 Maira Huerta-Reyes1
Maira Huerta-Reyes1 Guadalupe Maya-Núñez2
Guadalupe Maya-Núñez2 Marco Allán Pérez-Solis2
Marco Allán Pérez-Solis2 Eunice López-Muñoz2
Eunice López-Muñoz2 Nancy Guillén3
Nancy Guillén3 Jean-Christophe Olivo-Marin4,5
Jean-Christophe Olivo-Marin4,5 Arturo Aguilar-Rojas2,4*
Arturo Aguilar-Rojas2,4*- 1Unidad de Investigación Médica en Enfermedades Nefrológicas, Centro Médico Nacional Siglo XXI (CMN-SXXI), Instituto Mexicano del Seguro Social (IMSS), Hospital de Especialidades, Mexico City, Mexico
- 2Unidad de Investigación Médica en Medicina Reproductiva, IMSS, Unidad Médica de Alta Especialidad No. 4, Mexico City, Mexico
- 3Centre National de la Recherche Scientifique, CNRS-ERL9195, Paris, France
- 4Unité d'Analyse d'Images Biologiques, Institut Pasteur, Paris, France
- 5Centre National de la Recherche Scientifique, CNRS-UMR3691, Paris, France
Although significant progress has been made in the implementation of new breast cancer treatments over the last three decades, this neoplasm annually continues to show high worldwide rates of morbidity and mortality. In consequence, the search for novel therapies with greater effectiveness and specificity has not come to a stop. Among the alternative therapeutic targets, the human gonadotropin-releasing hormone type I and type II (hGnRH-I and hGnRH–II, respectively) and its receptor, the human gonadotropin-releasing hormone receptor type I (hGnRHR-I), have shown to be powerful therapeutic targets to decrease the adverse effects of this disease. In the present review, we describe how the administration of GnRH analogs is able to reduce circulating concentrations of estrogen in premenopausal women through their action on the hypothalamus–pituitary–ovarian axis, consequently reducing the growth of breast tumors and disease recurrence. Also, it has been mentioned that, regardless of the suppression of synthesis and secretion of ovarian steroids, GnRH agonists exert direct anticancer action, such as the reduction of tumor growth and cell invasion. In addition, we discuss the effects on breast cancer of the hGnRH-I and hGnRH-II agonist and antagonist, non-peptide GnRH antagonists, and cytotoxic analogs of GnRH and their implication as novel adjuvant therapies as antitumor agents for reducing the adverse effects of breast cancer. In conclusion, we suggest that the hGnRH/hGnRHR system is a promising target for pharmaceutical development in the treatment of breast cancer, especially for the treatment of advanced states of this disease.
Introduction
Breast cancer (BC) is the most common cancer in women around the world. This disease represents 33% of all female cancer cases and it is responsible for ~15% of all cancer deaths among women worldwide. The high incidence rate of this disease could be associated with several factors, such as age, gender, reproductive factors, weight, personal and family history, lifestyle conditions, infectious agents, tobacco and alcohol consumption, obesity, and diet, among others.
According to the World Health Organization (WHO), it is estimated that, by 2025, there will be more than 20 million new cases of BC around the world (1). Additionally, in the majority of countries, BC is one of the main causes of death in women (2), and unfortunately, survival rates after 5 years of treatment are much worse for less developed countries, such as Gambia, Algeria, India, and Brazil (12–58.4%) in comparison with developed countries such as Japan, Sweden, and the United States (81.6–83.9%) (1, 3).
BC is a complex disease with considerable variations in clinical development, morphological aspect, and gene expression patterns. Traditionally, clinical staging is based on extension of the primary tumor, regional lymph nodes, and distant metastases. In addition, clinical staging includes histopathological assessments that comprise histological type, tumor grade or proliferation status, and lymphovascular invasion, and these are used as prognostic variables that reflect tumor biology and guide therapeutic management. Likewise, expression of estrogen receptor (ER) and progesterone receptor (PR) and the overexpression and/or amplification of the human epidermal growth factor receptor 2 (HER2) have been included in routine clinical practice worldwide to predict the prognosis of and the response to endocrine or humanized monoclonal antibody therapy (4, 5). Additionally, the molecular classification of BC is based on the expression levels of ER, PR, and HER2, allowing the stratification of this disease into three major subtypes: luminal, HER2 overexpression, and triple-negative breast cancer (TNBC) tumors (6). Luminal is the most common tumor subtype, characterized by the expression of sex-steroid receptors (ER and PR). Recently, the expression levels of Ki67, a cellular marker of proliferation, have been employed to discriminate luminal A and luminal B subtypes (4). In general, luminal tumors respond well to hormone therapy but poorly to conventional chemotherapy. HER2-overexpressing tumors are characterized by the phenotype: ER negative (ER–), PR negative (PR–), and HER2 positive (HER2+). Although these types of tumors are associated with aggressive disease and decreased survival, the chemotherapy treatment in combination with novel anticancer agents, including monoclonal antibodies and small tyrosine kinase inhibitors, result in an improvement in disease-free survival (7). TNBC refers to a group of tumors that do not express either hormone receptors (ER–, PR–) or HER2 receptor (HER2–). This tumor type is of particular clinical interest because of its aggressive course, due to the deficient expression of potential therapeutic targets; thus, chemotherapy is the only option for these patients. Compared with the other subtypes, TNBC tumors are associated with younger patients, their frequency is more prevalent in African-American populations, and they are clinically more aggressive (8). Although TNBCs are sensitive to chemotherapy, response rates are low and, in addition, the prognosis remains poor. In patients with TNBC, recurrence occurs earlier, and the majority of the deaths occur within the first 5 years after diagnosis, underscoring the importance of identifying specific therapeutic targets for this cancer subtype.
Despite that the use of genomic/gene expression arrays allows the incorporation of prognostic or predictive markers that provide additional information for conventional clinical staging (9), the development of targeted drugs for BC treatment remains in evolution. Therefore, the characterization of specific tumor receptors or overexpressed receptors and the development of novel therapies based on new peptide hormone analogs as targets for cancer therapy have been proposed. Among these targets, the human gonadotropin-releasing hormone type I (hGnRH-I), also called the hypothalamic luteinizing hormone-releasing hormone (LHRH), and its receptor, the human gonadotropin-releasing hormone receptor type I (hGnRHR-I), also known as the hypothalamic luteinizing hormone-releasing hormone receptor (LHRH-R), have been predicted as potentially therapeutic agents for their antitumor activities (10–13).
Functions of hGnRH-I, hGnRH-II, and hGnRHR-I in Reproduction
GnRH-I is a hypothalamic hormone first isolated from porcine hypothalamus (14). On the other hand, its receptor was purified and characterized first from bovine anterior pituitaries (15). At present, it is well-established that both molecules comprise the signaling complex that controls sperm and ovary maturation, as well as steroidogenesis in gonads (16–18). Human GnRH-I (hGnRH-I) is a decapeptide hormone described for first time in 1971 by the group of Schally (Tables 1, 2) (19). Seeburg and Adelman cloned the encoding gene for the first time in 1984 (76). In humans, the GnRHI gene is located on chromosome 8p11.2-p21 and is composed of four exons separated by three introns (77). This hormone is produced in the hypothalamus by GnRH neurons for release in a pulsatile fashion into the hypophyseal portal circulation to act primarily on the anterior pituitary, where it binds its receptor, the hGnRHR-I, in gonadotropic cells to stimulate the synthesis and secretion of pituitary gonadotropic hormones: luteinizing hormone (LH) and follicle-stimulating hormone (FSH) (Figure 1) (18). In the gonads, gonadotropins trigger gametogenesis as well as the synthesis and release of steroid sex hormones in females and males (Figure 1). Likewise, gonadal steroids are able to regulate hGnRH-I secretion through both positive and negative feedback (78). hGnRH-I is synthesized as a prohormone. The full sequence is a 92-amino-acid (aa) peptide in which the first 23 aa are a signal peptide followed by the functional GnRH decapeptide. Later, an amidation/proteolytic processing signal (Gly-Lys-Arg) is found, and finally, there is a 56-aa region known as the GnRH-associated peptide (GAP), which is co-secreted with GnRH and which appears to be involved in the processing and packaging of the decapeptide (79, 80). In humans, there is a second subtype of GnRH denominated hGnRH-II (Tables 1, 2). It is also a decapeptide hormone that differs from hGnRH-I in three amino acids (GnRH-II; His5, Trp7, Tyr8-GnRH-I) and that is encoded by the gene GnRH2, which has been mapped in chromosome 20p13 (58). hGnRH-II is found in the midbrain region and serves principally as a neurotransmitter and stimulator of sexual behavior (81). However, it has been shown that hGnRH-II can stimulate gonadotropin release in vivo through its binding to hGnRHR-I (82). The expression of hGnRH-I and hGnRH-II is differential. By hGnRH-I, its expression is higher in the brain (18). In the opposite site, hGnRH-II is ubiquitously expressed in different systems, such as thoracic (heart, lung, and aorta), digestive (salivary gland, stomach, and intestine), endocrine (adrenal, pancreas, and thyroid), and immune (tonsil, leukocyte, and lymph node) (83, 84).
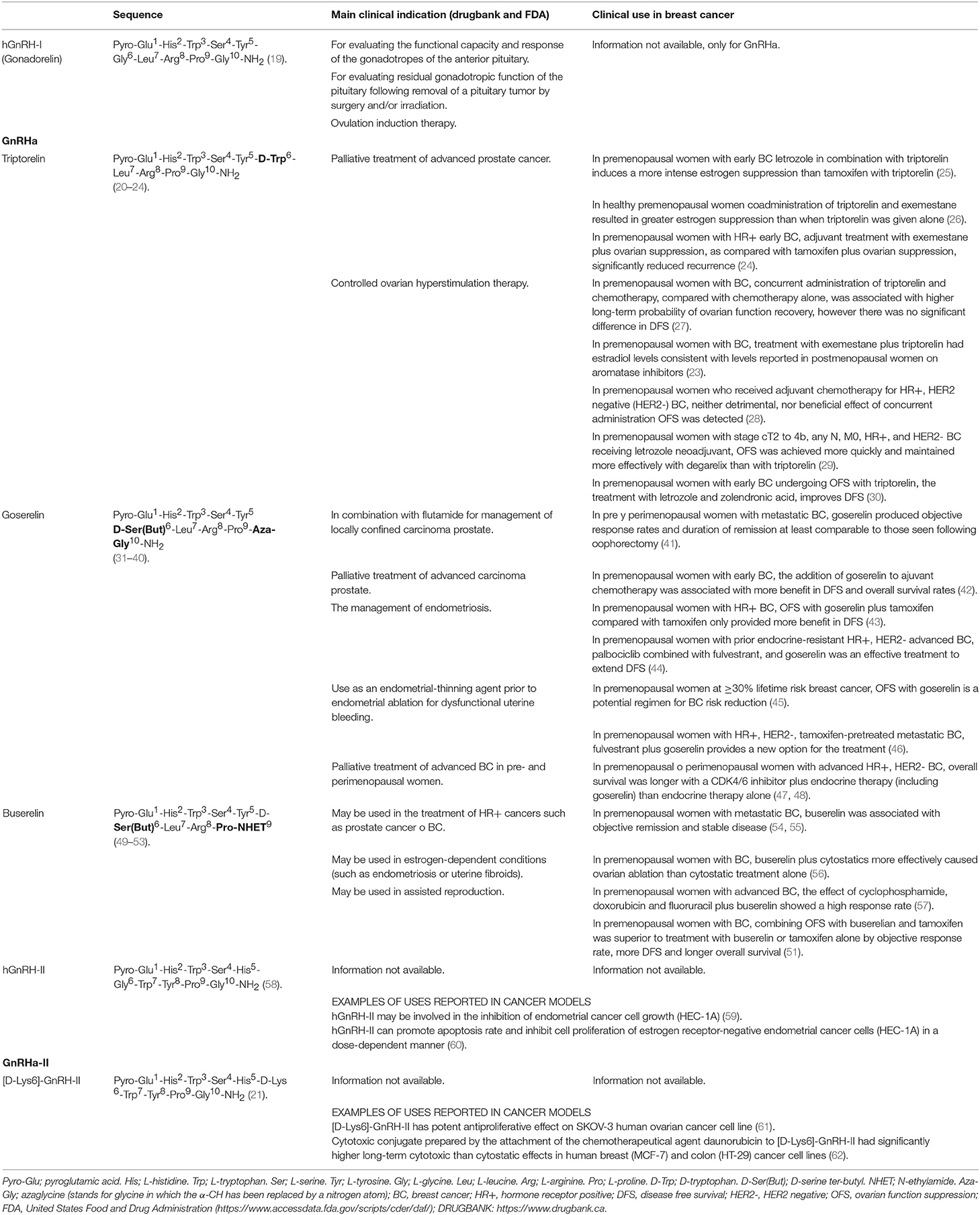
Table 1. Chemical structure of hGnRH-I agonists (GnRHa) and hGnRH-II agonists (GnRHa-II) evaluated against breast cancer.
