 Julian Biau
Julian Biau Emmanuel Chautard
Emmanuel Chautard Pierre Verrelle1,6,8,9
Pierre Verrelle1,6,8,9 Marie Dutreix
Marie Dutreix- 1Institut Curie, PSL Research University, Centre de Recherche, Paris, France
- 2UMR3347, CNRS, Orsay, France
- 3U1021, INSERM, Orsay, France
- 4Université Paris Sud, Orsay, France
- 5Université Clermont Auvergne, INSERM, U1240 IMoST, Clermont Ferrand, France
- 6Radiotherapy Department, Université Clermont Auvergne, Centre Jean Perrin, Clermont-Ferrand, France
- 7Pathology Department, Université Clermont Auvergne, Centre Jean Perrin, Clermont-Ferrand, France
- 8U1196, INSERM, UMR9187, CNRS, Orsay, France
- 9Radiotherapy Department, Institut Curie Hospital, Paris, France
Radiation therapy (RT) is widely used in cancer care strategies. Its effectiveness relies mainly on its ability to cause lethal damage to the DNA of cancer cells. However, some cancers have shown to be particularly radioresistant partly because of efficient and redundant DNA repair capacities. Therefore, RT efficacy might be enhanced by using drugs that can disrupt cancer cells' DNA repair machinery. Here we review the recent advances in the development of novel inhibitors of DNA repair pathways in combination with RT. A large number of these compounds are the subject of preclinical/clinical studies and target key enzymes involved in one or more DNA repair pathways. A totally different strategy consists of mimicking DNA double-strand breaks via small interfering DNA (siDNA) to bait the whole DNA repair machinery, leading to its global inhibition.
Introduction
Radiation therapy (RT), in conjunction with surgery and systemic therapies (chemotherapy, targeted therapies, immunotherapy…), is a cornerstone of cancer care. About 50% of cancer patients receive RT (1). The primary objective of RT is to increase the amount of radiation delivered to the tumor to ensure local control and reduce the amount of radiation in adjacent healthy tissues. Advanced developments such as image-guided RT (IGRT) or intensity-modulated RT (IMRT) have led to the enhancement of this therapeutic ratio (2). Despite such improvements, many patients still experience local recurrence of the disease after RT. Clinical factors such as tumor stage, frequently associated with increased hypoxia, can explain some of the failures, but it is clear that biological characteristics play a key part in successful treatment (3–5). RT-induced cell death is mostly due to DNA damage, especially to double-strand breaks (DSBs) (6, 7). Consequently, tumor cells with highly efficient DNA repair are radioresistant (8), whereas deficiencies in pathways that repair DSBs are particularly detrimental to the cells (9). Therefore, therapies that inhibit the DNA repair machinery have the potential to enhance RT efficacy (10, 11). Inhibiting DNA repair offers an opportunity to target genetic differences between tumor and normal cells, as DNA repair is often dysregulated in tumor cells (10, 12–14). Tumor cells divide rapidly because of unregulated cell cycle control. Thus, they have less time to repair DNA damage as compared to normal cells that are not dividing or will stop dividing after activation of key checkpoints induced by RT (15, 16). Beside altered cell cycle control, cancer cells may also present defects in their DNA repair system, inducing dependence on specific repair pathways and/or overexpression of alternative pathways (16, 17). Furthermore, cancer cells often develop under stress conditions, thus raising the frequency of endogenous DNA damage (18, 19). This review will firstly focus on the distinct categories of DNA lesions induced by RT and the DNA repair pathways required for their repair. Subsequently, it will present the approaches that are currently being developed to enhance RT efficacy by modulating DNA repair.
RT-Induced DNA Damage Response
DNA lesions induced by RT activate the DNA damage response (DDR), which essentially involves post-translational modifications of proteins to activate downstream signaling pathways (20). DDR is based on an intricate network of proteins that work together to manage DNA repair and cell cycle coordination. DDR interrupts the cell cycle, thereby inhibiting the spread of DNA damage to daughter cells and facilitating repair. Cell division arrest is mainly controlled by the checkpoint kinases CHK1 and CHK2, which are activated by the phosphatidylinsositol-3-kinases (PI3K) of the DDR machinery (15). DDR signaling is also essential for triggering apoptosis when repair is unsuccessful, notably through modifications to the p53 protein (20).
RT induces a variety of DNA lesions. Approximately 10,000 damaged bases, 1,000 single strand breaks (SSBs) and 40 DSBs are produced per gray per cell (21, 22). Such lesions, if not corrected, can lead to cell death by mitotic catastrophe and apoptosis. DSBs are the most lethal to the cells despite their low proportion, as one single unrepaired DSB can trigger cell death (7). DSBs are produced directly and indirectly by RT. Indirect DSBs most often occur during replication if the initial damage is unrepaired. As an example, when a replication fork encounters an unrepaired SSB, the fork is blocked and leads to the conversion of this SSB into a DSB (10, 23, 24). The resulting DSB can directly trigger cell death or activate DDR, which induces cell cycle arrest and promotes DNA repair. This repair is usually error-free, allowing the cell to survive without genetic consequences. It can also be error-prone, leading either to cell death if the error is not viable or mutation and chromosomic aberrations (25).
DNA Repair of RT-Induced Damage
Following RT, damaged bases induced by oxidative stress are repaired by the base excision repair pathway (BER) (26–31). In BER, damaged bases are excised by DNA glycosylases, resulting in apurinic (AP) sites. Subsequently; these AP sites are cleaved by apurinic endonuclease 1 (APE1) or an AP-lyase activity, leading to SSBs. SSBs are repaired by the part of the BER pathway called single strand break repair (SSBR) (Figure 1A) (12, 32). Either short-patch or long-patch SSBR can then proceed, depending on several factors such as type of lesion and cell cycle state. Single nucleotide insertion by DNA polymerase (Pol) β and ligation by DNA ligase III are described as short-patch SSBR, and interact with the protein X-ray repair cross-complementing 1 (XRCC1). Long-patch SSBR involves the removal of a larger DNA segment, which requires several DNA replication factors such as proliferating cell nuclear antigen (PCNA), Pol δ/ϵ, flap endonuclease 1 (FEN1), and DNA ligase I. Concerning SSBs detection, poly-ADP-ribose polymerase (PARP1 or PARP2) are required (28, 33–37). The binding of PARP to SSB activates its auto-PARylation, and leads to the recruitment of BER/SSBR proteins. PARP-1 was also reported as a regulator of DNA repair gene expression through the E2F1 pathway (38). Unrepaired SSB or a damaged base can block the replication forks, resulting in fork collapse and DSB (23). The great majority of oxidative damage induced by ionizing radiation is corrected by BER. However, under hypoxic conditions, IR causes the formation of cyclodeoxynucleosides that can be only removed by nucleotide excision repair (NER). Several results suggest that NER may be involved in the repair of oxidized DNA damage. In addition, ionizing-radiation breast cancer risk has been related to polymorphism in ERCC2 (one of the main NER enzymes) (39).
