 Sergi Cuartero1,2,3*
Sergi Cuartero1,2,3* Andrew J. Innes
Andrew J. Innes Matthias Merkenschlager
Matthias Merkenschlager- 1Faculty of Medicine, MRC London Institute of Medical Sciences, Institute of Clinical Sciences, Imperial College London, London, United Kingdom
- 2Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Barcelona, Spain
- 3Josep Carreras Leukaemia Research Institute (IJC), Barcelona, Spain
- 4Faculty of Medicine, Centre for Haematology, Imperial College London, London, United Kingdom
Classical driver mutations in acute myeloid leukemia (AML) typically affect regulators of cell proliferation, differentiation, and survival. The selective advantage of increased proliferation, improved survival, and reduced differentiation on leukemia progression is immediately obvious. Recent large-scale sequencing efforts have uncovered numerous novel AML-associated mutations. Interestingly, a substantial fraction of the most frequently mutated genes encode general regulators of transcription and chromatin state. Understanding the selective advantage conferred by these mutations remains a major challenge. A striking example are mutations in genes of the cohesin complex, a major regulator of three-dimensional genome organization. Several landmark studies have shown that cohesin mutations perturb the balance between self-renewal and differentiation of hematopoietic stem and progenitor cells (HSPC). Emerging data now begin to uncover the molecular mechanisms that underpin this phenotype. Among these mechanisms is a role for cohesin in the control of inflammatory responses in HSPCs and myeloid cells. Inflammatory signals limit HSPC self-renewal and drive HSPC differentiation. Consistent with this, cohesin mutations promote resistance to inflammatory signals, and may provide a selective advantage for AML progression. In this review, we discuss recent progress in understanding cohesin mutations in AML, and speculate whether vulnerabilities associated with these mutations could be exploited therapeutically.
Introduction
Hematopoietic homeostasis requires tight regulation to ensure production of sufficient numbers of blood cells at all stages of differentiation. This is achieved by a complex network of signaling pathways and gene regulatory mechanisms that control cell proliferation, differentiation, and survival of hematopoietic stem and progenitor cells (HSPC) and their progeny. Skewing of this balance in favor of excessive differentiation results in stem cell depletion, exhaustion and eventually, inability to replenish mature blood cells. In contrast, uncontrolled self-renewal, increased survival, and failure to differentiate are hallmarks of leukemia.
The homeostatic balance between self-renewal and differentiation of HSPC is sensitive to a broad range of perturbations. Mutations that disrupt it not only provide classifiers of clinical disease, but also offer insights into the molecular control of self-renewal, differentiation, and cell proliferation. Many AML-associated mutations are clearly linked to one of these categories, such as constitutive activation of RAS proteins or FLT3, that drive uncontrolled proliferation (1–3), mutations that prevent cell cycle arrest and apoptosis such as TP53 (4), and mutations that hinder differentiation such as in the transcription factors RUNX1 or C/EBPα (5, 6). The clonal advantage conferred by such mutations is immediately obvious.
Recent large-scale sequencing studies have shown that the mutational landscape of AML is highly enriched for mutations in general transcriptional regulators and chromatin modifiers, which are found in ~70% of patients (7). Examples of this group include mutations in proteins involved in chromatin modifications (ASXL1, EZH2), DNA methylation (DNMT3A, TET2), or transcriptional splicing (SRSF2, U2AF1) (7–9). Although we understand the biological functions of many of these molecules and pathways in exquisite detail, their selective advantage for AML cells remains largely unknown (10). Understanding the link between these novel AML mutations and the molecular mechanisms of self-renewal, differentiation and cell survival is critical for understanding the pathophysiology of AML, and for the identification of new therapeutic approaches to cancer (Figure 1).
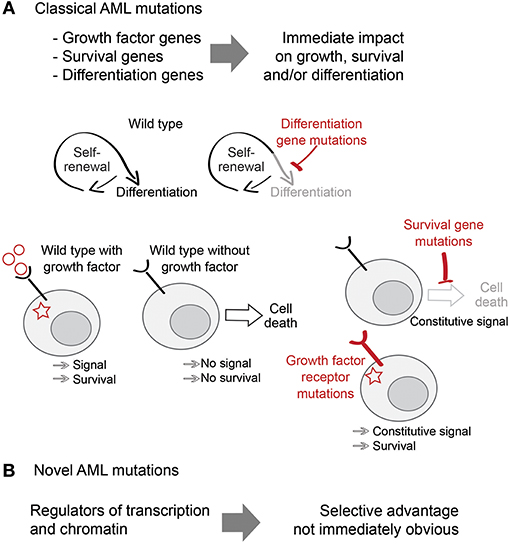
Figure 1. Classical and non-classical AML mutations. (A) Classical AML mutations deregulate proliferation, survival and differentiation pathways and provide an obvious selective advantage to AML. (B) Novel AML mutations include mutations in transcription and chromatin regulators and their selective advantage is less obvious.
A striking example are mutations in the subunits of the cohesin protein complex (SMC1, SMC3, RAD21, and STAG1/2). Cohesin forms a ring-shaped structure that can encircle DNA and hold sister chromatids together. This function of cohesin is essential for DNA replication (11–13), DNA repair (14–17), and chromosome segregation in mitosis (18–20). Despite this essential role in cell cycle progression, heterozygous or hypomorphic cohesin mutations are compatible with cell proliferation (21). This explains how leukemic cells can tolerate cohesin mutations, but fails to explain why cohesin mutations occur with high frequencies in AML.
In addition to essential functions in the cell cycle, cohesin has a major role in three-dimensional genome organization (22). Cohesin cooperates with the DNA-binding protein CTCF in the formation of topologically associated domains (TADs), which facilitate preferential interactions between genes and enhancers within the same CTCF-demarcated domain (23–26). Impaired formation of these structures randomizes the three-dimensional topology across single cells (27), thus exposing genes and enhancers to illegitimate interactions (28).