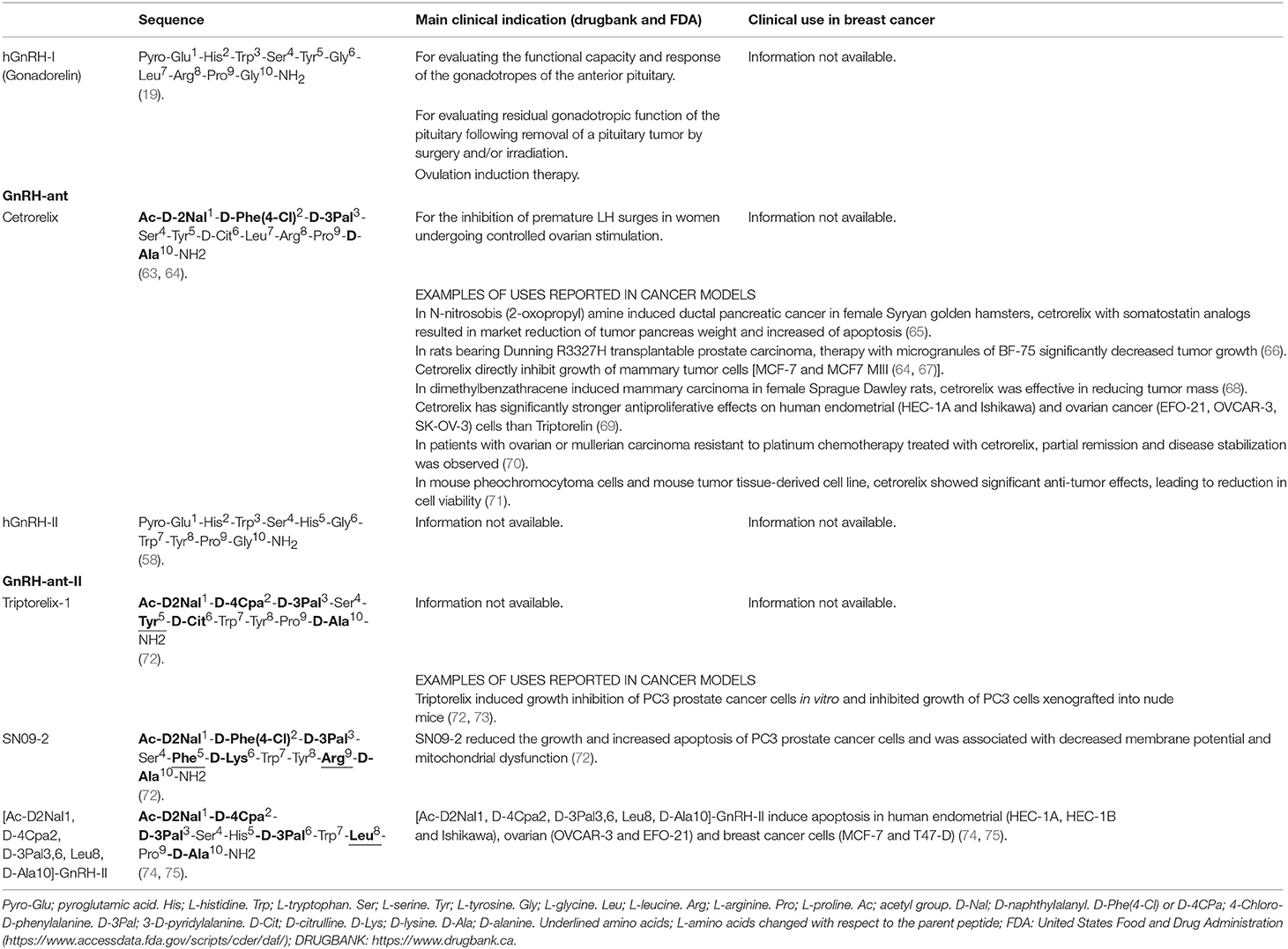
Table 2. Chemical structure of hGnRH-I antagonists (GnRH-ant) and hGnRH-II antagonists (GnRH-ant-II) evaluated against breast cancer.
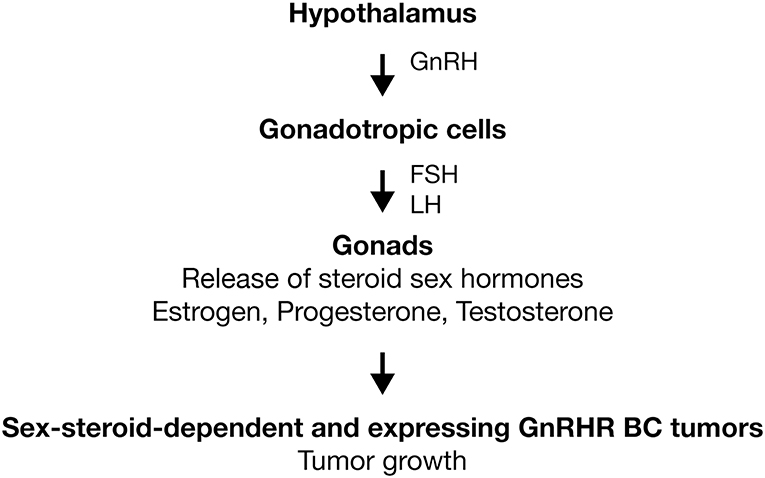
Figure 1. Activation of GnRHR in gonadotropic cells. GnRH is produced in hypothalamus and released in a pulsatile fashion to act primarily on the anterior pituitary. Here, the GnRHR is expressed in the membrane of gonadotropic cells (gonadotropes). Receptor activation stimulates the synthesis and secretion of LH and FSH. In gonads, gonadotropins trigger gametogenesis as well as the synthesis and release of steroid sex hormones (estrogen, progesterone, and testosterone). In sex-steroid-dependent BC tumors, sexual hormones promote the tumoral growth. In BC tumors that express GnRHR, analogs of GnRH could improve the treatments anticancer by the inhibition of tumoral growth. Activation ( ).
).
hGnRHR-I is a member of the superfamily of G protein-coupled receptor (GPCR) and, according to its aa sequence, this receptor belongs to the family of Rhodopsin. The structure of hGnRHR-I is similar to that of other GPCRs; it is a single amino-acid chain that shows an extracellular amino-terminal, seven transmembrane domains, and three extracellular and three intracellular loops (17). In the case of hGnRHR-I, this was cloned and characterized from human pituitary for the first time in 1992 (85). In comparison with other GPCRs, there are specific characteristics that distinguish the human GnRHR receptor, including the lack of a cytoplasmic carboxyl-terminal tail. This characteristic has been associated with inefficient desensitization on hGnRH-I stimulation and relatively slow internalization (86–88). Another feature of hGnRHR-I is the presence of a lysine at position 191. Research works have demonstrated the presence of this residue in human receptors and it is possibly implicated in reducing hGnRHR expression on the cellular surface (89, 90).
A second type of GnRHR, termed GnRHR-II, occurs in mammals. The search for this gene in human genome database revealed its presence as a putative gene (GnRHR2) on chromosome 1q12 (91, 92). Although the sequences display 40% of identity with hGnRHR-I (93), analysis of the corresponding reading frame shows the presence of a premature stop codon (94, 95). Nonetheless, the expression of mRNA and several alternatively spliced transcripts derived from GnRHR2 were demonstrated; in all of these, the premature stop codon was retained (91, 92). In conclusion, all of these observations support the notion that this gene is not functional and there is no GnRHR type II in humans.
Physiology of GnRH-I, GnRH-II, and hGnRHR-I in BC
The presence of hGnRH-I, hGnRH-II, and hGnRHR-I has been demonstrated in tumors of the reproductive tract, such as ovarian cancer (96–98), prostate cancer (99–101), and BC (102–104).
Numerous studies have shown the expression of hGnRH-I, hGnRH-II, and hGnRHR-I in BC tissue and provide a rationale for their use as a molecular target for the treatment of BC. hGnRH-I binding sites were reported in the biopsies of primary human carcinoma tissues and were localized in the cytoplasm of 64% of cases of invasive ductal carcinoma (102, 105). At the same time, nearly 50% of BC specimens possess hGnRH-I binding sites on their surfaces (106, 107). In TNBC tissues, GnRHR-I mRNA was detected in up to 70% (20, 107–110). Finally, in different human BC cell lines, including TNBC cell lines, either hGnRH-I-binding sites or hGnRH-II-binding sites were reported (20, 21, 63, 75, 107, 109, 111).
Effects of hGnRH-I, hGnRH-II, and hGnRHR-I on Cell Proliferation and Cell Migration in BC
The hGnRHR-I, hGnRH-I, and hGnRH-II systems have shown specific characteristics in cancer cells. For example, although the transcripts expressed in malignant tumor share coding region and protein from the pituitary, their expression levels are lower in comparison with those on gonadotropic cells (112, 113). In tumor cells, there are high-affinity binding sites or low-affinity binding sites by hGnRH-I and hGnRH-II; the dichotomy of the GnRH agonist and antagonist is not clearly present and the mechanisms activated by this receptor are different from those of the signaling pathways present in the pituitary–gonadal axis (96, 97, 102, 114–118). All of these aspects should be studied in greater depth in order to achieve the clinical implementation of hGnRHR-I, hGnRH-I, and hGnRH-II as therapeutic targets in human extra-pituitary tissue.
The antitumoral functions of hGnRH-I or GnRH-II and the hGnRHR-I in BC have been associated with the reduction of cell proliferation, invasion, and migration (10, 119–124). The molecular mechanisms employed have been explored in several research models. The reduction of cell proliferation in malignant cells is achieved through coupling between this receptor and Gi protein, as well as by crosstalk between hGnRHR-I and growth factor receptors (125). In the MCF-7 cell line, the overexpression of hGnRHR-I by adenoviruses was able to decrease cellular proliferation (117). The inhibitory cell growth effect promoted by hGnRHR-I activation in this cell line was mediated by the indirect inactivation of both the epidermal growth factor (EGF) receptor and the insulin-like growth factor (IGF) receptor (126, 127). Furthermore, it was additionally shown that hGnRH-I analogs were also able to suppress the EGF-induced activation of mitogen-activated protein kinase (MAPK) and the dephosphorylation of EGF receptors via the activation of phosphotyrosine phosphatase (128, 129). In a mouse model treated with GnRH agonist, the expression of EGF and IGF receptors was decreased (130). In 4OH-Tamoxifen-resistant MCF-7 and T47D-TR cell lines, GnRH agonists were able to abolish cell proliferation, blocking EGF-receptor autophosphorylation and ERK1/2 activation and reducing EGF-induced c-fos protein expression (131, 132).
The cellular mechanism that explains how the hGnRH-I/hGnRHR-I system inhibits cell invasion has been studied in the MDA-MB-231 cell line. The activation of hGnRHR-I by GnRH agonist results in decreased expression of the Rho GTPase-Activating Protein 18 (ARHGAP18) and the concomitant increase in the activation of RhoA (49, 124). The overactivity of RhoA triggers intracellular signaling pathways to produce more stress fibers, more focal adhesions, and major substrate adhesion, which in turn inhibit cellular migration. On the other hand, in MDA-MB-231 cells and in mesenchymal-transformed MCF-7 cells (MCF-7-EMT), the administration of GnRH agonist was also able to block the expression and activity of S100 calcium binding protein A4 (S100A4) and cysteine-rich angiogenic inducer 61 (CYR61). Considering that both proteins play important roles in cellular invasion and metastasis, their inhibition by GnRH analogs could be associated with the decrease of invasion observed (133).
In the case of hGnRH-II analogs, these exert an effect on the reduction of tumor growth rates by means of the induction of apoptosis, demonstrating a novel hGnRH-II signaling pathway that is active in tumors (69, 74, 75, 118). The molecular mechanism related with this antitumor effect mediated by the apoptotic pathway results in the activation of stress-induced MAPK or Bax via p38 and JNK. The deficiency of mitochondrial membrane potential and apoptotic cell death via the dose-dependent activation of caspase-3 was demonstrated by the use of several hGnRH-II analogs (69, 74, 75, 118). On the other hand, the ability to reduce metastases in BCs has also been exhibited by GnRH-II agonists (63).
All of the previously mentioned information supports the use of hGnRHR-I as a clinical asset against BC. Herein, in this review, we explored recent information on the development and use of GnRH analogs, such as agonists (defined in this work as GnRHa by hGnRH-I agonists and GnRHa-II by hGnRH-II agonists), antagonists (defined in this work as GnRH-ant by hGnRH-I antagonists and GnRH-ant-II by hGnRH-II antagonists), non-peptide GnRH antagonists, and cytotoxic analogs of GnRH and their possible implications as antitumor agents against BC.
