoplasms (68, 69) and forms part of the rationale for ongoing clinical trials investigating HMA combined with ICI. It may be necessary to disable multiple inhibitory pathways, as reported by a preclinical study in which inhibition of CTLA-4, PD-1, and LAG-3 was required for adoptive T cells to exert an anti-leukemia effect (70).
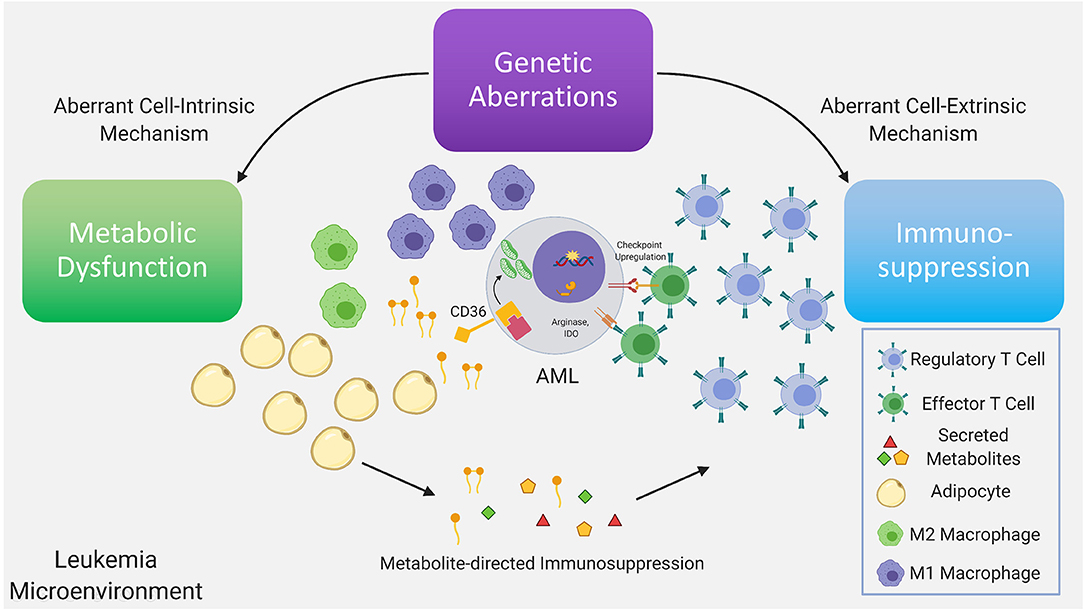
Figure 1. Genetic aberrations drive dysfunctional cell-intrinsic and -extrinsic signaling, with the former having direct metabolic consequences and the latter having direct immunologic consequences. Further dysregulation is driven via interactions between the altered metabolome and immune system as a consequence of the existing immunometabolic network. Recurrent genetic mutations found in AML have been demonstrated to promote T regulatory cell expansion, suppress proliferation of T effector cells, skew macrophage maturation toward the suppressive M2 phenotype. A subset of these immune suppressive mechanisms are mediated by metabolites. In AML, depletion of amino acids, such as tryptophan and asparagine via increased arginase-1 and IDO levels in leukemia cells, M2-macrophages and MDSC, limits T effector activity. Furthermore, mobilization of fatty acids in the leukemia microenvironment from adipocytes, a subset of which can function as pro-inflammatory lipid mediators, may simultaneously provide anti-apoptotic and immunosuppressive signals inhibiting T effector activity and promoting suppressive immune cell types. In leukemia cells the metabolic regulatory network that is stimulated by adipocytes may include upregulation of the lipid scavenger receptor CD36, the fatty acid activated receptor/transcription factor PPARG, the fatty acid binding protein (FABP4) along with BCL2. Upstream genetic determinants/mutations regulating specific metabolic adaptations in leukemia cells is the subject of ongoing research.
As noted above, soluble factors, such as arginase 2 have been implicated in the induction of a tolerogenic environment by AML blasts and M2 macrophages (9). Arginase 2 is the mitochondrial version of the enzyme and functions in wound repair together with nitric oxide synthase (NOS), with the two enzymes having the same substrate, L-arginine (71). Indoleamine 2,3 dioxygenase (IDO), that causes the breakdown of tryptophan, is another enzyme that has been linked to an immunosuppressive leukemia microenvironment. IDO may be involved in the increased Treg population and function observed in AML (9). Fatty acids and lipid mediators derived from fatty acids, are another class of metabolites that bridge cellular metabolism with immune signaling (72) (Figure 1). Their role in modulating the balance between an active and suppressed immune milieu in leukemia is currently under investigation (73). For example, the lipid mediator PGE2, has been demonstrated to promote tumor progression through induction and maintenance of MDSC (74, 75) with PGE2 inducing PD-L1 expression on tumor associated macrophages and MDSC (76). Strategies to target lipid mediator signaling include inhibition of PGE2 receptors and could potentially involve pharmacologic modulation of the PPAR nuclear receptor family whose natural ligands include lipid mediators (73). Targeting lipid mediators may represent a novel therapeutic approach for various malignancies (77) including AML.
Alongside the recognition that immune evasion is a hallmark of cancer (78) has emerged a paradigm in which tumor clearance by antineoplastic therapy by de facto must mobilize an effective anti-tumor immune response. Conventional therapies, like chemotherapy, have been revisited with renewed interest in their capacity to elicit immunogenic cell death and anti-tumor immunity (58). There is a growing literature on the mechanisms by which DNA demethylating/hypomethylating agents (HMA) reprogram leukemia cells and refashion the leukemia immune microenvironment. Likewise, there is increasing appreciation that targeted agents address cell-extrinsic mechanisms promoting an immune suppressive leukemia microenvironment. Below are highlights regarding the immune modulatory dimension of chemotherapy, HMAs and targeted agents. Notably, despite the general paucity of data on the genetic determinants of the leukemia immune microenvironment and response to immunotherapy, in the case of these three categories of therapy, there are clear links between therapy and AML genetics potentially suggesting the specific immunologic space in between.
A small subset of chemotherapeutic agents and fractionated radiation therapy can induce “immunogenic cell death” (79, 80). Immunogenic cell death (ICD) refers to apoptosis, that results in coordinated activation of the immune system and antigen-specific immunity (81), as compared to the “silent” cell death that occurs at steady state as part of the physiologic turnover of normal cells. Activation of the ER stress pathway/Unfolded Protein Response and ensuing emission of danger signals are defining characteristics of ICD. Damage associated molecular patterns (DAMPs) include surface–exposed ER chaperones (calreticulin, HSP70 and HSP90). DAMPs are recognized by pattern recognition receptors (PRRs) present on macrophages, monocytes and dendritic cells, leading to activation of the innate arm of the immune system. These chaperones, when exposed to the cell surface, can have antigenic peptides bound that are endocytosed by PRRs, leading to cross presentation of antigens to T cells (82–84). Notably, a number of anthracyclines, some of these a mainstay of AML therapy, have been demonstrated to induce ICD (85–90). Interestingly, type I interferon signaling downstream of toll-like receptor 3 (TLR3) signaling, is required for anthracycline-induced ICD in a murine breast cancer model.
Expression of DAMPs, PRRs has been explored as predictive biomarkers to ICD-inducing chemotherapy. In a prospective cohort of 50 patients with AML, Fucikova et al. addressed the effect of anthracycline-based chemotherapy on the surface expression of various DAMPs and their relation to leukemia-specific T cell immunity (91). They demonstrated that AML blasts, but not normal CD45+ CD33+ cells from healthy volunteers displayed exposed calreticulin, HSP70 and HSP90 independent of exposure to induction anthracycline-based chemotherapy suggesting a baseline level of ER-stress response in a subset of AML; in line with this, surface exposed calreticulin correlated with the expression of genes involved in ER stress response. Noting that surface calreticulin is best known for promoting phagocytosis by antigen-presenting cells and thus initiating anti-tumor immunity, the authors showed that upon recovery from chemotherapy, the normal PBMCs from patients expressing high surface calreticulin, as compared to low surface calreticulin, showed a gene expression pattern reflecting enrichment of TH1 polarization, T cell activation, CD8+ T-cell cytotoxicity and NK-related genes. Consistent with the gene expression data, surface calreticulinhi patients had increased circulating NK cells at the time of recovery from chemotherapy. Additionally, they had an increased fraction of CD8+ and CD4+ T cells responding to leukemia associated antigens both prior to and following chemotherapy as well as an increased percentage of T central memory cells, again both prior to and at the time of recovery from chemotherapy (91).
A cornerstone of therapy for patients unable to tolerate intensive induction chemotherapy has been DNA-demethylating agents, also called hypomethylating agents (HMA) i.e., azacitadine and decitabine. Both drugs are cytidine analogs and were originally developed as cytotoxic agents before being found, at low doses, to cause DNA demethylation by inhibition of DNA methyltransferase-1 (DNMT1) (92). DNA demethylation has been demonstrated in cell lines to cause reactivation of genes, leading to differentiation (93) and this is one of the proposed mechanisms of therapeutic activity in myeloid neoplasms including AML. However, DNA methylation/methylcytosine has not been found to correlate with response in patients with AML (94, 95). In addition to its effect on DNA, azacitadine has been reported to inhibit the RNA methyltransferase, DNMT2 (96) suggesting that it's mechanism of action in myeloid neoplasms may be linked to modification of RNA. Consistent with this possibility, in a recent report, distinct RNA 5-methylcytosine methyltransferase chromatin complexes were demonstrated to mediate sensitivity vs. resistance to azacitadine (97). Up-regulation of antigen presentation pathways, such as MHC expression, are part of the mechanism by which HMA are described to recalibrate the immune micoenvironment promoting anti-tumor responses (98–103).
Clinically, the optimal response to HMA has been noted to require multiple cycles of treatment in line with a cell-intrinsic mechanism involving slow kinetics, potentially related to the differentiation of leukemic cells (104) and akin to other differentiation therapies (105). Differentiation is associated with cell cycle arrest and apoptosis, but one could speculate that an additional benefit could be enhanced presentation of antigens, as is seen with the maturation of professional antigen presenting cells and/or diminished MDSC-like activity thus leading to more effective engagement of the immune system. Decitabine has also been demonstrated to alter Treg maturation and activate the expression of endogenous retroviruses resulting in engagement of the dsRNA response/interferon-beta pathway thus, suggesting a non-cell autonomous mechanism of action is also at play in AML (101, 102, 106–108). Indeed, preclinical data indicates that hypomethylating agents potentiate the response to anti-CTLA-4 immunotherapy (101) and a dendritic cell/AML fusion vaccine (109) with ongoing clinical trials in AML currently exploring the combination of HMA and checkpoint inhibitors (NCT02890329) or dendritic cell/AML fusion vaccine.
