 Roberta Roncarati
Roberta Roncarati Laura Lupini
Laura Lupini Ram C. Shankaraiah
Ram C. Shankaraiah Massimo Negrini1*
Massimo Negrini1*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 27 September 2019
Sec. Molecular and Cellular Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00988
This article is part of the Research Topic Oncogenic RAS-dependent Reprogramming of Cellular Plasticity View all 10 articles
microRNAs (miRNAs) regulate gene expression by modulating the translation of protein-coding RNAs. Their aberrant expression is involved in various human diseases, including cancer. Here, we summarize the experimental pieces of evidence that proved how dysregulated miRNA expression can lead to RAS (HRAS, KRAS, or NRAS) activation irrespective of their oncogenic mutations. These findings revealed relevant pathogenic mechanisms as well as mechanisms of resistance to target therapies. Based on this knowledge, potential approaches for the control of RAS oncogenic activation can be envisioned.
microRNAs (miRNAs) are small (19–24 nucleotides) non-coding RNAs discovered in 1993 in studies related to embryonic development of C. elegans (1, 2). Their importance significantly increased following the discovery of their existence in all eukaryotic organisms (3). Currently, 2,654 mature miRNAs, originating from 1917 precursors, are described in humans (http://www.mirbase.org/) (4, 5). Their main function is to negatively regulate gene expression at the post-transcriptional level through the interaction of their “seed” portion by sequence homology typically with the 3′ non-coding regions of messenger RNAs (mRNAs). Through this interaction, miRNAs limit translation, or promote degradation of target mRNAs (6, 7).
The modulation of target mRNAs by miRNAs is complex, considering that each mRNA is generally targeted by multiple miRNAs, and the strength of this interaction is variable (8). Classically, it has been thought that each miRNA can interact with hundreds of target mRNAs. However, recent reports have highlighted RNA transcripts inducing degradation of respective interacting miRNAs through a mechanism known as “target-directed miRNA degradation” (TDMD) (9, 10). Added to the complexity of these direct interactions is the fact that some long non-coding RNA (lncRNA) could function as “sponges,” that act as a buffer and prevent the action of miRNAs on target protein-coding mRNAs (11, 12). Lastly, it is also important to consider that cell co-localization of each miRNA with the target mRNAs is necessary and depends on the eventual tissue-specific expression of each of the interacting RNAs.
Thus, miRNAs, taken together, represent an essential phase in the regulation of gene expression by modulating the translation of the entire transcriptome (13, 14). Given their biological importance, their deregulation plays a significant role in pathogenic mechanisms, including the neoplastic transformation (15, 16). The first evidence associating miRNAs with human malignant diseases was the discovery of miR-15 and miR-16 in the minimal region of deletion at chromosome 13q14 in chronic lymphatic leukemia (17). Since this seminal study, a myriad of other studies has confirmed the role of miRNAs in tumorigenesis and other human diseases as well.
The first functional evidence to establish a molecular link between the deregulation of miRNAs with an explicit oncogenic pathway was published in 2005 when Slack and collaborators reported the importance of the downregulation of members of the let-7 miRNA family with the activation of oncogenes of the RAS family (18). The study demonstrated that the 3′ UTRs of KRAS, NRAS and HRAS mRNAs comprised multiple complementary let-7a binding sites. The enforced expression of let-7 could indeed reduce RAS protein levels (18). Conversely, let-7 downregulation could lead to the loss of its post-transcriptional control, causing RAS over-expression and activation. This study was decisive in proving that aberrant expression of miRNAs could play an important role in tumor initiation and progression, and paved the way for studies that extended miRNA involvement to all phases of neoplastic initiation and progression (19).
The involvement of RAS (KRAS, NRAS, HRAS) in human tumors is mainly associated with the presence of activating mutations at codons 12, 13 and 61, able to activate various molecular pathways, which play a key role in a large number of tumor traits, spanning from cell proliferation, cell survival, cytoskeleton organization, motility, and more (20). The demonstration of the role of miRNAs in the abnormal regulation of RAS thus represented another important mechanism involved in key steps of tumorigenesis.
Since then, quite a few other reports have demonstrated the modulation of RAS by miRNAs. In many cases, the interaction was only predicted by computer algorithms, but several studies have experimentally validated these interactions. Table 1 lists the microRNAs for which the ability to modulate the expression of KRAS, NRAS, or HRAS has been experimentally confirmed.

Table 1. Human microRNAs targeting RAS family members.
As mentioned, let-7 was the first, and probably the most important miRNA implicated in the regulation of genes of the RAS family (18). In the human genome, 12 loci are known to encode for members of the let-7 family: let-7a-1, -2, -3; let-7b; let-7c; let-7d; let-7e; let-7f-1, -2; let-7g; let-7i; miR-98. While it is described that members of the let-7 family are up-regulated in the course of cell differentiation, numerous studies have reported the reduction of let-7 expression in different tumor types (21, 22). Already in 2004, Takamizawa et al. demonstrated the downregulation of let-7 in non-small cell lung carcinoma (NSCLC) (23, 24) and documented its prognostic significance. Furthermore, in line with these observations, they proved that enforced expression of let-7 miRNA could inhibit in vitro cell growth of the lung adenocarcinoma A549 cells (23, 25–27). These studies were further confirmed in murine in vivo models of NSCLC (28, 29) and revealed that let-7 mimics could represent potential therapeutic molecules.
Given the proven interaction of let-7 with members of the RAS family, it is plausible that the observed effects were due to the modulation of RAS. However, let-7 can also regulate additional important oncogenes such as c-MYC, high-mobility group A (HMGA), Janus protein tyrosine kinase (JAK), signal transducer and activator of transcription 3 (STAT3) (30). Its action as a tumor suppressor gene is therefore achieved through the ability to interact with multiple oncogenes and inhibit the activation of their molecular pathways (18, 28).