Analogs of GnRH Used Against BC
After the discovery of the amino-acid sequence of hGnRH-I in 1971, the development of synthetic analogs began with the intent to synthesize stimulating and blocking variants. This process has been very intense due its clinical application, and as a consequence, ~5,000 analogs of this hormone were synthesized between 1972 and 2016 (134). In the case of stimulating variants or agonists, their design has been centered on improving receptor binding and subsequent activation. On the other hand, blocking variants or antagonists have been designed with strong receptor binding but without activation.
GnRH-I Agonists
The clinical applications of GnRH agonists were limited by their short half-life (2–4 min) promoted by the rapid cleavage among the amino acids at positions 5–6, 6–7, and 9–10 (134, 135). For this reason, the development of long-acting agonists was based on two principal criteria: first, they were derived from native hGnRH-I by adding D-amino acids at position number 6, 9, or 10 instead of L-amino acids. With this change, the agonists were resistant to degradation and also increased their receptor binding sites (134, 135). Second, because an essential role of NH2 and the COOH termini is played in binding to the receptor, usually Pro9- and/or Gly10-NH2 are modified by Pro9-NHET or Pro9-Aza-Gly10-NH2 to increase half-life and receptor occupancy time (136). All of these modifications have improved the ability of these agonists to activate their receptor by several hundreds of magnitude in comparison with the native hGnRH-I.
At present, it is clear that acute exposure to GnRHa produces adverse effects, such as hot flashes (“flares”) caused by the increase and sudden decrease of gonadotropin hormones and steroid hormones. This is due to the continuous administration of GnRHa producing early receptor activation and to its subsequent downregulation and desensitization in gonadotropic cells (Figure 2). These effects decrease the secretion of LH and FSH and the lack in sex-steroid hormones (estrogens, testosterone, and progesterone) with the concomitant appearance of “chemical castration” (Figure 2). This castration effect highlights the potential use of the treatment of early stage of endocrine-responsive tumors of BC, with GnRHa as adjuvant therapy in combination with Tamoxifen or aromatase inhibitors (Figure 2). Thus, it is important to bear in mind that the GnRH analogs employed in clinical practice commonly are administered in combination with estrogen antagonists or chemotherapy agents and not in direct form. Although this is primarily due to ethical reasons, it results in a limited understanding of the direct effects that GnRH analogs could exert on BC. Similarly, it is important to consider that, although the majority of the results obtained through studies carried out in animal models or in cell lines reflect or are in agreement with the effects observed in clinical studies, they must be interpreted in terms of their clinical application with caution and more studies are needed to generalize their use as therapeutic tools.
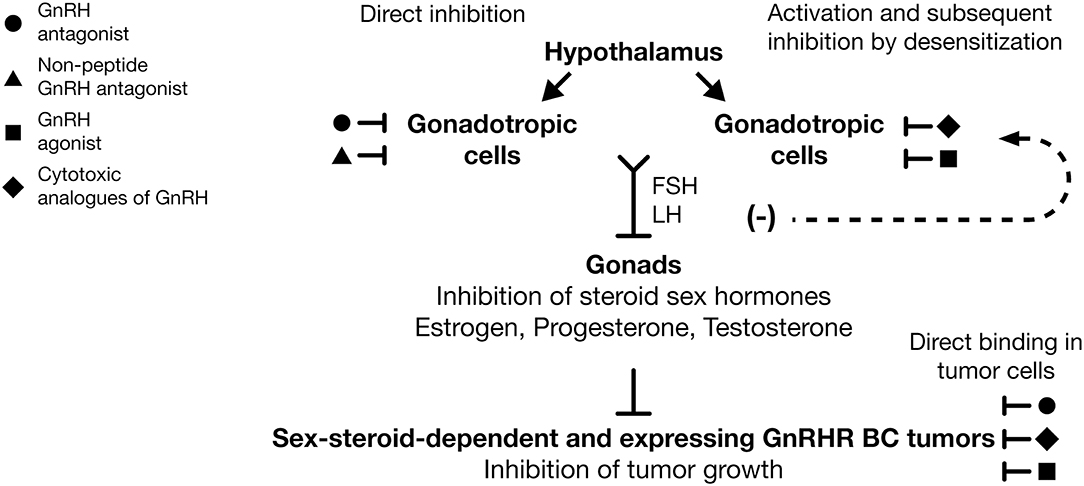
Figure 2. Use of GnRH analogs as adjuvant therapy against breast cancer. GnRH antagonists ( ) and non-peptide GnRH antagonists (
) and non-peptide GnRH antagonists ( ) are able to reduce FSH and LH expression by direct inhibition of GnRHR in gonadotropic cells. GnRH agonists (
) are able to reduce FSH and LH expression by direct inhibition of GnRHR in gonadotropic cells. GnRH agonists ( ) and cytotoxic analogs of GnRH (
) and cytotoxic analogs of GnRH ( ) are able to reduce gonadotropin hormones levels after GnRHR desensitization in gonadotropic cells. Suppression of FSH and LH evoke diminution of estrogen, progesterone, and testosterone levels and the subsequent inhibition of growth in dependent sex-steroid tumors. Likewise, GnRH agonists (), GnRH antagonists (), and cytotoxic analogs of GnRHR () have direct antitumor effects over cancer cells, promoting in those the inhibition of cell growth. The systemic and local effect of GnRH analogs could improve the clinical response in BC patients, principally those that are treated with combined therapies. Inhibition (⊥), desensitization (−).
) are able to reduce gonadotropin hormones levels after GnRHR desensitization in gonadotropic cells. Suppression of FSH and LH evoke diminution of estrogen, progesterone, and testosterone levels and the subsequent inhibition of growth in dependent sex-steroid tumors. Likewise, GnRH agonists (), GnRH antagonists (), and cytotoxic analogs of GnRHR () have direct antitumor effects over cancer cells, promoting in those the inhibition of cell growth. The systemic and local effect of GnRH analogs could improve the clinical response in BC patients, principally those that are treated with combined therapies. Inhibition (⊥), desensitization (−).
Administration of GnRHa as adjuvant therapy against BC is able to reduce, in a reversible manner, circulating concentrations of estrogen (ovarian suppression) in premenopausal women through its action on the hypothalamus–pituitary–ovarian axis (Figure 2). These effects reduce tumor growth that is sensitive to circulating estrogens either by preventing the development of distant metastases or by extending disease-free survival (DFS), and therefore overall survival (OS) (137, 138). The positive effect of GnRHa as adjuvant treatment in premenopausal women with ER+ tumors was shown by the analysis of 16 randomized trials. According to these data, when GnRHa was added to Tamoxifen or chemotherapy treatment or both, there was a reduction in the recurrence of BC and fewer people died after recurrence of the disease (139). On the other hand, the beneficial effect of GnRHa in TNBC was also shown. In this case, a meta-analysis of randomized controlled trials supports the use of GnRHa in the clinical setting as adjuvant therapy for the management of TNBC tumors (140).
Additionally, several research groups have demonstrated that GnRHa exerts direct anticancer activity on malignant tissue including mammary, which is independent from the suppression of the ovarian steroid synthesis and secretion (Figure 2) (50, 141). Based on the information mentioned previously, it is possible to propose that GnRHa exerts a systemic effect on breast tumors by ovarian suppression and a local effect by hGnRHR-I activation (Figure 2). Next, we describe the positive effects of specific GnRH-I and GnRH-II agonists employed as therapeutic agents against BC.
Triptorelin
Triptorelin (Table 1) is a GnRHa that has been approved in the European Union as an adjuvant endocrine therapy in combination with Tamoxifen or AI to treat endocrine-responsive, early-stage BC (22). This management blocks the action of estrogen in ER+ BC cells (Tamoxifen) or inhibits the production of estrogen (AI, GnRHa) in patients (Figure 2) (23, 142). The benefits of Triptorelin as adjuvant therapy were investigated by randomized, open-label, phase III trials (22, 24). In these studies, 2,359 premenopausal women with hormone-receptor-positive early BC received treatment with Exemestane plus ovarian suppression with Triptorelin and 2,358 received Tamoxifen plus ovarian suppression, also with Triptorelin. After a median follow-up of 68 months of treatment, DFS at 5 years was 91.1% in the Exemestane-ovarian suppression group and 87.3% in the Tamoxifen-ovarian suppression group. The rate of being free from BC at 5 years was 92.8% in the Exemestane-ovarian suppression group, as compared with 88.8% in the Tamoxifen-ovarian suppression group. Overall survival did not differ significantly between the two groups. These data show the favorable effect that Triptorelin as ovarian suppressor has over the BC treatment.
The antitumoral effects of Triptorelin, Cetrorelix, and the GnRHa-II [D-Lys6]-GnRHII also have been evaluated in in vitro and in vivo research models (21). In MCF-7, HCC 70, and T47-D cell lines and, in a mouse model, a reduction in metastasis and significant inhibition of bone metastasis formation were observed (21). On the other hand, Triptorelin and [D-Lys(6)]-GnRHII were able to inhibit EGF-receptor signaling transductional pathway and restored sensitivity to 4-OH-Tamoxifen in 4OH-Tamoxifen-resistant MCF-7 cells and T47D-TR cells [81. In TNBC cell lines, MDA-MB-231 and HCC1806, the administration of Triptorelin either individually or in combination with chemotherapeutic agents such as Cisplatin, Docetaxel, and AEZS-112, and PI3K/AKT inhibitors (Perifosine, AEZS-129), ERK inhibitor (AEZS-134), and dual PI3K/ERK inhibitor AEZS-136 showed antiproliferation activity. In both cell lines, synergistic effects took place when Triptorelin was combined with Cisplatin. In HCC1806 cells, synergy occurred when Triptorelin was applied with PI3K/AKT inhibitors Perifosine and AEZS-129. In MDA-MB-231 cells, synergy was observed after co-treatment with Triptorelin and the ERK inhibitor AEZS-134 and dual PI3K/ERK inhibitor AEZS-136 (20).
Goserelin
Goserelin (Table 1) is a GnRHa approved in 1989 by the Food and Drug Administration (FDA) for the treatment of BC as adjuvant endocrine therapy due to its abilities to reduce circulating concentrations of estrogen (Figure 2). One study demonstrated that, in premenopausal patients with ER-positive BC, Goserelin offers an equivalent and well-tolerated alternative to Cyclophosphamide, Methotrexate, and Fluorouracil (CMF) chemotherapy. In this case, Goserelin was also able to display equivalent DFS compared to cytotoxic chemotherapy, but with a more favorable safety profile (31, 32). Moreover, in a randomized trial performed in 2003 by the International Breast Cancer Study Group (IBCSG), it was shown that, in ER-negative patients, the course of the disease was improved if the patients received CMF in combination with Goserelin. Additionally, in women under the age of 39 years, the combination of chemotherapy followed by Goserelin resulted in superior DFS (33).
In a retrospective study performed in patients aged <40 years diagnosed with invasive BC, the administration of Goserelin as systemic adjuvant therapy following surgical removal of the primary tumor and a chemotherapy scheme with CMF demonstrated better results in comparison with the administration of CMF and Goserelin alone (34). In this study, the administration of Goserelin plus neoadjuvant chemotherapy exhibited higher rates of pathological Complete Response (pCR) and a markedly decreased proliferation index compared with the neochemotherapy-alone group, principally in ER- and/or in PR-negative tumors (34). Karlsson et al. reported similar results. These authors observed that the combination of CMF and Goserelin in premenopausal patients with ER-positive tumors showed a significant improvement of DFS and a BC-free interval in comparison with the administration of CMF and Goserelin alone (35). A positive effect on the patient's quality of life after the administration of Goserelin, in comparison with CMF chemotherapy alone, was demonstrated in premenopausal and perimenopausal patients with early BC (36). Finally, in a randomized study in patients with ER-negative BC, the administration of Goserelin during the course of chemotherapy protected ovarian function, reducing the risk of early menopause and improving prospects for fertility (37).
Beneficial effects due to the combination of Goserelin and AI or Goserelin and Tamoxifen also were revealed. In the former case, simultaneous administration of Goserelin and 4-hydroxyandrostenedione (4-OHA) improved the clinical response of patients with BC (38). Likewise, the combination of Goserelin and Tamoxifen in premenopausal patients with advanced BC increased progression-free survival time in comparison with the use of a GnRH agonist alone (39, 40).
Buserelin
Buserelin is another hGnRHa (Table 1). The use of 125I-Buserelin in 235 samples of biopsies from pre- and postmenopausal women demonstrated the presence of high-affinity binding sites for GnRH (50).
On the other hand, several studies have shown the beneficial effects in the use of combined therapies of Tamoxifen and Buserelin or AI and Buserelin in patients. In 161 premenopausal women with metastatic BC, the combined treatment with Buserelin and Tamoxifen evoked a significant improvement in patient survival in contrast with treatment with either drug alone (51). Furthermore, a couple of studies performed with metastatic male BC patients demonstrated that the treatment with an AI (Letrozole) or antiandrogens (cyproterone acetate) in combination with GnRHa (Leuprolide, Triptorelin, Buserelin, and Goserelin) could be associated with greater survival in comparison with the monotherapy (52, 53).