The combination of all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) for the treatment of APL is widely considered to constitute the first targeted therapy for cancer as well as the first example of differentiation therapy. ATRA targets the RARA moiety and ATO the PML moiety of the PML-RARA fusion protein for degradation in PML-RARA APL (110). The phenomenal success of this chemotherapy-free targeted combination therapy (111) was informed by genetic models providing insights into the underpinnings of resistance and response to ATRA and ATO (112). The road to this success is at once instructive and a reminder of the promise of targeted therapies. Remarkably, ATRA has well-documented immune modulatory activity with effects on CD4 and CD8 T cell development and dendritic cells (113–119) including in preclinical murine models of APL, as a single agent and in combination with DNA vaccines (120, 121). ATRA's potential as a chemical adjuvant, has been explored in various studies (118, 120, 121). In a cohort of patients with metastatic renal cell carcinoma treated with 7 days of ATRA followed by interleukin 2, ATRA was noted to reduce the number of immature myeloid suppressor cells by day 7, approaching the levels of these cells seen in normal controls; this effect or lack thereof was noted to correlate with plasma concentration of ATRA. In this study, ATRA also improved the ability of peripheral blood mononuclear cells to stimulate allogeneic T cells (122). As regards ATO, this drug has been shown to selectively deplete Treg in vitro and in vivo by inducing the expression of pro-oxidative genes in Treg with concomitant induction of increased levels of nitric oxide and reactive oxygen species (ROS). ATO-induced Treg depletion appeared to be dependent on “oxidative and nitrosative bursts.” Consistent with these observations, the authors found that in a model of colon cancer, a reduction in tumor volume was observed in immunocompetent but not athymic NUDE mice (123). Xu et al. examined the effect of ATO treatment on Treg from APL patients and likewise found that the number and function of Treg was attenuated by ATO treatment (124). ATO has also been reported to up-regulate NK ligands on tumor cells (125) modulate NK cell receptors toward enhanced NK cell mediated cytolytic activity (126). Taken together, the data on the immune modulatory activities of ATRA and ATO raise the possibility that these targeted agents could be employed in the treatment of subsets of AML, beyond APL, because of their immune modulatory activity. In this regard, ATRA and ATO could be considered together or in novel combinations as recently suggested by preclinical studies in IDH1/2-mutated AML (127, 128). As discussed in the next section, targeted agents against recurrent genetic mutations in non-APL AML, likewise display significant affects against components of the suppressive leukemia immune microenvironment.
It has become apparent that all categories of anti-neoplastic drugs can modulate the immune response. Approved targeted therapies possess highly specific immune modulatory activities pertinent to their potential roles in the induction, consolidation and maintenance phases of therapy. Ongoing delineation of the immunologic mechanisms of action of targeted therapies is expected to inform the rationale combination of targeted therapies, as well as, novel combinations with chemotherapy, HMA, cellular therapies and biologics.
Recurrent genetic mutations found in AML have been intensely studied from a cell intrinsic perspective. By comparison the literature on cell extrinsic mechanisms of action of these mutations is still building and is heavily based on studies of solid tumors. Nonetheless, the mechanisms by which recurrent genetic mutations in AML, such as NPM1, FLT3-ITD, IDH1/2, and TP53 can modulate the immune microenvironment in AML, are steadily being elucidated (Figure 1).
NPM1 is among the three most frequently mutated genes in AML, along with FLT3-ITD and DNMT3A, occurring in 30–35% of de novo AML (6, 15). The NPM1 protein shuttles between the nucleus and cytoplasm (129) and is most prominently located in the nucleolus (130). More recently, in an elegant series of experiments Nachmani et al. demonstrated that NPM1 regulates 2′-O-methylation of rRNA through snoRNA binding which is required for normal hematopoietic stem cell maintenance; Npm1-deletion in hematopoietic stem cells leads to a bone marrow failure syndrome and proclivity to develop leukemia (131). In 2005 Falini et al. discovered NPM1-mutated AML reporting that in 35.2% of 591 bone marrow specimens of patients with de novo AML, NPM1 was located in the cytoplasm as a result of a mutation in the cytoplasmic portion of the protein (NPM1c) (132). NPM1c AML was noted to be associated with a normal karyotype and response to induction chemotherapy. FLT3-ITD co-occurs with NPM1 mutation in ~40% of cases (132, 133). FLT3-ITD (VAF of >50%) negates the positive prognosis associated with NPM1c (38, 134–137). NPM1-mutated AML in the absence of FLT3-ITD in CR1 does not derive RFS or OS benefit from allogeneic stem cell transplantation (7, 136) underscoring the chemosensitivity and likely immunogenicity (as discussed below), of this subtype of AML. Notably, in the Phase 1b dose escalation and expansion study of venetoclax combined with HMA in elderly patients with AML, NPM1-mutated AML was associated with a remarkably high CR + CRi [91% (138)]. This result remains to be confirmed in larger studies, but is suggestive a decreased threshold for apoptosis. Given multiple publications indicating that NPM1c elicits endogenous T cell immunity, another pending question is whether the high rates of CR and the duration of response following venetoclax combined with HMA, is linked to tumor-specific immunity. Finally, the persistence of NPM1-mutated transcripts has been demonstrated to be a powerful biomarker for disease relapse (139).
The favorable prognosis of NPM1-mutated AML in the absence of FLT3-ITD has been posited to be at least partially mediated by engagement of the immune system (140). In particular, the unique C-terminal sequences of mutated NPM1 in NPM1-mutant AML have been investigated as potential leukemia specific antigens that may elicit endogenous T cell responses and could also serve as targets for immunotherapy. Noting that aberrant localization of mutant NPM1 in the cytoplasm could lead to processing by the class I degradation pathway and subsequent presentation by HLA, Liso et al. tested and confirmed that mutated-NPM1 peptides can be presented by HLA molecules (141). Greiner et al. demonstrated that mutant NPM1 is immunogenic with T cells from the peripheral blood of patients with NPM1-mutant AML reacting in vitro to specific peptides derived from the mutant NPM1 (142). Notably, in a patient with molecular relapse of NPM1-mutant AML, donor lymphocyte infusion displayed mutant-NPM1 specific T cell responses that were linked to molecular remission (140). Subsequently, Forghieri and colleagues investigated NPM1-mutated specific T cell response in NPM1-mutant AML patients over time. They observed that a robust response could be identified following attainment of CR post-induction chemotherapy and was sustained in patients with a durable remission compared to a low number or absence of NPM1-mutated specific T cells in patients with leukemia relapse suggesting that a mutated NPM1-specific T cell response is inversely correlated with MRD and relapse (143). Van der Lee et al. characterized the HLA 1 ligandome of primary AMLs to investigate whether mutant NPM1 is a neoantigen in AML. Multiple peptides were identified from mutant NPM1 in the precipitated ligandome. Mutant NPM1 is characterized by a 4 bp hotspot insertion in exon 12 that results in a mutant protein that is 4 amino acids (aa) longer than the wildtype protein with the c-terminal 11 aa, CLAVEEVSLRK, translated in an alternative reading frame. 5 mutant NPM1-derived peptides were identified in the HLA 1 ligandome in NPM1-mutant but not NPM1-wildtype AML. The CLAVEEVSL peptide, predicted to bind to HLA-A*02:01, was chosen for further characterization. As tumor-specific T cells have previously been described in healthy individuals, the authors utilized PBMCs from healthy HLA-A*02:01–positive individuals to isolate CD8+ T cell specific for CLAVEEVSL and then further identified clones that were reactive against HLA-A*02:01–positive primary AMLs with mutant NPM1. Finally, the authors tested the potential of CLAVEEVSL as a target for TCR gene transfer and found that T cells transduced with the TCR for mutant NPM1 resulted in killing of NPM1-mutant AML cells in vitro and in vivo, notably, with a survival benefit observed in the latter experiments (144).
FMS-like Tyrosine Kinase 3 (FLT-3) is a receptor tyrosine kinase, normally involved in hematopoietic stem and progenitor cell proliferation and survival (145). It is mutated in ~30% of AML cases and is known to confer adverse prognosis (146). Mutations in FLT3 are observed as either point mutations in the tyrosine kinase domain (TKD) or as an internal tandem duplication of the juxtamembrane domain (ITD), with the latter more frequently occurring than the former (147). It is known that the direct consequence of FLT3-ITD mutations is strong, constitutive activation of signaling, and that this activation is insensitive to negative feedback via SOCS1 (148). It is appreciated that while FLT3-ITD utilizes downstream pathways active during normal FLT3 signaling, downstream signaling becomes deregulated, as observed with the aberrant activation of STAT5 signaling (149). In addition to being less frequent, TKD mutations confer a less striking phenotype clinically, being less strongly associated with poor prognosis and are even associated with a favorable prognosis in combination with NPM1 mutations (150).
Constitutive activation of FLT3 via internal tandem duplication (ITD) mutation or point mutations in the tyrosine kinase domain (TKD) has been shown to mediate cell-extrinsic changes in the immune landscape of AML. Mice with a total body ITD mutation demonstrate expanded populations of dendritic cells (DCs), dendritic cell precursors (pre-DCs), and regulatory T cells (Treg) as a result of the ITD mutation (151). The expansion of dendritic cells and dendritic cell precursors was demonstrated to be intrinsic to ITD-mutant hematopoietic cells in experiments utilizing bone marrow transplant (BMT) chimera mice. The subsequent expansion of Treg observed in total body ITD-mutant mice was corroborated in BMT chimeras and shown to be a cell-extrinsic effect given that the T cells were of Flt3 wild-type origin. Interestingly, effector T cell (Teff) populations were expanded as well, although the ratio of Treg to Teff within the total T cell population was higher as compared to wild-type chimeras. Scurfy mice, which possess a truncated, non-functional variant of FOXP3, rapidly develop a lethal autoimmune condition due to insufficient immunoregulation caused by a deficiency of Tregs. Immunologically relevant genotypes crossed with these mice either exasperate or ameliorate this condition, resulting in increased or decreased survival of these mice. In their study, Lau et al. crossed ITD and scurfy mice, and observed significantly more rapid lethality, due to accelerated autoimmunity. This strongly suggests that the cell-extrinsically expanded Teff population is able to perform effector functions in the absence of the enriched Treg population.
These results mirror similar observations in patients with AML, where expanded populations of dendritic cells and dendritic cell precursors containing the ITD mutation have been observed (152). A prospective study tracking these populations in patients at first diagnosis and in remission following chemotherapy demonstrated that the populations of expanded dendritic cell precursors persist after patients enter remission (153). The group noted that persistence of pre-DCs was associated with increased risk of relapse in patients with the ITD mutation. Midostaurin, a tyrosine kinase inhibitor that potently targets both wildtype and mutated FLT3 has demonstrated immune-modulatory effects with implications on patient prognosis following remission (154). A reduction in CD4+, CD25+ cells, and in the mRNA level of FOXP3, consistent with a decrease in Treg, was observed following treatment in AML patients treated with Midostaurin, a possible consequence of ITD inhibition (155).
Since FLT3 mutations are associated with increased risk of relapse following treatment with chemotherapy, this patient subset is often treated with allogeneic hematopoietic stem cell transplant (HSCT) in first remission (156). Several groups have noted that the reduced risk of relapse following HSCT is directly linked to the graft-vs.-leukemia (GVL) effect (157, 158). A recent study demonstrated production of IL-15 by leukemia cells in patients with FLT3 mutations treated with Sorafenib, a tyrosine kinsase inhibitor which targets mutant FLT3, similar to Midostaurin (159, 160). The production of IL-15 was specific to patients with the ITD mutation, as non-ITD AML cells or ITD cells not treated with Sorafenib did not express the same level of IL-15. The effect was abrogated by depletion of CD8+ T cells but not NK cells, indicating that the increased alloreactivity is due to changes in the Teff population. Antibody-based depletion of IL-15 or transfer of Il15ra-deficient T cells to recipient mice blunted the GVL effect and subsequently resulted in failure to control disease. Mechanistically, the increase in IL-15 was due to decreased ATF4 activity, resulting in an increase in IRF7 phosphorylation. Knockdown of IRF7 or overexpression of ATF4 resulted in abrogated IL-15 production. Competitive Kinobead pulldown of Sorafenib binding targets in patient samples implicated FLT3 as a possible mediator of ATF4 expression. Accordingly, the authors proposed a mechanism by which FLT3 activity in ITD-mutated patients suppresses IL-15 production in AML blasts via ATF4 inhibition of IRF7. These results were corroborated in patients treated with Sorafenib, as responders achieving complete remission demonstrated increased levels of IL-15, p-IRF7, and IFN-g following treatment as compared to non-responders, for whom remission was not attained, who did not mirror this effect.