Here we review recent progress that links impaired cohesin function to the regulation of inflammatory gene expression, self-renewal, and differentiation of hematopoietic progenitors (29–34), revealing potential explanations for why cohesin is recurrently mutated in AML. We speculate about the role of inflammatory gene expression in AML and its potential therapeutic implications.
Cohesin Mutations in AML
Mutations in members of the cohesin complex are found in 6–13% of AML cases (7, 35–37). In addition to AML, cohesin mutations are also found in other myeloid malignancies, including Down syndrome acute megakaryoblastic leukemia (DS-AMKL), myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPN), chronic myelomonocytic leukemia (CMML), and chronic myelogenous leukemia (CML) (38, 39). Cohesin mutations are frequent in myeloid but not in lymphoid malignancies, suggesting that the pro-leukemic effect of reduced cohesin function may be connected to myeloid-specific traits.
The majority of RAD21 and STAG2 mutations cause non-sense, frame-shift, or splice-site changes, presumably leading to protein truncation or exon skipping. On the other hand, mutations in SMC1A and SMC3 are missense, causing amino acid substitutions in different protein domains (38). The effect of each of these mutations on the formation of the cohesin complex is still largely unexplored. Some of the mutant transcripts can give rise to dominant-negative proteins in cord blood progenitors (32) while others result in the degradation of the mutant transcript (38).
Most cohesin mutations are heterozygous, consistent with the idea that complete loss of the complex is incompatible with cell cycle progression. This has been confirmed by studies showing that partial cohesin loss in AML cells is not linked to increased aneuploidy (29, 36, 38, 40). However, since the Stag2 and Smc1a genes are on the X chromosome, male cases with mutations in these genes are not heterozygous. In the case of STAG2 mutant cells it has been shown that STAG1 becomes essential (41), suggesting that loss of STAG2 can be at least partially compensated by STAG1.
Cohesin mutations in patients appear to be mutually exclusive, indicating that a mutation in just one member of the complex is sufficient to reduce cohesin activity to the point where it provides a clonal advantage. Cohesin mutations often co-occur with mutations in other genes, such as NPM1, TET2, ASXL1, and EZH2 (36, 38). Nonetheless, it is thought that the majority of cohesin mutations are early events in leukemogenesis (38, 42). The prognostic significance of cohesin mutations in myeloid malignancies is not yet fully clear. In MDS, STAG2 mutations are associated with significantly reduced survival (37). However, no significant association between cohesin mutations and survival in AML was found in an early study (36) while a more recent study reports a significant association with increased overall survival and disease-free survival (43).
The Role of Cohesin Early Hematopoiesis
The frequency of cohesin mutations in AML prompted several groups to investigate the contribution of cohesin to early hematopoiesis and myeloid differentiation (Table 1). A mouse model of conditional Smc3 heterozygosity (31) presented an altered composition of the hematopoietic stem cell (HSC) compartment. Short-term HSCs and multipotent progenitor populations (MPP) were increased, while in long-term HSCs were decreased (31). In competitive repopulation assays, cohesin-deficient cells outcompeted wild-type cells. An important aspect of this study was the demonstration that Smc3 heterozygosity on its own is not sufficient to trigger leukemic transformation. However, the combination of Smc3 heterozygosity and an internal duplication in the FLT3 receptor (one of the most common mutations in AML) induced acute myeloid leukemia in mice. This indicates that SMC3 mutations must cooperate with other mutations to cause leukemia in this model. However, a mouse model of conditional Stag2 deletion presented features of myeloid dysplasia (44). Also, these mice had increased frequencies of both long-term and short-term HSC, indicating that mutations in different cohesin subunits do not always cause the same phenotypes.
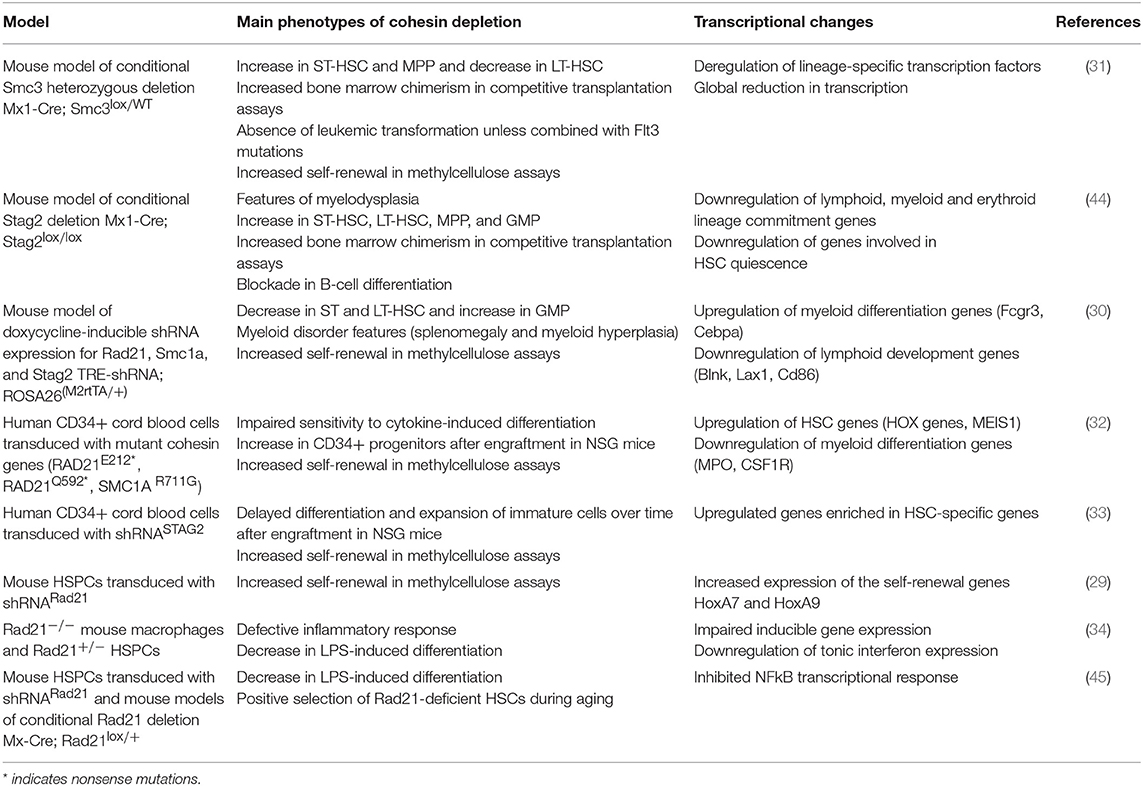
Table 1. Main phenotypes and transcriptional changes in models of cohesin depletion in hematopoiesis.