Several groups have reported that hGnRHR-I activation by Buserelin in cell lines is also able to evoke the antitumor activities observed in malignant tissues. In MCF-7 cells, Buserelin promotes a potent antiproliferative effect (114, 143). On the other hand, in the TNBC cell line MDA-MB-231, Buserelin inhibited cell proliferation, cell migration, and invasion (49, 144).
GnRH-II Agonists
Few reports show the use of GnRHa-II against BC. In this case, [D-Lys6]-GnRHII (Table 1) induced the inhibition of cell migration in human BC cell lines HCC 70, MCF-7, MDA-MB-453, and T47-D (21).
GnRH-I Antagonist
GnRH-ant do not induce an initial stimulation of the receptor; they compete for receptor occupancy, causing a rapid and reversible suppression of the hGnRHR-I response, reducing the onset time of therapeutic effects and also eliminating clinical “flare” (Figure 2). These two points comprise the major advantage of these molecules over the agonists. Initially, early GnRH-ant were developed by the replacement of aa residues in position 2 and position 3 due to the importance of these aa in receptor binding and functional effects (145). Later, substitutions of D-aa instead of L-aa in several positions, such as 2, 3, 6, and 10, further increased antagonistic activity. However, their clinical use was restricted by solubility limitations and anaphylactic reactions (146, 147). For these reasons, other modifications, such as the use of various substituents in the benzene ring of the D-Phe residue in position 2, the addition of groups such as Ac-D-2Nal, D-3Pal in the N-terminal part of the peptide, and the substitution by D-Ala in position 10 or different aa derivatives, mostly in positions 5, 6, and 8, improved the inhibitory activities of several antagonists (148).
Cetrorelix
Cetrorelix is a GnRH-ant that has been shown as safe and effective in inhibiting the secretion of gonadotrophins (Table 2). Due to its capability to promote the suppression of LH and sex-steroid hormones, this GnRH-ant has been employed in the treatment of hormone-dependent cancers such as prostate and ovarian cancer (Figure 2) (149). Currently, the FDA indicated the use of this antagonist for inhibiting the premature LH surge in controlled ovarian hyperstimulation (COH) (150).
The direct antitumor effects of Cetrorelix in BC have been reported (Figure 2). In samples from patients diagnosed with TNBC, in cellular models such as TNBC cell lines (HCC1806 and HCC1937), estrogen-responsive tumor cell line MCF-7, and in xenografts carried out with the MCF-7 cell line in nude mice, the administration of Cetrorelix resulted in a significant decrease of cell proliferation (64, 67, 107). On the other hand, the antimetastatic activity associated with hGnRHR-I activation in breast tumors by the administration of Cetrorelix was confirmed. In this case, this antagonist reduced the formation of metastasis by the MDA-MB-231 cell line in a nude mouse model (63).
GnRH-II Antagonists
Several works have revealed that other GnRH-ant-II exert a direct effect on malignant tumors (Figure 2). For example, Trptorelix-1 and SN09-2 (Table 2) induced cell death via the apoptotic process in cancer cells (72, 151). On the other hand, in cellular models of human BC such as MCF-7, MDA-MB-231, and T47-D, the administration of [Ac-D2Nal1, D-4Cpa2, D-3Pal3,6, Leu8, D-Ala10]-GnRHII (Table 2) induced the activation of apoptosis and promoted significant inhibition of tumor growth (74, 75).
Non-peptide GnRH Antagonists and Their Use as Therapeutic Agents in BC
Several GnRHa peptides (Goserelin and Triptorelin) have been approved by their incorporation into the market. On the other hand, various GnRH-ant peptides, such as Cetrorelix, have been utilized clinically for several diseases, including in malignant tumors, such as BC. However, the clinical use of both types of GnRH analogs has been limited because they must be administered frequently by subcutaneous injection or sustained release formulations. For these reasons, several research groups have been developing non-peptide GnRH antagonists for clinical use (152). These are small non-peptidic molecules characterized by their oral administration and that exhibit better patient compliance during their clinical administration in comparison with injectable antagonists. They have titratable doses to accomplish the partial inhibition of sex-steroid hormones according to the disorder treated (Figure 2). They do not present, as do other GnRH-ant, the phenomenon of “flare,” and there is no risk of bone loss, such as that observed during long-term administration of GnRH agonists (78, 153).
Many classes of non-peptide GnRH antagonists have been described. They belong to different chemical families, such as Bicyclicpyrimidones/Pyrimidinediones, Uracils, Indoles, Quinolones, Quinolines and Phthalazines, Furan-2-Carboxamides, and Benzimidazoles; however, the majority of these have not achieved clinical application to date (154–157). The first non-peptidic compound evaluated in rat pituitary membranes was Ketoconazole (158, 159). Takeda Pharmaceutical developed the next non-peptide GnRH antagonist. This was called T-98475 and was described as the first highly potent and orally active non-peptidic GnRH antagonist (160). Later, a derivative from this molecule was synthesized, that is, the first non-peptide GnRH antagonist, which was evaluated in humans and is known as TAK-013 or Sufugolix (161). This compound was evaluated in a phase II study in patients with endometriosis, but it was not able to advance to phase III studies. Recently, a new compound was reported that is derived from TAK-013 and is denominated TAK-385 or Relugolix (162). This molecule was evaluated in a humanized mouse model, showing its ability to work as an antagonist of GnRH with the capacity to suppress, in a powerful, continuous, and reversible manner, the way that hGnRHR-I functions (Figure 2). Due the potent effect of TAK-385 as a non-peptide GnRH antagonist, it is currently employed in different clinical trials to determine its therapeutic use against several diseases including BC (163).
Cytotoxic Analogs of GnRH Employed Against BC
Although the principal clinical effects of GnRH analogs have been associated with their ability to suppress estrogen action, as previously mentioned, in human clinical specimens of BC (49%) and in cell lines derived from malignant breast tumors, there are binding sites for hGnRH-I and hGnRH-II that could be successfully employed as therapeutic targets (Figure 2). With this in mind, molecules known as cytotoxic analogs have been designed. They are GnRH agonists conjugated to chemotherapeutic agents that combine hormonal and cytotoxic activity.
Clinical Use of AN-152 and AN-207 Conjugates
AN-152 (also known as AEZS-108) and AN-207 are two cytotoxic analogs developed in 1996. In them, the GnRHa [D-Lys6]-GnRH is linked with Doxorubicin (DOX), a widely used anticancer agent. AN152 (AEZS-108) is conjugated to DOX and AN-207 is conjugated to 2-pyrrolino-DOX, a 500 ± 1,000 times more active derivative of DOX (164). Although the antitumor effect of both cytotoxic analogs as inhibitors of tumor growth has been demonstrated in an estrogen-independent and MXT mouse mammary cancer model (Figure 2), stronger effects of AN-207 as an inhibitor of tumor growth, as a promoter of the apoptotic index, and in terms of necrosis in tumors were also determined (165–167). On the other hand, the cellular targets of these conjugate molecules are specific for cancer cells that express hGnRHR-I, as demonstrated in MCF-7 (Figure 2). In this case, the expression of this receptor on cell surfaces promotes the conjugate-internalization process and metabolism, and evoked the cytotoxic effect of AN-152 (AEZS-108) (168). Similar results were observed in TNBC cell lines MDA-MB-231 and HCC-1806 (109, 110, 169, 170). In in vivo models, AN-207 gave rise to the regression of tumor growth in an MX-1 hormone-independent Doxorubicin-resistant human BC cell line (166, 171).
Emons et al. select AN-152 (AEZS-108) for a clinical trial because of its lower toxic adverse effects, and the authors also showed that the half-life of this compound in human serum is 2 h (172). Later, this same research group designed the first phase I study with 17 women who have a confirmed diagnosis of epithelial cancer of the ovary, endometrium, or breast. The AN-152 (AEZS-108) dose used in this trial was 267 mg/m2. This dose exhibited maximal tolerance without supportive medication and decrease in LH and FSH after administration of AESZ-108 (Figure 2) (173). Evaluation of AN152 (AEZS-108) in a phase II study and subsequent phase III clinical trials (https://clinicaltrials.gov/ct2/show/NCT01767155) was performed in patients with ovarian cancer and in patients with endometrial cancer, confirming in both of these the anticancer activity of this cytotoxic analog (174).
Cytotoxic Analogs of GnRH That Employ Specific Spacers/Linkers Such as Oxime Bond and Hydrazone and Their Evaluation Against BC
Other conjugated anticancer GnRH agonists have been designed. In order to increase stability in the human serum of these molecules, GnRH peptides were attached to anticancer drugs through several spacers/linkers, such as oxime bond and hydrazone bond (175, 176). Daunorubicin was conjugated via oxime bond to GnRH-III; this modification increased the stability of the GnRH bioconjugates in human serum at least for 24 h and did not affect its antitumor properties in the MCF-7 cell line (175). Incorporation of anticancer drugs such as the anthracyclines, Daunorubicin and Doxorubicin, to GnRH-III and [D-Lys6]-GnRH analogs through oxime, hydrazine, or ester bonds as spacers showed potent anticancer action in the MCF-7 cell line and increased their half-life in human serum (176). New cytotoxic compounds include two GnRH analogs ([D-Lys6] GnRH-I and [D-Lys6] GnRH-II), with a link between them and the chemotherapeutic agent, Daunorubicin (Dau), through an enzyme-labile spacer using oxime bond. In this work, it was shown that both conjugates possess similar receptor-binding affinity and antitumoral properties, such as an apoptotic effect in MCF-7 cells and in the colon cell line, HT-29 (Figure 2) (62). Five conjugates of Paclitaxel (PTX) were linked to the GnRH antagonist Degarelix as targeting moiety and were evaluated in MCF-7 and HT-29 cell lines as anticancer agent. The half-lives of all of the conjugates in human serum were close to 10 h, and high-affinity GnRH-receptor binding and an antiproliferative effect on MCF-7 cells were shown (177).
Conclusions
All of the information reported in the present review supports the use of hGnRH type I and hGnRH type II analogs, non-peptide GnRH antagonists, and the cytotoxic analog complex to GnRH as adjuvants in the therapy against BC. However, it is important to consider that, for the use of GnRH analogs in clinical practice, it is necessary to have an understanding of the actions of these molecules in breast tumors, such as the pharmacology of agonists on the breast tumor, defining in detail their potency, and the signaling pathways involved, as well as possible adverse effects, among others. Similarly, extrapolation of the data obtained through studies carried out in animal models or in cell lines must be performed with caution. Even so, many clinical studies have shown that hGnRH agonists have the ability to reduce BC growth and disease recurrence, principally in patients with TNBC. In addition, these agonists are able to reduce tumor migration, including in advanced cases. Another important point to consider is that GnRHa might exert systemic and local action on BC. The use of GnRH analogs as adjuvant therapy against BC has demonstrated their ability to reduce circulating concentrations of estrogen, with the concomitant reduction of tumor growth. Likewise, agonists might exert a local effect by direct activation of the GnRHR-I and the concomitant reduction in tumor growth and prevention of distant metastasis. This dual effect could improve the clinical response, principally in combined therapies. Finally, although the use of GnRH analogs, non-peptide GnRH antagonists, and cytotoxic analogs of GnRH requires more clinical trials for their clinical implementation, the hGnRH/hGnRHR system comprises a very promising target for further pharmaceutical development in the treatment of BC, especially for the treatment of advanced stages of this disease.
Author Contributions
AA-R and MH-R conceptualized and wrote the first and final versions of the article. NG and J-CO-M contributed to refining the article. GM-N improved the GnRH and GnRH receptor section. MP-S and EL-M wrote and improved the clinical section. All of the authors read and approved the final version of the article.
Funding
This work was supported by the Fondo de Investigación en Salud (FIS)-Instituto Mexicano del Seguro Social (IMSS)-México (Grant No: FIS/IMSS/PROT/EMER18/1848) to AA-R. AA-R is a recipient of Research Career Development Awards from Fundación IMSS, México.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rivera-Franco MM, Leon-Rodriguez E. Delays in breast cancer detection and treatment in developing countries. Breast Cancer. (2018) 12:1178223417752677. doi: 10.1177/1178223417752677
2. Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, et al. The global burden of cancer 2013. JAMA Oncol. (2015) 1:505–27. doi: 10.1001/jamaoncol.2015.0735
3. Coleman MP, Quaresma M, Berrino F, Lutz JM, De Angelis R, Capocaccia R, et al. Cancer survival in five continents: a worldwide population-based study (CONCORD). Lancet Oncol. (2008) 9:730–56. doi: 10.1016/S1470-2045(08)70179-7
4. Vuong D, Simpson PT, Green B, Cummings MC, Lakhani SR. Molecular classification of breast cancer. Virchows Arch. (2014) 465:1–14. doi: 10.1007/s00428-014-1593-7
5. Rakha EA, Green AR. Molecular classification of breast cancer: what the pathologist needs to know. Pathology. (2017) 49:111–9. doi: 10.1016/j.pathol.2016.10.012
6. Dai X, Li T, Bai Z, Yang Y, Liu X, Zhan J, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res. (2015) 10:2929–43.