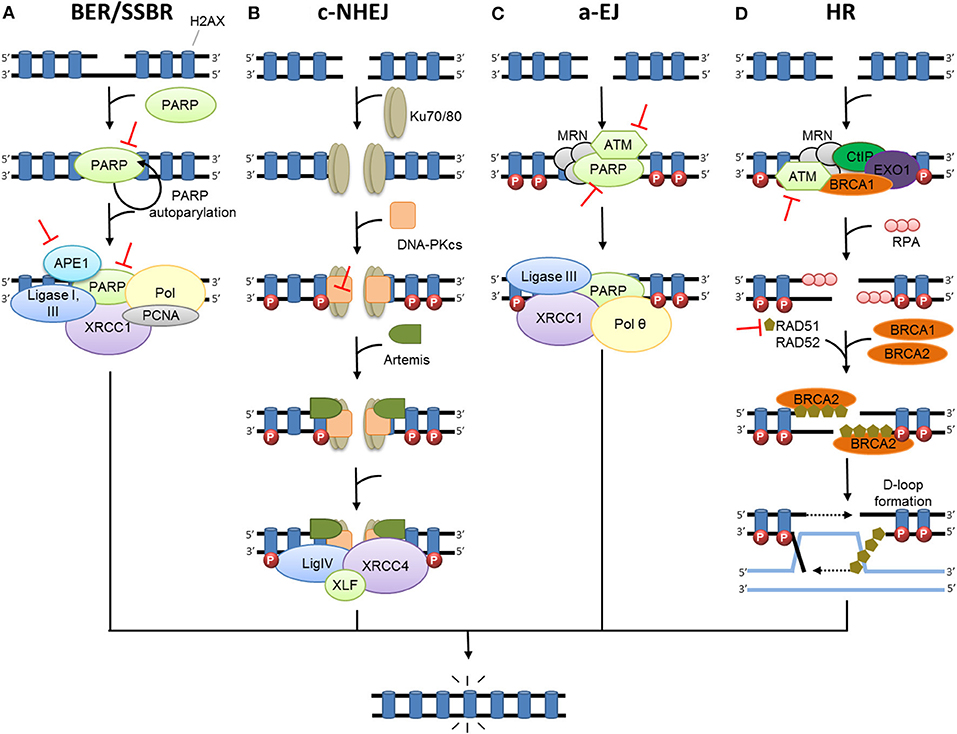
Figure 1. DNA damage repair after radiation therapy. In irradiated cells, a number of DNA lesions are induced including single (SSB) and double-strand breaks (DSB). (A) SSBs are corrected by the part of base excision repair (BER) known as single-strand break repair (SSBR). The binding of PARP to SSB activates its auto-PARylation and leads to the recruitment of BER/SSBR proteins including AP endonucleases (APE1), XRCC1 (helper protein), PCNA, FEN, PNK, and DNA polymerases (Pol; damage processing) and DNA ligases. (B) In c-NHEJ, DSB is recognized by the Ku80-Ku70 heterodimer, which leads to DNA-dependent protein kinase catalytic subunit DNA-PKcs recruitment, gathering the DNA-PK complex and activating its kinase activity. This leads to the involvement of repair proteins (XRCC4, DNA ligase IV and others), which perform the processing and final junction reaction. (C) When c-NHEJ is impaired, an alternative pathway called a-EJ (alternative EJ) takes place and involves mainly PARP1, XRCC1, ligase III, MRN complex and the DNA polymerase θ. (D) In HR, after ATM activation, the DSB site is bounded by the MRE11-RAD50-NBS1 complex (MRN). The consequence is the phosphorylation of a set of targets including H2AX (γ-H2AX), localized at the site of DSB. HR uses the sister chromatid as a model to repair DSB. First, the resection of the DNA at DSB results in a 3′ single-strand DNA, which is then coated by proteins of replication A (RPA). Subsequently, proteins of the RAD family are recruited and mediate the invasion of the homologous strand of the sister chromatid, leading to the formation of Holliday junctions. DNA polymerases can then synthetize across the missing regions. The Holliday junctions are finally resolved by cleavage and followed by ligation of adjacent ends.  Represents inhibitors of DNA repair in preclinical or clinical development.
Represents inhibitors of DNA repair in preclinical or clinical development.
Two major pathways repair DSBs: homologous recombination (HR) and non-homologous end joining (NHEJ) (40). However, both mismatch repair (MMR) and NER pathways have been reported to affect both HR- and NHEJ-mediated DSB repair efficacy to a lesser extent (41). The formation of DSBs triggers the activation of three key enzymes from the PIKK family: ataxia telangiectasia mutated kinase (ATM), ATM-related kinase (ATR), and DNA-dependent protein kinase (DNA-PK). This leads to the phosphorylation of many proteins, signaling damage and initiating DNA repair. One of the early steps is the phosphorylation of histone H2AX (γ-H2AX), which signals the presence of DSB to repair proteins where they aggregate in ionizing radiation-induced foci (IRIF) (42). Besides their crucial roles in DDR signaling, the kinases ATR and ATM are also involved in maintaining replication fork stability (14) and fork reversal in case of fork-stalling lesions, notably through SMARCAL1 (43).
In mammalian cells, c-NHEJ (classical NHEJ, Figure 1B) is the most efficient DSB repair mechanism. It acts by directly ligating the broken DNA ends (44). c-NHEJ can occur during the entire cell cycle. It is frequently accompanied by small deletions at the repair break site and is considered to be the main cause of DSB error-prone repair. The first step of NHEJ is the binding of the heterodimer Ku70/Ku80 at the end of the DSB (45), allowing the recruitment of catalytic subunit DNA-PKcs forming the protein complex DNA-PK (46). DNA-PK, bounded to DNA, is activated and phosphorylates numerous proteins including H2AX (47), Artemis (48), X-ray repair cross- complementing 4 (XRCC4) and ligase IV complex (49), and XLF (XRCC4-like factor) (50) that are recruited on the site of the DSB and participate in its repair. When c-NHEJ is impaired, an alternative pathway called a-EJ (alternative EJ) or MHEJ (Micro Homology End Joining) (Figure 1C) is activated (51). At the initial breaking site, a deletion of 5–25 nucleotides is necessary to reveal micro-homologies to realize a-EJ (52), while a maximum of 4 deleted nucleotides is necessary for c-NHEJ (44). The micro-homologies that are slightly longer in the case of a-EJ could partly explain the higher number of large deletions and other genomic rearrangements that occur (53, 54). The a-EJ pathway is differentiated from c-NHEJ by the fact that it is independent of Ku proteins (52). a-EJ involves mainly PARP1, XRCC1, ligase III (LIGIII), and the MRE11/RAD50/NBS1 (MRN) complex (55, 56). DNA polymerase theta (Pol θ or PolQ) is specifically involved in nucleotide incorporation in the a-EJ mechanism through the TMEJ (theta-mediated end joining) pathway (57).
HR is an alternative pathway for repairing DSBs that uses the sister chromatid as a model, restricting this mechanism to the S and G2 cell cycle phases (Figure 1D) (40). HR is the most conservative and least error-prone repair mechanism. It necessitates the presence of BRCA proteins, defects of which increase susceptibility to breast or ovarian cancer. The DSB site is bounded by several factors such as the MRN complex, EXO1 (exonuclease 1), DNA2-BLM (Bloom syndrome), BRCA1 and CTIP (CtBP-interacting protein) that contribute to DNA resection and formation of a 3′ single-strand DNA (58–60), which is then coated by proteins of replication A (RPA). After the RPA protein's displacement by RAD51, BRCA2 together with the localizer of BRCA2 (PALB2), RAD54, and BARD1 (BRCA1-associated RING domain protein 1) mediates the nucleoprotein filament invasion of the homologous strand of the sister chromatid and creates the “D-loop” (61). DNA polymerases can then synthetize across the missing regions. The resulting Holliday junctions are finally resolved by cleavage and followed by ligation of adjacent ends (62). However, HR can sometimes be error-prone, especially if template switching occurs, e.g., in repeat sequences (63).