Mouse models of shRNA-mediated knock-down of different cohesin subunits developed similar, but not identical alterations in stem cell compartments (30). In the bone marrow there was a marked increase in granulocyte-macrophage progenitors (GMP), accompanied by a decrease in long-term and short-term HSCs. These models of cohesin deficiency did not develop acute myeloid leukemia. However, the mice displayed several features resembling a myeloid disorder, including splenomegaly and myeloid hyperplasia. In addition, cohesin-mutated mouse cells acquire increased self-renewal capacity in in vitro methylcellulose colony formation assays.
Importantly, similar results were obtained with human cells (32, 33). Cohesin-deficient cord blood progenitors or AML cell lines displayed reduced sensitivity to the differentiation-inducing effects of cytokines. The same effect was observed by over-expressing cohesin genes carrying mutations identified in AML, indicating that these can act as dominant-negative mutants. These cells were also characterized by a higher frequency of CD34+ progenitors and increased self-renewal capacity in methylcellulose (32). In line with these findings, cohesin-deficient human blood progenitors have increased in vivo reconstitution capacity after transplantation into immunodeficient mice (33).
Transcriptional Consequences of Cohesin Mutations in HSPCs
Given that AML-associated cohesin mutations do not affect genome integrity (38), the observed resistance to differentiation has been ascribed to the gene regulatory role of cohesin. In all models tested, transcriptional changes were mild (31, 32). This is expected, as even complete removal of cohesin only changes the expression of 10% of genes (23).
Previous reports suggested a role for cohesin in facilitating chromatin remodeling in human and mouse cells (46, 47). Consistent with this, cohesin-deficient HSPCs present genome-wide alterations in chromatin accessibility (30–33, 45). These changes broadly correlate with altered gene expression. Therefore, it has been proposed that defective chromatin accessibility impacts on normal dynamics of transcription factor binding, which leads to transcriptional deregulation and abnormal differentiation.
An extreme case of chromatin alterations was observed in human cord blood cells, where dominant negative cohesin mutations reduced chromatin accessibility genome-wide (32). Interestingly, a minority of sites displayed increased accessibility, specifically binding sites for the transcription factors GATA2 and RUNX1. This has been proposed to result in an upregulation of HSPC transcriptional programs and obstruct differentiation. A role for cohesin in regulating RUNX1 expression has also been described in model organisms (48).
How is cohesin linked to chromatin accessibility? Cohesin binding sites are highly accessible (49). In yeast, cohesin cooperates with the chromatin structure remodeling complex (RSC) to actively evict nucleosomes and generate nucleosome-free DNA (50–52), which is required for cohesin loading (53). In mouse embryonic stem cells, depletion of a member of the PBAF complex (a vertebrate ortholog of RSC) results in sister-chromatid cohesion defects (54). In humans, cohesin is found in a complex with the ATP-dependent chromatin remodeling enzyme SNF2H (55). SNF2H, but not cohesin, is required for the establishment of arrays of phased nucleosomes around CTCF binding sites (56).
Reduced cohesin dosage can also alter the frequency of chromatin interactions near transcription factor genes. One example is the transcriptional regulation of the lymphoid transcription factor Ebf1, that has four STAG2 binding sites in hematopoietic progenitors. In Stag2−/− mice, cis-interactions at this locus are lost, leading to abrogation of Ebf1 expression and failure to differentiate into lymphoid progenitors (44).
Another mechanism that has been proposed to explain the increased self-renewal capacity of cohesin-deficient HSPCs is the derepression of the self-renewal transcription factor HOXA9 (29, 57). HOXA9 is normally silenced by the Polycomb complex, which represses Hox loci in HSPCs by H3K27 trimethylation. In cohesin-deficient mouse HSPCs, this repressive chromatin mark is lost and Hoxa9 is upregulated, leading to increased self-renewal. This finding suggests that cohesin cooperates with Polycomb to silence Hox genes in HSPCs. Consistently, in mouse embryonic stem cells, cohesin complexes containing STAG2 (but not STAG1) contribute to the maintenance of chromatin interactions within Polycomb domains (58).
Cohesin in the Control of the Inflammatory Response
As discussed in the previous section, all studies that have compared wild-type and cohesin-deficient HSPCs found clear changes in chromatin accessibility and gene expression. These changes may indicate a direct effect of cohesin, or, alternatively, they may reflect the less mature state of cohesin-deficient progenitor populations. In order to rule out this possibility and determine what genes are directly controlled by cohesin in myeloid cells, a recent study used terminally differentiated macrophages to allow a like-for-like comparison of transcriptional and chromatin state between wild-type and cohesin-deficient cells (34). This strategy uncovered a key role for cohesin in the regulation of inflammatory gene expression. Consistent with a body of knowledge demonstrating that cohesin is required for interactions within topological domains (23–26), interactions between upstream key transcriptional regulators of the inflammatory response and their surrounding enhancers were decreased after acute cohesin depletion. As the organization of the inflammatory response is hierarchical, reduced levels of upstream regulators impact on the network, and deregulation spreads to the majority of inducible genes. Importantly, re-analysis of HSPC gene expression data showed that cohesin also controls inflammatory gene expression in progenitor cells (34).