Essentially all types of human cancer present a general down-regulation of let-7 (21). Among others, the modulation of RAS by let-7 was demonstrated in colorectal cancer (CRC) where let-7 is strongly down-regulated in tumor tissues compared to adjacent healthy tissues. Similar to the study on NSCLC cells, let-7 was also shown to act as a growth suppressor in human CRC cells (31).
Confirming the importance of RAS regulation by let-7, the discovery of the LCS6 polymorphism (Let-7 Complementary Sites 6, rs61764370) in the KRAS 3′ UTR region further demonstrated let-7 expression altering interaction. This polymorphism has been associated with a greater risk of developing tumors and worse prognosis in lung, oral, and colorectal cancer (32–34).
An understanding of a mechanism leading to let-7 down-regulation in cancer came from studies on LIN28 in mammals. Lin28 and Lin28b act as RNA binding proteins that are able to associate with the terminal loop of the precursors of let-7 family miRNAs and block their processing into mature miRNAs (35–38). Since LIN28 is over-expressed in human cancer, this mechanism causes let-7 down-regulation, which establishes a connection with RAS and other cancer-associated signalings.
Let-7 is not the only miRNA involved in the regulation of RAS (HRAS, KRAS, or NRAS) (39). Among the miRNAs involved in the regulation of members of the RAS family, miR-143 and miR-145, co-expressed in the same primary transcript, can target both KRAS and NRAS, and have been found to be down-regulated in numerous human tumors (40–42). Already in 2003 Michael et al. documented a significant reduction of miR-145 in CRC compared to normal mucosa (43) and in 2014, Pagliuca et al. confirmed that the miR-143/miR-145 cluster, highly expressed in normal colon, was significantly decreased in CRC (44). Their reduced expression has been correlated with p53 mutations capable of reducing the maturation process of these miRNAs (45).
Very similar to let-7, members of the miR-181 family were shown to target all the RAS family members (HRAS, KRAS, and NRAS). They were found downregulated in different types of cancer, such as oral squamous cell carcinoma (46, 47), gastric cancer (48), and gliomas (49). These findings suggest that miR-181 down-regulation is one of the mechanisms leading to oncogenic RAS activation in these tumors.
It is notable that in spite of KRAS activation by gene mutation in 90% of the cases in pancreatic cancer, various miRNAs capable of directly targeting KRAS are simultaneously downregulated. Specifically, miR-96, miR-126, and miR-217 (50–53). Since the reduced expression of these miRNAs correlates with higher KRAS expression, these alterations likely represent a mechanism for strengthening the already activated RAS signaling.
Another noteworthy miRNA capable of targeting KRAS is miR-134. It was found downregulated in glioblastoma and renal cell carcinoma (54, 55). miR-134 downregulation correlated with the activation of the MAPK pathway and its enforced expression in renal cancer cells could inhibit in vitro migration and invasive traits.
Oncogenic mutations resulting in RAS activation are prevalent in most human tumors, but there are exceptions. RAS mutations in HCC are rare events but paradoxical wild-type RAS activation is common (56). Dietrich et al. (57) discovered that wild-type KRAS expression was increased in HCC compared to non-tumor liver and revealed an inverse correlation with miR-622 expression.
In addition to the above-mentioned examples, several other miRNAs were proven to target and inhibit the expression of RAS oncoproteins (Table 1). These miRNAs are generally downregulated in tumors, thus concurring with reciprocal overexpression and activation of RAS, irrespective of activating gene mutations.
The interplay between miRNAs and RAS is not only represented by miRNAs acting as negative modulators of RAS but also includes downstream miRNA effectors. The most significant is undoubtedly miR-21, which is up-regulated by KRAS oncogenic mutants in non-small-cell lung cancer (58), laryngeal squamous cell carcinoma (59), and pancreatic adenocarcinoma (60) as well as many other human cancers. miR-21 is a known oncomiR capable of blocking the expression of tumor suppressor genes antagonists of the PI3K-AKT pathway, such as PTEN, or of the RAS-MAPK pathway, such as PDCD4 or RASA1 (61–63) (Figure 1).
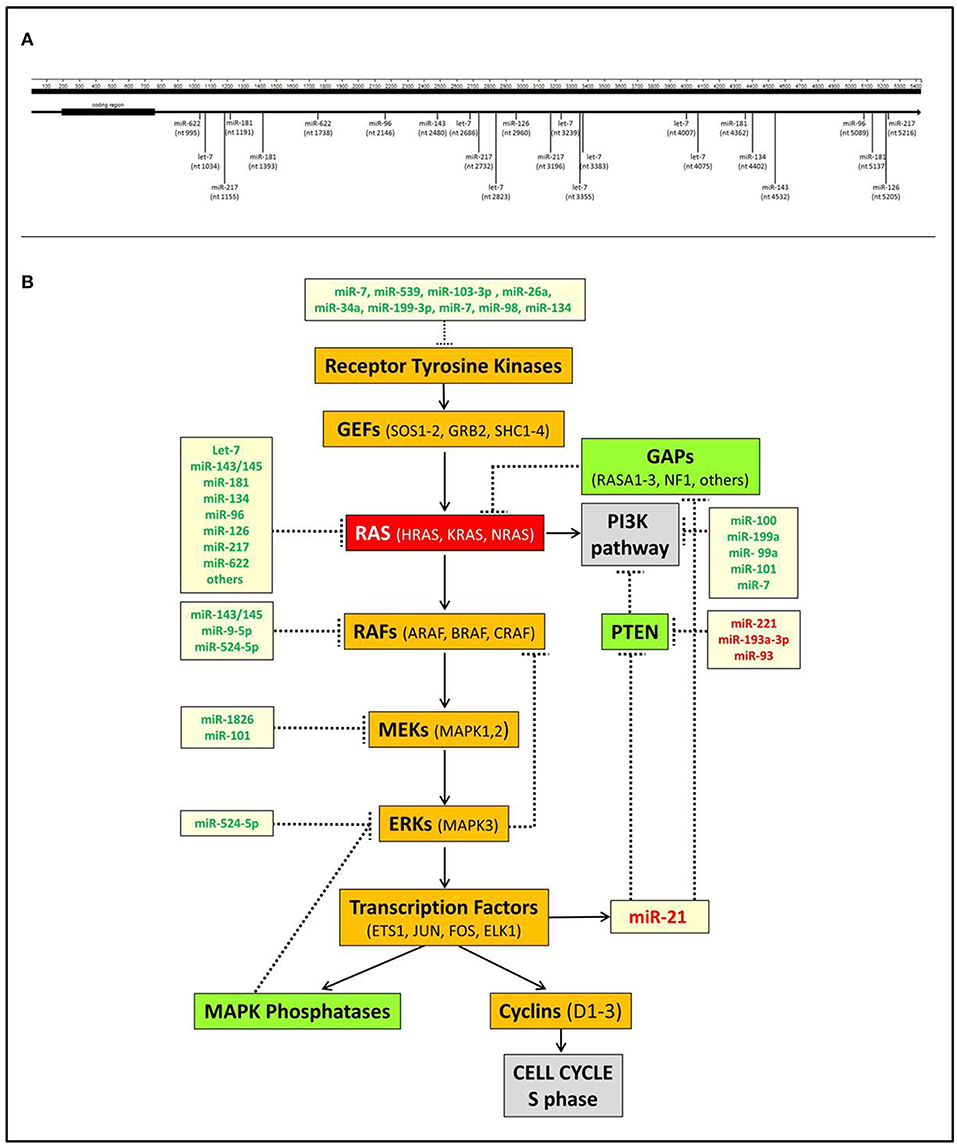