7. Baselga J. Treatment of HER2-overexpressing breast cancer. Ann Oncol. (2010) 21:vii36–40. doi: 10.1093/annonc/mdq421
8. Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. (2008) 52:108–18. doi: 10.1111/j.1365-2559.2007.02889.x
9. Hortobagyi GN, Edge SB, Giuliano A. New and important changes in the TNM staging system for breast cancer. Am Soc Clin Oncol Educ Book. (2018) 38:457–67. doi: 10.1200/EDBK_201313
10. Schally AV, Comaru-Schally AM, Nagy A, Kovacs M, Szepeshazi K, Plonowski A, et al. Hypothalamic hormones and cancer. Front Neuroendocrinol. (2001) 22:248–91. doi: 10.1006/frne.2001.0217
11. Signore A, Annovazzi A, Chianelli M, Corsetti F, Van De Wiele C, Watherhouse R, et al. Peptide radiopharmaceuticals for diagnosis and therapy. Eur J Nucl Med. (2001) 28:1555–65. doi: 10.1007/s00259-001-0663-7
12. Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. (2003) 24:389–427. doi: 10.1210/er.2002-0007
13. Weiner RE, Thakur ML. Radiolabeled peptides in oncology: role in diagnosis and treatment. BioDrugs. (2005) 19:145–63. doi: 10.2165/00063030-200519030-00002
14. Schally AV, Nair RM, Redding TW, Arimura A. Isolation of the luteinizing hormone and follicle-stimulating hormone-releasing hormone from porcine hypothalami. J Biol Chem. (1971) 246:7230–6.
15. Zolman JC, Valenta LJ. Gonadotropin-releasing hormone receptor binding in bovine anterior pituitary. Biochim Biophys Acta. (1980) 627:172–89. doi: 10.1016/0304-4165(80)90319-0
16. Conn PM, Staley D, Harris C, Andrews WV, Gorospe WC, McArdle CA, et al. Mechanism of action of gonadotropin releasing hormone. Annu Rev Physiol. (1986) 48:495–513. doi: 10.1146/annurev.ph.48.030186.002431
17. Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR. Gonadotropin-releasing hormone receptors. Endocr Rev. (2004) 25:235–75. doi: 10.1210/er.2003-0002
18. Stamatiades GA, Kaiser UB. Gonadotropin regulation by pulsatile GnRH: signaling and gene expression. Mol Cell Endocrinol. (2018) 463:131–41. doi: 10.1016/j.mce.2017.10.015
19. Matsuo H, Baba Y, Nair RM, Arimura A, Schally AV. Structure of the porcine LH-and FSH-releasing hormone. I The proposed amino acid sequence. Biochem Biophys Res Commun. (1971) 43:1334–9. doi: 10.1016/S0006-291X(71)80019-0
20. Kwok CW, Treeck O, Buchholz S, Seitz S, Ortmann O, Engel JB. Receptors for luteinizing hormone-releasing hormone (GnRH) as therapeutic targets in triple negative breast cancers (TNBC). Target Oncol. (2015) 10:365–73. doi: 10.1007/s11523-014-0340-y
21. von Alten J, Fister S, Schulz H, Viereck V, Frosch KH, Emons G, et al. GnRH analogs reduce invasiveness of human breast cancer cells. Breast Cancer Res Treat. (2006) 100:13–21. doi: 10.1007/s10549-006-9222-z
22. Frampton JE. Triptorelin: a review of its use as an adjuvant anticancer therapy in early breast cancer. Drugs. (2017) 77:2037–48. doi: 10.1007/s40265-017-0849-3
23. Bellet M, Gray KP, Francis PA, Láng I, Ciruelos E, Lluch A, et al. Twelve-month estrogen levels in premenopausal women with hormone receptor-positive breast cancer receiving adjuvant triptorelin plus exemestane or tamoxifen in the Suppression of Ovarian Function Trial (SOFT): the SOFT-EST Substudy. J Clin Oncol. (2016) 34:1584–93. doi: 10.1200/JCO.2015.61.2259
24. Pagani O, Regan MM, Walley BA, Fleming GF, Colleoni M, Láng I, et al. Adjuvant exemestane with ovarian suppression in premenopausal breast cancer. N Engl J Med. (2014) 371:107–18. doi: 10.1056/NEJMoa1404037
25. Rossi E, Morabito A, De Maio E, Di Rella F, Esposito G, Gravina A, et al. Endocrine effects of adjuvant letrozole + triptorelin compared with tamoxifen + triptorelin in premenopausal patients with early breast cancer. J Clin Oncol. (2008) 26:264–70. doi: 10.1200/JCO.2007.13.5319
26. Jannuzzo MG, Di Salle E, Spinelli R, Pirotta N, Buchan P, Bello A. Estrogen suppression in premenopausal women following 8 weeks of treatment with exemestane and triptorelin versus triptorelin alone. Breast Cancer Res Treat. (2009) 113:491–9. doi: 10.1007/s10549-008-9949-9
27. Lambertini M, Boni L, Michelotti A, Gamucci T, Scotto T, Gori S, et al. Ovarian suppression with triptorelin during adjuvant breast cancer chemotherapy and long-term ovarian function, pregnancies, and disease-free-survival: a randomized clinical trial. JAMA. (2015) 314:2632–40. doi: 10.1001/jama.2015.17291
28. Regan MM, Walley BA, Francis PA, Fleming GF, Láng I, Gómez HL, et al. Concurrent and sequential initiation of ovarian function suppression with chemotherapy in premenopausal women with endocrine-responsive early breast cancer: an exploratory analysis of TEXT and SOFT. Ann Oncol. (2017) 28:2225–32. doi: 10.1093/annonc/mdx285
29. Dellapasqua S, Gray KP, Munzone E, Rubino D, Gianni L, Johansson H, et al. Neoadjuvant degarelix vs. triptorelin in premenopausal patients who receive letrozole for locally advanced endocrine-responsive breast cancer: a randomized phase II trial. J Clin Oncol. (2019) 37:386–95. doi: 10.1200/JCO.18.00296
30. Perrone F, De Laurentiis M, De Placido S, Ortidura M, Cinieri S, Riccardi F, et al. Adjuvant zolendronic acid and letrozole plus ovarian function suppression in premenopausal breast cancer: HOBOE phase 3 randomised trial. Eur J Cancer. (2019) 118:178–86. doi: 10.1016/j.ejca.2019.05.004
31. Jakesz R, Hausmaninger H, Kubista E, Gnant M, Menzel C, Bauernhofer T, et al. Randomized adjuvant trial of tamoxifen and goserelin versus cyclophosphamide, methotrexate, and fluorouracil: evidence for the superiority of treatment with endocrine blockade in premenopausal patients with hormone-responsive breast cancer-Austrian Breast and Colorectal Cancer Study Group Trial 5. J Clin Oncol. (2002) 20:4621–7. doi: 10.1200/JCO.2002.09.112
32. Jonat W, Kaufmann M, Sauerbrei W, Blamey R, Cuzick J, Namer M, et al. Goserelin versus cyclophosphamide, methotrexate, and fluorouracil as adjuvant therapy in premenopausal patients with node-positive breast cancer: the Zoladex Early Breast Cancer Research Association Study. J Clin Oncol. (2002) 20:4628–35. doi: 10.1200/JCO.2002.05.042
33. International Breast Cancer Study Group, Castiglione-Gertsch M, O'Neill A, Price KN, Goldhirsch A, Coates AS, et al. (IBCSG)Adjuvant chemotherapy followed by goserelin versus either modality alone for premenopausal lymph node-negative breast cancer: a randomized trial. J Natl Cancer Inst. (2003) 95:1833–46. doi: 10.1093/jnci/djg119
34. Kim HJ, Yoon TI, Chae HD, Kim JE, Chae EY, Yu JH, et al. Concurrent gonadotropin-releasing hormone agonist administration with chemotherapy improves neoadjuvant chemotherapy responses in young premenopausal breast cancer patients. J Breast Cancer. (2015) 18:365–70. doi: 10.4048/jbc.2015.18.4.365
35. Karlsson P, Sun Z, Braun D, Price KN, Castiglione-Gertsch M, Rabaglio M, et al. Long-term results of International Breast Cancer Study Group Trial VIII: adjuvant chemotherapy plus goserelin compared with either therapy alone for premenopausal patients with node-negative breast cancer. Ann Oncol. (2011) 22:2216–26. doi: 10.1093/annonc/mdq735
36. de Haes H, Olschewski M, Kaufmann M, Schumacher M, Jonat W, Sauerbrei W, et al. Quality of life in goserelin-treated versus cyclophosphamide + methotrexate + fluorouracil-treated premenopausal and perimenopausal patients with node-positive, early breast cancer: the Zoladex Early Breast Cancer Research Association Trialists Group. J Clin Oncol. (2003) 21:4510–6. doi: 10.1200/JCO.2003.11.064
37. Moore HC, Unger JM, Phillips KA, Boyle F, Hitre E, Porter D, et al. Goserelin for ovarian protection during breast-cancer adjuvant chemotherapy. N Engl J Med. (2015) 372:923–32. doi: 10.1056/NEJMoa1413204
38. Dowsett M, Stein RC, Coombes RC. Aromatization inhibition alone or in combination with GnRH agonists for the treatment of premenopausal breast cancer patients. J Steroid Biochem Mol Biol. (1992) 43:155–9. doi: 10.1016/0960-0760(92)90201-S
39. Boccardo F, Rubagotti A, Perrotta A, Amoroso D, Balestrero M, De Matteis A, et al. Ovarian ablation versus goserelin with or without tamoxifen in pre-perimenopausal patients with advanced breast cancer: results of a multicentric Italian study. Ann Oncol. (1994) 5:337–42. doi: 10.1093/oxfordjournals.annonc.a058837
40. Jonat W, Kaufmann M, Blamey RW, Howell A, Collins JP, Coates A, et al. A randomised study to compare the effect of the luteinising hormone releasing hormone (LHRH) analogue goserelin with or without tamoxifen in pre-and perimenopausal patients with advanced breast cancer. Eur J Cancer. (1995) 31:137–42. doi: 10.1016/0959-8049(94)00415-2
41. Kaufmann M, Jonat W, Kleeberg U, Eiermann W, Jänicke F, Hilfrich J, et al. Goserelin, a depot gonadotrophin-releasing hormone agonist in the treatment of premenopausal patients with metastatic breast cancer. German Zoladex Trial Group J Cli Oncol. (1989) 7:1113–9. doi: 10.1200/JCO.1989.7.8.1113
42. Recchia F, Necozione S, Bratta M, Rosselli M, Guerriero G, Rea S. LH-RH analogues in the treatment of young women with early breast cancer: long term follow-up of a phase II study. Int J Oncol. (2015) 46:1354–60. doi: 10.3892/ijo.2014.2811
43. Kim HA, Ahn SH, Nam SJ, Park S, Ro J, Im SA, et al. The role of the addition of ovarian suppression to tamoxifen in young women with hormone-sensitive breast cancer who remain premenopausal or regain menstruation after chemotherapy (ASTRRA): a study protocol for a randomized controlled trial and progress. BMC Cancer. (2016) 16:319. doi: 10.1186/s12885-016-2354-6
44. Loibl S, Turner NC, Ro J, Cristofanilli M, Iwata H, Im SA, et al. Palbociclib combined with fulvestrant in premenopausal women with advanced breast cancer and prior progression on endocrine therapy: PALOMA-3 results. Oncologist. (2017) 22:1028–38. doi: 10.1634/theoncologist.2017-0072
45. Howell A, Ashcroft L, Fallowfield L, Eccles DM, Eeles RA, Ward A, et al. RAZOR: a phase II open randomized trial of screening plus goserelin and raloxifene versus screening aline in premenopausal women at increased risk of breast cancer. Cancer Epidemiol Biomarkers Prev. (2018) 27:58–66. doi: 10.1158/1055-9965.EPI-17-0158
46. Kim JY, Im SA, Jung KH, Ro J, Sohn J, Kim JH, et al. Fulvestrant plus goserelin versus anastrozole plus goserelin versus goserelin alone for hormone receptor-positive, HER2-negative tamoxifen-pretreated premenopausal women with recurrent or metastatic breast cancer (KCSG BR10-04): a multicenter, open-label, three-arm, randomised phase II trial (FLAG study). Eur J Cancer. (2018) 103:127–36. doi: 10.1016/j.ejca.2018.08.004
47. Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med. (2019) 381:307–16. doi: 10.1056/NEJMoa1903765
48. Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. (2018) 19:904–15. doi: 10.1016/S1470-2045(18)30292-4
49. Aguilar-Rojas A, Huerta-Reyes M, Maya-Núñez G, Arechavaleta-Velásco F, Conn PM, Ulloa-Aguirre A, et al. Gonadotropin-releasing hormone receptor activates GTPase RhoA and inhibits cell invasion in the breast cancer cell line MDA-MB-231. BMC Cancer. (2012) 12:550. doi: 10.1186/1471-2407-12-550