Inflammatory signals not only mediate cross-talk between immune cells to coordinate the immune response, but also regulate the balance between HSPC self-renewal and differentiation. This function, known as emergency hematopoiesis, is normally activated during infection in order to regenerate mature myeloid cell populations (59). Several inflammatory cytokines and ligands, including interferons, are involved in the activation of emergency hematopoiesis. Type I interferon induces HSC exit from quiescence, entry into the cell cycle and differentiation. Importantly, chronic exposure to type I interferon is detrimental to HSCs (60, 61). Type II interferon—or IFNγ–also regulates HSC activity both in homeostasis and during infection (62). IFNγ acts on a subset of HSCs to induce myeloid differentiation by activating transcription factors like C/EBPβ (63). The interleukin IL-1 brings about myeloid differentiation through activation of a NF-κB-PU.1 axis (64). Activation of Toll-like receptor (TLR) signaling in HSPCs, which activates both the NF-κB and the interferon pathways, also promotes myeloid differentiation (65–69). As in the case of chronic interferon exposure, sustained TLR activation becomes detrimental and impairs the repopulating capacity of HSPCs.
As cohesin is required to induce expression of inflammatory response genes, cohesin-deficient HSPCs are less prone to differentiate in inflammatory conditions [(34, 45); Figure 2]. This acquired resistance to differentiation allows increased proliferation of immature progenitors, providing a possible explanation for some of the phenotypes displayed by cohesin-deficient mice. Therefore, cohesin mutations in AML illustrate a mechanistic connection between the control of transcriptional regulation and the responsiveness to differentiation-inducing stimuli in myeloid cells. The selective advantage of mutations in other transcriptional regulators may potentially be explained by similar mechanisms involving the control of inflammatory signaling.
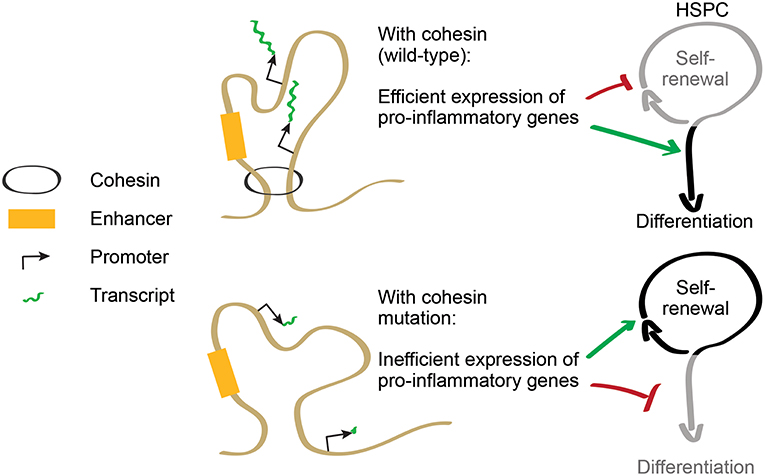
Figure 2. Cohesin regulates the balance between self-renewal and differentiation. Cohesin controls expression of pro-inflammatory genes that promote HSPC differentiation. In cohesin-mutant AML, inflammatory gene expression is downregulated, increasing resistance to differentiation and favoring HSPC self-renewal.
Inflammatory Gene Expression in AML
Consistent with the finding that cohesin regulates inflammatory gene expression in hematopoietic progenitors and myeloid cells, AML patient cells with cohesin mutations show a striking reduction of inflammatory and interferon pathways. This is the case when comparing AML with and without cohesin mutations across all samples in The Cancer Genome Atlas (TCGA), as well as within a specific histological subtype (34). These data suggest that the same mechanism that favors self-renewal in cohesin-deficient HSPCs through impaired sensitivity to inflammatory signals may operate in cohesin-deficient AMLs. The implication is that in settings with increased inflammatory signaling, cohesin mutations could confer resistance to inflammatory signals and increased self-renewal and clonal expansion.
Constitutively increased inflammatory signals are a hallmark of aging. Basal levels of pro-inflammatory cytokines such as IL6 or TNFα increase with age in healthy individuals (70). This leads to alterations in hematopoietic differentiation, which are reminiscent of emergency hematopoiesis: myelopoiesis-biased differentiation and reduced HSC self-renewal (71, 72). Consistent with its role in conferring resistance to inflammatory signals, competitive assays show that cohesin mutant HSCs become dominant over wild-type HSCs in aged mice (45). As clonal hematopoiesis is a feature of aging (73), it has been suggested that cohesin mutations could be positively selected during aging, eventually promoting a pre-leukemic state (45). This is in line with a report showing that cohesin mutations are early events, considered to be pre-leukemic (42). However, cohesin subunits are not among the top frequently mutated genes in cases where clones of hematopoietic cells carrying somatic mutations are found in the absence of any hematologic dysplasia, known as clonal hematopoiesis of indeterminate potential (CHIP) (73). Therefore, the emergence of cohesin mutations associated to aging may immediately lead to pre-leukemic dysplasias rather than CHIP. Further investigation is required to understand the role of cohesin mutations during aging.
A number of previous studies have reported altered expression of cytokines and other inflammatory mediators in myeloid disorders (74–76). For example, FLT3-ITD+ AML show increased expression of microRNA miR-155, which is known for its anti-inflammatory effects, its ability to inhibit interferon signaling, and to increase HSPC self-renewal in mouse models (77). Sensitivity of human AML cells to IFNγ is inversely related to RIP1/3 signaling. High levels of RIP1/3 signaling stabilize SOCS1, and SOCS1 antagonizes IFNγ signaling, effectively protecting AML cells from the differentiation-inducing effects of IFNγ (78). On the other hand, reduced RIPK3 expression is thought to reduce differentiation and TNFR-driven death of AML cells (79). Interleukin-1 (IL-1) inhibits growth of normal hematopoietic progenitors, but promotes expansion of AML cells by increasing p38 MAPK phosphorylation. This effect can be reversed by blocking IL-1 with p38 MAPK inhibitors (80).