Figure 1. Interactions of miRNAs with RAS. (A) Scheme of the direct interactions of miRNAs with the 3′UTR of KRAS. Data were derived from TargetScan v7.2 (http://www.targetscan.org) and from Johnson et al. (18), Chen et al. (40), Jiao et al. (53), Liu et al. (55), and Dietrich et al. (57). (B) A simplified scheme of the interplay between miRNAs and RAS pathways. It shows that several miRNAs negatively regulate the MAPK and PI3K RAS-linked pathways at different points. Conversely, miR-21, which is transcriptionally induced by the transcription factor ELK1, inhibits the MAPK and PI3K suppressors GAPs and PTEN, thereby further promoting RAS activation. miRNAs indicated in green are downregulated in tumors, miRNAs indicated in red are upregulated.
RAS is a crucial node that connects receptor tyrosine kinases (RTKs) with downstream molecular pathways (Figure 1). Hence, miRNAs can affect RAS activity by acting on RTKs as well as MAPK, PI3K, or other pathways.
It is a well-known notion that RAS activation is physiologically triggered by RTKs, a category of transmembrane receptors that become activated in response to growth factors. Several miRNAs are known to target RTK mRNAs and their dysregulation can lead to inappropriate activation of the targeted RTK. Just to mention a few examples, miR-7, miR-539 and miR-103-3p can target and modulate the expression of the epidermal growth factor receptor (EGFR) (64–66); miR-26a was shown to target c-MET, the hepatocyte growth factor receptor (67); miR-199-3p can target the vascular endothelial growth factor receptors 1 and 2 and the VEGFA ligand (68); miR-7 and miR-98 can target the insulin growth factor receptor gene (64, 69).
All the above-mentioned miRNAs were found dysregulated in a variety of human cancers. miR-539 is downregulated in breast cancer (BC) tissues and cell lines. miR-539 enforced expression could inhibit BC cells proliferation and tumor growth in vitro and in vivo (65). miR-7 is downregulated in breast and colorectal cancer (CRC) cells (64, 66) and its reduced expression in BC patients correlated with higher stage, grade, and poor prognosis (64). The tumor suppressor activity of miR-103-3p was confirmed by the anti-proliferative effects after its enforced expression in lung cancer cell lines; furthermore, the downregulation of miR-103a-3p in NSCLC was associated with poor prognosis (66). miR-26a reduced levels were associated with poor prognosis in Hepatocellular carcinoma (HCC) (67). MiR-26a can also control the expression of VEGFA in HCC cells and impairs VEGFR2-signaling thereby controlling angiogenesis. miR-199-3p, another miRNA that can target VEGFR1, VEGFR2, and the ligand VEGFA (68), is frequently down-regulated in HCC and it has been shown to have in vitro and in vivo anti-tumor activity in HCC models (68, 70). MiR-98 is down-regulated in retinoblastoma, where it also represents a prognostic marker (69).
The above-reported miRNAs are just a few examples to show how their deregulation can lead to RTKs overexpression and consequently activation of RAS and its downstream pathways. The latter are themselves regulated by miRNAs, whose deregulation may directly cause the activation of RAS downstream effectors independently from RAS triggering.
The MAPK pathway is a well-studied pathway that promotes cell proliferation and is controlled by RAS activation. It includes several effectors with oncogenic function, widely studied in different types of tumors and whose mutations also represent tumorigenic mechanisms.
BRAF is probably the most studied element of the MAPK pathway. BRAF activation has been associated with a missense mutation V600E, commonly found in melanoma and thyroid cancer, but also present at low frequency in several other types of human cancer (71). As expected, various miRNAs can target and regulate BRAF expression. KRAS targeting miR-143 and miR-145, that we have mentioned above, can also target BRAF, indicating a very important role of these miRNAs in regulating the MAPK pathway at several levels (44). As mentioned earlier, these miRNAs are frequently downregulated in various types of cancer. miR-9-5p is another miRNA targeting BRAF, which was shown to be down-regulated in papillary thyroid carcinoma (65).
Further downstream of MAPK pathway cascade, MEK1/MEK2 (also called (MAP2K1 and MAP2K2) as well as ERK1/ERK2, are also targets of miRNAs. miR-1826 can target MEK1. It is down-regulated in bladder cancer and its reduced expression is associated with more severe pathological traits (pT and grade) (72). miR-101 can also target MEK1. This miRNA exhibits reduced expression in diffuse large B cell lymphoma (DLBCL) and it is associated with a worse prognosis (73). miR-665 has been also shown to indirectly activate MEK in BC cells by targeting the nuclear receptor subfamily 4 group A member 3 (NR4A3) gene. This miRNA is upregulated in breast cancer where its upregulation is associated with metastasis and poor survival (74).
