
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Oncol. , 06 September 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00850
This article is part of the Research Topic Spectrum and clinical implications of chromatin structure and epigenetic changes in leukemia and lymphoma View all 8 articles
 Bettina Wingelhofer
Bettina Wingelhofer Tim C. P. Somervaille*
Tim C. P. Somervaille*Acute myeloid leukemia (AML) is a genetically heterogeneous malignancy for which treatment options have been largely limited to cytotoxic chemotherapy for the past four decades. Next-generation sequencing and other approaches have identified a spectrum of genomic and epigenomic alterations that contribute to AML initiation and maintenance. The key role of epigenetic modifiers and the reversibility of epigenetic changes have paved the way for evaluation of a new set of drug targets, and facilitated the design of novel candidate treatment strategies. More recently, seven new targeted therapies have been FDA-approved demonstrating successful implementation of the past decades' research. In this review, we will summarize the most recent advances in targeted therapeutics designed for a focused group of key epigenetic regulators in AML, outline their mechanism of action and their current status in clinical development. Furthermore, we will discuss promising new approaches for epigenetic targeted treatment in AML which are currently being tested in pre-clinical trials.
Epigenetic regulation of the state of a cell involves various dynamic and reversible post-translational modifications of DNA and histone proteins. These modifications in their totality regulate the accessibility of DNA for the transcription machinery, thereby determining which specific genomic loci are transcriptionally active or repressed (1). The best-researched chromatin modifications include lysine acetylation, lysine mono-, di-, or tri-methylation, and arginine methylation. In addition, DNA methylation is an important regulator of gene expression and other DNA-dependent processes.
Normal hematopoiesis is regulated by the cooperative action of various transcription factors and epigenetic modulators that drive cell type-specific transcriptional programs. Recent advances in next-generation sequencing-based approaches and global projects, such as the Encyclopedia of DNA Elements (ENCODE, 2003), The Cancer Genome Atlas (TCGA, 2006), the International Cancer Genome Consortium (ICGC, 2008), and the European Community initiative BLUEPRINT (2011) have been critical in defining the regulatory networks in different normal hematopoietic cell types as well as how they are deregulated in myeloid malignancies (2–4). The key role of epigenetic modifiers in diseases, such as leukemia and the reversibility of epigenetic changes create an opportunity for development of targeted therapies with significant implications for clinical prevention and treatment. Indeed, a plethora of preclinical and clinical studies covering several hematologic malignancies show that targeting these epigenetic regulators can restore normal epigenetic and transcriptional programs (5, 6). Acute myeloid leukemia (AML) represents a group of genetically heterogeneous malignant clonal disorders which share the common feature of a block to normal myeloid differentiation. Various genetic and epigenetic mechanisms regulating the pathophysiology of AML have been identified many of which cluster in particular categories of genes including those coding for signaling molecules (such as FLT3 and KIT), transcription factors (such as CEBPA and RUNX1), chromatin modifiers (such as MLL and ASXL1) or direct and indirect regulators of DNA methylation (such as DNMT3A, IDH1, IDH2, and TET2) (7, 8). Although the number of potential targets for novel therapeutics has expanded in the last decade, a major challenge in AML is the genetic heterogeneity; there remains a substantial lack of understanding as to how mutations and their associated aberrant patterns of epigenetic modification interact with one another to confer malignant transformation. Perhaps as a result, with some notable exceptions, certain clinical studies of candidate epigenetic therapies have yielded disappointing results.