As an illustration of the complexity of the pathways involved in the regulation of inflammation and differentiation, TNF activates NFκB via JNK in AML cells (81) and can dampen interferon signaling via SOCS1 (78). NFκB is constitutively activated in CD34+CD38− AML cells (82), promoting leukemia stem cell survival and proliferation (77). Although exogenous interferon reduces in vitro self-renewal induced by RUNX1-ETO and RUNX1-ETO9a, interferon and interferon-stimulated genes are elevated by RUNX1-ETO in human and in murine models (83). Finally, the chromatin modifier TET2 is required for emergency myelopoiesis (84), and TET2 mutants show greater fitness in inflammatory environments partially due to increased resistance to TNF, which triggers IL6 overproduction and activation of an anti-apoptotic lncRNA (85, 86). Taken together, these studies link myeloid disorders with inflammation and indicate that AML may use a spectrum of different strategies for managing inflammatory signals.
Interferon Treatment in AML
The interferon pathway is central to the inflammatory gene expression network, and it is heavily deregulated in cohesin-deficient macrophages (34). The deregulation of upstream interferon regulators like STAT1 and IRF7 disrupts basal interferon secretion, which maintains anti-viral transcriptional responsiveness by auto- and paracrine feed-forward signaling (87, 88). In the absence of cohesin, STAT and IRF-dependent enhancers fail to be induced, and consequently most interferon-induced genes are deregulated. Importantly, both enhancer activation and constitutive interferon gene expression can be partially rescued with exogenous interferon (34). These findings provide grounds to speculate that cohesin-mutated AMLs could be particularly vulnerable to interferon treatment. Mechanistically, supplying exogenous interferon could partially rescue expression of upstream transcription factors and regulators of the pathway, enabling normal enhancer activation, and transcription of downstream effectors. This would in turn increase the inflammatory responsiveness of cohesin-mutant AML cells, potentially restoring the balance between self-renewal and differentiation and restricting their selective advantage.
There is a long history of using type I interferons to treat hematological malignancies, with varying degrees of success. While early studies were hampered by treatment limiting side-effects, more recent recombinant and pegylated preparations are tolerated much better and can be used with feasible dosing regimens. There are currently established roles for interferon in the myeloproliferative disorders (89, 90), hypereosinophilic syndromes (91), and chronic myeloid leukemia (CML) (92). Intriguingly in CML, interferon treatment appears to preferentially target the leukemic stem cell population, and can induce cytogenetic remissions, some of which are durable upon treatment withdrawal, suggesting that it can cure some patients (92). Similar effects are observed in the JAK2 myeloproliferative disorders, with reduction or clearance of the mutant clones in up to 50% of patients (93).
In acute leukemia, interferon treatment impairs proliferation of AML cell lines in vitro, and has anti-leukemic effects in patient-derived xenograft models (PDX) in a dose dependent manner (94). This has been explained by cell intrinsic effects of interferon on leukemic blasts (reduced proliferation, increased apoptosis, and reduced secretion of growth-promoting cytokines), increased immunogenicity of interferon-treated leukemic blasts, as well as immunomodulatory effects on the residual normal hematopoietic cells, and increased clearance by the host immune system. However, despite the encouraging pre-clinical data, the clinical outcomes in interferon trials in AML have been disappointing, with durable responses seen in only small percentage of patients. While patients with secondary AML arising from a myeloproliferative disorder seem most susceptible, this is not exclusively the case. However, much of the clinical experience pre-dates the availability of current sequencing technologies and so stratification of AML by mutation may reveal genetic susceptibilities to interferon treatment.
Conclusions
Many recently identified AML mutations are in genes encoding regulators of transcription and chromatin state. Understanding how these mutations are beneficial to cancer cell fitness is a major challenge (10). Regulators of transcription and chromatin state usually regulate the expression of hundreds or thousands of genes, which complicates the task of pinpointing the target genes that are responsible for the increased fitness of mutated cells.
Mutations in subunits of the cohesin complex result in clear alterations in the hematopoietic stem cell compartment and in HSPC function (29–33, 45). However, the specificity of cohesin control on HSPC gene expression has been difficult to accommodate with current models of cohesin function.
The transcriptional control of inducible gene expression provides a possible explanation for the high frequency of cohesin mutations in myeloid malignancies. Inflammatory signaling promotes the differentiation of HSPCs toward a myeloid fate (59), and cohesin-deficient cells show increased resistance to these differentiation-inducing stimuli (34, 45). In bone marrow microenvironments with alterations in cytokine levels such as those found in aging (70), myelodysplastic syndrome (MDS) (95) or leukemia (75, 76), mutations that confer reduced responsiveness to differentiation-inducing signals are likely to be positively selected and clonally expanded (45). This is consistent with observations that cohesin mutations appear early in the history of AML (42), and that cohesin mutations by themselves alter the composition of the HSPC compartment but are insufficient to trigger AML (31).