Among the several miRNAs that regulate elements of the RAS-centered pathways, some miRNAs target multiple genes belonging to the pathway thus reinforcing their role in modulating MAPK pathway activation.
In this respect, miR-134 is a typical example, as its target genes not only include KRAS (75), but also EGFR (76), HER2 (77), STAT5B (54), and PIK3CA (78), which are upstream and downstream elements of the RAS-centered pathways. This miRNA is downregulated in numerous types of human cancers, where it affects cell proliferation, survival, invasiveness, metastasis, and apoptosis [reviewed in (79)]. This miRNA exemplifies the deregulatory action of single miRNA and consequent wide effects on tumorigenic signals by acting on multiple elements of the RAS pathways (79). Other miRNAs targeting multiple RAS effectors include miR-143 / miR-145, previously mentioned to target all RAS genes and BRAF; miR-524-5p that can target both BRAF and ERK2. In melanoma, miR-524-5p is downregulated and affects cell migration and proliferation both in vitro and in vivo (80).
These miRNAs are potentially very important, as they can represent useful molecules to effectively restore the normal expression of multiple proteins belonging to RAS pathways.
Therapeutic interventions in advanced cancers include traditional chemotherapy as well as targeted/immuno-therapies. Targeted therapies make use of molecules capable of blocking aberrantly activated oncogenes that act as tumorigenic drivers. Oncogenic RAS proteins would represent outstanding targets for such therapies. But, no drug targeting RAS has been yet validated for clinical use. At present, most available targeted therapies are instead designed to block the activity of several elements of RAS-centered pathways. These include a large number of tyrosine kinase inhibitors (TKIs) or antibodies against RTKs; drugs that target BRAF V600E mutation (vemurafenib and dabrafenib), MEK (trametinib, cobimetinib and binimetinib), PI3K mutations (alpelisib), and mTOR (everolimus). The RAS pathways are therefore targeted by several drugs, with the RAS itself being a major exception.
Even more disappointing is the fact that mutant activated RAS often reduces the efficacy of targeted drugs and patients become resistant to therapies. One of the best-known mechanisms associated with the emergence of TKI resistance is indeed KRAS mutation. It is known that tumors with KRAS mutations at codons 12, 13, 61, or 146 do not respond to treatment with anti- EGFR antibodies or TKIs and therefore mutational analyses on all RAS genes are carried out on tumor biopsies before a therapeutic regimen is chosen.
Albeit not implemented for clinical use, given their important role in regulating RAS and linked pathways, it is reasonable to believe that altered miRNA expression could also affect the development of resistance to targeted therapies. To this effect, a number of experimental evidences exist (81–95).
Among miRNAs that target KRAS, the reduced expression of miR-181a was shown to be associated with gefitinib resistance in lung cancer (96, 97); in CRC patients treated with cetuximab, it was reported that low levels of miR-181a were associated with a lower overall survival, indicating a reduced efficacy of anti-EGFR therapy (98). While miR-145 was shown to synergize with cetuximab activity (99), high levels of let-7 could predict the efficacy of cetuximab therapy even in CRC patients carrying mutant KRAS (100).
Dietrich et al. (57) not only revealed an inverse correlation of KRAS and miR-622 expression but, additionally, they could attribute KRAS-miR-622 interplay to therapy resistance since sorafenib induced further KRAS augmentation and down-regulation of miR-622. These few examples suggest that the miRNA-mediated modulation of RAS protein levels can indeed affect the response to TKIs or anti-EGFR targeted therapies.
In addition to RAS, the dysregulation of miRNAs responsible for the activation of elements of the MAPK or the PI3K pathways can also reduce the efficacy of TKIs. For example, the reduction of PTEN protein level by up-regulated miRNAs, like miR-21, miR-221, miR-23a and miR-214, can reduce efficacy of TKIs in lung cancer by activating the PI3K pathway (83, 101–106). miRNAs have also been associated with resistance to the BRAF inhibitor vemurafenib (107–110). In short, several studies have shown that the dysregulation of miRNAs has an important role in the efficacy of target therapies, thus suggesting that their levels of expression can be useful to guide the choice of therapy, alongside the more conventional mutational investigations. Furthermore, they also provide suggestions for potential therapeutic approaches useful to restore or improve sensitivity to treatments.
Taken together, published data provides a strong indication that altered miRNA expression represents an important mechanism for RAS activation, with various implications. First, it represents a mechanism of pathogenic relevance, responsible for the promotion of several tumor traits, irrespective of RAS oncogenic mutations. Second, considering that the activation of RAS represents a frequent mechanism of resistance for drugs directed against RTKs, it is possible that miRNA dysregulation represents a relevant aspect to consider when assessing the proper management of patients on target therapies. Third, miRNAs may represent potentially useful molecules for the control of RAS oncogenic activation, aimed at overcoming the lack of drugs targeting RAS and possibly improving the efficacy of target therapies.
RR, LL, RS, and MN contributed to the writing and editing of the manuscript.