50. Baumann KH, Kiesel L, Kaufmann M, Bastert G, Runnebaum B. Characterization of binding sites for a GnRH-agonist (Buserelin) in human breast cancer biopsies and their distribution in relation to tumor parameters. Breast Cancer Res Treat. (1993) 25:37–46. doi: 10.1007/BF00662399
51. Klijn JG, Beex LV, Mauriac L, van Zijl JA, Veyret C, Wildiers J, et al. Combined treatment with buserelin and tamoxifen in premenopausal metastatic breast cancer: a randomized study. J Natl Cancer Inst. (2000) 92:903–11. doi: 10.1093/jnci/92.11.903
52. Di Lauro L, Pizzuti L, Barba M, Sergi D, Sperduti I, et al. Role of gonadotropin-releasing hormone analogues in metastatic male breast cancer: results from a pooled analysis. J Hematol Oncol. (2015) 8:53. doi: 10.1186/s13045-015-0147-z
53. Di Lauro L, Vici P, Barba M, Pizzuti L, Sergi D, Rinaldi M, et al. Antiandrogen therapy in metastatic male breast cancer: results from an updated analysis in an expanded case series. Breast Cancer Res Treat. (2014) 148:73–80. doi: 10.1007/s10549-014-3138-9
54. Klijn JG, de Jong FH, Blankestein MA, Docter R, Alexieva-Figusch J, Blonk-van der Wijst J, et al. Anti-tumor and endocrine effects of chronic LHRH agonist treatment (Buserelin) with or without tamoxifen in premenopausal metastatic breast cancer. Breast Cancer Res Treat. (1984) 4:209–20. doi: 10.1007/BF01806487
55. Klijn JG, de Jong FH, Lamberts SW, Blankestein MA. LHRH-agonist treatment in clinical and experimental human breast cancer. J Steroid Biochem. (1985) 23:867–73. doi: 10.1016/S0022-4731(85)80029-7
56. Falkson CI, Falkson HC, Falkson G. Effect of chemotherapy with or without buserelin on serum hormone levels in premenopausal women with breast cancer. Eur J Cancer. (1991) 27:1208–11. doi: 10.1016/0277-5379(91)90082-O
57. Falkson CI, Falkson HC, Falkson G. C yclophosphamide, doxorubicin and fluorouracil (CAF) plus depo-buserelion in the treatment of premenopausal women with metastatic breast cancer. Ann Oncol. (1992) 3:849–53. doi: 10.1093/oxfordjournals.annonc.a058109
58. Chen A, Yahalom D, Ben-Aroya N, Kaganovsky E, Okon E, Koch Y. A second isoform of gonadotropin-releasing hormone is present in the brain of human and rodents. FEBS Lett. (1998) 435:199–203. doi: 10.1016/S0014-5793(98)01064-3
59. Park DW, Choi KC, MacCalman CD, Leung PC. Gonadotropin-releasing hormone (GnRH)-I and GnRH-II induce cell growth inhibition in human endometrial cancer cells: involvement of integrin neta3 and focal adhesion kinase. Reprod Biol Endocrinol. (2009) 7:81. doi: 10.1186/1477-7827-7-81
60. Zhao LJ, Liu N, Li XP, Wang JL, Wei LH. Phosphatase and tensin homolog gene inhibits the effect induced by gonadotropin-releasing hormone subtypes in human endometrial carcinoma cells. Chin Med J. (2010) 123:1170–5. doi: 10.3760/cma.j.issn.0366-6999.2010.09.013
61. Gründker C, Schlotawa L, Viereck V, Eicke N, Horst A, Kairies B, et al. Antiproliferative effects of the GnRH antagonist cetrorelix and of GnRH-II on human endometrial and ovarian cancer cells are not mediated through the GnRH type I receptor. Eur J Endocrinol. (2004) 151:141–9. doi: 10.1530/eje.0.1510141
62. Szabó I, Bosze S, Orbán E, Sipos É, Halmos G, Kovács M, et al. Comparative in vitro biological evaluation of daunorubicin containing GnRH-I and GnRH-II conjugates developed for tumor targeting. J Pept Sci. (2015) 21:426–35. doi: 10.1002/psc.2775
63. Schubert A, Hawighorst T, Emons G, Gründker C. Agonists and antagonists of GnRH-I and -II reduce metastasis formation by triple-negative human breast cancer cells in vivo. Breast Cancer Res Treat. (2011) 130:783–90. doi: 10.1007/s10549-011-1358-9
64. Yano T, Korkut E, Pinski J, Szepeshazi K, Milovanovic S, Groot K, et al. Inhibition of growth of MCF-7 MIII human breast carcinoma in nude mice by treatment with agonists or antagonists of LH-RH. Breast Cancer Res Treat. (1992) 21:35–45. doi: 10.1007/BF01811962
65. Szepeshazi K, Lapis K, Schally AV. Effect of combination treatment with analogs of luteinizing hormone-releasing hormone (LH-RH) or somatostatin and 5-fluorouracil on pancreatic cancer hamsters. Int J Cancer. (1991) 49:260–6. doi: 10.1002/ijc.2910490220
66. Korkut E, Bokser L, Comaru-Schally AM, Groot K, Schally AV. Inhibition of growth of experimental prostate cancer with sustained delivery systems (microcapsules and microgranules) of luteinizing hormone-releasing hormone antagonist SB-75. Proc Natl Acad Sci USA. (1991) 88:844–8. doi: 10.1073/pnas.88.3.844
67. Segal-Abramson T, Kitroser H, Levy J, Schally AV, Sharoni Y. Direct effects of luteinizing hormone-releasing hormone agonists and antagonists on MCF-7 mammary cancer cells. Proc Natl Acad Sci USA. (1992) 89:2336–9. doi: 10.1073/pnas.89.6.2336
68. Reissmann T, Hilgard P, Harlema JH, Engel J, Comaru-Schally AM, Schally AV. Treatment of experimental DMBA induced mammary carcinoma with Cetrorelix (SB-75): a potent antagonist of luteinizing hormone-releasing hormone. J Can Res Clin Oncol. (1992) 118:44–9. doi: 10.1007/BF01192310
69. Gründker C, Günthert AR, Millar RP, Emons G. Expression of gonadotropin-releasing hormone II (GnRH-II) receptor in human endometrial and ovarian cancer cells and effects of GnRH-II on tumor cell proliferation. J Clin Endocrinol Metab. (2002) 87:1427–30. doi: 10.1210/jcem.87.3.8437
70. Verschraegen CF, Westphalen S, Hu W, Loyer E, Kudelka A, Völver P, et al. Phase II study of cetrorelix, a luteinizing hoirmone-releasing hormone antagonist in patients with platinum-resistant ovarian cancer. Gynecol Oncol. (2003) 90:552–9. doi: 10.1016/S0090-8258(03)00408-6
71. Ziegler CG, Ullrich M, Schally AV, Bergmann R, Pietzsch J, Gebaur L, et al. Anti-tumor effects of peptide analogs targeting neuropeptide hormone receptors on mouse pheochromocytoma cells. Mol Cell Endocrinol. (2013) 371:189–1994. doi: 10.1016/j.mce.2012.12.011
72. Park S, Han JM, Cheon J, Hwang JI, Seong JY. Apoptotic death of prostate cancer cells by a gonadotropin-releasing hormone-II antagonist. PLoS ONE. (2014) 9:e99723. doi: 10.1371/journal.pone.0099723
73. Kim DK, Yang JS, Maiti K, Hwang JI, Kim K, Seen D, et al. A gonadotropin-releasing hormone-II antagonist induces autophagy of prostate cancer cells. Cancer Res. (2009) 69:923–31. doi: 10.1158/0008-5472.CAN-08-2115
74. Fister S, Günthert AR, Aicher B, Paulini KW, Emons G, Gründker C. GnRH-II antagonists induce apoptosis in human endometrial, ovarian, and breast cancer cells via activation of stress-induced MAPKs p38 and JNK and proapoptotic protein Bax. Cancer Res. (2009) 69:6473–81. doi: 10.1158/0008-5472.CAN-08-4657
75. Gründker C, Föst C, Fister S, Nolte N, Günthert AR, Emons G. Gonadotropin-releasing hormone type II antagonist induces apoptosis in MCF-7 and triple-negative MDA-MB-231 human breast cancer cells in vitro and in vivo. Breast Cancer Res. (2010) 12:R49. doi: 10.1186/bcr2606
76. Seeburg PH, Adelman JP. Characterization of cDNA for precursor of human luteinizing hormone releasing hormone. Nature. (1984) 311:666–8. doi: 10.1038/311666a0
77. Lee VH, Lee LT, Chow BK. Gonadotropin-releasing hormone: regulation of the GnRH gene. FEBS J. (2008) 275:5458–78. doi: 10.1111/j.1742-4658.2008.06676.x
78. Tukun FL, Olberg DE, Riss PJ, Haraldsen I, Kaass A, Klaveness J. Recent development of non-peptide GnRH antagonists. Molecules. (2017) 22:E2188. doi: 10.3390/molecules22122188
79. Ikemoto T, Park MK. Molecular and evolutionary characterization of the GnRH-II gene in the chicken: distinctive genomic organization, expression pattern, and precursor sequence. Gene. (2006) 368:28–36. doi: 10.1016/j.gene.2005.10.004
80. Kaprara A, Huhtaniemi IT. The hypothalamus–pituitary–gonad axis: tales of mice and men. Metabolism. (2018) 86:3–17. doi: 10.1016/j.metabol.2017.11.018
81. Millar RP. GnRHs and GnRH receptors. Anim Reprod Sci. (2005) 88:5–28. doi: 10.1016/j.anireprosci.2005.05.032
82. Okada Y, Murota-Kawano A, Kakar SS, Winters SJ. Evidence that gonadotropin-releasing hormone (GnRH) II stimulates luteinizing hormone and follicle-stimulating hormone secretion from monkey pituitary cultures by activating the GnRH I receptor. Biol Reprod. (2003) 69:1356–61. doi: 10.1095/biolreprod.103.016162
83. Neill JD, Duck LW, Sellers JC, Musgrove LC. A gonadotropin-releasing hormone (GnRH) receptor specific for GnRH II in primates. Biochem Biophys Res Commun. (2001) 282:1012–8. doi: 10.1006/bbrc.2001.4678
84. Desaulniers AT, Cederberg RA, Lents CA, White BR. Expression and role of gonadotropin-releasing hormone 2 and its receptor in mammals. Front Endocrinol. (2017) 8:269. doi: 10.3389/fendo.2017.00269
85. Kakar SS, Musgrove LC, Devor DC, Sellers JC, Neill JD. Cloning, sequencing, and expression of human gonadotropin releasing hormone (GnRH) receptor. Biochem Biophys Res Commun. (1992) 189:289–95. doi: 10.1016/0006-291X(92)91556-6
86. Pawson AJ, Faccenda E, Maudsley S, Lu ZL, Naor Z, Millar RP. Mammalian type I gonadotropin-releasing hormone receptors undergo slow, constitutive, agonist-independent internalization. Endocrinology. (2007) 149:1415–22. doi: 10.1210/en.2007-1159
87. Finch AR, Caunt CJ, Armstrong SP, McArdle CA. Agonist-induced internalization and downregulation of gonadotropin-releasing hormone receptors. Am J Physiol Cell Physiol. (2009) 297:C591–600. doi: 10.1152/ajpcell.00166.2009
88. Flanagan CA, Manilall A. Gonadotropin-releasing hormone (GnRH) receptor structure and GnRH binding. Front Endocrinol. (2017) 8:274. doi: 10.3389/fendo.2017.00274
89. Arora KK, Chung HO, Catt KJ. Influence of a species-specific extracellular amino acid on expression and function of the human gonadotropin-releasing hormone receptor. Mol Endocrinol. (1999) 13:890–6. doi: 10.1210/mend.13.6.0291
90. Jardón-Valadez E, Aguilar-Rojas A, Maya-Núñez G, Leaños-Miranda A, Piñeiro Á, Conn PM, et al. Conformational effects of Lys191 in the human GnRH receptor: mutagenesis and molecular dynamics simulations studies. J Endocrinol. (2009) 201:297–307. doi: 10.1677/JOE-08-0527
91. Millar R, Conklin D, Lofton-Day C, Hutchinson E, Troskie B, Illing N, et al. A novel human GnRH receptor homolog gene: abundant and wide tissue distribution of the antisense transcript. J Endocrinol. (1999) 162:117–26. doi: 10.1677/joe.0.1620117
92. Morgan K, Conklin D, Pawson AJ, Sellar R, Ott TR, Millar RP. A transcriptionally active human type II gonadotropin-releasing hormone receptor gene homolog overlaps two genes in the antisense orientation on chromosome 1q.12. Endocrinology. (2003) 144:423–36. doi: 10.1210/en.2002-220622
93. Cheng CK, Leung PC. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr Rev. (2005) 26:283–306. doi: 10.1210/er.2003-0039
94. Faurholm B, Millar RP, Katz AA. The genes encoding the type II gonadotropin-releasing hormone receptor and the ribonucleoprotein RBM8A in humans overlap in two genomic loci. Genomics. (2001) 78:15–8. doi: 10.1006/geno.2001.6650
95. Neill JD. GnRH and GnRH receptor genes in the human genome. Endocrinology. (2002) 143:737–43. doi: 10.1210/endo.143.3.8705
96. Srkalovic G, Wittliff JL, Schally A. Detection and partial characterization of receptors for [D-Trp6]-luteinizing hormone-releasing hormone and epidermal growth factor in human endometrial carcinoma. Cancer Res. (1990) 50:1841–6.