AML has an inherently poor prognosis, and even with intensive chemotherapy or hematopoietic stem cell transplantation the risk of relapse remains high. AML is a highly heterogeneous disease by morphological, clinical, and genetic criteria, underlining the need for targeted approaches. For a subset of recurrent mutations, such as FLT3, specific inhibitors are in clinical use (2). For others, like cohesin mutations, greater understanding of the molecular circuitries involved in the increased fitness of mutated cells is necessary to find vulnerabilities and new therapeutic approaches.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was funded by Wellcome Investigator Award 099276/Z/12/Z (MM). AI was supported by an NIHR Clinical Lectureship, and acknowledges support from the NIHR and Imperial Biomedical Research Center (BRC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Saultz J, Garzon R. Acute myeloid leukemia: a concise review. J Clin Med. (2016) 5:33. doi: 10.3390/jcm5030033
2. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. (2019) 33:299–312. doi: 10.1038/s41375-018-0357-9
3. Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. (2012) 120:3397–406. doi: 10.1182/blood-2012-05-378596
4. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
5. Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-α (C/EBPα), in acute myeloid leukemia. Nat Genet. (2001) 27:263–70. doi: 10.1038/85820
6. Ito Y. Oncogenic potential of the RUNX gene family: ‘overview.’ Oncogene. (2004) 23:4198–208. doi: 10.1038/sj.onc.1207755
7. The Cancer Genome Atlas Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
8. Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. (2012) 26:934–42. doi: 10.1038/leu.2011.326
9. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. (2018) 562:526–31. doi: 10.1038/s41586-018-0623-z
10. Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. (2012) 12:599–612. doi: 10.1038/nrc3343
11. Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner K-P, Shirahige K, et al. Establishment of sister chromatid cohesion at the S. cerevisiae replication. Fork Mol Cell. (2006) 23:787–99. doi: 10.1016/j.molcel.2006.08.018
12. Terret M-E, Sherwood R, Rahman S, Qin J, Jallepalli PV. Cohesin acetylation speeds the replication fork. Nature. (2009) 462:231–4. doi: 10.1038/nature08550
13. Guillou E, Ibarra A, Coulon V, Casado-Vela J, Rico D, Casal I, et al. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. (2010) 24:2812–22. doi: 10.1101/gad.608210
14. Ünal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, et al. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. (2004) 16:991–1002. doi: 10.1016/j.molcel.2004.11.027
15. Ström L, Lindroos HB, Shirahige K, Sjögren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. (2004) 16:1003–15. doi: 10.1016/j.molcel.2004.11.026
16. McAleenan A, Clemente-Blanco A, Cordon-Preciado V, Sen N, Esteras M, Jarmuz A, et al. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature. (2012) 493:250–4. doi: 10.1038/nature11630
17. Ladstätter S, Tachibana-Konwalski K. A surveillance mechanism ensures repair of DNA lesions during zygotic reprogramming. Cell. (2016) 167:1774–1787.e13. doi: 10.1016/j.cell.2016.11.009
18. Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. (1997) 91:35–45.
19. Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. (1997) 91:47–57.
20. Haering CH, Löwe J, Hochwagen A, Nasmyth K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol Cell. (2002) 9:773–88. doi: 10.1016/S1097-2765(02)00515-4
21. Heidinger-Pauli JM, Mert O, Davenport C, Guacci V, Koshland D. Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr Biol. (2010) 20:957–63. doi: 10.1016/j.cub.2010.04.018
22. Merkenschlager M, Nora EP. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu Rev Genomics Hum Genet. (2016) 17:17–43. doi: 10.1146/annurev-genom-083115-022339
23. Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, et al. Cohesin loss eliminates all loop domains. Cell. (2017) 171:305–320.e24. doi: 10.1016/j.cell.2017.09.026
24. Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature. (2017) 551:51. doi: 10.1038/nature24281
25. Wutz G, Várnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. (2017) 36:3573–99. doi: 10.15252/embj.201798004
26. Haarhuis JHI, van der Weide RH, Blomen VA, Yáñez-Cuna JO, Amendola M, van Ruiten MS, et al. The cohesin release factor WAPL restricts chromatin loop extension. Cell. (2017) 169:693–707.e14. doi: 10.1016/j.cell.2017.04.013
27. Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science. (2018) 362:eaau1783. doi: 10.1126/science.aau1783
28. Seitan VC, Faure AJ, Zhan Y, McCord RP, Lajoie BR, Ing-Simmons E, et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. (2013) 23:2066–77. doi: 10.1101/gr.161620.113
29. Fisher JB, Peterson J, Reimer M, Stelloh C, Pulakanti K, Gerbec ZJ, et al. The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia. (2017) 31:712–9. doi: 10.1038/leu.2016.240
30. Mullenders J, Aranda-Orgilles B, Lhoumaud P, Keller M, Pae J, Wang K, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med. (2015) 212:1833–50. doi: 10.1084/jem.20151323
31. Viny AD, Ott CJ, Spitzer B, Rivas M, Meydan C, Papalexi E, et al. Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J Exp Med. (2015) 212:1819–32. doi: 10.1084/jem.20151317
32. Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, et al. Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell. (2015) 17:675–88. doi: 10.1016/j.stem.2015.09.017
33. Galeev R, Baudet A, Kumar P, Rundberg Nilsson A, Nilsson B, Soneji S, et al. Genome-wide RNAi screen identifies cohesin genes as modifiers of renewal and differentiation in human HSCs. Cell Rep. (2016) 14:2988–3000. doi: 10.1016/j.celrep.2016.02.082