This work was supported by funding from the University of Ferrara (FAR 2018 and 2019), the Consorzio Futuro in Ricerca (donation from friends of Arianna, project Yume, Ferrara) and the Italian Association for Cancer Research to MN.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-Y
2. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans Cell. (1993) 75:855–62. doi: 10.1016/0092-8674(93)90530-4
3. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. (2004) 303:83–6. doi: 10.1126/science.1091903
4. Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. (2006) 34:D140–4. doi: 10.1093/nar/gkj112
5. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. (2019) 47:D155–62. doi: 10.1093/nar/gky1141
6. Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. (2002) 297:2056–60. doi: 10.1126/science.1073827
7. Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA. (2006) 103:4034–9. doi: 10.1073/pnas.0510928103
9. de la Mata M, Gaidatzis D, Vitanescu M, Stadler MB, Wentzel C, Scheiffele P, et al. Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep. (2015) 16:500–11. doi: 10.15252/embr.201540078
10. Bitetti A, Mallory AC, Golini E, Carrieri C, Carreno Gutierrez H, Perlas E, et al. MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat Struct Mol Biol. (2018) 25:244–51. doi: 10.1038/s41594-018-0032-x
11. Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. (2016) 17:272–83. doi: 10.1038/nrg.2016.20
12. Gaiti F, Degnan BM, Tanurdzic M. Long non-coding regulatory RNAs in sponges and insights into the origin of animal multicellularity. RNA Biol. (2018) 15:696–702. doi: 10.1080/15476286.2018
13. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. (2005) 120:15–20. doi: 10.1016/j.cell.2004.12.035
14. Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. (2009) 19:92–105. doi: 10.1101/gr.082701.108
15. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/s0092-8674(04)00045-5
16. Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. (2009) 4:199–227. doi: 10.1146/annurev.pathol.4.110807.092222
17. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. (2002) 99:15524–9. doi: 10.1073/pnas.242606799
18. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 MicroRNA family. Cell. (2005) 120:635–47. doi: 10.1016/j.cell.2005.01.014
19. Negrini M, Ferracin M, Sabbioni S, Croce CM. MicroRNAs in human cancer: from research to therapy. J Cell Sci. (2007) 120:1833–40. doi: 10.1242/jcs.03450
20. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. (2003) 3:459–65. doi: 10.1038/nrc1097
21. Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. (2010) 17:F19–36. doi: 10.1677/ERC-09-0184
22. Ambros V. MicroRNAs and developmental timing. Curr Opin Genet Dev. (2011) 21:511–7. doi: 10.1016/j.gde.2011.04.003
23. Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. (2004) 64:3753–6. doi: 10.1158/0008-5472.CAN-04-0637
24. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. (2006) 9:189–98. doi: 10.1016/j.ccr.2006.01.025
25. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. (2001) 15:3243–8. doi: 10.1101/gad.943001
26. Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. (2008) 105:3903–8. doi: 10.1073/pnas.0712321105
27. Trang P, Medina PP, Wiggins JF, Ruffino L, Kelnar K, Omotola M, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. (2010) 29:1580–7. doi: 10.1038/onc.2009.445
28. Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. (2007) 67:7713–22. doi: 10.1158/0008-5472.CAN-07-1083
29. Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. (2008) 7:759–64. doi: 10.4161/cc.7.6.5834
30. Wang X, Cao L, Wang Y, Wang X, Liu N, You Y. Regulation of let-7 and its target oncogenes (Review). Oncol Lett. (2012) 3:955–60. doi: 10.3892/ol.2012.609
31. Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. (2006) 29:903–6. doi: 10.1248/bpb.29.903
32. Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. (2008) 68:8535–40. doi: 10.1158/0008-5472.CAN-08-2129
33. Smits KM, Paranjape T, Nallur S, Wouters KA, Weijenberg MP, Schouten LJ, et al. A let-7 microRNA SNP in the KRAS 3′UTR is prognostic in early-stage colorectal cancer. Clin Cancer Res. (2011) 17:7723–31. doi: 10.1158/1078-0432.CCR-11-0990
34. De Ruyck K, Duprez F, Ferdinande L, Mbah C, Rios-Velazquez E, Hoebers F, et al. A let-7 microRNA polymorphism in the KRAS 3′-UTR is prognostic in oropharyngeal cancer. Cancer Epidemiol. (2014) 38:591–8. doi: 10.1016/j.canep.2014.07.008
35. Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. (2008) 32:276–84. doi: 10.1016/j.molcel.2008.09.014
36. Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. (2008) 14:1539–49. doi: 10.1261/rna.1155108
37. Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, et al. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol. (2008) 10:987–93. doi: 10.1038/ncb1759
38. Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. (2008) 320:97–100. doi: 10.1126/science.1154040
39. Masliah-Planchon J, Garinet S, Pasmant E. RAS-MAPK pathway epigenetic activation in cancer: miRNAs in action. Oncotarget. (2016) 7:38892–907. doi: 10.18632/oncotarget.6476
40. Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene. (2009) 28:1385–92. doi: 10.1038/onc.2008.474
41. Gao JS, Zhang Y, Tang X, Tucker LD, Tarwater PM, Quesenberry PJ, et al. The Evi1, microRNA-143, K-Ras axis in colon cancer. FEBS Lett. (2011) 585:693–9. doi: 10.1016/j.febslet.2011.01.033
42. Wang L, Shi ZM, Jiang CF, Liu X, Chen QD, Qian X, et al. MiR-143 acts as a tumor suppressor by targeting N-RAS and enhances temozolomide-induced apoptosis in glioma. Oncotarget. (2014) 5:5416–27. doi: 10.18632/oncotarget.2116