Mutations in Isocitrate Dehydrogenase 1 and 2 (IDH1 and IDH2, respectively) account for between 9 and 20% of mutations in AML (15). These mutations result in production of the oncometabolite R-2-Hydroxyglutarate (R-2-HG) from alpha-Ketoglutarate (a-KG) (161). In the context of AML, R-2-HG accumulation is known to be responsible for a blockade in terminal differentiation (162), epigenetic rewiring of AML blasts (163, 164), and increasing genome instability resulting in acquisition of subsequent mutations (165). Recently, patients with relapsed, refractory IDH1- and IDH2-mutant AML were demonstrated to have significant ORR and CR rates to the novel, selective IDH1- and IDH2-mutant inhibitors, ivosidenib and enasidenib, with evidence of differentiation of blasts, in a subset of patients leading to a differentiation syndrome akin to the previously described differentiation syndrome described in patients with APL treated with ATRA and ATO, in accordance with the predicted mechanisms of actions of these novel inhibitors (105, 166). Interestingly, preclinical models also indicate that IDH1/2- mutant AML also demonstrate sensitivity to, ATRA and ATO, the targeted combination therapy employed in APL (128).
The production of the oncometabolite R-2-Hydroxyglutarate (R-2-HG) mediates the proto-oncogenic effects of mutant IDH1 and IDH2. Human T cells express multiple transporters capable of importing R-2HG, including SLC13A3 and SLC22A6 (167). The impact of R-2-HG on the immune response has been characterized largely in gliomas and it remains to be seen whether there are cancer-specific mechanisms of R-2-HG immune suppression. In gliomas, levels of R-2-HG present in the extracellular space can be as great as 5-fold higher than within tumor cells and can be transported intracellularly to T cells via these receptors (168). Extracellular R-2-HG is taken in rapidly regardless of activation status of T cells. This enantiomer demonstrated dose-dependent inhibition of T lymphocyte proliferation, while S-2HG had no such effect (166). This inhibition of T cell proliferation was rescued by addition of the SLC13A3 inhibitor NAA. Mechanistically, R-2-HG caused reduced calcium influx and subsequently suppressed NFAT translocation and proliferation. R-2-HG treated T lymphocytes were also shown to have reduced mitochondrial fitness and ATP production. A syngeneic glioma model of wild-type and IDH1-mutated cancers demonstrated no change in survival in RAG2 knockout mice, indicating that suppression of anti-tumor response mediates the competitive advantage of R-2-HG production in vivo. Interestingly, T lymphocyte polarization was shown to be affected in the context of AML as compared to gliomas, with exogenous addition of R-2-HG interfering with HIF-1a stability and skewing the development of Th17 cells toward Treg cells (169). Subsequent work has demonstrated that IDH1-mutant gliomas correlated with lower expression of the T cell attracting chemokines CXCL9 and CXCL10, and subsequent reduction in number of CD3+, CD8+ tumor infiltrating lymphocytes (166, 170, 171). Both total and phosphorylated STAT1 protein was shown to be reduced in IDH1 mutant gliomas, and knockdown of STAT1 in wildtype IDH1 tumors resulted in reduced expression of these chemokines, suggesting that R-2-HG can potentially abrogate cytokine signaling through this pathway. The fraction of Foxp3+/CD4+ cells within gliomas was not observed to change with IDH status (166). Interestingly, the R172K mutation in IDH2 has the unique functional ability to impact T cell development, increasing the amount of double negative (CD4− CD8−) and decreasing the fraction of double positive thymocytes, unlike that of other mutations in IDH1 and IDH2, as a result of the exceptionally high levels of intracellular R-2-HG this mutation produces (172).
R-2-HG has also been shown to modulate the bone marrow microenvironment and the innate immune system. In addition to reduced T lymphocyte chemotaxis, mutant IDH gliomas also exhibit reduced influx of neutrophils in murine models, as well as a reduced expression of innate immune chemotactic molecules including CCL2, CCL3, CXCL1, CXCL2, CXCL4, CXCL16, GM-CSF, IL1RA, IL-2, IL-6 (166). In the microenvironment of gliomas, R-2HG is able to suppress activation of the C5 component of the complement system and subsequent complement-mediated phagocytosis, although it does not directly affect dendritic cell differentiation from bone marrow mononuclear cells and subsequent antigen-presenting function to T lymphocytes. However, reduced levels of CD83 were observed on human monocyte-derived dendritic cells in addition to differential metabolic changes and decreased IL-12 secretion in response to LPS stimulation. Natural Killer cell ligands and subsequent tumor cell lysis has also been shown to be reduced in IDH1-mutated gliomas (173). Within the bone marrow microenvironment, R-2-HG is taken up by stromal cells, where it inhibits TETs as well as induces activation of the transcription factors NF-kB and GATA (174).
The p53 tumor suppressor is a transcription factor that regulates multiple, critical cellular functions including cell division, DNA damage and repair, apoptosis, senescence, metabolism (175–179). It is one of the most commonly mutated genes in cancer (180). Though best known for its role in regulating the response to DNA damage and apoptosis thus functioning as a guardian of the genome, studies indicate that p53 may function as a tumor suppressor in a non-cell autonomous fashion via modulation of the immune system and inflammation.
AML with TP53 mutations are present in ~6% of patients with AML and can be combined into a genomic category with chromosomal aneuploidy (13% of AML) as the two types of genetic alterations are closely correlated (12). The incidence of TP53 mutation in AML with complex karyotype (3 or more chromosomal abnormalities) has been reported as 69–78% (181). Missense point mutations and small deletions occur (15) in addition to allelic loss through deletion of the short arm of chromosome 17 (182). TP53 mutated AML, particularly in combination with adverse cytogenetics, has a dismal prognosis following chemotherapy (30, 183). Among driver mutations, mutations in TP53 are second only to inv (3), GATA2 and MECOM(EVI1) mutations in terms of the increased risk of death associated with the mutation (12) likely reflecting, at least in part, the chemoresistance associated with TP53 mutations (184). TP53 mutations are also associated with decreased survival and an increased risk of relapse following allogeneic stem cell transplantation (185–187) with TP53 mutations detected at relapse indicating that the TP53 mutated clone was involved in relapse (187). The poor outcomes associated with TP53 mutated AML following allogeneic stem cell transplantation could be mediated by the persistence of minimal residual disease and/or resistance to graft vs. leukemia effect and thus negative modulation of the anti-leukemia immune response.
Patients with TP53 mutated AML tend to be older (188) and as such, more likely to be intolerant of intensive induction chemotherapy, making them more likely to be treated with hypomethylating agents. Interestingly, in a single institution study of patients with AML treated with the hypomethylating agent, decitabine, that sought to understand whether specific somatic mutations predicted response to therapy, TP53 mutation was highly correlated with bone marrow blast clearance albeit with incomplete mutation clearance. In this study, a post-hoc analysis indicated that overall survival was not negatively affected by unfavorable risk cytogenetic abnormalities nor by the presence of TP53 mutations and TP53 status did not affect outcome following transplantation (95). In patients ineligible for intensive cytotoxic therapy and/or allogeneic stem cell transplantation, the addition of venetoclax to hypomethylating agents represents one of the most exciting and promising novel combination therapies. In the phase 1b dose-escalation and expansion study of the BCL2 inhibitor, venetoclax, combined with azacitadine or decitabine, in elderly patients with AML, the rate of CR + CRi, the median duration of CR + CRi and the median overall survival in patients with TP53 mutation compared to all comers was 47%, 5.6, 7.2 months vs. 67%, 11.3 and 17.5 months (138).
P53 mediates the cellular response to multiple stressors including DNA damage, oncogene activation and viral infection and there is evidence of interplay among these three insults mediated by p53. It has been suggested that in addition to guarding genomic integrity, tumor suppressors, such as p53, also guard the integrity of tumor immunosurveillance (189, 190). Many viruses, including oncoviruses, have developed mechanisms to directly or indirectly disrupt p53 function underscoring the critical role of p53 in antiviral immunity and the suppression of virus-associated cancers. In line with this, in a mouse model bearing an extra copy of TP53, enhanced resistance to viral infection was observed (191–193). P53 activates transcription of critical regulators of the innate immune response, including genes involved in pathogen sensing, such as Toll-like receptors 3 and 8 (194) and type I IFN. Notably, upregulation of Toll-like receptors is induced by p53 in response to DNA-damaging agents in human primary cells and cell lines (192) and differential regulation of TLRs is observed between wildtype and mutants of p53 which retain transactivation capacity (193). The immunostimulatory effects of radiation therapy and certain chemotherapeutic agents has been proposed to be mediated via p53- activated TLR and IFN type 1 pathway (195) and by extension, dysregulation of these pathways downstream of mutated p53 may mediate resistance to these therapies in multiple malignancies including TP53-mutated AML. P53 can also activate natural killer cell ligands, ULBP1 and 2 on cancer cells leading to enhanced recognition and killing by NK cells (196, 197).
P53 senescence programs have been implicated in the resolution of inflammation (198) with chronic inflammation being a well-established risk factor for inflammation-associated epithelial malignancies. A non-cell autonomous mechanism of tumor suppression by p53 has been described through skewing of macrophage polarization toward the tumor-inhibiting M1-subtype; selective p53 deletion in the myeloid lineage led to elevated levels of inflammatory cytokines and a significant increase in tumor initiation whereas mild activation of p53 in the myeloid lineage restricted tumor progression (199, 200). It remains to be seen whether p53 inactivation or mutation in AML recapitulates the non-cell autonomous mechanisms observed in epithelial tumor models leading to increased inflammatory cytokines and M2-macrophages associated with disease initiation and progression or whether impaired regulation of TLR signaling and interferon pathways is implicated in the initiation or maintenance of TP53-mutated AML. Another element of p53 function is regulation of metabolism with promotion of OXPHOS (201). Interestingly mutations in mitochondrial-encoded electron transport genes, is associated with mutated p53 in patients with AML (202). Additional studies are needed to clarify whether mutated p53 is a driver of aberrant mitochondrial metabolism in this subset of AML contributing to remodeling of the leukemia microenvironment and chemoresistance. p53 reactivating therapies are currently in clinical trials in AML and may shed light on the role of p53 in the anti-leukemia immune response.