34. Cuartero S, Weiss FD, Dharmalingam G, Guo Y, Ing-Simmons E, Masella S, et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat Immunol. (2018) 19:932–41. doi: 10.1038/s41590-018-0184-1
35. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. (2012) 150:264–78. doi: 10.1016/j.cell.2012.06.023
36. Thol F, Bollin R, Gehlhaar M, Walter C, Dugas M, Suchanek KJ, et al. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood. (2014) 123:914–20. doi: 10.1182/blood-2013-07-518746
37. Thota S, Viny AD, Makishima H, Spitzer B, Radivoyevitch T, Przychodzen B, et al. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood. (2014) 124:1790–8. doi: 10.1182/blood-2014-04-567057
38. Kon A, Shih L-Y, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. (2013) 45:1232–7. doi: 10.1038/ng.2731
39. Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A, et al. The landscape of somatic mutations in Down syndrome–related myeloid disorders. Nat Genet. (2013) 45:1293–9. doi: 10.1038/ng.2759
40. Balbás-Martínez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Márquez M, Vazquez M, et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet. (2013) 45:1464–69. doi: 10.1038/ng.2799
41. van der Lelij P, Lieb S, Jude J, Wutz G, Santos CP, Falkenberg K, et al. Synthetic lethality between the cohesin subunits STAG1 and STAG2 in diverse cancer contexts. Elife. (2017) 6:e26980. doi: 10.7554/eLife.26980
42. Corces-Zimmerman MR, Hong W-J, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. (2014) 111:2548–53. doi: 10.1073/pnas.1324297111
43. Tsai C-H, Hou H-A, Tang J-L, Kuo Y-Y, Chiu Y-C, Lin C-C, et al. Prognostic impacts and dynamic changes of cohesin complex gene mutations in de novo acute myeloid leukemia. Blood Cancer J. (2017) 7:663. doi: 10.1038/s41408-017-0022-y
44. Viny AD, Bowman RL, Liu Y, Lavallée V-P, Eisman SE, Xiao W, et al. Stag1 and Stag2 regulate cell fate decisions in hematopoiesis through non-redundant topological control. bioRxiv. (2019) 2019:581868. doi: 10.1101/581868
45. Chen Z, Amro EM, Becker F, Hölzer M, Rasa SMM, Njeru SN, et al. Cohesin-mediated NF-κB signaling limits hematopoietic stem cell self-renewal in aging and inflammation. J Exp Med. (2019) 216:152–75. doi: 10.1084/jem.20181505
46. Yan J, Enge M, Whitington T, Dave K, Liu J, Sur I, et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. (2013) 154:801–13. doi: 10.1016/j.cell.2013.07.034
47. Faure AJ, Schmidt D, Watt S, Schwalie PC, Wilson MD, Xu H, et al. Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res. (2012) 22:2163–75. doi: 10.1101/gr.136507.111
48. Horsfield JA, Anagnostou SH, Hu JK-H, Cho KHY, Geisler R, Lieschke G, et al. Cohesin-dependent regulation of Runx genes. Development. (2007) 134:2639–49. doi: 10.1242/dev.002485
49. Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. (2008) 132:422–33. doi: 10.1016/j.cell.2008.01.011
50. Baetz KK, Krogan NJ, Emili A, Greenblatt J, Hieter P. The ctf13-30/CTF13 genomic haploinsufficiency modifier screen identifies the yeast chromatin remodeling complex RSC, which is required for the establishment of sister chromatid cohesion. Mol Cell Biol. (2004) 24:1232–44. doi: 10.1128/mcb.24.3.1232-1244.2003
51. Huang J, Hsu J-M, Laurent BC. The RSC nucleosome-remodeling complex is required for Cohesin's association with chromosome arms. Mol Cell. (2004) 13:739–50. doi: 10.1016/s1097-2765(04)00103-0
52. Lopez-Serra L, Kelly G, Patel H, Stewart A, Uhlmann F. The Scc2–Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome-free regions. Nat Genet. (2014) 46:1147–51. doi: 10.1038/ng.3080
53. Muñoz S, Minamino M, Casas-Delucchi CS, Patel H, Uhlmann F. A role for chromatin remodeling in cohesin loading onto chromosomes. Mol Cell. (2019) 74:664–673.e5. doi: 10.1016/j.molcel.2019.02.027
54. Brownlee PM, Chambers AL, Cloney R, Bianchi A, Downs JA. BAF180 promotes cohesion and prevents genome instability and aneuploidy. Cell Rep. (2014) 6:973–81. doi: 10.1016/j.celrep.2014.02.012
55. Hakimi M-A, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Speicher DW, et al. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature. (2002) 418:994–8. doi: 10.1038/nature01024
56. Wiechens N, Singh V, Gkikopoulos T, Schofield P, Rocha S, Owen-Hughes T. The chromatin remodelling enzymes SNF2H and SNF2L position nucleosomes adjacent to CTCF and other transcription factors. PLOS Genet. (2016) 12:e1005940. doi: 10.1371/journal.pgen.1005940
57. Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. (2002) 99:121–9. doi: 10.1182/blood.v99.1.121
58. Cuadrado A, Giménez-Llorente D, Kojic A, Rodríguez-Corsino M, Cuartero Y, Martín-Serrano G, et al. Specific contributions of cohesin-SA1 and cohesin-SA2 to TADs and polycomb domains in embryonic stem cells. Cell Rep. (2019) 27:3500–3510.e4. doi: 10.1016/j.celrep.2019.05.078
59. Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. (2017) 130:1693–8. doi: 10.1182/blood-2017-06-780882
60. Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNα activates dormant haematopoietic stem cells in vivo. Nature. (2009) 458:904–8. doi: 10.1038/nature07815
61. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon–dependent exhaustion. Nat Med. (2009) 15:696–700. doi: 10.1038/nm.1973
62. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature. (2010) 465:793–7. doi: 10.1038/nature09135
63. Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, et al. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. (2016) 17:2584–95. doi: 10.1016/j.celrep.2016.11.031
64. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. (2016) 18:607–18. doi: 10.1038/ncb3346
65. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. (2006) 24:801–12. doi: 10.1016/j.immuni.2006.04.008
66. Zhao JL, Ma C, O'Connell RM, Mehta A, DiLoreto R, Heath JR, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell. (2014) 14:445–59. doi: 10.1016/j.stem.2014.01.007
67. Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. (2017) 21:225–240.e5. doi: 10.1016/j.stem.2017.06.013