43. Michael MZ, Sm OC, Van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. (2003) 1:882–91.
44. Pagliuca A, Valvo C, Fabrizi E, Di Martino S, Biffoni M, Runci D, et al. Analysis of the combined action of miR-143 and miR-145 on oncogenic pathways in colorectal cancer cells reveals a coordinate program of gene repression. Oncogene. (2013) 32:4806–13. doi: 10.1038/onc.2012.495
45. Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. (2009) 460:529–33. doi: 10.1038/nature08199
46. Shin KH, Bae SD, Hong HS, Kim RH, Kang MK, Park NH. miR-181a shows tumor suppressive effect against oral squamous cell carcinoma cells by downregulating K-ras. Biochem Biophys Res Commun. (2011) 404:896–902. doi: 10.1016/j.bbrc.2010.12.055
47. India Project Team of the International Cancer Genome C. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun. (2013) 4:2873. doi: 10.1038/ncomms3873
48. Hashimoto Y, Akiyama Y, Otsubo T, Shimada S, Yuasa Y. Involvement of epigenetically silenced microRNA-181c in gastric carcinogenesis. Carcinogenesis. (2010) 31:777–84. doi: 10.1093/carcin/bgq013
49. Wang XF, Shi ZM, Wang XR, Cao L, Wang YY, Zhang JX, et al. MiR-181d acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2. J Cancer Res Clin Oncol. (2012) 138:573–84. doi: 10.1007/s00432-011-1114-x
50. Szafranska AE, Davison TS, John J, Cannon T, Sipos B, Maghnouj A, et al. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. (2007) 26:4442–52. doi: 10.1038/sj.onc.1210228
51. Yu S, Lu Z, Liu C, Meng Y, Ma Y, Zhao W, et al. miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. (2010) 70:6015–25. doi: 10.1158/0008-5472.CAN-09-4531
52. Zhao WG, Yu SN, Lu ZH, Ma YH, Gu YM, Chen J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis. (2010) 31:1726–33. doi: 10.1093/carcin/bgq160
53. Jiao LR, Frampton AE, Jacob J, Pellegrino L, Krell J, Giamas G, et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PLoS ONE. (2012) 7:e32068. doi: 10.1371/journal.pone.0032068
54. Zhang Y, Kim J, Mueller AC, Dey B, Yang Y, Lee DH, et al. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ. (2014) 21:720–34. doi: 10.1038/cdd.2013.196
55. Liu Y, Zhang M, Qian J, Bao M, Meng X, Zhang S, et al. miR-134 functions as a tumor suppressor in cell proliferation and epithelial-to-mesenchymal Transition by targeting KRAS in renal cell carcinoma cells. DNA Cell Biol. (2015) 34:429–36. doi: 10.1089/dna.2014.2629
56. Delire B, Starkel P. The Ras/MAPK pathway and hepatocarcinoma: pathogenesis and therapeutic implications. Eur J Clin Invest. (2015) 45:609–23. doi: 10.1111/eci.12441
57. Dietrich P, Koch A, Fritz V, Hartmann A, Bosserhoff AK, Hellerbrand C. Wild type Kirsten rat sarcoma is a novel microRNA-622-regulated therapeutic target for hepatocellular carcinoma and contributes to sorafenib resistance. Gut. (2018) 67:1328–41. doi: 10.1136/gutjnl-2017-315402
58. Frezzetti D, De Menna M, Zoppoli P, Guerra C, Ferraro A, Bello AM, et al. Upregulation of miR-21 by Ras in vivo and its role in tumor growth. Oncogene. (2011) 30:275–86. doi: 10.1038/onc.2010.416
59. Ren J, Zhu D, Liu M, Sun Y, Tian L. Downregulation of miR-21 modulates Ras expression to promote apoptosis and suppress invasion of Laryngeal squamous cell carcinoma. Eur J Cancer. (2010) 46:3409–16. doi: 10.1016/j.ejca.2010.07.047
60. Du Rieu MC, Torrisani J, Selves J, Al Saati T, Souque A, Dufresne M, et al. MicroRNA-21 is induced early in pancreatic ductal adenocarcinoma precursor lesions. Clin Chem. (2010) 56:603–12. doi: 10.1373/clinchem.2009.137364
61. Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, Van Rooij E, et al. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell. (2010) 18:282–93. doi: 10.1016/j.ccr.2010.08.013
62. Pan X, Wang ZX, Wang R. MicroRNA-21: a novel therapeutic target in human cancer. Cancer Biol Ther. (2010) 10:1224–32. doi: 10.4161/cbt.10.12.14252
63. Pfeffer SR, Yang CH, Pfeffer LM. The role of miR-21 in cancer. Drug Dev Res. (2015) 76:270–7. doi: 10.1002/ddr.21257
64. Cui YX, Bradbury R, Flamini V, Wu B, Jordan N, Jiang WG. MicroRNA-7 suppresses the homing and migration potential of human endothelial cells to highly metastatic human breast cancer cells. Br J Cancer. (2017) 117:89–101. doi: 10.1038/bjc.2017.156
65. Guo J, Gong G, Zhang B. miR-539 acts as a tumor suppressor by targeting epidermal growth factor receptor in breast cancer. Sci Rep. (2018) 8:2073. doi: 10.1038/s41598-018-20431-z
66. Fan X, Liu M, Tang H, Leng D, Hu S, Lu R, et al. MicroRNA-7 exerts antiangiogenic effect on colorectal cancer via ERK signaling. J Surg Res. (2019) 240:48–59. doi: 10.1016/j.jss.2019.02.035
67. Yang X, Zhang XF, Lu X, Jia HL, Liang L, Dong QZ, et al. MicroRNA-26a suppresses angiogenesis in human hepatocellular carcinoma by targeting hepatocyte growth factor-cMet pathway. Hepatology. (2014) 59:1874–85. doi: 10.1002/hep.26941
68. Ghosh A, Dasgupta D, Ghosh A, Roychoudhury S, Kumar D, Gorain M, et al. MiRNA199a-3p suppresses tumor growth, migration, invasion and angiogenesis in hepatocellular carcinoma by targeting VEGFA, VEGFR1, VEGFR2, HGF and MMP2. Cell Death Dis. (2017) 8:e2706. doi: 10.1038/cddis.2017.123