Until recently, FDA-approved targeted therapies in myeloid malignancies were limited to all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) for treatment of acute promyelocytic leukemia (9) and the DNA hypomethylating agents decitabine and 5-azacitidine targeting DNA methyltransferases (DNMTs) for the treatment of myelodysplasia (10, 11). However, since 2017 seven new targeted therapies have been FDA-approved in AML. These are the mutant IDH1 inhibitor ivosidenib and the mutant IDH2 inhibitor enasidenib, for patients with relapsed or refractory AML with the appropriate mutation; the BCL2 inhibitor venetoclax in combination with azacitidine or decitabine or low-dose cytarabine for newly-diagnosed AML in the elderly; the smoothened receptor inhibitor glasdegib in combination with low-dose cytarabine for newly-diagnosed AML in the elderly; gemtuzumab ozagamicin for newly-diagnosed CD33+ AML alone or in combination with conventional chemotherapy; the multi-kinase inhibitor midostaurin for newly diagnosed FLT3-mutated AML in combination with conventional chemotherapy; and the FLT3, AXL, and ALK inhibitor gilteritinib for relapsed or refractory FLT3-mutated AML (12–19). IDH1 and IDH2 inhibitors are excellent examples of what are presumed to be epigenetic therapies, but with an indirect mechanism of action. Through blockade of production of the putative oncometabolite D-2-hydroxyglutarate, which is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases, such as the TET family of 5-methylcytosine hydroxylases, the Jumonji family of lysine demethylases and prolyl hydroxylases, they target altered transcriptional programs in AML caused by global changes in DNA methylation and histone modifications (12). As a general principle these compounds have only moderate activity as single agents (3, 20) and so research into their combinatorial use remains an intense and active area of interest. As an aside, it is worth noting that histone deacetylase (HDAC) inhibitors vorinostat and panobinostat are approved for use in cutaneous T-cell lymphoma and multiple myeloma, respectively, and that the oral HDAC inhibitor pracinostat is currently being tested in a phase 3 setting in combination with azacitidine in elderly patients with AML (NCT03151408).
While these new developments in FDA approval are welcome, there remains much to do to improve the outcome of patients with myeloid malignancies. In this perspective, we will discuss a discrete set of candidate epigenetic therapeutic targets currently under evaluation in AML: the lysine demethylase LSD1, the protein methyltransferases EZH2, DOT1L, and PRMT5, and the BET bromodomain proteins. We will describe the importance of these transcriptional activators and repressors in different AML subtypes as well as their targeting potential, possible limitations and potential toxicities. We will summarize their current status in clinical development. For detailed review of other equally important targets, such as DNMTs and HDACs the reader is referred to recent comprehensive reviews (20–22). Finally, we will discuss a number of novel epigenetic targets currently undergoing pre-clinical evaluation.
Histone methylation and demethylation are tightly regulated, dynamic processes that regulate transcriptional activation or repression depending on the location of the modification. Methylation is generated by specific histone methyltransferases (HMTs), such as MLL, DOT1L, and EZH2. As for other histone modifications, methylation can be reversed by two classes of demethylases (KDM): the larger Jumonji domain family and the smaller lysine-specific demethylase (LSD) family.
LSD1/KDM1A is a flavin-adenine dinucleotide (FAD)-dependent histone demethylase (23, 24) with activity vs. mono- and dimethyl-H3K4 and H3K9 marks as well as non-histone proteins, such as DNMT1 and TP53 (25). LSD1 is typically found as a component of repressive multi-subunit complexes, such as CoREST and NuRD (26–30). More recent studies have revealed that LSD1 also binds with high affinity to N-terminal sequences of SNAG domain transcription factor family members, an interaction facilitated by molecular mimicry of the histone H3 tail by the SNAG domain (31–33). Indeed physical association of LSD1 with the SNAG domain of GFI1 is essential for the activity of GFI1 as a transcription repressor (34). In keeping with these observations, LSD1 has a critical role in normal hematopoiesis as well as in hematological malignancies (25, 35). In MLL-rearranged AMLs, LSD1 is critical for maintenance of leukemic stem cell (LSC) potential by sustaining an oncogenic transcriptional program and blocking differentiation and apoptosis (36). Inhibition of LSD1 by tranylcypromine sensitizes AML cells to differentiation induction by all-trans-retinoic acid (ATRA) (35). An essential point is that inhibitors of LSD1 both inhibit the demethylase activity of the enzyme and block the physical interaction of LSD1 with GFI1, thus impairing enzymatic and scaffolding functions of the protein, and inactivating the transcription repressor activity of SNAG domain transcription factors (Figure 1A).