The choice between the two major mechanisms for DSB repair (NHEJ and HR pathways) seems to be linked to several factors such as cell-cycle phase, chromatin context, or availability of key actors such as the Ku complex, 53BP1 or RAD51 (64, 65).
Current Strategies Involved in DDR Inhibition in Combination with RT
Targeting Key Enzymes Involved in a Specific DNA Repair Pathway
Inhibiting BER/SSBR
BER and SSBR pathways repair damaged bases and SSBs. Inhibiting BER/SSBR may lead to unrepaired damages that are converted to DSBs when encountering a replication fork (23). Therefore, in cells already defective for HR, such as BRCA−/− breast or ovarian cancer tumors, the inhibition of BER by PARP inhibitors leads to unrepaired DSBs and cell death (14). This effect, called synthetic lethality, has been extensively described (17, 66, 67) and studied in numerous clinical trials (68–70). Since the majority of RT-induced damages are repaired by BER/SSBR, inhibition of this pathway should highly sensitize cells to RT even in HR-proficient cells (29). The preclinical evaluation of PARP inhibitors has shown enhanced RT efficacy both in vitro and in vivo (71–73). Several PARP inhibitors have already been tested in or entered into numerous clinical trials in association with RT for brain metastases, ovarian cancer, breast cancer, rectal cancer, or glioblastoma, among others (Table 1). However, early data did not demonstrate convincing and coherent proof of synergy, although neither did they demonstrate unexpected toxic effects (74, 75). Another strategy for the inhibition of BER/SSBR is the development of APE1 inhibitors. APE1 is crucial for BER/SSBR and is commonly overexpressed in cancer cells (80, 81), giving to this strategy some tumor specificity. APE1 inhibitors have shown efficacy in combination with RT in preclinical studies (82). Lucanthone, an APE1 inhibitor, combined with temozolomide, has recently been tested in a phase 2 clinical trial in glioblastoma (Table 1). The results are not yet published.
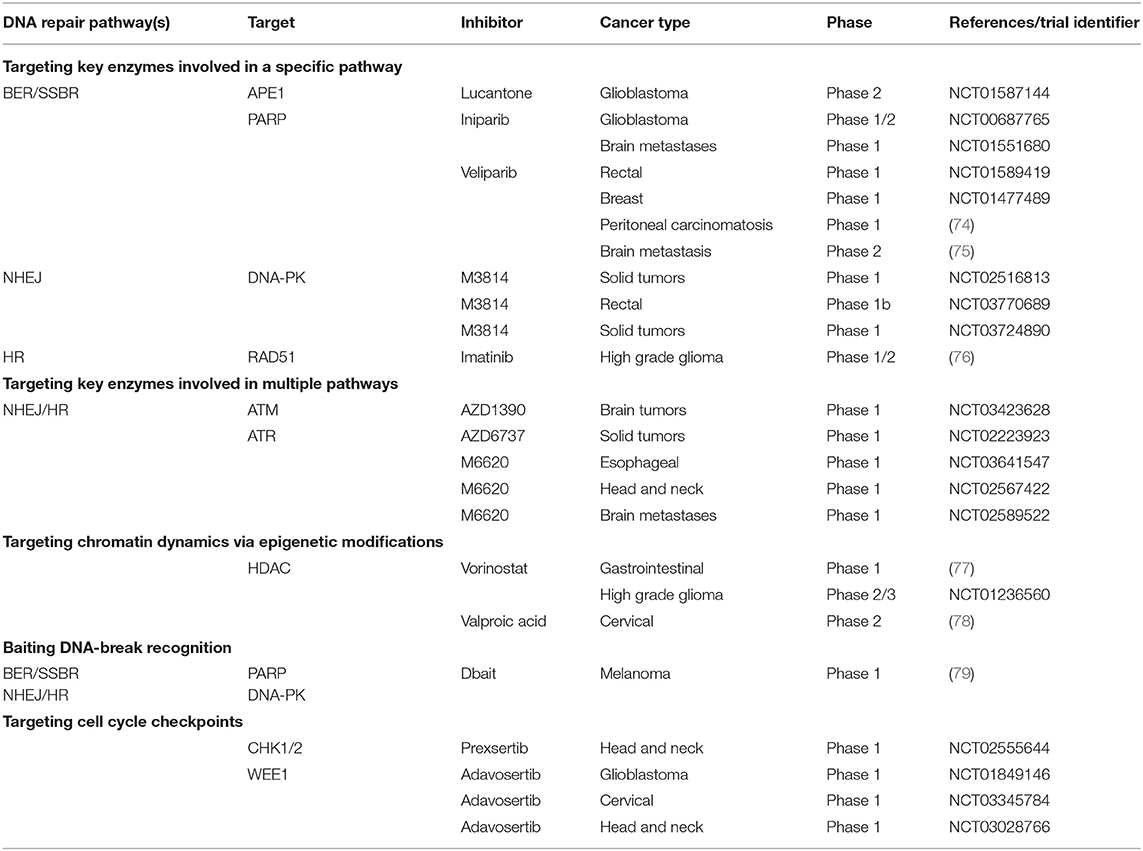
Table 1. Examples of clinical trials of inhibitors of the DNA damage response in combination with radiation therapy.
Inhibiting NHEJ
DNA-PK, a key enzyme in NHEJ, is a member of the PI3K family that performs a central role in many cellular functions (83). Selective DNA-PK inhibitors have led to radiosensitization in preclinical studies (84–86). Three phase 1 trials are currently testing the safety and tolerability of a DNA-PK inhibitor (M3814) in combination with palliative RT +/- immunotherapy in advanced solid tumors (NCT02516813 and NCT03724890) and curative-intent radiotherapy in locally advanced rectal cancer (NCT03770689) (Table 1). Such strategies, which are not based on a selective effect on the tumor, are considered promising by some (14) though they have been criticized by others (87). Early reports of the clinical combination of M3814 and palliative RT showed enhanced normal tissue reactions including dysphagia, prolonged mucosal inflammation/stomatitis, and skin injury (87, 88). Inhibition of Ku subunits could also result in reduced DNA-PK activity and NHEJ. This is consistent with the existing data reporting that shRNA depletion of Ku70 or Ku80 showed cytotoxicity and radiosensitization in pancreatic cancer cells (89, 90). CC-115, a dual inhibitor of DNA-PK and mammalian target of rapamycin (mTOR), is being tested; preliminary anti-tumor activity has been reported, although whether these responses are attributable to activity against DNA-PK or mTOR is unclear (14, 91). A phase 1 trial testing CC-115 in combination with RT and temozolomide in the treatment of glioblastoma is ongoing (NCT02977780). NHEJ can also be indirectly inhibited via the EGFR pathway (see below).
Inhibiting HR
Cancer cells are known to be more proliferative than normal cells (92). Inhibitors of replication-associated processes such as HR exploit this specificity to enhance the therapeutic ratio. Nevertheless, there are few specific inhibitors of HR. It has been shown that RAD51 expression and functional HR can be reduced using imatinib during experimental RT, leading to increased radiosensitization (13, 93). Indirect inhibition of HR can also be obtained via cell cycle checkpoint targeting (see below).