P53 and PML share multiple overlapping functions, such as the response to various stressors (oxidative, DNA damage, viral infection), regulation of senescence and apoptosis, regulation of fatty acid oxidation (203–205). Moreover, the two proteins have been shown to functionally and physically interact (206). PML is a key cofactor for p53 transcriptional function in part through recruitment of p53 into PML nuclear bodies (NB) where p53 undergoes post-translational modification (207–209). In APL, where PML-RARA disrupts normal NB formation and function, one would predict that p53 functions, including immunoregulatory activities, would be compromised. Indeed, it has been demonstrated that engagement of a PML-p53 checkpoint is key to the efficacy of ATRA and ATO in APL, downstream of NB re-formation (210). Hence, APL serves as an example of p53 dysfunction in the wildtype p53 setting, which has been well-documented in non-APL AML (211, 212) and raises the question of whether immune dysregulation secondary to inactivation of p53 pathways plays also a role in the pathogenesis of APL, and in its profound response to therapy upon the restoration of the p53-PML crosstalk.
While the above mutations have been studied most extensively with respect to their immunomodulatory properties, a modicum of data exists for a handful of additional, recurrently altered genes in AML. For example, the JAK2V617F mutation, most commonly associated with the Myeloproliferative Neoplasm (MPN) Polycythemia Vera (PV) which can progress to AML, has been shown to regulate expression of PD-L1 cell-intrinsically (213). The authors demonstrated that this was dependent on constitutively active JAK2/STAT3/STAT5 signaling as a result of the V617F mutation. Secondary AML evolving from an MPN has a dismal prognosis with a median overall survival of 2.5–7 months (214). The JAK1/2 inhibitor, Ruxolitinib, is currently in clinical trials in combination with Decitabine (NCT02257138) or chemotherapy (NCT03878199) in an effort to improve outcomes for this subset of AML patients. The results of the phase II study of Ruxolitinib plus Decitabine reported an ORR of 61%, CRi 11%, and 17% of patients (3/18) proceeding to transplant (215). Ruxolitinib is also currently under investigation in combination with venetoclax for the treatment of relapsed, refractory AML (NCT03874052). Jak2V617F was shown to cooperate with loss of Dnmt3a, one of the top three mutated genes in AML, to induce myelofibrosis (MF) and inflammatory signaling in bone marrow HSPCs (216). Similarly, mutated RUNX1, estimated to occur in ~13% of human AML cases, has been demonstrated to modulate NF-kb signaling in a cell intrinsic manner, and has been proposed to be able to promote inflammatory signaling in the bone marrow microenvironment (217). Finally, in metastatic melanoma (MM), mutations in NRAS are associated with poor response to immune checkpoint blockade, suggesting yet another mechanism of immune dysfunction that could result from this mutation in AML (218, 219).
The genetic determinants of the leukemia immune microenvironment are starting to be elucidated. Forthcoming reports may yield insight as to whether favorable risk AML subgroups, currently defined in genetic terms, are enriched for a favorable immune profile as complement, as one might imagine for NPM1-mutated AML with wildtype FLT3. By comparison, the current high-risk genetic subgroups of AML may have in common the enrichment of a suppressive immunologic milieu supported by non-cell autonomous mechanisms as has been suggested for FLT3-ITD.
The altered lipid composition of AML cells was first reported in the 1970s (220). Recent publications further substantiated an altered lipidome in the plasma and bone marrow of patients with AML (221, 222). Lipids function as signaling molecules for a variety of immune-related processes. Multiple studies support a dual role for fatty acids and fatty acid derived lipid mediators in sustaining malignant cells and modulating antitumor immune responses toward tolerance (73).
Among the reported metabolic changes in AML are increased mitochondrial mass and oxygen consumption, but decreased spare reserve relative to normal hematopoietic cells (223) and thus, increased vulnerability to metabolic perturbations. Within the mitochondria, the Krebs cycle and fatty acid oxidation (FAO) generate metabolites that feed into the electron transport chain, donating electrons to form a gradient which powers the production of ATP, with oxygen acting as the electron acceptor through a process called, oxidative phosphorylation (OXPHOS). Fatty acids are obtained by cells from the extracellular environment or catabolized from intracellular sources. Within the mitochondria, during FAO, fatty acids undergo iterative cycles of catabolism producing FADH2 and NADH and acetyl CoA during each cycle; FADH2 and NADH feed directly into the electron transport chain and thus OXPHOS (224). In solid tumors, FAO has been reported to support energy requirements during times of stress (224, 225) and promote chemoresistance (226). In AML, FAO has been reported to modulate the threshold for apoptosis and regulate quiescence in leukemic progenitors (227). In a model of blast crisis CML, Ye and colleagues found that leukemia stem cells (LSC) enriched in visceral adipose tissue, display a pro-inflammatory phenotype that drives lipolysis. They further demonstrated that LSC in adipose tissue have high FAO with fatty acid uptake regulated by the scavenger receptor, CD36. CD36+ LSC were shown to be chemoresistant consistent with the findings by Farge et al. on increased FAO, up-regulated CD36 and an OXPHOS signature in pre-existing, residual AML cells that survive cytarabine treatment in a cohort of patient derived xenografts (228). LSC from patients with relapsed AML, but not de novo AML, employ FAO as a means of sustaining OXPHOS to avoid cell death induced by combination therapy with HMA and the BCL2 inhibitor, venetoclax (229). Transcriptomics analysis of chemoresistant leukemia cells reveals an immune/inflammatory stress response gene signature highlighting the dynamic between mitochondrial metabolism/fatty acids and the immune component of the microenvironment (228).
Metabolic reprogramming in cancer cells is achieved by genetic mutations and altered signaling (230). One gap that is starting to be addressed in this field is which genetic mutations central to the pathogenesis of AML are able to reprogram the lipidome. Stuani et al. reported that in a cell line model of IDH1R132H AML, proteomic, lipidomic and isotope-labeled glucose and glutamine approaches to profile the metabolic status of IDH1-mutated vs. wildtype cells, revealed that mutant IDH1 directs high lipid anabolism (231). Similarly, the FLT3-ITD mutation is associated with specific metabolic perturbations including promotion of the Warburg effect (232, 233). Mutations in mitochondrial-encoded electron transport genes, are associated with mutated p53 (202). Interestingly, localization of leukemia to adipose tissue was observed by Ye et al. in the blast crisis BCR-ABL CML model, but not an MLL-AF9 murine model, indicating that genetics and perhaps cell of origin may regulate the metabolic phenotype of leukemia (234). In normal hematopoietic stem cells, we previously reported that FAO is critical in cell fate determination and regulated by Ppard downstream of Pml (235). The Andreef group used a bone marrow-derived adipocyte and acute monocytic leukemia co-culture system, to interrogate the genetic and metabolic networks that are stimulated in monocytic leukemia by adipocytes under conditions of limiting nutrients. They found that adipocytes activate a transcriptional network that includes PPARG and its target genes CD36, FABP4, and BCL2 (236). The PPAR family of nutrient-activated steroid hormone receptors thus, appears to represent one family of transcription factors regulating fatty acid metabolism that carries over from normal to malignant hematopoiesis. There remains a paucity of information on the genetic determinants of metabolic reprogramming and the apparent concomitant changes in inflammation/immune response. A substantial body of literature exists delineating the interaction between AML metabolism, leukemia cell survival, response to existing therapies and novel therapeutic strategies, extensively reviewed elsewhere (237, 238). Mounting evidence indicates that identifying the upstream regulators and genetic determinants of AML metabolism will have clear implications for understanding how the genetic and immune landscapes of AML are interconnected as well as inform risk stratification and therapeutic algorithms in patients with AML (Figure 2).
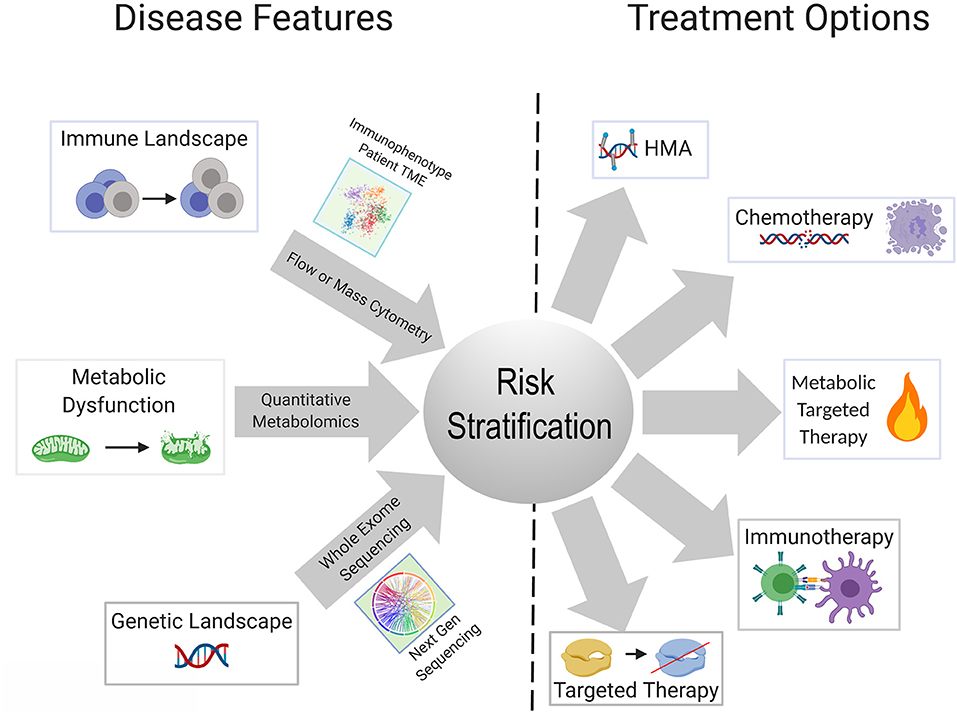
Figure 2. Schematic showing possible integration of available tools for improved risk stratification to inform therapeutic strategy for an individual patient. Advances in next gen sequencing technology, metabolite quantification methods, and widespread use of cytometry-based immunophenotyping, will continue to improve the feasibility of these techniques as diagnostic tools. Better understanding of the relationships and dependencies between the immunologic, metabolic, and genetic perturbations seen in AML can allow for refined usage of available and emerging therapeutics and combinations thereof.
The genetic landscape of AML has come into focus through large scale sequencing efforts. Together with intensive study of cell-intrinsic mechanisms of leukemia initiation and maintenance driven by recurrent mutations, this has led to the development and approval of multiple targeted agents for the treatment of AML. Approximately 50 years after the observation that “adoptive immunotherapy,” that is allogeneic stem cell transplantation, is effective in the treatment of leukemia, a myriad of biologics and cellular therapies are under investigation for the treatment of AML. Following in the steps of solid tumor biology, we have learned that immune modulation is not limited to biologics and cellular therapy and rather is also seen with targeted agents consistent with a paradigm in which eliciting anti-tumor immunity is a cornerstone of effective antineoplastic therapy.
The explosive growth in the armamentarium of approved and investigational agents for the treatment of AML has outstripped our current classifications systems for AML. Though there are snapshots of cell-extrinsic mechanisms of immune remodeling induced by recurrent AML mutations, which we have described in this review, a panoramic view of the AML immune landscape awaits the results of ongoing large scale profiling efforts of AML patient samples. Collaborative efforts have the potential to integrate the genomic and immunologic landscapes of large numbers of patients with AML with clinical outcomes. Combined with transcriptomic, and perhaps metabolic data, this will undoubtedly lead to revised classification systems for the diagnosis, risk stratification and treatment of patients with AML (Figure 2). Diagnostic procedures at the time of initial diagnosis and in the event of relapse, will almost certainly evolve to include assessment of the immune milieu and potentially metabolic profiling, in addition to existing morphologic, immunophenotypic and cytogenetic/molecular genetic testing. Concomitant assessment of leukemia and the immune milieu is accessible via flow cytometry based immunophenotyping (239) while the application of technologies, such as mass cytometry has yet to be incorporated into clinical practice. Metabolic characterization of leukemia and its niche can be performed through metabolomics analysis employing NMR spectroscopy or mass spectrometry or through the measurement of single metabolites of interest (240). Evaluation of the metabolic profile of the leukemic microenvironment could identify actionable metabolic targets ultimately resulting in immunomodulation that complements cytotoxic, targeted or immunotherapy.