69. Guo L, Bai Y, Ji S, Ma H. MicroRNA98 suppresses cell growth and invasion of retinoblastoma via targeting the IGF1R/kRas/Raf/MEK/ERK signaling pathway. Int J Oncol. (2019) 54:807–20. doi: 10.3892/2Fijo.2019.4689
70. Callegari E, D'abundo L, Guerriero P, Simioni C, Elamin BK, Russo M, et al. miR-199a-3p modulates MTOR and PAK4 pathways and inhibits tumor growth in a hepatocellular carcinoma transgenic mouse model. Mol Ther Nucleic Acids. (2018) 11:485–93. doi: 10.1016/j.omtn.2018.04.002
71. Vakiani E, Solit DB. KRAS and BRAF: drug targets and predictive biomarkers. J Pathol. (2011) 223:219–29. doi: 10.1002/path.2796
72. Hirata H, Hinoda Y, Ueno K, Shahryari V, Tabatabai ZL, Dahiya R. MicroRNA-1826 targets VEGFC, beta-catenin (CTNNB1) and MEK1 (MAP2K1) in human bladder cancer. Carcinogenesis. (2012) 33:41–8. doi: 10.1093/carcin/bgr239
73. Huang Y, Zou Y, Lin L, Ma X, Zheng R. miR101 regulates the cell proliferation and apoptosis in diffuse large Bcell lymphoma by targeting MEK1 via regulation of the ERK/MAPK signaling pathway. Oncol Rep. (2019) 41:377–86. doi: 10.3892/or.2018.6821
74. Zhao XG, Hu JY, Tang J, Yi W, Zhang MY, Deng R, et al. miR-665 expression predicts poor survival and promotes tumor metastasis by targeting NR4A3 in breast cancer. Cell Death Dis. (2019) 10:479. doi: 10.1038/s41419-019-1705-z
75. Zhao Y, Pang D, Wang C, Zhong S, Wang S. MicroRNA-134 modulates glioma cell U251 proliferation and invasion by targeting KRAS and suppressing the ERK pathway. Tumour Biol. (2016) 37:11485–93. doi: 10.1007/s13277-016-5027-9
76. Qin Q, Wei F, Zhang J, Wang X, Li B. miR-134 inhibits non-small cell lung cancer growth by targeting the epidermal growth factor receptor. J Cell Mol Med. (2016) 20:1974–83. doi: 10.1111/jcmm.12889
77. Leivonen SK, Sahlberg KK, Makela R, Due EU, Kallioniemi O, Borresen-Dale AL, et al. High-throughput screens identify microRNAs essential for HER2 positive breast cancer cell growth. Mol Oncol. (2014) 8:93–104. doi: 10.1016/j.molonc.2013.10.001
78. El-Daly SM, Abba ML, Patil N, Allgayer H. miRs-134 and−370 function as tumor suppressors in colorectal cancer by independently suppressing EGFR and PI3K signalling. Sci Rep. (2016) 6:24720. doi: 10.1038/srep24720
79. Pan JY, Zhang F, Sun CC, Li SJ, Li G, Gong FY, et al. miR-134: a human cancer suppressor? Mol Ther Nucleic Acids. (2017) 6:140–9. doi: 10.1016/j.omtn.2016.11.003
80. Liu SM, Lu J, Lee HC, Chung FH, Ma N. miR-524-5p suppresses the growth of oncogenic BRAF melanoma by targeting BRAF and ERK2. Oncotarget. (2014) 5:9444–59. doi: 10.18632/oncotarget.2452
81. Rai K, Takigawa N, Ito S, Kashihara H, Ichihara E, Yasuda T, et al. Liposomal delivery of MicroRNA-7-expressing plasmid overcomes epidermal growth factor receptor tyrosine kinase inhibitor-resistance in lung cancer cells. Mol Cancer Ther. (2011) 10:1720–7. doi: 10.1158/1535-7163.MCT-11-0220
82. Gao Y, Fan X, Li W, Ping W, Deng Y, Fu X. miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem Biophys Res Commun. (2014) 446:179–86. doi: 10.1016/j.bbrc.2014.02.073
83. Shen H, Zhu F, Liu J, Xu T, Pei D, Wang R, et al. Alteration in Mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer. PLoS ONE. (2014) 9:e103305. doi: 10.1371/journal.pone.0103305
84. Wang Y, Xia H, Zhuang Z, Miao L, Chen X, Cai H. Axl-altered microRNAs regulate tumorigenicity and gefitinib resistance in lung cancer. Cell Death Dis. (2014) 5:e1227. doi: 10.1038/cddis.2014.186
85. Ahsan A. Mechanisms of resistance to EGFR tyrosine kinase inhibitors and therapeutic approaches: an update. Adv Exp Med Biol. (2016) 893:137–53. doi: 10.1007/978-3-319-24223-1_7
86. Han J, Zhao F, Zhang J, Zhu H, Ma H, Li X, et al. miR-223 reverses the resistance of EGFR-TKIs through IGF1R/PI3K/Akt signaling pathway. Int J Oncol. (2016) 48:1855–67. doi: 10.3892/ijo.2016.3401
87. Lukamowicz-Rajska M, Mittmann C, Prummer M, Zhong Q, Bedke J, Hennenlotter J, et al. MiR-99b-5p expression and response to tyrosine kinase inhibitor treatment in clear cell renal cell carcinoma patients. Oncotarget. (2016) 7:78433–47. doi: 10.18632/oncotarget.12618
88. Xu S, Wang T, Yang Z, Li Y, Li W, Wang T, et al. miR-26a desensitizes non-small cell lung cancer cells to tyrosine kinase inhibitors by targeting PTPN13. Oncotarget. (2016) 7:45687–701. doi: 10.18632/oncotarget.9920
89. Xu Y, Huang J, Ma L, Shan J, Shen J, Yang Z, et al. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. (2016) 371:171–81. doi: 10.1016/j.canlet.2015.11.034
90. Zhao FY, Han J, Chen XW, Wang J, Wang XD, Sun JG, et al. miR-223 enhances the sensitivity of non-small cell lung cancer cells to erlotinib by targeting the insulin-like growth factor-1 receptor. Int J Mol Med. (2016) 38:183–91. doi: 10.3892/ijmm.2016.2588
91. Migliore C, Morando E, Ghiso E, Anastasi S, Leoni VP, Apicella M, et al. miR-205 mediates adaptive resistance to MET inhibition via ERRFI1 targeting and raised EGFR signaling. EMBO Mol Med. (2018) 10:e8746. doi: 10.15252/emmm.201708746