Targeting Key Enzymes Involved in Multiple Repair Pathways
DNA damage detection and signaling is the first step common to all DNA repair pathways. Acting on this step will alter several pathways. Therefore, several approaches have been tested to disable part or all of the DNA damage recognition/signaling steps.
Inhibiting ATM
ATM is one of the key enzymes in DNA damage signaling of DSBs for HR but also NHEJ (94). Defective cells in ATM are extremely radiosensitive, independent of their p53 status (95). ATM inhibitors have shown radiosensitization in preclinical studies (96–98). A single 15Gy RT dose suppressed tumor growth in a preclinical model when ATM was deleted in cancer cells vs. when deleted in endothelial cells (99), underlining the interest in testing ATM inhibitors in combination with highly conformal RT. Like DNA-PK, ATM is part of the PI3K family and has many cellular functions. A phase 1 study is currently testing an ATM inhibitor (AZD1390) in combination with RT in brain tumors including glioblastoma and brain metastases (NCT03423628). Indirectly, inhibition of the TGFβ-signaling or mitogen-activated protein kinase (MAPK) pathway can lead to reduced ATM activation and increased tumor cell radiosensitivity through reduced DSB repair (100–102).
Inhibiting ATR
ATR is a critical kinase that is activated in reaction to replication stress and blocked replication forks. ATR is one of the key enzymes in DNA damage signaling of DSBs (103). Cancer cells, which exhibit relatively elevated levels of replication stress, are more susceptible to dependence on ATR signaling for survival (104). An ATR inhibitor (AZD6738) has given encouraging preclinical results (67, 105) and is currently in phase I trials as monotherapy or in combination with olaparib, RT (NCT02223923), carboplatin and immunotherapy agents. Another ATR inhibitor (M6620) is being tested in three phase 1 trials with radiotherapy in esophageal cancer (NCT03641547), locally advanced head and neck squamous cell carcinoma (NCT02567422) and brain metastases (NCT02589522).
Inhibiting MRN Complex
Mirin is an inhibitor of MRE11 endonuclease and thus of HR function. However, MRE11 is also upstream of NHEJ, and so mirin inhibits NHEJ and its effects are not specific to HR (16, 106, 107). It might be of particular interest in combination with RT.
Baiting DNA Breaks Signaling
This approach developed recently is represented by the molecules called Dbait/AsiDNA™. Dbait/AsiDNA™ consist of double-strand oligonucleotides that mimic DNA strand breaks and consequently bind and trap the signaling and repair proteins DNA-PK (24, 108, 109) and PARP (110), leading to inhibition of both SSB and DSB repair. In preclinical studies, the proof of concept that a RT-Dbait association could be used in treating melanoma (24) and high-grade glioma (111) has been established. A first-in-man phase 1 trial was conducted combining Dbait/AsiDNA™ in combination with palliative RT in in-transit metastases of melanoma (79) (Table 1). In this trial, no dose-limiting toxicity was reported and the maximum tolerated dose was not met.
Targeting Chromatin Dynamics via Epigenetic Modifications
Epigenetics is an emerging field in cancer biology (112). It focuses on functionally relevant genome modifications that do not modify the nucleotide sequence. Such modifications include DNA methylation or histone modifications that may regulate gene expression but do not alter the associated DNA sequence. These modifications could also affect DNA repair ability. The loss of ARID1A, a piece of the SWI/SNF chromatin remodeling complex, was recently reported to induce a selective vulnerability to combined RT and PARP inhibitor therapy (113).
Inhibiting Histone Deacetylases (HDACs)
HDAC inhibitors are epigenetic therapeutics. They have the capacity to lower RT-induced damage repair both at the level of damage signaling, via inhibition of the ATM or MRN complex, and by directly affecting proteins involved in NHEJ and HR (112, 114–117). Several clinical trials have been carried out for various cancer types (77, 78) (Table 1).
Inhibition of Kinases Involved in DDR-Related Survival Pathways
Sorafenib, a multi-kinase inhibitor, is currently utilized in the clinic for the treatment of hepatocellular and renal cancers. It inhibits MAPK signaling together with additional intracellular Ser/Thr kinases, leading to both NHEJ and HR inhibition. Sorafenib has shown a radiosensitization effect in preclinical studies (118). Dasatinib is another multi-kinase inhibitor inhibiting ABL and SRC tyrosine kinases. In preclinical studies, it has shown a radiosensitization effect partly due to blocking of DNA repair pathways involved in DSB repair (119). Sorafenib and dasatinib are clinically evaluated in association with RT. Because of their large spectrum of targets, most of these inhibitors may show high toxicity, which prevents them from being used at the dosage required to be efficient in combination with RT in the management of aggressive tumors that overexpress some of their targets (120, 121).
After RT, EGFR has been found to translocate into the nucleus and modulate DNA repair (especially NHEJ) through association with DNA-PKcs (122, 123). A current in-clinic approach is using the monoclonal antibody cetuximab to inhibit this nuclear translocation of EGFR. Cetuximab combined with RT has improved patients' overall survival in a phase III trial in head and neck cancer (124). Furthermore, EGFR signaling may be interrupted by small-molecule tyrosine kinase inhibitors such as erlotinib or gefitinib, especially in the case of specific EGFR mutation; these are currently being tested in combination with RT (125, 126).
Targeting Cell Cycle Checkpoints
Checkpoint dysfunction represents a common molecular defect acquired during tumorigenesis (15, 127), underlying the importance of its regulation in cancer development. Interfering with cell cycle checkpoint signaling is an alternative approach to modulating DNA repair activity and potentially improving the therapeutic ratio. The induction of DNA lesions by RT in normal cells stops their progression in the cell cycle, thereby avoiding the accumulation of other lesions and their damaging effects (20). This cell cycle arrest is subtly correlated with DNA repair to fine-tune cell cycle restart with the disappearance of damage. In cancer cells with an altered G1 checkpoint, cell cycle progression goes on relentlessly and, as a result, the removal of the G2 block increases unrepaired damage and its transfer to the daughter cells. This finally causes the loss of essential genetic material and cell death, a process that strengthens checkpoint inhibition strategies. Combination of RT with a dual CHK1 and CHK2 inhibitors (AZD7762 and prexsertib) showed a radiosensitization effect with an increase in mitotic catastrophe in different cancer cell lines and xenografts (128–130). A phase 1b trial was completed that combined prexsertib with RT and cisplatin or cetuximab in locally advanced head and neck cancer (NCT02555644), the results of which are not yet published. However, in addition to checkpoint activation, CHK1 is also involved in HR (131, 132) and it is uncertain if this is only a result of checkpoint inhibition or if it is partly due to HR inhibition (133).
Another target is the WEE1 kinase, which has been shown to be a major regulator of the G2-M checkpoint (134). This tyrosine kinase inhibits the entrance in mitosis by adding an inhibitory phosphorylation to Cdc2 (the human homolog of tyrosine kinase 1[Cdk1]) to tyrosine 15 (Y15). As a consequence, the Cdc2/cyclin B complex becomes inactivated, which stops the cells in G2-M and allows DNA repair. Preclinical studies have shown the potential use of WEE 1 inhibitors as radiosentizers (135, 136). Several ongoing clinical trials are testing WEE1 inhibitors with RT. In addition, several phase 1 trials are currently testing the WEE1 inhibitor adavosertib (AZD1775) in combination with RT and temozolomide in the treatment of glioblastoma (NCT01849146), with RT and cisplatin in cervical, vaginal or uterine cancer (NCT03345784), or in combination with RT and cisplatin in advanced head and neck cancer (NCT03028766 and NCT02585973).