The crosstalk between recurrent genetic alterations in AML and the immune system and the net outcome following treatment may be significantly influenced by leukemia metabolism modulating the threshold for cell death and effecting changes in the leukemia microenvironment promoting leukemia survival. Successfully employing the spectrum of approved and emerging therapies in AML, in rational combinations and sequences toward personalized medicine, will require algorithms incorporating “immunologic” risk alongside genetic risk. Ongoing, mechanistic insights into the links between the genetics and immunologic status of subtypes of AML are expected to inform selection of therapy(s) that can converge on vulnerabilities, including metabolic, of a given subtype, resulting in elimination of leukemia.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported in part by the Ludwig Center at Harvard and the NIH/NCI grant R35CA197529 to PP.
PP serves in the Scientific Advisory Board of Agios Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Jessica Liegel, Jonathan Lee, and Anish Sharda for critical comments on the manuscript. Figures were created with Biorender.com.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.01162/full#supplementary-material
1. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. (1997) 3:730–7. doi: 10.1038/nm0797-730
2. Noone AM, Howlader N, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review. Bethesda, MD: National Cancer Institute (2017).
3. Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. (2010) 115:453–74. doi: 10.1182/blood-2009-07-235358
4. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. (2015) 373:1136–52. doi: 10.1056/NEJMra1406184
5. Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. (2016) 127:53–61. doi: 10.1182/blood-2015-08-604520
6. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. (2017) 129:424–47. doi: 10.1182/blood-2016-08-733196
7. Koreth J, Schlenk R, Kopecky KJ, Honda S, Sierra J, Djulbegovic BJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA. (2009) 301:2349–61. doi: 10.1001/jama.2009.813
8. Meijer E, Cornelissen JJ. Allogeneic stem cell transplantation in acute myeloid leukemia in first or subsequent remission: weighing prognostic markers predicting relapse and risk factors for non-relapse mortality. Semin Oncol. (2008) 35:449–57. doi: 10.1053/j.seminoncol.2008.04.015
9. Lamble AJ, Lind EF. Targeting the immune microenvironment in acute myeloid leukemia: a focus on T cell immunity. Front Oncol. (2018) 8:213. doi: 10.3389/fonc.2018.00213
10. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. (2019) 19:133–50. doi: 10.1038/s41568-019-0116-x
11. Mrózek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. (2012) 30:4515–23. doi: 10.1200/JCO.2012.43.4738
12. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
13. Byrd JC, Mrózek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. (2002) 100:4325. doi: 10.1182/blood-2002-03-0772
14. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
15. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
16. Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. (2009) 94:131–4. doi: 10.3324/haematol.13299
17. Valk PJM, Verhaak RGW, Beijen AM, Erpelinck CAJ, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. (2004) 350:1617–28. doi: 10.1056/NEJMoa040465
18. Paschka P, Du J, Schlenk RF, Gaidzik VI, Bullinger L, Corbacioglu A, et al. Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): a study of the German-Austrian AML Study Group (AMLSG). Blood. (2013) 121:170–7. doi: 10.1182/blood-2012-05-431486
19. Corces-Zimmerman RM, Hong W-J, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. (2014) 111:2548. doi: 10.1073/pnas.1324297111
20. Gröschel S, Sanders MA, Hoogenboezem R, Zeilemaker A, Havermans M, Erpelinck C, et al. Mutational spectrum of myeloid malignancies with inv(3)/t(3;3) reveals a predominant involvement of RAS/RTK signaling pathways. Blood. (2015) 125:133–9. doi: 10.1182/blood-2014-07-591461
21. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. (2018) 562:526–31. doi: 10.1038/s41586-018-0623-z
22. Quek L, Otto GW, Garnett C, Lhermitte L, Karamitros D, Stoilova B, et al. Genetically distinct leukemic stem cells in human CD34− acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J Exp Med. (2016) 213:1513–35. doi: 10.1084/jem.20151775
23. van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-Cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell. (2019) 176:1265–81.e24. doi: 10.1016/j.cell.2019.01.031
24. Mathe G, Amiel JL, Schwarzenberg L, Cattan A, Schneider M. Adoptive immunotherapy of acute leukemia: experimental and clinical results. Cancer Res. (1965) 25:1525–31.
25. Austin R, Smyth MJ, Lane SW. Harnessing the immune system in acute myeloid leukaemia. Crit Rev Oncol Hematol. (2016) 103:62–77. doi: 10.1016/j.critrevonc.2016.04.020
26. Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P. Harnessing the immune system to improve cancer therapy. Ann Transl Med. (2016) 4:261–61. doi: 10.21037/atm.2016.04.01
27. Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. (2018) 379:2330–41. doi: 10.1056/NEJMoa1808777
28. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. (2015) 350:207–11. doi: 10.1126/science.aad0095
29. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. doi: 10.1126/science.aaf1490
30. Bowen D, Groves MJ, Burnett AK, Patel Y, Allen C, Green C, et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. (2009) 23:203–6. doi: 10.1038/leu.2008.173
31. Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, Loré K. Neutrophils acquire antigen presentation capacity to memory CD4+ T cells in vitro and ex vivo. Blood. (2017) 129:1991–2001. doi: 10.1182/blood-2016-10-744441
32. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
33. Takashima A, Yao Y. Neutrophil plasticity: acquisition of phenotype and functionality of antigen-presenting cell. J Leukoc Biol. (2015) 98:489–96. doi: 10.1189/jlb.1MR1014-502R
34. Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol. (2014) 14:719–30. doi: 10.1038/nri3754
35. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
36. Linette GP, Carreno BM. Neoantigen vaccines pass the immunogenicity test. Trends Mol Med. (2017) 23:869–71. doi: 10.1016/j.molmed.2017.08.007
37. Kremser A, Dressig J, Grabrucker C, Liepert A, Kroell T, Scholl N, et al. Dendritic cells (DCs) can be successfully generated from leukemic blasts in individual patients with AML or MDS: an evaluation of different methods. J Immunother. (2010) 33:185–99. doi: 10.1097/CJI.0b013e3181b8f4ce
38. Ho VT, Vanneman M, Kim H, Sasada T, Kang YJ, Pasek M, et al. Biologic activity of irradiated, autologous, GM-CSF-secreting leukemia cell vaccines early after allogeneic stem cell transplantation. Proc Natl Acad Sci USA. (2009) 106:15825. doi: 10.1073/pnas.0908358106
39. Rosenblatt J, Stone RM, Uhl L, Neuberg D, Joyce R, Levine JD, et al. Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci Transl Med. (2016) 8:368ra171. doi: 10.1126/scitranslmed.aag1298
40. Avigan D, Rosenblatt J. Vaccine therapy in hematologic malignancies. Blood. (2018) 134:2640–50. doi: 10.1182/blood-2017-11-785873
41. Wang Z, Celis E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunol Immunother. (2015) 64:1057–66. doi: 10.1007/s00262-015-1713-5
42. Chen MF, Kuan FC, Yen TC, Lu MS, Lin PY, Chung YH, et al. IL-6-stimulated CD11b+ CD14+ HLA-DR- myeloid-derived suppressor cells, are associated with progression and poor prognosis in squamous cell carcinoma of the esophagus. Oncotarget. (2014) 5:8716–28. doi: 10.18632/oncotarget.2368
43. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
44. Pyzer AR, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. (2017) 129:1791. doi: 10.1182/blood-2016-07-730614
45. Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, et al. Acute myeloid leukaemia creates an arginase-dependent immunosuppressive microenvironment. Blood. (2013) 122:749–58. doi: 10.1182/blood-2013-01-480129
46. Mussai F, Egan S, Higginbotham-Jones J, Perry T, Beggs A, Odintsova E, et al. Arginine dependence of acute myeloid leukaemia blast proliferation: a novel therapeutic target. Blood. (2015) 125:2386–96. doi: 10.1182/blood-2014-09-600643
47. Al-Matary YS, Botezatu L, Opalka B, Hönes JM, Lams RF, Thivakaran A, et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a growth factor independence 1 dependent manner. Haematologica. (2016) 101:1216–27. doi: 10.3324/haematol.2016.143180
48. Yang X, Feng W, Wang R, Yang F, Wang L, Chen S, et al. Repolarizing heterogeneous leukemia-associated macrophages with more M1 characteristics eliminates their pro-leukemic effects. Oncoimmunology. (2018) 7:e1412910. doi: 10.1080/2162402X.2017.1412910
49. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
50. Niedzwiecki M, Budziło O, Adamkiewicz-Drozynska E, Pawlik-Gwozdecka D, Zielinski M, Maciejka-Kembłowska L, et al. CD4(+)CD25(high)CD127(low/-)FoxP(3) (+) regulatory T-cell population in acute leukemias: a review of the literature. J Immunol Res. (2019) 2019:2816498–98. doi: 10.1155/2019/2816498
51. Miyara M, Sakaguchi S. Human FoxP3(+)CD4(+) regulatory T cells: their knowns and unknowns. Immunol Cell Biol. (2011) 89:346–51. doi: 10.1038/icb.2010.137
52. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med. (2007) 13:108–16. doi: 10.1016/j.molmed.2007.01.003
53. Aggarwal N, Swerdlow SH, TenEyck SP, Boyiadzis M, Felgar RE. Natural killer cell (NK) subsets and NK-like T-cell populations in acute myeloid leukemias and myelodysplastic syndromes. Cytometry B. (2016) 90:349–57. doi: 10.1002/cyto.b.21349
54. Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. (2009) 15:3325. doi: 10.1158/1078-0432.CCR-08-3010
55. Wang X, Zheng J, Liu J, Yao J, He Y, Li X, et al. Increased population of CD4(+)CD25(high), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur J Haematol. (2005) 75:468–76. doi: 10.1111/j.1600-0609.2005.00537.x
56. Ersvaer E, Liseth K, Skavland J, Gjertsen BT, Bruserud O. Intensive chemotherapy for acute myeloid leukemia differentially affects circulating TC1, TH1, TH17 and TREG cells. BMC Immunol. (2010) 11:38. doi: 10.1186/1471-2172-11-38
57. Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, et al. Elevated frequencies of CD4+CD25+CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer. (2011) 129:1373–81. doi: 10.1002/ijc.25791
58. Wang Y-J, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. (2018) 5:194–203. doi: 10.1016/j.gendis.2018.05.003
59. Hofmann S, Schubert ML, Wang L, He B, Neuber B, Dreger P, et al. Chimeric antigen receptor (CAR) T cell therapy in acute myeloid leukemia (AML). J Clin Med. (2019) 8:E200. doi: 10.3390/jcm8020200
60. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. (2016) 375:1767–78. doi: 10.1056/NEJMra1514296
61. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
62. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
63. Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol. (2007) 19:309–14. doi: 10.1016/j.coi.2007.04.012
64. Berthon C, Driss V, Liu J, Kuranda K, Leleu X, Jouy N, et al. In acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitors. Cancer Immunol Immunother. (2010) 59:1839–49. doi: 10.1007/s00262-010-0909-y
65. Kronig H, Kremmler L, Haller B, Englert C, Peschel C, Andreesen R, et al. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol. (2014) 92:195–203. doi: 10.1111/ejh.12228
66. Dolen Y, Esendagli G. Myeloid leukemia cells with a B7-2(+) subpopulation provoke Th-cell responses and become immuno-suppressive through the modulation of B7 ligands. Eur J Immunol. (2013) 43:747–57. doi: 10.1002/eji.201242814
67. Ansell SM. Targeting immune checkpoints in lymphoma. Curr Opin Hematol. (2015) 22:337–42. doi: 10.1097/MOH.0000000000000158
68. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. (2014) 28:1280–8. doi: 10.1038/leu.2013.355
69. Orskov AD, Treppendahl MB, Skovbo A, Holm MS, Friis LS, Hokland M, et al. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: a rationale for combined targeting of PD-1 and DNA methylation. Oncotarget. (2015) 6:9612–26. doi: 10.18632/oncotarget.3324
70. Berrien-Elliott MM, Jackson SR, Meyer JM, Rouskey CJ, Nguyen TL, Yagita H, et al. Durable adoptive immunotherapy for leukemia produced by manipulation of multiple regulatory pathways of CD8+ T-cell tolerance. Cancer Res. (2013) 73:605–16. doi: 10.1158/0008-5472.CAN-12-2179
71. Caldwell RB, Toque HA, Narayanan SP, Caldwell RW. Arginase: an old enzyme with new tricks. Trends Pharmacol Sci. (2015) 36:395–405. doi: 10.1016/j.tips.2015.03.006
72. de Jong AJ, Kloppenburg M, Toes REM, Ioan-Facsinay A. Fatty acids, lipid mediators, and T-cell function. Front Immunol. (2014) 5:483. doi: 10.3389/fimmu.2014.00483
73. Loew A, Köhnke T, Rehbeil E, Pietzner A, Weylandt K-H. A role for lipid mediators in acute myeloid leukemia. Int J Mol Sci. (2019) 20:E2425. doi: 10.3390/ijms20102425
74. Obermajer N, Wong JL, Edwards RP, Odunsi K, Moysich K, Kalinski P. PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunol Invest. (2012) 41:635–57. doi: 10.3109/08820139.2012.695417
75. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. (2007) 67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174
76. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci USA. (2017) 114:1117. doi: 10.1073/pnas.1612920114
77. Sulciner ML, Gartung A, Gilligan MM, Serhan CN, Panigrahy D. Targeting lipid mediators in cancer biology. Cancer Metastasis Rev. (2018) 37:557–72. doi: 10.1007/s10555-018-9754-9
78. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
79. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
80. Golden EB, Frances D, Pellicciotta I, Demaria SM, Barcellos-Hoff H, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. (2014) 3:e28518. doi: 10.4161/onci.28518
81. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. (2005) 202:1691–701. doi: 10.1084/jem.20050915
82. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. (2011) 334:1573–7. doi: 10.1126/science.1208347
83. Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science. (2015) 350:972–8. doi: 10.1126/science.aad0779
84. Serrano-del Valle A, Anel A, Naval J, Marzo I. Immunogenic cell death and immunotherapy of multiple myeloma. Front Cell Dev Biol. (2019) 7:50. doi: 10.3389/fcell.2019.00050
85. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. (2011) 71:4821–33. doi: 10.1158/0008-5472.CAN-11-0950
86. Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. (2011) 30:1147–58. doi: 10.1038/onc.2010.500
87. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13:54–61. doi: 10.1038/nm1523
88. Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. (2011) 71:768–78. doi: 10.1158/0008-5472.CAN-10-2788
89. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. (2010) 29:482–91. doi: 10.1038/onc.2009.356
90. Gebremeskel S, Johnston B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: impact on clinical studies and considerations for combined therapies. Oncotarget. (2015) 6:41600–19. doi: 10.18632/oncotarget.6113
91. Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood. (2016) 128:3113–24. doi: 10.1182/blood-2016-08-731737
92. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. (2008) 123:8–13. doi: 10.1002/ijc.23607
93. Heller G, Schmidt WM, Ziegler B, Holzer S, Mullauer L, Bilban M, et al. Genome-wide transcriptional response to 5-aza-2'-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. (2008) 68:44–54. doi: 10.1158/0008-5472.CAN-07-2531
94. Treppendahl MB, Kristensen LS, Grønbæk K. Predicting response to epigenetic therapy. J Clin Invest. (2014) 124:47–55. doi: 10.1172/JCI69737
95. Welch JS, Petti AA, Miller CA, Fronick CC, O'Laughlin M, Fulton RS, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med. (2016) 375:2023–36. doi: 10.1056/NEJMoa1605949
96. Schaefer M, Hagemann S, Hanna K, Lyko F. Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res. (2009) 69:8127–32. doi: 10.1158/0008-5472.CAN-09-0458
97. Cheng JX, Chen L, Li Y, Cloe A, Yue M, Wei J, et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat Commun. (2018) 9:1163. doi: 10.1038/s41467-018-04518-9
98. Li J, Wang J, Chen R, Bai Y, Lu X. The prognostic value of tumor-infiltrating T lymphocytes in ovarian cancer. Oncotarget. (2017) 8:15621–31. doi: 10.18632/oncotarget.14919
99. Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. (2012) 21:430–46. doi: 10.1016/j.ccr.2011.12.029
100. Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget. (2013) 4:2067–79. doi: 10.18632/oncotarget.1542
101. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. (2015) 162:974–86. doi: 10.1016/j.cell.2015.07.011
102. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. (2015) 162:961–73. doi: 10.1016/j.cell.2015.07.056
103. Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun. (2018) 9:248. doi: 10.1038/s41467-017-02630-w
104. Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. (2002) 21:5483–95. doi: 10.1038/sj.onc.1205699
105. Stein EM, Tallman MS. Emerging therapeutic drugs for AML. Blood. (2016) 127:71. doi: 10.1182/blood-2015-07-604538
106. Klco JM, Spencer DH, Lamprecht TL, Sarkaria SM, Wylie T, Magrini V, et al. Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood. (2013) 121:1633–43. doi: 10.1182/blood-2012-09-459313
107. Choi J, Ritchey J, Prior JL, Holt M, Shannon WD, Deych E, et al. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood. (2010) 116:129–39. doi: 10.1182/blood-2009-12-257253
108. Nahas MR, Stroopinsky D, Rosenblatt J, Cole L, Pyzer AR, Anastasiadou E, et al. Hypomethylating agent alters the immune microenvironment in acute myeloid leukaemia (AML) and enhances the immunogenicity of a dendritic cell/AML vaccine. Br J Haematol. (2019) 185:679–90. doi: 10.1111/bjh.15818
109. Nahas MR, Soiffer RJ, Kim HT, Alyea E, Arnason J, Joyce R, et al. Phase I clinical trial evaluating abatacept in patients with steroid-refractory chronic graft-versus-host disease. Blood. (2018) 131:2836–45. doi: 10.1182/blood-2017-05-780239
110. de The H, Pandolfi PP, Chen Z. Acute promyelocytic leukemia: a paradigm for oncoprotein-targeted cure. Cancer Cell. (2017) 32:552–60. doi: 10.1016/j.ccell.2017.10.002
111. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. (2013) 369:111–21. doi: 10.1056/NEJMoa1300874
112. He L-Z, Merghoub T, Pandolfi PP. In vivo analysis of the molecular pathogenesis of acute promyelocytic leukemia in the mouse and its therapeutic implications. Oncogene. (1999) 18:5278–92. doi: 10.1038/sj.onc.1203088
113. Semba R, Hussey G, Mackerras D, Stansfield S. Vitamin A supplementation and childhood morbidity. Lancet. (1993) 342:1176–78. doi: 10.1016/0140-6736(93)92162-M
114. Stephensen CB, Rasooly R, Jiang X, Ceddia MA, Weaver CT, Chandraratna RAS, et al. Vitamin A enhances in vitro Th2 development via retinoid X receptor pathway. J Immunol. (2002) 168:4495. doi: 10.4049/jimmunol.168.9.4495
115. Westervelt P, Pollock JL, Oldfather KM, Walter MJ, Ma MK, Williams A, et al. Adaptive immunity cooperates with liposomal all-trans-retinoic acid (ATRA) to facilitate long-term molecular remissions in mice with acute promyelocytic leukemia. Proc Natl Acad Sci USA. (2002) 99:9468. doi: 10.1073/pnas.132657799
116. Geissmann F, Revy P, Brousse N, Lepelletier Y, Folli C, Durandy A, et al. Retinoids regulate survival and antigen presentation by immature dendritic cells. J Exp Med. (2003) 198:623–34. doi: 10.1084/jem.20030390
117. Dawson HD, Collins G, Pyle R, Key M, Weeraratna A, Deep-Dixit V, et al. Direct and indirect effects of retinoic acid on human Th2 cytokine and chemokine expression by human T lymphocytes. BMC Immunol. (2006) 7:27. doi: 10.1186/1471-2172-7-27
118. Darmanin S, Chen J, Zhao S, Cui H, Shirkoohi R, Kubo N, et al. All-trans retinoic acid enhances murine dendritic cell migration to draining lymph nodes via the balance of matrix metalloproteinases and their inhibitors. J Immunol. (2007) 179:4616–25. doi: 10.4049/jimmunol.179.7.4616
119. Allie SR, Zhang W, Tsai CY, Noelle RJ, Usherwood EJ. Critical role for all-trans retinoic acid for optimal effector and effector memory CD8 T cell differentiation. J Immunol. (2013) 190:2178–87. doi: 10.4049/jimmunol.1201945
120. Padua RA, Larghero J, Robin M, le Pogam C, Schlageter MH, Muszlak S, et al. PML-RARA-targeted DNA vaccine induces protective immunity in a mouse model of leukemia. Nat Med. (2003) 9:1413–7. doi: 10.1038/nm949
121. Furugaki K, Pokorna K, Le Pogam C, Aoki M, Reboul M, Bajzik V, et al. DNA vaccination with all-trans retinoic acid treatment induces long-term survival and elicits specific immune responses requiring CD4+ and CD8+ T-cell activation in an acute promyelocytic leukemia mouse model. Blood. (2010) 115:653–6. doi: 10.1182/blood-2007-08-109009
122. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. (2006) 66:9299–307. doi: 10.1158/0008-5472.CAN-06-1690
123. Thomas-Schoemann A, Batteux F, Mongaret C, Nicco C, Chereau C, Annereau M, et al. Arsenic trioxide exerts antitumor activity through regulatory T cell depletion mediated by oxidative stress in a murine model of colon cancer. J Immunol. (2012) 189:5171–7. doi: 10.4049/jimmunol.1103094