97. Emons G, Ortmann O, Becker M, Irmer G, Springer B, Laun R, et al. High affinity binding and direct antiproliferative effects of LHRH analogues in human ovarian cancer cell lines. Cancer Res. (1993) 53:5439–46.
98. Gründker C, Emons G. Role of gonadotropin-releasing hormone (GnRH) in ovarian cancer. Reprod Biol Endocrinol. (2003) 1:65. doi: 10.1186/1477-7827-1-65
99. Tieva Å, Stattin P, Wikström P, Bergh A, Damber J. Gonadotropin-releasing hormone receptor expression in the human prostate. Prostate. (2001) 47:276–84. doi: 10.1002/pros.1072
100. Bahk JY, Hyun JS, Lee BH, Kim MO, Cho GJ, Choi WS. Expression of gonadotropin-releasing hormone (GnRH) and GnRH receptor mRNA in prostate cancer cells and effect of GnRH on the proliferation of prostate cancer cells. Urol Res. (1998) 26:259–64. doi: 10.1007/s002400050054
101. Limonta P, Moretti RM, Marelli MM, Dondi D, Parenti M, Motta M. The luteinizing hormone-releasing hormone receptor in human prostate cancer cells: messenger ribonucleic acid expression, molecular size, and signal transduction pathway. Endocrinology. (1999) 140:5250–6. doi: 10.1210/endo.140.11.7087
102. Eidne KA, Flanagan CA, Millar RP. Gonadotropin-releasing hormone binding sites in human breast carcinoma. Science. (1985) 229:989–91. doi: 10.1126/science.2992093
103. Kottler M, Starzec A, Carre M, Lagarde J, Martin A, Counis R. The genes for gonadotropin-releasing hormone and its receptor are expressed in human breast with fibrocystic disease and cancer. Int J Cancer. (1997) 71:595–9. doi: 10.1002/(SICI)1097-0215(19970516)71:4<595::AID-IJC14>3.0.CO;2-B
104. Pazaitou-Panayiotou K, Chemonidou C, Poupi A, Koureta M, Kaprara A, Lambropoulou M, et al. Gonadotropin-releasing hormone neuropeptides and receptor in human breast cancer: correlation to poor prognosis parameters. Peptides. (2013) 42:15–24. doi: 10.1016/j.peptides.2012.12.016
105. Moriya T, Suzuki T, Pilichowska M, Ariga N, Kimura N, Ouchi N, et al. Immunohistochemical expression of gonadotropin releasing hormone receptor in human breast carcinoma. Pathol Int. (2001) 51:333–7. doi: 10.1046/j.1440-1827.2001.01210.x
106. Fekete M, Wittliff JL, Schally AV. Characteristics and distribution of receptors for [d-trp6]-luteinizing hormone-releasing hormone, somatostatin, epidermal growth factor, and sex steroids in 500 biopsy samples of human breast cancer. J Clin Lab Anal. (1989) 3:137–47. doi: 10.1002/jcla.1860030302
107. Buchholz S, Seitz S, Schally AV, Engel JB, Rick FG, Szalontay L, et al. Triple-negative breast cancers express receptors for luteinizing hormone-releasing hormone (LHRH) and respond to LHRH antagonist cetrorelix with growth inhibition. Int J Oncol. (2009) 35:789–96. doi: 10.3892/ijo_00000391
108. Köster F, Engel JB, Schally AV, Hönig A, Schröer A, Seitz S, et al. Triple-negative breast cancers express receptors for growth hormone-releasing hormone (GHRH) and respond to GHRH antagonists with growth inhibition. Breast Cancer Res Treat. (2009) 116:273–9. doi: 10.1007/s10549-008-0120-4
109. Föst C, Duwe F, Hellriegel M, Schweyer S, Emons G, Gründker C. Targeted chemotherapy for triple-negative breast cancers via LHRH receptor. Oncol Rep. (2011) 25:1481–7. doi: 10.3892/or.2011.1188
110. Engel J, Emons G, Pinski J, Schally AV. AEZS-108: A targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin Investig Drugs. (2012) 21:891–9. doi: 10.1517/13543784.2012.685128
111. Eidne KA, Flanagan CA, Harris NS, Millar RP. Gonadotropin-releasing hormone (GnRH)-binding sites in human breast cancer cell lines and inhibitory effects of GnRH antagonists. J Clin Endocrinol Metab. (1987) 64:425–32. doi: 10.1210/jcem-64-3-425
112. Finch AR, Sedgley KR, Caunt CJ, McArdle CA. Plasma membrane expression of GnRH receptors: regulation by antagonists in breast, prostate, and gonadotrope cell lines. J Endocrinol. (2008) 196:353–67. doi: 10.1677/JOE-07-0471
113. Cheung LW, Wong AS. Gonadotropin-releasing hormone: GnRH receptor signaling in extrapituitary tissues. FEBS J. (2008) 275:5479–95. doi: 10.1111/j.1742-4658.2008.06677.x
114. Miller WR, Scott WN, Morris R, Fraser HM, Sharpe RM. Growth of human breast cancer cells inhibited by a luteinizing hormone-releasing hormone agonist. Nature. (1985) 313:231–3. doi: 10.1038/313231a0
115. Chatzaki E, Bax CM, Eidne KA, Anderson L, Grudzinskas JG, Gallagher CJ. The expression of gonadotropin-releasing hormone and its receptor in endometrial cancer, and its relevance as an autocrine growth factor. Cancer Res. (1996) 56:2059–65.
116. Emons G, Ortmann O, Schulz KD, Schally AV. Growth-inhibitory actions of analogues of luteinizing hormone releasing hormone on tumor cells. Trends Endocrinol Metab. (1997) 8:355–62. doi: 10.1016/S1043-2760(97)00155-0
117. Everest HM, Hislop JN, Harding T, Uney JB, Flynn A, Millar RP, et al. Signaling and antiproliferative effects mediated by GnRH receptors after expression in breast cancer cells using recombinant adenovirus. Endocrinology. (2001) 142:4663–72. doi: 10.1210/endo.142.11.8503
118. Finch AR, Green L, Hislop JN, Kelly E, McArdle CA. Signaling and antiproliferative effects of type I and II gonadotropin-releasing hormone receptors in breast cancer cells. J Clin Endocrinol Metab. (2004) 89:1823–32. doi: 10.1210/jc.2003-030787
119. Schally AV, Halmos G, Rekasi Z, Arencibia-Jimenez JM. The actions of luteinizing hormone-releasing hormone agonists, antagonists, and cytotoxic analogues on the luteinizing hormone-releasing hormone receptors on the pituitary and tumors. In: Devroey P, editor. Infertility and Reproductive Medicine Clinics of North America: GnRH Analogs. Philadelphia, PA: Saunders (2001). p. 17–44.
120. Limonta P, Moretti RM, Marelli MM, Motta M. The biology of gonadotropin hormone-releasing hormone: role in the control of tumor growth and progression in humans. Front Neuroendocrinol. (2003) 24:279–95. doi: 10.1016/j.yfrne.2003.10.003
121. Moretti RM, Marelli MM, Van Groeninghen JC, Motta M, Limonta P. Inhibitory activity of luteinizing hormone-releasing hormone on tumor growth and progression. Endocr Relat Cancer. (2003) 10:161–7. doi: 10.1677/erc.0.0100161
122. Aguilar-Rojas A, Pérez-Solis MA, Maya-Núñez G. The gonadotropin releasing hormone system: perspectives from reproduction to cancer. Int J Oncol. (2016) 48:861–8. doi: 10.3892/ijo.2016.3346
123. Gründker C, Emons G. The role of gonadotropin-releasing hormone in cancer cell proliferation and metastasis. Front Endocrinol. (2017) 8:187. doi: 10.3389/fendo.2017.00187
124. Aguilar-Rojas A, Maya-Núñez G, Huerta-Reyes M, Pérez-Solis MA, Silva-García R, Guillén N, et al. Activation of human gonadotropin-releasing hormone receptor promotes down regulation of ARHGAP18 and regulates the cell invasion of MDA-MB-231 cells. Mol Cell Endocrinol. (2018) 460:94–103. doi: 10.1016/j.mce.2017.07.009
125. Imai A, Takagi H, Horibe S, Fuseya T, Tamaya T. Coupling of gonadotropin-releasing hormone receptor to Gi protein in human reproductive tract tumors. J Clin Endocrinol Metab. (1996) 81:3249–53. doi: 10.1210/jcem.81.9.8784077
126. Scott WN, Mullen P, Miller WR. Factors influencing the response of MCF-7 cells to an agonist of luteinising hormone-releasing hormone. Eur J Cancer. (1991) 27:1458–61. doi: 10.1016/0277-5379(91)90031-8
127. Hershkovitz E, Marbach M, Bosin E, Levy J, Roberts CT Jr, Leroith D, et al. Luteinizing hormone-releasing hormone antagonists interfere with autocrine and paracrine growth stimulation of MCF-7 mammary cancer cells by insulin-like growth factors. J Clin Endocrinol Metab. (1993) 77:963–8. doi: 10.1210/jcem.77.4.8408472
128. Emons G, Ortmann O, Teichert H, Fassl H, Löhrs U, Kullander S, et al. Luteinizing hormone-releasing hormone agonist triptorelin in combination with cytotoxic chemotherapy in patients with advanced ovarian carcinoma: a prospective double blind randomized trial. Decapeptyl Ovarian Cancer Study Group. Cancer. (1996) 78:1452–60. doi: 10.1002/(SICI)1097-0142(19961001)78:7<1452::AID-CNCR12>3.0.CO;2-Z
129. Gründker C, Völker P, Emons G. Antiproliferative signaling of luteinizing hormone-releasing hormone in human endometrial and ovarian cancer cells through G proteinα I-mediated activation of phosphotyrosine phosphatase. Endocrinology. (2001) 142:2369–80. doi: 10.1210/endo.142.6.8190
130. Szepeshazi K, Milovanovic S, Lapis K, Groot K, Schally AV. Growth inhibition of estrogen independent MXT mouse mammary carcinomas in mice treated with an agonist or antagonist of LH-RH, an analog of somatostatin, or a combination. Breast Cancer Res Treat. (1992) 21:181–92. doi: 10.1007/BF01975001
131. Gründker C, Völker P, Schulz KD, Emons G. Luteinizing hormone–releasing hormone agonist triptorelin and antagonist cetrorelix inhibit EGF-induced c-fos expression in human gynecological cancers. Gynecol Oncol. (2000) 78:194–202. doi: 10.1006/gyno.2000.5863
132. Günthert AR, Gründker C, Olota A, Läsche J, Eicke N, Emons G. Analogs of GnRH-I and GnRH-II inhibit epidermal growth factor-induced signal transduction and resensitize resistant human breast cancer cells to 4OH-tamoxifen. Eur J Endocrinol. (2005) 153:613–25. doi: 10.1530/eje.1.01996
133. Gründker C, Bauerschmitz G, Schubert A, Emons G. Invasion and increased expression of S100A4 and CYR61 in mesenchymal transformed breast cancer cells is downregulated by GnRH. Int J Oncol. (2016) 48:2713–21. doi: 10.3892/ijo.2016.3491
134. Schally AV, Block NL, Rick FG. Discovery of LHRH and development of LHRH analogs for prostate cancer treatment. Prostate. (2017) 77:1036–54. doi: 10.1002/pros.23360
135. Magon N. Gonadotropin releasing hormone agonists: expanding vistas. Indian J Endocrinol Metab. (2011) 15:261–7. doi: 10.4103/2230-8210.85575
136. Karten MJ, Rivier JE. Gonadotropin-releasing hormone analog design. Structure–function studies toward the development of agonists and antagonists: Rationale and perspective. Endocr Rev. (1986) 7:44–66. doi: 10.1210/edrv-7-1-44
137. Jonat W. Goserelin (Zoladex)—Its role in early breast cancer in pre- and perimenopausal women. Br J Cancer. (2001) 85(Suppl. 2):1–5. doi: 10.1054/bjoc.2001.1981
138. Noguchi S, Kim HJ, Jesena A, Parmar V, Sato N, Wang HC, et al. Phase 3, open-label, randomized study comparing 3-monthly with monthly goserelin in pre-menopausal women with estrogen receptor-positive advanced breast cancer. Breast Cancer. (2016) 23:771–9. doi: 10.1007/s12282-015-0637-4