Combining DNA Repair Targeting, Immunotherapy, and Radiotherapy
DNA repair proteins preserve the integrity of the genome; therefore, DNA repair targeting may enhance the tumor mutational burden, which may lead to the production of neoantigens and increased activity of anti-cancer T cells. Some clinical trials have been set to investigate the use of immune checkpoint inhibitors, notably by combining Dravulumab (anti-PD-L1) with PARP (NCT02484404), ATR (NCT02264678) or WEE1 inhibition (NCT02617277). To date, the interplay between radiation and the immune system is far from being completely deciphered, but several interesting facts have been reported. The cytotoxic action of radiotherapy on tumor cells provides T lymphocytes with tumor neoantigens and releases pro-inflammatory cytokines, thus promoting the immune response (137). The cell death inducing this type of immune response is called immunogenic cell death. Combining immunotherapy with radiotherapy (several recent trials: NCT02707588, NCT02952586, NCT02999087) could increase the ability to cause immunogenic cell death by removing locks that block the immune system (138). The non-overlapping toxicities of DNA repair targeting and immune checkpoint inhibitors render the use of combinations of these agents with radiotherapy appealing (14, 139).
Conclusion
Targeted therapies are beginning to demonstrate activity across a number of tumor types. The most promising approach toward improving the efficiency of a treatment and gaining a reliable response is to develop therapy combinations that decrease the chance of resistance and to treat resistance when it emerges. There has been a considerable renewed emphasis on new targeted treatments such as radiosensitizers that do not cause overlapping dose-limiting toxicities. Selection of appropriate targeted agents represents a challenge. As predicted, during preclinical and clinical trials, particular attention was paid to proteins involved in DNA repair pathways. Various strategies have been explored, ranging from specific protein targeting to global inhibition, and many DNA repair inhibitors have been developed. Up until now, only a few of them have reached the clinical stage, while even fewer have been tested in combination with RT. The several clinical trials currently underway will tell whether these new compounds can be tolerable and efficient.
RT-induced lesions can be corrected by various DNA repair pathways. The intricacy of crosstalk between DNA repair pathways suggests that biomarker assays to determine the status of multiple DNA repair pathways could provide essential information on the sensitivity and resistance of cancer cells to repair inhibitors. Understanding these DNA repair pathways and identifying effective stratification biomarkers from the various DNA repair pathways that are specifically altered in some tumors would be required to characterize patients' responses to specific DNA repair inhibitors.
Author Contributions
JB and EC analyzed the literature and wrote the manuscript. PV and MD gave important intellectual input and carefully revised the manuscript. All authors approved the final manuscript for submission.
Conflict of Interest
MD is the cofounder of DNA Therapeutics/Onxeo, which is involved in DT01 development.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. (2005) 104:1129–37. doi: 10.1002/cncr.21324
2. Elshaikh M, Ljungman M, Ten Haken R, Lichter AS. Advances in radiation oncology. Annu Rev Med. (2006) 57:19–31. doi: 10.1146/annurev.med.57.121304.131431
3. Fertil B, Malaise EP. Intrinsic radiosensitivity of human cell lines is correlated with radioresponsiveness of human tumors: analysis of 101 published survival curves. Int J Radiat Oncol Biol Phys. (1985) 11:1699–707. doi: 10.1016/0360-3016(85)90223-8
4. Taghian A, Ramsay J, Allalunis-Turner J, Budach W, Gioioso D, Pardo F, et al. Intrinsic radiation sensitivity may not be the major determinant of the poor clinical outcome of glioblastoma multiforme. Int J Radiat Oncol Biol Phys. (1993) 25:243–9. doi: 10.1016/0360-3016(93)90345-V
5. Gerweck LE, Vijayappa S, Kurimasa A, Ogawa K, Chen DJ. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer Res. (2006) 66:8352–5. doi: 10.1158/0008-5472.CAN-06-0533
6. Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. (2001) 27:247–54. doi: 10.1038/85798
7. Radford IR. The level of induced DNA double-strand breakage correlates with cell killing after X-irradiation. Int J Radiat Biol Relat Stud Phys Chem Med. (1985) 48:45–54. doi: 10.1080/09553008514551051
8. Jeggo P, Lavin MF. Cellular radiosensitivity: how much better do we understand it? Int J Radiat Biol. (2009) 85:1061–81. doi: 10.3109/09553000903261263
9. O'Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. (2006) 7:45–54. doi: 10.1038/nrg1746
10. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. (2008) 8:193–204. doi: 10.1038/nrc2342
11. Stover EH, Konstantinopoulos PA, Matulonis UA, Swisher EM. Biomarkers of response and resistance to DNA repair targeted therapies. Clin Cancer Res. (2016) 22:5651–60. doi: 10.1158/1078-0432.CCR-16-0247
12. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. (2011) 11:239–53. doi: 10.1038/nrc3007
13. Thoms J, Bristow RG. DNA repair targeting and radiotherapy: a focus on the therapeutic ratio. Semin Radiat Oncol. (2010) 20:217–22. doi: 10.1016/j.semradonc.2010.06.003
14. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. (2019) 16:81–104. doi: 10.1038/s41571-018-0114-z
15. Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. (2004) 432:316–23. doi: 10.1038/nature03097
16. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. (2012) 12:801–17. doi: 10.1038/nrc3399
17. Shaheen M, Allen C, Nickoloff JA, Hromas R. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood. (2011) 117:6074–82. doi: 10.1182/blood-2011-01-313734
18. Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res. (2011) 711:193–201. doi: 10.1016/j.mrfmmm.2010.12.016
19. Nowsheen S, Wukovich RL, Aziz K, Kalogerinis PT, Richardson CC, Panayiotidis MI, et al. Accumulation of oxidatively induced clustered DNA lesions in human tumor tissues. Mutat Res. (2009) 674:131–6. doi: 10.1016/j.mrgentox.2008.09.010
20. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071–8. doi: 10.1038/nature08467
21. Ward JF. DNA damage and repair. Basic Life Sci. (1991) 58:403–15; discussion 415–21. doi: 10.1007/978-1-4684-7627-9_15
22. Burkart W, Jung T, Frasch G. Damage pattern as a function of radiation quality and other factors. C R Acad Sci III, Sci Vie. (1999) 322:89–101. doi: 10.1016/S0764-4469(99)80029-8
23. Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci USA. (2001) 98:8241–6. doi: 10.1073/pnas.131009198
24. Biau J, Devun F, Jdey W, Kotula E, Quanz M, Chautard E, et al. A preclinical study combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of melanoma. Neoplasia. (2014) 16:835–44. doi: 10.1016/j.neo.2014.08.008
25. Jalal S, Earley JN, Turchi JJ. DNA repair: from genome maintenance to biomarker and therapeutic target. Clin Cancer Res. (2011) 17:6973–84. doi: 10.1158/1078-0432.CCR-11-0761
26. Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. (2013) 5:a012583. doi: 10.1101/cshperspect.a012583
27. van Loon B, Markkanen E, Hübscher U. Oxygen as a friend and enemy: how to combat the mutational potential of 8-oxo-guanine. DNA Repair. (2010) 9:604–16. doi: 10.1016/j.dnarep.2010.03.004
28. Visnes T, Grube M, Hanna BMF, Benitez-Buelga C, Cázares-Körner A, Helleday T. Targeting BER enzymes in cancer therapy. DNA Repair. (2018) 71:118–26. doi: 10.1016/j.dnarep.2018.08.015
29. Vens C, Begg AC. Targeting base excision repair as a sensitization strategy in radiotherapy. Semin Radiat Oncol. (2010) 20:241–9. doi: 10.1016/j.semradonc.2010.05.005
30. Hyun J-W, Cheon G-J, Kim H-S, Lee Y-S, Choi E-Y, Yoon B-H, et al. Radiation sensitivity depends on OGG1 activity status in human leukemia cell lines. Free Radic Biol Med. (2002) 32:212–20. doi: 10.1016/S0891-5849(01)00793-6
31. Yang N, Galick H, Wallace SS. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair. (2004) 3:1323–34. doi: 10.1016/j.dnarep.2004.04.014
32. Fortini P, Dogliotti E. Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair. (2007) 6:398–409. doi: 10.1016/j.dnarep.2006.10.008
33. Carter RJ, Parsons JL. Base excision repair, a pathway regulated by posttranslational modifications. Mol Cell Biol. (2016) 36:1426–37. doi: 10.1128/MCB.00030-16
34. Wallace SS. Base excision repair: a critical player in many games. DNA Repair. (2014) 19:14–26. doi: 10.1016/j.dnarep.2014.03.030
35. Limpose KL, Corbett AH, Doetsch PW. BERing the burden of damage: pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair. (2017) 56:51–64. doi: 10.1016/j.dnarep.2017.06.007
36. Jacobs AL, Schär P. DNA glycosylases: in DNA repair and beyond. Chromosoma. (2012) 121:1–20. doi: 10.1007/s00412-011-0347-4
37. Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutat Res Rev Mutat Res. (2015) 763:212–45. doi: 10.1016/j.mrrev.2014.11.002
38. Schiewer MJ, Mandigo AC, Gordon N, Huang F, Gaur S, de Leeuw R, et al. PARP-1 regulates DNA repair factor availability. EMBO Mol Med. (2018) 10:e8816. doi: 10.15252/emmm.201708816
39. Rajaraman P, Bhatti P, Doody MM, Simon SL, Weinstock RM, Linet MS, et al. Nucleotide excision repair polymorphisms may modify ionizing radiation-related breast cancer risk in US radiologic technologists. Int J Cancer. (2008) 123:2713–6. doi: 10.1002/ijc.23779
40. Wyman C, Kanaar R. DNA double-strand break repair: all's well that ends well. Annu Rev Genet. (2006) 40:363–83. doi: 10.1146/annurev.genet.40.110405.090451
41. Zhang Y, Rohde LH, Wu H. Involvement of nucleotide excision and mismatch repair mechanisms in double strand break repair. Curr Genomics. (2009) 10:250–8. doi: 10.2174/138920209788488544
42. Belyaev IY. Radiation-induced DNA repair foci: spatio-temporal aspects of formation, application for assessment of radiosensitivity and biological dosimetry. Mutat Res. (2010) 704:132–41. doi: 10.1016/j.mrrev.2010.01.011
43. Cortez D. Replication-coupled DNA repair. Mol Cell. (2019) 74:866–76. doi: 10.1016/j.molcel.2019.04.027
44. Pannunzio NR, Watanabe G, Lieber MR. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J Biol Chem. (2018) 293:10512–23. doi: 10.1074/jbc.TM117.000374
45. Britton S, Coates J, Jackson SP. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol. (2013) 202:579–95. doi: 10.1083/jcb.201303073
46. Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. (1993) 72:131–42. doi: 10.1016/0092-8674(93)90057-W
47. Park E-J, Chan DW, Park J-H, Oettinger MA, Kwon J. DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res. (2003) 31:6819–27. doi: 10.1093/nar/gkg921
48. Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. (2002) 108:781–94. doi: 10.1016/S0092-8674(02)00671-2
49. Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol Cell Biol. (2000) 20:2996–3003. doi: 10.1128/MCB.20.9.2996-3003.2000
50. Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. (2006) 124:301–13. doi: 10.1016/j.cell.2005.12.031
51. Sallmyr A, Tomkinson AE. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem. (2018) 293:10536–46. doi: 10.1074/jbc.TM117.000375
52. McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. (2008) 24:529–38. doi: 10.1016/j.tig.2008.08.007
53. Boboila C, Jankovic M, Yan CT, Wang JH, Wesemann DR, Zhang T, et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc Natl Acad Sci USA. (2010) 107:3034–9. doi: 10.1073/pnas.0915067107
54. Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. (2007) 449:478–82. doi: 10.1038/nature06020
55. Haince J-F, McDonald D, Rodrigue A, Déry U, Masson J-Y, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. (2008) 283:1197–208. doi: 10.1074/jbc.M706734200
56. El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. (2003) 31:5526–33. doi: 10.1093/nar/gkg761
57. Kent T, Chandramouly G, McDevitt SM, Ozdemir AY, Pomerantz RT. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat Struct Mol Biol. (2015) 22:230–7. doi: 10.1038/nsmb.2961
58. Lavin MF. The Mre11 complex and ATM: a two-way functional interaction in recognising and signaling DNA double strand breaks. DNA Repair. (2004) 3:1515–20. doi: 10.1016/j.dnarep.2004.07.001
59. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, et al. Human CtIP promotes DNA end resection. Nature. (2007) 450:509–14. doi: 10.1038/nature06337
60. Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. (2015) 7:a016600. doi: 10.1101/cshperspect.a016600
61. Heyer W-D, Li X, Rolfsmeier M, Zhang X-P. Rad54: the Swiss Army knife of homologous recombination? Nucleic Acids Res. (2006) 34:4115–25. doi: 10.1093/nar/gkl481
62. Solinger JA, Heyer WD. Rad54 protein stimulates the postsynaptic phase of Rad51 protein-mediated DNA strand exchange. Proc Natl Acad Sci USA. (2001) 98:8447–53. doi: 10.1073/pnas.121009898
63. Rodgers K, McVey M. Error-prone repair of DNA double-strand breaks. J Cell Physiol. (2016) 231:15–24. doi: 10.1002/jcp.25053
64. Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. (2019). doi: 10.1038/s41580-019-0152-0. [Epub ahead of print].