92. Wu DW, Wang YC, Wang L, Chen CY, Lee H. A low microRNA-630 expression confers resistance to tyrosine kinase inhibitors in EGFR-mutated lung adenocarcinomas via miR-630/YAP1/ERK feedback loop. Theranostics. (2018) 8:1256–69. doi: 10.7150/thno.22048
93. Yue J, Lv D, Wang C, Li L, Zhao Q, Chen H, et al. Epigenetic silencing of miR-483-3p promotes acquired gefitinib resistance and EMT in EGFR-mutant NSCLC by targeting integrin β3. Oncogene. (2018) 37:4300–12. doi: 10.1038/s41388-018-0276-2
94. Lai Y, Kacal M, Kanony M, Stukan I, Jatta K, Kis L, et al. miR-100-5p confers resistance to ALK tyrosine kinase inhibitors Crizotinib and Lorlatinib in EML4-ALK positive NSCLC. Biochem Biophys Res Commun. (2019) 511:260–5. doi: 10.1016/j.bbrc.2019.02.016
95. Leonetti A, Assaraf YG, Veltsista PD, El Hassouni B, Tiseo M, Giovannetti E. MicroRNAs as a drug resistance mechanism to targeted therapies in EGFR-mutated NSCLC: Current implications and future directions. Drug Resist Updat. (2019) 42:1–11. doi: 10.1016/j.drup.2018.11.002
96. Wang P, Chen D, Ma H, Li Y. LncRNA SNHG12 contributes to multidrug resistance through activating the MAPK/Slug pathway by sponging miR-181a in non-small cell lung cancer. Oncotarget. (2017) 8:84086–101. doi: 10.18632/oncotarget.20475
97. Ping W, Gao Y, Fan X, Li W, Deng Y, Fu X. MiR-181a contributes gefitinib resistance in non-small cell lung cancer cells by targeting GAS7. Biochem Biophys Res Commun. (2018) 495:2482–9. doi: 10.1016/j.bbrc.2017.12.096
98. Pichler M, Winter E, Ress AL, Bauernhofer T, Gerger A, Kiesslich T, et al. miR-181a is associated with poor clinical outcome in patients with colorectal cancer treated with EGFR inhibitor. J Clin Pathol. (2014) 67:198–203. doi: 10.1136/jclinpath-2013-201904
99. Gomes SE, Simoes AE, Pereira DM, Castro RE, Rodrigues CM, Borralho PM. miR-143 or miR-145 overexpression increases cetuximab-mediated antibody-dependent cellular cytotoxicity in human colon cancer cells. Oncotarget. (2016) 7:9368–87. doi: 10.18632/oncotarget.7010
100. Ruzzo A, Graziano F, Vincenzi B, Canestrari E, Perrone G, Galluccio N, et al. High let-7a microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapy-refractory metastatic disease. Oncologist. (2012) 17:823–9. doi: 10.1634/theoncologist.2012-0081
101. Garofalo M, Romano G, Di Leva G, Nuovo G, Jeon YJ, Ngankeu A, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. (2011) 18:74–82. doi: 10.1038/nm.2577
102. Garofalo M, Quintavalle C, Romano G, Croce CM, Condorelli G. miR221/222 in cancer: their role in tumor progression and response to therapy. Curr Mol Med. (2012) 12:27–33. doi: 10.2174/156652412798376170
103. Wang YS, Wang YH, Xia HP, Zhou SW, Schmid-Bindert G, Zhou CC. MicroRNA-214 regulates the acquired resistance to gefitinib via the PTEN/AKT pathway in EGFR-mutant cell lines. Asian Pac J Cancer Prev. (2012) 13:255–60. doi: 10.7314/APJCP.2012.13.1.255
104. Li B, Ren S, Li X, Wang Y, Garfield D, Zhou S, et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer. (2014) 83:146–53. doi: 10.1016/j.lungcan.2013.11.003
105. Han Z, Zhou X, Li S, Qin Y, Chen Y, Liu H. Inhibition of miR-23a increases the sensitivity of lung cancer stem cells to erlotinib through PTEN/PI3K/Akt pathway. Oncol Rep. (2017) 38:3064–70. doi: 10.3892/or.2017.5938
106. Liao J, Lin J, Lin D, Zou C, Kurata J, Lin R, et al. Down-regulation of miR-214 reverses erlotinib resistance in non-small-cell lung cancer through up-regulating LHX6 expression. Sci Rep. (2017) 7:781. doi: 10.1038/s41598-017-00901-6
107. Peng W, Hu J, Zhu XD, Liu X, Wang CC, Li WH, et al. Overexpression of miR-145 increases the sensitivity of vemurafenib in drug-resistant colo205 cell line. Tumour Biol. (2014) 35:2983–8. doi: 10.1007/s13277-013-1383-x
108. Fattore L, Mancini R, Acunzo M, Romano G, Lagana A, Pisanu ME, et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc Natl Acad Sci USA. (2016) 113:E5005–13. doi: 10.1073/pnas.1607753113
109. Sun X, Li J, Sun Y, Zhang Y, Dong L, Shen C, et al. miR-7 reverses the resistance to BRAFi in melanoma by targeting EGFR/IGF-1R/CRAF and inhibiting the MAPK and PI3K/AKT signaling pathways. Oncotarget. (2016) 7:53558–70. doi: 10.18632/oncotarget.10669
Keywords: microRNA, RAS, cancer, MAPK, target therapies
Citation: Roncarati R, Lupini L, Shankaraiah RC and Negrini M (2019) The Importance of microRNAs in RAS Oncogenic Activation in Human Cancer. Front. Oncol. 9:988. doi: 10.3389/fonc.2019.00988
Received: 23 July 2019; Accepted: 16 September 2019;
Published: 27 September 2019.
Edited by:
Georgia Konstantinidou, University of Bern, SwitzerlandReviewed by:
George Calin, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2019 Roncarati, Lupini, Shankaraiah and Negrini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimo Negrini, bWFzc2ltby5uZWdyaW5pQHVuaWZlLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.