139. LHRH-agonists in Early Breast Cancer Overview Group, Cuzick J, Ambroisine L, Davidson N, Jakesz R, Kaufmann M, et al. Use of luteinising-hormone-releasing hormone agonists as adjuvant treatment in premenopausal patients with hormone-receptor-positive breast cancer: a meta-analysis of individual patient data from randomised adjuvant trials. Lancet. (2007) 369:1711–23. doi: 10.1016/S0140-6736(07)60778-8
140. Corona SP, Roviello G, Strina C, Milani M, Allevi G, Aguggini S, et al. Could gonadotropin-releasing hormone analogs be helpful in the treatment of triple-negative breast cancer. Future Oncol. (2017) 13:2473–7. doi: 10.2217/fon-2017-0272
142. Rossi L, Pagani O. Adjuvant endocrine therapy in breast cancer: evolving paradigms in premenopausal women. Curr Treat Options Oncol. (2017) 18:28. doi: 10.1007/s11864-017-0473-1
143. Blankenstein MA, Henkelman MS, Klijn JG. Direct inhibitory effect of a luteinizing hormone-releasing hormone agonist on MCF-7 human breast cancer cells. Eur J Cancer Clin Oncol. (1985) 21:1493–9. doi: 10.1016/0277-5379(85)90244-5
144. Kéri G, Balogh A, Szöke B, Teplán I, Csuka O. Gonadotropin-releasing hormone analogues inhibit cell proliferation and activate signal transduction pathways in MDA-MB-231 human breast cancer cell line. Tumour Biol. (1991) 12:61–7. doi: 10.1159/000217689
145. Schally AV, Coy DH, Arimura A. LH-RH agonists and antagonists. Int J Gynaecol Obstet. (1980) 18:318–24. doi: 10.1002/j.1879-3479.1980.tb00507.x
146. Herbst KL. Gonadotropin-releasing hormone antagonists. Curr Opin Pharmacol. (2003) 3:660–6. doi: 10.1016/j.coph.2003.06.009
147. Limonta P, Marelli Marelli M, Mai S, Motta M, Martini L, Moretti RM. GnRH receptors in cancer: from cell biology to novel targeted therapeutic strategies. Endocr Rev. (2012) 33:784–811. doi: 10.1210/er.2012-1014
148. Erchegyi J, Coy DH, Nekola MV, Coy EJ, Schally AV, Mezo I, et al. Luteinizing hormone-releasing hormone analogs with increased anti-ovulatory activity. Biochem Biophys Res Commun. (1981) 100:915–20. doi: 10.1016/0006-291X(81)91910-0
149. Reissmann T, Schally AV, Bouchard P, Riethmiiller H, Engel J. The LHRH antagonist cetrorelix: a review. Hum Reprod Update. (2000) 6:322–31. doi: 10.1093/humupd/6.4.322
150. Tan O, Bukulmez O. Biochemistry, molecular biology and cell biology of gonadotropin-releasing hormone antagonists. Curr Opin Obstet Gynecol. (2011) 23:238–44. doi: 10.1097/GCO.0b013e328348a3ce
151. Maiti K, Oh DY, Moon JS, Acharjee S, Li JH, Bai DG, et al. Differential effects of gonadotropin-releasing hormone (GnRH)-I and GnRH-II on prostate cancer cell signaling and death. J Clin Endocrinol Metab. (2005) 90:4287–98. doi: 10.1210/jc.2004-1894
152. Millar RP, Zhu YF, Struthers RS. Progress towards the development of non-peptide orally-active gonadotropin-releasing hormone (GnRH) antagonists: therapeutic implications. Br Med Bull. (2000) 56:761–72. doi: 10.1258/0007142001903346
153. Millar RP, Newton CL. Current and future applications of GnRH, kisspeptin and neurokinin B analogues. Nat Rev Endocrinol. (2013) 9:451–66. doi: 10.1038/nrendo.2013.120
154. Anderes KL, Luthin DR, Castillo R, Kraynov EA, Castro M, Nared-Hood K, et al. Biological characterization of a novel, orally active small molecule gonadotropin-releasing hormone (GnRH) antagonist using castrated and intact rats. J Pharmacol Exp Ther. (2003) 305:688–95. doi: 10.1124/jpet.102.046656
155. Armer RE, Smelt KH. Non-peptidic GnRH receptor antagonists. Curr Med Chem. (2004) 11:3017–28. doi: 10.2174/0929867043363983
156. Randolph JT, Waid P, Nichols C, Sauer D, Haviv F, Diaz G, et al. Nonpeptide luteinizing hormone-releasing hormone antagonists derived from erythromycin A: design, synthesis, and biological activity of cladinose replacement analogues. J Med Chem. (2004) 47:1085–97. doi: 10.1021/jm030418i
157. Sarma PKS, Tiwari A, Kondaskar A, Cliffe IA. Peptidomimetic GnRH receptor antagonists for the treatment of reproductive and proliferative diseases. Expert Opinin Ther Pat. (2006) 16:733–51. doi: 10.1517/13543776.16.6.733
158. Trachtenberg J, Pont A. Ketoconazole therapy for advanced prostate cancer. Lancet. (1984) 324:433–5. doi: 10.1016/S0140-6736(84)92909-X
159. De B, Plattner JJ, Bush EN, Jae HS, Diaz G, Johnson ES, et al. LH-RH antagonists: design and synthesis of a novel series of peptidomimetics. J Med Chem. (1989) 32:2036–8. doi: 10.1021/jm00129a003
160. Cho N, Harada M, Imaeda T, Imada T, Matsumoto H, Hayase Y, et al. Discovery of a novel, potent, and orally active non-peptide antagonist of the human luteinizing hormone-releasing hormone (LHRH) receptor. J Med Chem. (1998) 41:4190–5. doi: 10.1021/jm9803673
161. Hara T, Araki H, Kusaka M, Harada M, Cho N, Suzuki N, et al. Suppression of a pituitary-ovarian axis by chronic oral administration of a novel non-peptide gonadotropin-releasing hormone antagonist, TAK-013, in cynomolgus monkeys. J Clin Endocrinol Metab. (2003) 88:1697–704. doi: 10.1210/jc.2002-021065
162. Miwa K, Hitaka T, Imada T, Sasaki S, Yoshimatsu M, Kusaka M, et al. Discovery of 1-{4-[1-(2, 6-difluorobenzyl)-5-[(dimethylamino) methyl]-3-(6-methoxypyridazin-3-yl)-2, 4-dioxo-1, 2, 3, 4-tetrahydrothieno [2, 3-d] pyrimidin-6-yl] phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor. J Med Chem. (2011) 54:4998–5012. doi: 10.1021/jm200216q
163. Nakata D, Masaki T, Tanaka A, Yoshimatsu M, Akinaga Y, Asada M, et al. Suppression of the hypothalamic–pituitary–gonadal axis by TAK-385 (relugolix), a novel, investigational, orally active, small molecule gonadotropin-releasing hormone (GnRH) antagonist: studies in human GnRH receptor knock-in mice. Eur J Pharmacol. (2014) 723:167–74. doi: 10.1016/j.ejphar.2013.12.001
164. Nagy SAV, Armatis P, Szepeshazi K, Halmos G, Kovacs M, Zarandi M, et al. Cytotoxic analogs of luteinizing hormone-releasing hormone containing doxorubicin or 2-pyrrolinodoxorubicin, a derivative 500–1,000 times more potent. Proc Natl Acad Sci USA. (1996) 93:7269–73. doi: 10.1073/pnas.93.14.7269
165. Szepeshazi K, Schally AV, Nagy A. Effective treatment of advanced estrogen-independent MXT mouse mammary cancers with targeted cytotoxic LH-RH analogs. Breast Cancer Res Treat. (1999) 56:267–76. doi: 10.1023/A:1006267327007
166. Schally AV, Nagy A. Chemotherapy targeted to cancers through tumoral hormone receptors. Trends Endocrinol Metab. (2004) 15:300–10. doi: 10.1016/j.tem.2004.07.002
167. Emons G, Sindermann H, Engel J, Schally AV, Gründker C. Luteinizing hormone-releasing hormone receptor-targeted chemotherapy using AN-152. Neuroendocrinology. (2009) 90:15–8. doi: 10.1159/000225410
168. Krebs LJ, Wang X, Pudavar HE, Bergey EJ, Schally AV, Nagy A, et al. Regulation of targeted chemotherapy with cytotoxic lutenizing hormone-releasing hormone analogue by epidermal growth factor. Cancer Res. (2000) 60:4194–9.
169. Kahán Z, Nagy A, Schally AV, Halmos G, Arencibia JM, Groot K. Administration of a targeted cytotoxic analog of luteinizing hormone-releasing hormone inhibits growth of estrogen-independent MDA-MB-231 human breast cancers in nude mice. Breast Cancer Res Treat. (2000) 59:255–62. doi: 10.1023/A:1006352401912
170. Seitz S, Buchholz S, Schally AV, Weber F, Klinkhammer-Schalke M, Inwald EC, et al. Triple negative breast cancers express receptors for LHRH and are potential therapeutic targets for cytotoxic LHRH-analogs, AEZS 108 and AEZS 125. BMC Cancer. (2014) 14:847. doi: 10.1186/1471-2407-14-847
171. Bajo AM, Schally AV, Halmos G, Nagy A. Targeted doxorubicin-containing luteinizing hormone-releasing hormone analogue AN-152 inhibits the growth of doxorubicin-resistant MX-1 human breast cancers. Clin Cancer Res. (2003) 9:3742–8.
172. Nagy A, Plonowski A, Schally AV. Stability of cytotoxic luteinizing hormone-releasing hormone conjugate (AN-152) containing doxorubicin 14-O-hemiglutarate in mouse and human serum in vitro: implications for the design of preclinical studies. Proc Natl Acad Sci USA. (2000) 97:829–34. doi: 10.1073/pnas.97.2.829
173. Emons G, Kaufmann M, Gorchev G, Tsekova V, Gründker C, Günthert AR, et al. Dose escalation and pharmacokinetic study of AEZS-108 (AN-152), an LHRH agonist linked to doxorubicin, in women with LHRH receptor-positive tumors. Gynecol Oncol. (2010) 119:457–61. doi: 10.1016/j.ygyno.2010.08.003
174. Emons G, Gorchev G, Sehouli J, Wimberger P, Stähle A, Hanker L, et al. Efficacy and safety of AEZS-108 (INN: zoptarelin doxorubicin acetate) an LHRH agonist linked to doxorubicin in women with platinum refractory or resistant ovarian cancer expressing LHRH receptors: a multicenter phase II trial of the ago-study group (AGO GYN 5). Gynecol Oncol. (2014) 133:427–32. doi: 10.1016/j.ygyno.2014.03.576
175. Orbán E, Mezo G, Schlage P, Csík G, Kulić Z, Ansorge P, et al. in vitro degradation and antitumor activity of oxime bond-linked daunorubicin–GnRH-III bioconjugates and DNA-binding properties of daunorubicin-amino acid metabolites. Amino Acids. (2011) 41:469–83. doi: 10.1007/s00726-010-0766-1
176. Schlage P, Mezo G, Orbán E, Bosze S, Manea M. Anthracycline-GnRH derivative bioconjugates with different linkages: synthesis, in vitro drug release and cytostatic effect. J Control Release. (2011) 156:170–8. doi: 10.1016/j.jconrel.2011.08.005
Keywords: gonadotropin-releasing hormone (GnRH), gonadotropin-releasing hormone receptor (GnRHR), breast cancer, breast cancer adjuvant therapy, GnRH analogs, GnRH agonist, GnRH antagonist
Citation: Huerta-Reyes M, Maya-Núñez G, Pérez-Solis MA, López-Muñoz E, Guillén N, Olivo-Marin J-C and Aguilar-Rojas A (2019) Treatment of Breast Cancer With Gonadotropin-Releasing Hormone Analogs. Front. Oncol. 9:943. doi: 10.3389/fonc.2019.00943
Received: 16 July 2019; Accepted: 09 September 2019;
Published: 01 October 2019.
Edited by:
Chun Hei Antonio Cheung, National Cheng Kung University, TaiwanReviewed by:
Barbara Pasculli, Casa Sollievo della Sofferenza (IRCCS), ItalyGiovanni Porta, University of Insubria, Italy
Copyright © 2019 Huerta-Reyes, Maya-Núñez, Pérez-Solis, López-Muñoz, Guillén, Olivo-Marin and Aguilar-Rojas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arturo Aguilar-Rojas, YV9hZ3VpbGFyQHVuYW0ubXg=