124. Xu W, Li X, Quan L, Yao J, Mu G, Guo J, et al. Arsenic trioxide decreases the amount and inhibits the function of regulatory T cells, which may contribute to its efficacy in the treatment of acute promyelocytic leukemia. Leuk Lymphoma. (2018) 59:650–59. doi: 10.1080/10428194.2017.1346253
125. Kim J-Y, Bae J-H, Lee S-H, Lee E-Y, Chung B-S, Kim S-H, et al. Induction of NKG2D ligands and subsequent enhancement of NK cell-mediated lysis of cancer cells by arsenic trioxide. J Immunother. (2008) 31:475–86. doi: 10.1097/CJI.0b013e3181755deb
126. Alex AA, Ganesan S, Palani HK, Balasundaram N, David S, Lakshmi KM, et al. Arsenic trioxide enhances the NK cell cytotoxicity against acute promyelocytic leukemia while simultaneously inhibiting its bio-genesis. Front Immunol. (2018) 9:1357. doi: 10.3389/fimmu.2018.01357
127. Boutzen H, Saland E, Larrue C, de Toni F, Gales L, Castelli FA, et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. J Exp Med. (2016) 213:483. doi: 10.1084/jem.20150736
128. Mugoni V, Panella R, Cheloni G, Chen M, Pozdnyakova O, Stroopinsky D, et al. Vulnerabilities in mIDH2 AML confer sensitivity to APL-like targeted combination therapy. Cell Res. (2019) 29:446–59. doi: 10.1038/s41422-019-0162-7
129. Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. (1989) 56:379–90. doi: 10.1016/0092-8674(89)90241-9
130. Cordell JL, Pulford KA, Bigerna B, Roncador G, Banham A, Colombo E, et al. Detection of normal and chimeric nucleophosmin in human cells. Blood. (1999) 93:632–42. doi: 10.1182/blood.V93.2.632
131. Nachmani D, Bothmer AH, Grisendi S, Mele A, Bothmer D, Lee JD, et al. Germline NPM1 mutations lead to altered rRNA 2′-O-methylation and cause dyskeratosis congenita. Nat Genet. (2019) 51:1518–29. doi: 10.1038/s41588-019-0502-z
132. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. (2005) 352:254–66. doi: 10.1056/NEJMoa041974
133. Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. (2016) 127:29–41. doi: 10.1182/blood-2015-07-604496
134. Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. (2008) 111:2776–84. doi: 10.1182/blood-2007-08-109090
135. Pratcorona M, Brunet S, Nomdedeu J, Ribera JM, Tormo M, Duarte R, et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood. (2013) 121:2734–8. doi: 10.1182/blood-2012-06-431122
136. Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. (2008) 358:1909–18. doi: 10.1056/NEJMoa074306
137. Ho AD, Schetelig J, Bochtler T, Schaich M, Schafer-Eckart K, Hanel M, et al. Allogeneic stem cell transplantation improves survival in patients with acute myeloid leukemia characterized by a high allelic ratio of mutant FLT3-ITD. Biol Blood Marrow Transplant. (2016) 22:462–9. doi: 10.1016/j.bbmt.2015.10.023
138. DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. (2018) 133:7–17. doi: 10.1182/blood-2018-08-868752
139. Ivey A, Hills RK, Simpson MA, Jovanovic JV, Gilkes A, Grech A, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. (2016) 374:422–33. doi: 10.1056/NEJMoa1507471
140. Greiner J, Schneider V, Schmitt M, Götz M, Döhner K, Wiesneth M, et al. Immune responses against the mutated region of cytoplasmatic NPM1 might contribute to the favorable clinical outcome of AML patients with NPM1-mutations (NPM1mut). Blood. (2013) 122:1087–8. doi: 10.1182/blood-2013-04-496844
141. Liso A, Colau D, Benmaamar R, De Groot A, Martin W, Benedetti R, et al. Nucleophosmin leukaemic mutants contain C-terminus peptides that bind HLA class I molecules. Leukemia. (2008) 22:424–6. doi: 10.1038/sj.leu.2404887
142. Greiner J, Ono Y, Hofmann S, Schmitt A, Mehring E, Gotz M, et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood. (2012) 120:1282–9. doi: 10.1182/blood-2011-11-394395
143. Forghieri F, Riva G, Lagreca I, Barozzi P, Vallerini D, Morselli M, et al. Characterization and dynamics of specific T cells against nucleophosmin-1 (NPM1)-mutated peptides in patients with NPM1-mutated acute myeloid leukemia. Oncotarget. (2019) 10:869–82. doi: 10.18632/oncotarget.26617
144. van der Lee DI, Reijmers RM, Honders MW, Hagedoorn RS, de Jong RCM, Kester MGD, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Invest. (2019) 129:774–85. doi: 10.1172/JCI97482
145. Small D. FLT3 mutations: biology and treatment. Hematol Am Soc Hematol Educ Program. (2006) 2006:178–84. doi: 10.1182/asheducation-2006.1.178
146. Whitman SP, Maharry K, Radmacher MD, Becker H, Mrozek K, Margeson D, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. (2010) 116:3622–6. doi: 10.1182/blood-2010-05-283648
147. Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematol Am Soc Hematol Educ Program. (2013) 2013:220–6. doi: 10.1182/asheducation-2013.1.220
148. Reddy PN, Sargin B, Choudhary C, Stein S, Grez M, Muller-Tidow C, et al. SOCS1 cooperates with FLT3-ITD in the development of myeloproliferative disease by promoting the escape from external cytokine control. Blood. (2012) 120:1691–702. doi: 10.1182/blood-2010-08-301416
149. Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Muller C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. (2000) 96:3907–14. doi: 10.1182/blood.V96.12.3907
150. Boddu P, Kantarjian HM, Garcia-Manero G, Ravandi F, Verstovsek S, Jabbour E, et al. Treated secondary acute myeloid leukemia: a distinct high-risk subset of AML with adverse prognosis. Blood Adv. (2017) 1:1312–23. doi: 10.1182/bloodadvances.2017008227
151. Lau CM, Nish SA, Yogev N, Waisman A, Reiner SL, Reizis B. Leukemia-associated activating mutation of Flt3 expands dendritic cells and alters T cell responses. J Exp Med. (2016) 213:415–31. doi: 10.1084/jem.20150642
152. Rickmann M, Krauter J, Stamer K, Heuser M, Salguero G, Mischak-Weissinger E, et al. Elevated frequencies of leukemic myeloid and plasmacytoid dendritic cells in acute myeloid leukemia with the FLT3 internal tandem duplication. Ann Hematol. (2011) 90:1047–58. doi: 10.1007/s00277-011-1231-2
153. Rickmann M, Macke L, Sundarasetty BS, Stamer K, Figueiredo C, Blasczyk R, et al. Monitoring dendritic cell and cytokine biomarkers during remission prior to relapse in patients with FLT3-ITD acute myeloid leukemia. Ann Hematol. (2013) 92:1079–90. doi: 10.1007/s00277-013-1744-y
154. Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. (2002) 1:433–43. doi: 10.1016/S1535-6108(02)00069-7
155. Gutierrez L, Jang M, Zhang T, Akhtari M, Alachkar H. Midostaurin reduces regulatory T cells markers in acute myeloid leukemia. Sci Rep. (2018) 8:17544–44. doi: 10.1038/s41598-018-35978-0
156. Li GX, Wang L, Yaghmour B, Ramsingh G, Yaghmour G. The role of FLT3 inhibitors as maintenance therapy following hematopoietic stem cell transplant. Leuk Res Rep. (2018) 10:26–36. doi: 10.1016/j.lrr.2018.06.003
157. Metzelder SK, Schroeder T, Finck A, Scholl S, Fey M, Gotze K, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia. (2012) 26:2353–9. doi: 10.1038/leu.2012.105
158. Lange A, Jaskula E, Lange J, Dworacki G, Nowak D, Simiczyjew A, et al. The sorafenib anti-relapse effect after alloHSCT is associated with heightened alloreactivity and accumulation of CD8+PD-1+ (CD279+) lymphocytes in marrow. PLoS ONE. (2018) 13:e0190525. doi: 10.1371/journal.pone.0209108
159. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. (2008) 7:3129–40. doi: 10.1158/1535-7163.MCT-08-0013
160. Mathew NR, Baumgartner F, Braun L, O'Sullivan D, Thomas S, Waterhouse M, et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat Med. (2018) 24:282–91. doi: 10.1038/nm.4484
161. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. (2010) 17:225–34. doi: 10.1016/j.ccr.2010.01.020
162. Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. (2014) 14:329–41. doi: 10.1016/j.stem.2013.12.016
163. Melnick AM, Levine RL, Figueroa ME, Thompson CB, Abdel-Wahab O. Epigenetic effects of IDH1/IDH2 mutations. Blood. (2011) 118:SCI–33. doi: 10.1182/blood.V118.21.SCI-33.SCI-33
164. Shih AH, Shank KR, Meydan C, Intlekofer AM, Ward P, Thompson CB, et al. AG-221, a small molecule mutant IDH2 inhibitor, remodels the epigenetic state of IDH2-mutant cells and induces alterations in self-renewal/differentiation in IDH2-mutant AML model in vivo. Blood. (2014) 124:437. doi: 10.1182/blood.V124.21.437.437
165. Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell. (2016) 30:337–48. doi: 10.1016/j.ccell.2016.05.018
166. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. (2018) 378:2386–98. doi: 10.1056/NEJMoa1716984
167. Muhlhausen C, Burckhardt BC, Hagos Y, Burckhardt G, Keyser B, Lukacs Z, et al. Membrane translocation of glutaric acid and its derivatives. J Inherit Metab Dis. (2008) 31:188–93. doi: 10.1007/s10545-008-0825-x
168. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. (2018) 24:1192–203. doi: 10.1038/s41591-018-0095-6
169. Böttcher M, Renner K, Berger R, Mentz K, Thomas S, Cardenas-Conejo ZE, et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology. (2018) 7:e1445454. doi: 10.1080/2162402X.2018.1445454
170. Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. (2017) 31:774–86. doi: 10.1101/gad.294991.116
171. Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. (2017) 127:1425–37. doi: 10.1172/JCI90644
172. Lemonnier F, Cairns RA, Inoue S, Li WY, Dupuy A, Broutin S, et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci USA. (2016) 113:15084–89. doi: 10.1073/pnas.1617929114
173. Zhang X, Rao A, Sette P, Deibert C, Pomerantz A, Kim WJ, et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. (2016) 18:1402–12. doi: 10.1093/neuonc/now061
174. Chen JY, Lai YS, Tsai HJ, Kuo CC, Yen BL, Yeh SP, et al. The oncometabolite R-2-hydroxyglutarate activates NF-kappaB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci Rep. (2016) 6:32428. doi: 10.1038/srep32428
175. Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. (2013) 18:617–33. doi: 10.1016/j.cmet.2013.06.019
176. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. (2005) 18:283–93. doi: 10.1016/j.molcel.2005.03.027
177. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. (2015) 16:393–405. doi: 10.1038/nrm4007
178. Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. (2010) 2:a000893. doi: 10.1101/cshperspect.a000893
179. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
180. Pfeifer GP, Holmquist GP. Mutagenesis in the P53 gene. Biochim Biophys Acta. (1997) 1333:M1–8. doi: 10.1016/S0304-419X(97)00004-8
181. Ferrari A, Papayannidis C, Baldazzi C, Iacobucci I, Paolini S, Padella A, et al. Leukemia associated TP53 mutations in AML patients ARE strongly associated with complex karyotype and poor outcome. Blood. (2014) 124:2379. doi: 10.1182/blood.V124.21.484.484
182. Seifert H, Mohr B, Thiede C, Oelschlagel U, Schakel U, Illmer T, et al. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia. (2009) 23:656–63. doi: 10.1038/leu.2008.375
183. Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. (2012) 119:2114–21. doi: 10.1182/blood-2011-08-375758
184. Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F, et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood. (2012) 120:2963–72. doi: 10.1182/blood-2012-03-419622
185. Middeke JM, Fang M, Cornelissen JJ, Mohr B, Appelbaum FR, Stadler M, et al. Outcome of patients with abnl(17p) acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. Blood. (2014) 123:2960–7. doi: 10.1182/blood-2013-12-544957
186. Middeke JM, Herold S, Rucker-Braun E, Berdel WE, Stelljes M, Kaufmann M, et al. TP53 mutation in patients with high-risk acute myeloid leukaemia treated with allogeneic haematopoietic stem cell transplantation. Br J Haematol. (2016) 172:914–22. doi: 10.1111/bjh.13912
187. Getta BM, Ghosh A, Coombs CC, Devlin S, Arcila M, Mohanty A, et al. TP53 mutations in AML predict adverse outcome in patients undergoing allogeneic hematopoietic stem cell transplant. Blood. (2016) 128:3481. doi: 10.1182/blood.V128.22.3481.3481
188. Klepin HD. Myelodysplastic syndromes and acute myeloid leukemia in the elderly. Clin Geriatr Med. (2016) 32:155–73. doi: 10.1016/j.cger.2015.08.010
189. Munoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol. (2016) 16:741–50. doi: 10.1038/nri.2016.99
190. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. (2007) 445:656–60. doi: 10.1038/nature05529
191. Munoz-Fontela C, Garcia MA, Garcia-Cao I, Collado M, Arroyo J, Esteban M, et al. Resistance to viral infection of super p53 mice. Oncogene. (2005) 24:3059–62. doi: 10.1038/sj.onc.1208477
192. Menendez D, Shatz M, Azzam K, Garantziotis S, Fessler MB, Resnick MA. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. (2011) 7:e1001360. doi: 10.1371/journal.pgen.1001360
193. Shatz M, Menendez D, Resnick MA. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. (2012) 72:3948–57. doi: 10.1158/0008-5472.CAN-11-4134
194. Taura M, Eguma A, Suico MA, Shuto T, Koga T, Komatsu K, et al. p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol. (2008) 28:6557–67. doi: 10.1128/MCB.01202-08
195. Cui Y, Guo G. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int J Mol Sci. (2016) 17:E1942. doi: 10.3390/ijms17111942
196. Li H, Lakshmikanth T, Garofalo C, Enge M, Spinnler C, Anichini A, et al. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle. (2011) 10:3346–58. doi: 10.4161/cc.10.19.17630
197. Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. (2011) 71:5998–6009. doi: 10.1158/0008-5472.CAN-10-3211
198. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. (2008) 134:657–67. doi: 10.1016/j.cell.2008.06.049
199. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, et al. Non-cell-autonomous tumor suppression by p53. Cell. (2013) 153:449–60. doi: 10.1016/j.cell.2013.03.020
200. He XY, Xiang C, Zhang CX, Xie YY, Chen L, Zhang GX, et al. p53 in the myeloid lineage modulates an inflammatory microenvironment limiting initiation and invasion of intestinal tumors. Cell Rep. (2015) 13:888–97. doi: 10.1016/j.celrep.2015.09.045
201. Napoli M, Flores ER. The p53 family orchestrates the regulation of metabolism: physiological regulation and implications for cancer therapy. Br J Cancer. (2017) 116:149–55. doi: 10.1038/bjc.2016.384
202. Wu S, Akhtari M, Alachkar H. Characterization of mutations in the mitochondrial encoded electron transport chain complexes in acute myeloid leukemia. Sci Rep. (2018) 8:13301. doi: 10.1038/s41598-018-31489-0
203. Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. (2002) 108:165–70. doi: 10.1016/S0092-8674(02)00626-8
204. Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. (2007) 8:1006. doi: 10.1038/nrm2277
205. Bernardi R, Pandolfi PP. A dialog on the first 20 years of PML research and the next 20 ahead. Front Oncol. (2014) 4:23. doi: 10.3389/fonc.2014.00023
206. Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, et al. The function of PML in p53-dependent apoptosis. Nat Cell Biol. (2000) 2:730–6. doi: 10.1038/35036365
207. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. (2000) 406:207–10. doi: 10.1038/35018127
208. Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. Embo J. (2000) 19:6185–95. doi: 10.1093/emboj/19.22.6185
209. Matt S, Hofmann TG. Crosstalk between p53 modifiers at PML bodies. Mol Cell Oncol. (2018) 5:e1074335. doi: 10.1080/23723556.2015.1074335
210. Ablain J, Rice K, Soilihi H, de Reynies A, Minucci S, de The H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med. (2014) 20:167–74. doi: 10.1038/nm.3441
211. Abramowitz J, Neuman T, Perlman R, Ben-Yehuda D. Gene and protein analysis reveals that p53 pathway is functionally inactivated in cytogenetically normal acute myeloid leukemia and acute promyelocytic leukemia. BMC Med Genomics. (2017) 10:18. doi: 10.1186/s12920-017-0249-2
212. Prokocimer M, Molchadsky A, Rotter V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood. (2017) 130:699. doi: 10.1182/blood-2017-02-763086
213. Prestipino A, Emhardt AJ, Aumann K, O'Sullivan D, Gorantla SP, Duquesne S, et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Transl Med. (2018) 10:eaam7729. doi: 10.1126/scitranslmed.aam7729
214. Iurlo A, Cattaneo D, Gianelli U. Blast transformation in myeloproliferative neoplasms: risk factors, biological findings, and targeted therapeutic options. Int J Mol Sci. (2019) 20:1839. doi: 10.3390/ijms20081839
215. Rampal RK, Mascarenhas JO, Kosiorek HE, Price L, Berenzon D, Hexner E, et al. Safety and efficacy of combined ruxolitinib and decitabine in accelerated and blast-phase myeloproliferative neoplasms. Blood Adv. (2018) 2:3572–80. doi: 10.1182/bloodadvances.2018019661
216. Jacquelin S, Straube J, Cooper L, Vu T, Song A, Bywater M, et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood. (2018) 132:2707–21. doi: 10.1182/blood-2018-04-846220
217. Nakagawa M, Shimabe M, Watanabe-Okochi N, Arai S, Yoshimi A, Shinohara A, et al. AML1/RUNX1 functions as a cytoplasmic attenuator of NF-κB signaling in the repression of myeloid tumors. Blood. (2011) 118:6626–37. doi: 10.1182/blood-2010-12-326710
218. Mann GJ, Pupo GM, Campain AE, Carter CD, Schramm SJ, Pianova S, et al. BRAF mutation, NRAS mutation, and the absence of an immune-related expressed gene profile predict poor outcome in patients with stage III melanoma. J Invest Dermatol. (2013) 133:509–17. doi: 10.1038/jid.2012.283
219. Johnson DB, Lovly CM, Flavin M, Ayers GD, Zhao Z, Iams WT, et al. NRAS mutation: a potential biomarker of clinical response to immune-based therapies in metastatic melanoma (MM). J Clin Oncol. (2013) 31:9019–19. doi: 10.1200/jco.2013.31.15_suppl.9019
220. Klock JC, Pieprzyk JK. Cholesterol, phospholipids, and fatty acids of normal immature neutrophils: comparison with acute myeloblastic leukemia cells and normal neutrophils. J Lipid Res. (1979) 20:908–11.
221. Pabst T, Kortz L, Fiedler GM, Ceglarek U, Idle JR, Beyoglu D. The plasma lipidome in acute myeloid leukemia at diagnosis in relation to clinical disease features. BBA Clin. (2017) 7:105–14. doi: 10.1016/j.bbacli.2017.03.002
222. Stefanko A, Thiede C, Ehninger G, Simons K, Grzybek M. Lipidomic approach for stratification of acute myeloid leukemia patients. PLoS ONE. (2017) 12:e0168781. doi: 10.1371/journal.pone.0168781
223. Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. (2015) 125:2120–30. doi: 10.1182/blood-2014-08-594408
224. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. (2013) 13:227–32. doi: 10.1038/nrc3483
225. Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VCJ, Laurent G, et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest. (2012) 122:3088–100. doi: 10.1172/JCI62129
226. Harper ME, Antoniou A, Villalobos-Menuey E, Russo A, Trauger R, Vendemelio M, et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. Faseb J. (2002) 16:1550–7. doi: 10.1096/fj.02-0541com
227. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. (2010) 120:142–56. doi: 10.1172/JCI38942
228. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. (2017) 7:716–35. doi: 10.1158/2159-8290.CD-16-0441
229. Jones CL, Stevens BM, D'Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. (2018) 34:724–40.e4. doi: 10.1016/j.ccell.2018.10.005
230. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. (2017) 168:657–69. doi: 10.1016/j.cell.2016.12.039
231. Stuani L, Riols F, Millard P, Sabatier M, Batut A, Saland E, et al. Stable isotope labeling highlights enhanced fatty acid and lipid metabolism in human acute myeloid leukemia. Int J Mol Sci. (2018) 19:E3325. doi: 10.3390/ijms19113325
232. Ju HQ, Zhan G, Huang A, Sun Y, Wen S, Yang J, et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia. (2017) 31:2143–50. doi: 10.1038/leu.2017.45
233. Stockard B, Garrett T, Guingab-Cagmat J, Meshinchi S, Lamba J. Distinct metabolic features differentiating FLT3-ITD AML from FLT3-WT childhood acute myeloid leukemia. Sci Rep. (2018) 8:5534. doi: 10.1038/s41598-018-23863-9
234. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. (2016) 19:23–37. doi: 10.1016/j.stem.2016.06.001
235. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. (2012) 18:1350–8. doi: 10.1038/nm.2882
236. Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, et al. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. (2017) 77:1453–64. doi: 10.1158/0008-5472.CAN-16-1645
237. Chapuis N, Poulain L, Birsen R, Tamburini J, Bouscary D. Rationale for targeting deregulated metabolic pathways as a therapeutic strategy in acute myeloid leukemia. Front Oncol. (2019) 9:405. doi: 10.3389/fonc.2019.00405
238. Castelli G, Pelosi E, Testa U. Emerging therapies for acute myelogenus leukemia patients targeting apoptosis and mitochondrial metabolism. Cancers. (2019) 11:E260. doi: 10.3390/cancers11020260
239. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. (2019) 9:370–383. doi: 10.1158/2159-8290.CD-18-0774
Keywords: acute myeloid leukemia, genetics, immune landscape, metabolism, targeted (selective) treatment, immunotherapy
Citation: Mendez LM, Posey RR and Pandolfi PP (2019) The Interplay Between the Genetic and Immune Landscapes of AML: Mechanisms and Implications for Risk Stratification and Therapy. Front. Oncol. 9:1162. doi: 10.3389/fonc.2019.01162
Received: 10 July 2019; Accepted: 17 October 2019;
Published: 07 November 2019.
Edited by:
Antonio Curti, University of Bologna, ItalyReviewed by:
Kavita Raj, Guy's and St Thomas' NHS Foundation Trust, United KingdomCopyright © 2019 Mendez, Posey and Pandolfi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pier Paolo Pandolfi, cHBhbmRvbGZAYmlkbWMuaGFydmFyZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.