68. Laouedj M, Tardif MR, Gil L, Raquil M-A, Lachhab A, Pelletier M, et al. S100A9 induces differentiation of acute myeloid leukemia cells through TLR4. Blood. (2017) 129:1980–90. doi: 10.1182/blood-2016-09-738005
69. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol. (2011) 186:5367–75. doi: 10.4049/jimmunol.1003438
70. Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front Immunol. (2016) 7:502. doi: 10.3389/fimmu.2016.00502
71. Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. (2000) 192:1273–80. doi: 10.1084/jem.192.9.1273
72. Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci. (2010) 107:5465–70. doi: 10.1073/pnas.1000834107
73. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. (2014) 371:2488–98. doi: 10.1056/NEJMoa1408617
74. Sanchez-Correa B, Bergua JM, Campos C, Gayoso I, Arcos MJ, Bañas H, et al. Cytokine profiles in acute myeloid leukemia patients at diagnosis: survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine. (2013) 61:885–91. doi: 10.1016/j.cyto.2012.12.023
75. Kornblau SM, McCue D, Singh N, Chen W, Estrov Z, Coombes KR. Recurrent expression signatures of cytokines and chemokines are present and are independently prognostic in acute myelogenous leukemia and myelodysplasia. Blood. (2010) 116:4251–61. doi: 10.1182/blood-2010-01-262071
76. Hemmati S, Haque T, Gritsman K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front Oncol. (2017) 7:265. doi: 10.3389/fonc.2017.00265
77. Wallace JA, Kagele DA, Eiring AM, Kim CN, Hu R, Runtsch MC, et al. miR-155 promotes FLT3-ITD–induced myeloproliferative disease through inhibition of the interferon response. Blood. (2017) 129:3074–86. doi: 10.1182/blood-2016-09-740209
78. Xin J, You D, Breslin P, Li J, Zhang J, Wei W, et al. Sensitizing acute myeloid leukemia cells to induced differentiation by inhibiting the RIP1/RIP3 pathway. Leukemia. (2017) 31:1154–65. doi: 10.1038/leu.2016.287
79. Höckendorf U, Yabal M, Jost PJ. RIPK3-dependent cell death and inflammasome activation in FLT3-ITD expressing LICs. Oncotarget. (2016) 7:57483–4. doi: 10.18632/oncotarget.11195
80. Carey A, Edwards DK, Eide CA, Newell L, Traer E, Medeiros BC, et al. Identification of interleukin-1 by functional screening as a key mediator of cellular expansion and disease progression in acute myeloid leukemia. Cell Rep. (2017) 18:3204–18. doi: 10.1016/j.celrep.2017.03.018
81. Volk A, Li J, Xin J, You D, Zhang J, Liu X, et al. Co-inhibition of NF-κB and JNK is synergistic in TNF-expressing human AML. J Exp Med. (2014) 211:1093–108. doi: 10.1084/jem.20130990
82. Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. (2001) 98:2301–7. doi: 10.1182/blood.v98.8.2301
83. DeKelver RC, Lewin B, Weng S, Yan M, Biggs J, Zhang D-E. RUNX1–ETO induces a type I interferon response which negatively effects t(8;21)-induced increased self-renewal and leukemia development. Leuk Lymphoma. (2014) 55:884–91. doi: 10.3109/10428194.2013.815351
84. Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature. (2018) 554:123–7. doi: 10.1038/nature25434
85. Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. (2018) 23:833–849.e5. doi: 10.1016/j.stem.2018.10.013
86. Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An inflammatory environment containing TNFα favors Tet2 -mutant clonal hematopoiesis. Exp Hematol. (2018) 59:60–5. doi: 10.1016/j.exphem.2017.11.002
87. Mostafavi S, Yoshida H, Moodley D, LeBoité H, Rothamel K, Raj T, et al. Parsing the interferon transcriptional network and its disease associations. Cell. (2016) 164:564–78. doi: 10.1016/J.CELL.2015.12.032
88. Gough DJ, Messina NL, Clarke CJP, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. (2012) 36:166–74. doi: 10.1016/j.immuni.2012.01.011
89. Reilly JT, McMullin MF, Beer PA, Butt N, Conneally E, Duncombe A, et al. Guideline for the diagnosis and management of myelofibrosis. Br J Haematol. (2012) 158:453–71. doi: 10.1111/j.1365-2141.2012.09179.x
90. McMullin MF, Harrison CN, Ali S, Cargo C, Chen F, Ewing J, et al. A guideline for the diagnosis and management of polycythaemia vera. A british society for haematology guideline. Br J Haematol. (2019) 184:176–91. doi: 10.1111/bjh.15648
91. Butt NM, Lambert J, Ali S, Beer PA, Cross NCP, Duncombe A, et al. Guideline for the investigation and management of eosinophilia. Br J Haematol. (2017) 176:553–72. doi: 10.1111/bjh.14488
92. Talpaz M, Hehlmann R, Quintás-Cardama A, Mercer J, Cortes J. Re-emergence of interferon-α in the treatment of chronic myeloid leukemia. Leukemia. (2013) 27:803–12. doi: 10.1038/leu.2012.313
93. Quintás-Cardama A, Kantarjian H, Manshouri T, Luthra R, Estrov Z, Pierce S, et al. Pegylated interferon Alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. (2009) 27:5418–24. doi: 10.1200/JCO.2009.23.6075
94. Benjamin R, Khwaja A, Singh N, McIntosh J, Meager A, Wadhwa M, et al. Continuous delivery of human type I interferons (/) has significant activity against acute myeloid leukemia cells in vitro and in a xenograft model. Blood. (2006) 109:1244–7. doi: 10.1182/blood-2006-02-002915
Keywords: cohesin, leukemia, interferon, inflammation, hematopoiesis, AML
Citation: Cuartero S, Innes AJ and Merkenschlager M (2019) Towards a Better Understanding of Cohesin Mutations in AML. Front. Oncol. 9:867. doi: 10.3389/fonc.2019.00867
Received: 29 March 2019; Accepted: 21 August 2019;
Published: 09 September 2019.
Edited by:
Cyrus Khandanpour, University Hospital Münster, GermanyReviewed by:
Jisha Antony, University of Otago, New ZealandManja Wobus, Dresden University of Technology, Germany
Copyright © 2019 Cuartero, Innes and Merkenschlager. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergi Cuartero, c2VyZ2kuY3VhcnRlcm9AY3JnLmV1; Matthias Merkenschlager, bWF0dGhpYXMubWVya2Vuc2NobGFnZXJAbG1zLm1yYy5hYy51aw==