65. Fattah F, Lee EH, Weisensel N, Wang Y, Lichter N, Hendrickson EA. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. (2010) 6:e1000855. doi: 10.1371/journal.pgen.1000855
66. Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. (2011) 10:351–64. doi: 10.1038/nrd3374
67. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. (2017) 7:20–37. doi: 10.1158/2159-8290.CD-16-0860
68. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. (2011) 12:852–61. doi: 10.1016/S1470-2045(11)70214-5
69. Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. (2012) 30:372–9. doi: 10.1200/JCO.2011.36.9215
70. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. (2012) 366:1382–92. doi: 10.1056/NEJMoa1105535
71. Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. (2004) 96:56–67. doi: 10.1093/jnci/djh005
72. Dungey FA, Löser DA, Chalmers AJ. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys. (2008) 72:1188–97. doi: 10.1016/j.ijrobp.2008.07.031
73. Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. (2007) 13:2728–37. doi: 10.1158/1078-0432.CCR-06-3039
74. Reiss KA, Herman JM, Armstrong D, Zahurak M, Fyles A, Brade A, et al. A final report of a phase I study of veliparib (ABT-888) in combination with low-dose fractionated whole abdominal radiation therapy (LDFWAR) in patients with advanced solid malignancies and peritoneal carcinomatosis with a dose escalation in ovarian and fallopian tube cancers. Gynecol Oncol. (2017) 144:486–90. doi: 10.1016/j.ygyno.2017.01.016
75. Chabot P, Hsia T-C, Ryu J-S, Gorbunova V, Belda-Iniesta C, Ball D, et al. Veliparib in combination with whole-brain radiation therapy for patients with brain metastases from non-small cell lung cancer: results of a randomized, global, placebo-controlled study. J Neurooncol. (2017) 131:105–15. doi: 10.1007/s11060-016-2275-x
76. Pollack IF, Jakacki RI, Blaney SM, Hancock ML, Kieran MW, Phillips P, et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a pediatric brain tumor consortium report. Neuro-oncology. (2007) 9:145–60. doi: 10.1215/15228517-2006-031
77. Ree AH, Dueland S, Folkvord S, Hole KH, Seierstad T, Johansen M, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. (2010) 11:459–64. doi: 10.1016/S1470-2045(10)70058-9
78. Candelaria M, Cetina L, Pérez-Cárdenas E, de la Cruz-Hernández E, González-Fierro A, Trejo-Becerril C, et al. Epigenetic therapy and cisplatin chemoradiation in FIGO stage IIIB cervical cancer. Eur J Gynaecol Oncol. (2010) 31:386–91.
79. Le Tourneau C, Dreno B, Kirova Y, Grob JJ, Jouary T, Dutriaux C, et al. First-in-human phase I study of the DNA-repair inhibitor DT01 in combination with radiotherapy in patients with skin metastases from melanoma. Br J Cancer. (2016) 114:1199–205. doi: 10.1038/bjc.2016.120
80. Tell G, Fantini D, Quadrifoglio F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell Mol Life Sci. (2010) 67:3589–608. doi: 10.1007/s00018-010-0486-4
81. Naidu MD, Mason JM, Pica RV, Fung H, Peña LA. Radiation resistance in glioma cells determined by DNA damage repair activity of Ape1/Ref-1. J Radiat Res. (2010) 51:393–404. doi: 10.1269/jrr.09077
82. Xiang D-B, Chen Z-T, Wang D, Li M-X, Xie J-Y, Zhang Y-S, et al. Chimeric adenoviral vector Ad5/F35-mediated APE1 siRNA enhances sensitivity of human colorectal cancer cells to radiotherapy in vitro and in vivo. Cancer Gene Ther. (2008) 15:625–35. doi: 10.1038/cgt.2008.30
83. Mohiuddin IS, Kang MH. DNA-PK as an emerging therapeutic target in cancer. Front Oncol. (2019) 9:635. doi: 10.3389/fonc.2019.00635
84. Ismail IH, Mårtensson S, Moshinsky D, Rice A, Tang C, Howlett A, McMahon G, Hammarsten O. SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene. (2004) 23:873–82. doi: 10.1038/sj.onc.1207303
85. Shinohara ET, Geng L, Tan J, Chen H, Shir Y, Edwards E, et al. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res. (2005) 65:4987–992. doi: 10.1158/0008-5472.CAN-04-4250
86. Davidson D, Amrein L, Panasci L, Aloyz R. Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front Pharmacol. (2013) 4:5. doi: 10.3389/fphar.2013.00005
87. Brown JM. beware of clinical trials of DNA repair inhibitors. Int J Radiat Oncol Biol Phys. (2019) 103:1182–3. doi: 10.1016/j.ijrobp.2018.11.063
88. Mau-Sorensen M, van Bussel M, Kuipers M, Nielsen DL, Verheul HM, Aftimos P, et al. 1845PSafety, clinical activity and pharmacological biomarker evaluation of the DNA-dependent protein kinase (DNA-PK) inhibitor M3814: results from two phase I trials. Ann Oncol. (2018) 29. doi: 10.1093/annonc/mdy303.015
89. Li Y-H, Wang X, Pan Y, Lee D-H, Chowdhury D, Kimmelman AC. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS ONE. (2012) 7:e39588. doi: 10.1371/journal.pone.0039588
90. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, et al. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol Ther. (2016) 160:65–83. doi: 10.1016/j.pharmthera.2016.02.003
91. Munster PN, Mahipal A, Nemunaitis JJ, Mita MM, Paz-Ares LG, Massard C, et al. Phase I trial of a dual TOR kinase and DNA-PK inhibitor (CC-115) in advanced solid and hematologic cancers. JCO. (2016) 34:2505. doi: 10.1200/JCO.2016.34.15_suppl.2505
92. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
93. Qiao B, Kerr M, Groselj B, Teo MTW, Knowles MA, Bristow RG, et al. Imatinib radiosensitizes bladder cancer by targeting homologous recombination. Cancer Res. (2013) 73:1611–20. doi: 10.1158/0008-5472.CAN-12-1170
94. Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. (2008) 18:134–47. doi: 10.1038/cr.2007.111
95. Westphal CH, Hoyes KP, Canman CE, Huang X, Kastan MB, Hendry JH, et al. Loss of atm radiosensitizes multiple p53 null tissues. Cancer Res. (1998) 58:5637–9.
96. Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. (2008) 68:7466–74. doi: 10.1158/0008-5472.CAN-08-0763
97. Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NMB, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. (2004) 64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727
98. Golding SE, Rosenberg E, Adams BR, Wignarajah S, Beckta JM, O'Connor MJ, et al. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle. (2012) 11:1167–73. doi: 10.4161/cc.11.6.19576
99. Torok JA, Oh P, Castle KD, Reinsvold M, Ma Y, Luo L, et al. Deletion of Atm in tumor but not endothelial cells improves radiation response in a primary mouse model of lung adenocarcinoma. Cancer Res. (2019) 79:773–82. doi: 10.1158/0008-5472.CAN-17-3103
100. Andarawewa KL, Paupert J, Pal A, Barcellos-Hoff MH. New rationales for using TGFbeta inhibitors in radiotherapy. Int J Radiat Biol. (2007) 83:803–11. doi: 10.1080/09553000701711063
101. Kirshner M, Rathavs M, Nizan A, Essers J, Kanaar R, Shiloh Y, et al. Analysis of the relationships between ATM and the Rad54 paralogs involved in homologous recombination repair. DNA Repair. (2009) 8:253–61. doi: 10.1016/j.dnarep.2008.11.005
102. Golding SE, Rosenberg E, Neill S, Dent P, Povirk LF, Valerie K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res. (2007) 67:1046–53. doi: 10.1158/0008-5472.CAN-06-2371
103. Minchom A, Aversa C, Lopez J. Dancing with the DNA damage response: next-generation anti-cancer therapeutic strategies. Ther Adv Med Oncol. (2018) 10:1758835918786658. doi: 10.1177/1758835918786658
104. Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 axis in cancer therapy. Cancers. (2017) 9:E41. doi: 10.3390/cancers9050041
105. Vendetti FP, Lau A, Schamus S, Conrads TP, O'Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. (2015) 6:44289–305. doi: 10.18632/oncotarget.6247
106. Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee J-H, Nicolette ML, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. (2008) 4:119–25. doi: 10.1038/nchembio.63
107. Kuroda S, Urata Y, Fujiwara T. Ataxia-telangiectasia mutated and the Mre11-Rad50-NBS1 complex: promising targets for radiosensitization. Acta Med Okayama. (2012) 66:83–92. doi: 10.18926/AMO/48258
108. Quanz M, Berthault N, Roulin C, Roy M, Herbette A, Agrario C, et al. Small-molecule drugs mimicking DNA damage: a new strategy for sensitizing tumors to radiotherapy. Clin Cancer Res. (2009) 15:1308–16. doi: 10.1158/1078-0432.CCR-08-2108
109. Quanz M, Chassoux D, Berthault N, Agrario C, Sun J-S, Dutreix M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS ONE. (2009) 4:e6298. doi: 10.1371/journal.pone.0006298
110. Croset A, Cordelières FP, Berthault N, Buhler C, Sun J-S, Quanz M, et al. Inhibition of DNA damage repair by artificial activation of PARP with siDNA. Nucleic Acids Res. (2013) 41:7344–55. doi: 10.1093/nar/gkt522
111. Biau J, Chautard E, Berthault N, de Koning L, Court F, Pereira B, et al. Combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of high grade glioma: efficacy and protein biomarkers of resistance in preclinical models. Front Oncol. (2019) 9:549. doi: 10.3389/fonc.2019.00549
112. Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer. (2013) 108:748–54. doi: 10.1038/bjc.2013.21
113. Park Y, Chui MH, Suryo Rahmanto Y, Yu Z-C, Shamanna RA, Bellani MA, et al. Loss of ARID1A in tumor cells renders selective vulnerability to combined ionizing radiation and PARP inhibitor therapy. Clin Cancer Res. (2019) 25:5584–94. doi: 10.1158/1078-0432.CCR-18-4222
114. Blattmann C, Oertel S, Ehemann V, Thiemann M, Huber PE, Bischof M, et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys. (2010) 78:237–45. doi: 10.1016/j.ijrobp.2010.03.010
115. Lee J-H, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci USA. (2010) 107:14639–44. doi: 10.1073/pnas.1008522107
116. Konsoula Z, Cao H, Velena A, Jung M. Adamantanyl-histone deacetylase inhibitor H6CAHA exhibits favorable pharmacokinetics and augments prostate cancer radiation sensitivity. Int J Radiat Oncol Biol Phys. (2011) 79:1541–8. doi: 10.1016/j.ijrobp.2010.11.057
117. Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther. (2006) 5:1967–74. doi: 10.1158/1535-7163.MCT-06-0022
118. Yu W, Gu K, Yu Z, Yuan D, He M, Ma N, et al. Sorafenib potentiates irradiation effect in hepatocellular carcinoma in vitro and in vivo. Cancer Lett. (2013) 329:109–17. doi: 10.1016/j.canlet.2012.10.024
119. Raju U, Riesterer O, Wang Z-Q, Molkentine DP, Molkentine JM, Johnson FM, et al. Dasatinib, a multi-kinase inhibitor increased radiation sensitivity by interfering with nuclear localization of epidermal growth factor receptor and by blocking DNA repair pathways. Radiother Oncol. (2012) 105:241–9. doi: 10.1016/j.radonc.2012.08.010
120. Khurshid H, Dipetrillo T, Ng T, Mantripragada K, Birnbaum A, Berz D, et al. A phase I study of dasatinib with concurrent chemoradiation for stage III non-small cell lung cancer. Front Oncol. (2012) 2:56. doi: 10.3389/fonc.2012.00056
121. Kroeze SGC, Fritz C, Hoyer M, Lo SS, Ricardi U, Sahgal A, et al. Toxicity of concurrent stereotactic radiotherapy and targeted therapy or immunotherapy: a systematic review. Cancer Treatment Rev. (2017) 53:25–37. doi: 10.1016/j.ctrv.2016.11.013
122. Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. (2011) 71:1103–14. doi: 10.1158/0008-5472.CAN-10-2384
123. Ang KK, Berkey BA, Tu X, Zhang H-Z, Katz R, Hammond EH, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. (2002) 62:7350–6.
124. Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. (2006) 354:567–78. doi: 10.1056/NEJMoa053422
125. Martins RG, Parvathaneni U, Bauman JE, Sharma AK, Raez LE, Papagikos MA, et al. Cisplatin and radiotherapy with or without erlotinib in locally advanced squamous cell carcinoma of the head and neck: a randomized phase II trial. J Clin Oncol. (2013) 31:1415–21. doi: 10.1200/JCO.2012.46.3299
126. Sato Y, Ebara T, Sunaga N, Takahashi T, Nakano T. Interaction of radiation and gefitinib on a human lung cancer cell line with mutant EGFR gene in vitro. Anticancer Res. (2012) 32:4877–81.
127. Aziz K, Nowsheen S, Pantelias G, Iliakis G, Gorgoulis VG, Georgakilas AG. Targeting DNA damage and repair: embracing the pharmacological era for successful cancer therapy. Pharmacol Ther. (2012) 133:334–50. doi: 10.1016/j.pharmthera.2011.11.010
128. Mitchell JB, Choudhuri R, Fabre K, Sowers AL, Citrin D, Zabludoff SD, et al. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin Cancer Res. (2010) 16:2076–84. doi: 10.1158/1078-0432.CCR-09-3277
129. Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. (2008) 7:2955–66. doi: 10.1158/1535-7163.MCT-08-0492
130. Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. (2017) 16:591–600. doi: 10.1158/1535-7163.MCT-16-0352
131. Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. (2015) 66:129–43. doi: 10.1146/annurev-med-081313-121208
132. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. (2010) 40:179–204. doi: 10.1016/j.molcel.2010.09.019
133. Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. (2010) 70:4972–81. doi: 10.1158/0008-5472.CAN-09-3573
134. Mueller S, Haas-Kogan DA. WEE1 kinase as a target for cancer therapy. JCO. (2015) 12:3159–64. doi: 10.1200/JCO.2015.62.2290
135. Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, et al. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. (2011) 17:5638–48. doi: 10.1158/1078-0432.CCR-11-0650
136. Caretti V, Hiddingh L, Lagerweij T, Schellen P, Koken PW, Hulleman E, et al. WEE1 kinase inhibition enhances the radiation response of diffuse intrinsic pontine gliomas. Mol Cancer Ther. (2013) 12:141–50. doi: 10.1158/1535-7163.MCT-12-0735
137. Ko EC, Formenti SC. Radiation therapy to enhance tumor immunotherapy: a novel application for an established modality. Int J Radiat Biol. (2019) 95:936–9. doi: 10.1080/09553002.2019.1623429
138. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. (2015) 25:11–7. doi: 10.1016/j.semradonc.2014.07.005
Keywords: DNA damage, repair systems, radiotherapy, radioresistance, inhibition
Citation: Biau J, Chautard E, Verrelle P and Dutreix M (2019) Altering DNA Repair to Improve Radiation Therapy: Specific and Multiple Pathway Targeting. Front. Oncol. 9:1009. doi: 10.3389/fonc.2019.01009
Received: 15 July 2019; Accepted: 19 September 2019;
Published: 10 October 2019.
Edited by:
Paul N. Span, Radboud University Nijmegen Medical Centre, NetherlandsReviewed by:
Michael Wayne Epperly, University of Pittsburgh, United StatesBevin P. Engelward, Massachusetts Institute of Technology, United States
Copyright © 2019 Biau, Chautard, Verrelle and Dutreix. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julian Biau, SnVsaWFuLmJpYXVAY2xlcm1vbnQudW5pY2FuY2VyLmZy