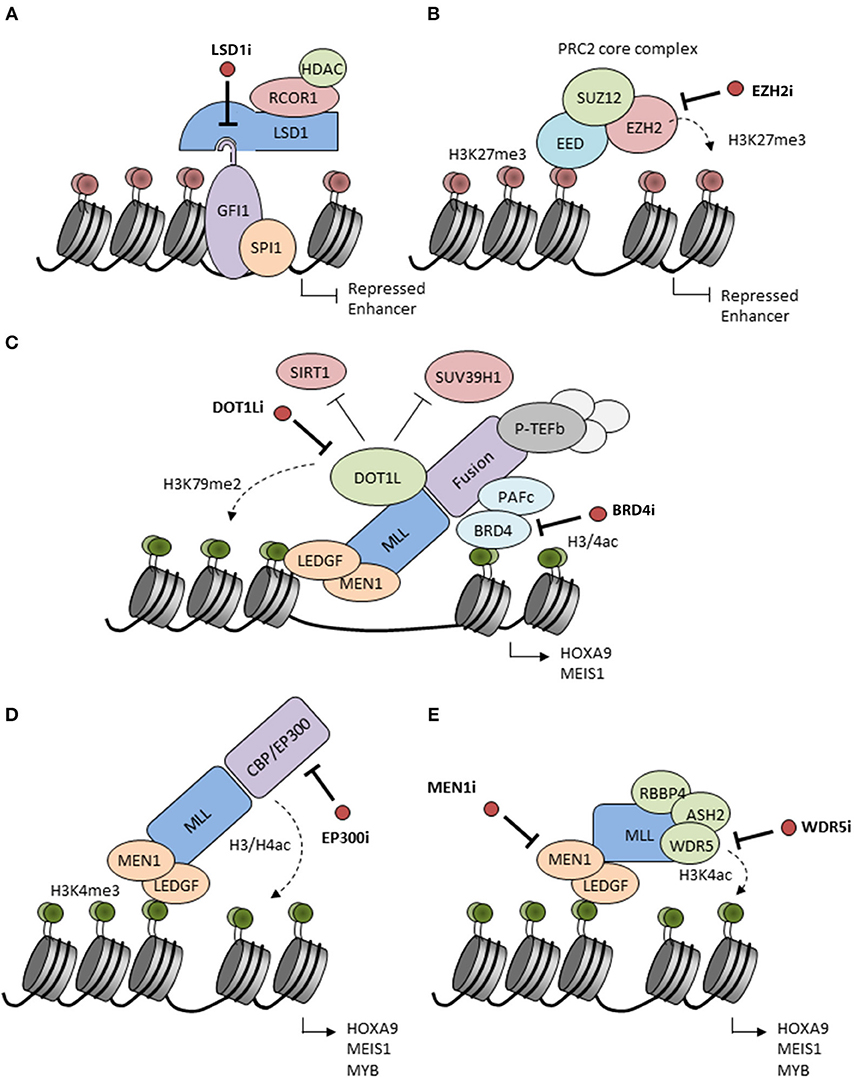
Figure 1. Putative mechanisms of action of candidate epigenetic inhibitors. (A) LSD1 interacts with the SNAG domain of GFI1 recruiting repressors to chromatin. Inhibitors of LSD1 (LSD1i) disrupt the interaction and inactivate GFI1 leading to enhancer acetylation and activation. LSD1 inhibitors also inactivate the histone demethylase activity of LSD1 (not shown). (B) EZH2 catalyzes H3K27 methylation inducing transcriptional repression. This activity is blocked by S-adenosyl-methionine (SAM)-competitive inhibitors of EZH2 (EZH2i). (C) MLL fusion proteins form complexes on chromatin with Polymerase Associated Factor complex (PAFc) (which recruits Super Elongation Complex components), Positive Transcription Elongation Factor b (pTEFb) and other factors to facilitate the expression of MLL-driven target genes, such as HOXA9 and MEIS1. DOT1L is ectopically recruited by MLL fusions and adds activating H3K79me2 marks while reducing H3K9me2 repressive marks by inhibition of SUV39H1 and SIRT1. BRD4 recognizes H3K27ac marks and is essential for recruitment and stabilization of the MLL complex on chromatin. Inhibitors of the enzymatic activities of DOT1L (DOT1Li) or BRD4 (BRD4i) are considered to disrupt the MLL fusion protein complexes leading to the release of the differentiation block. (D) MLL may be fused to the histone acetyltransferases CBP or EP300 which are associated with H3/H4 acetylation and active gene transcription. CBP/EP300 bromodomain inhibition (EP300i) decreases H3K27 acetylation and chromatin accessibility at target promoters and enhancers. (E) The N-terminal part of the MLL complex associates with different proteins, such as LEDGF and Menin which stabilize the complex on chromatin. Proteins, such as RBBP5, ASH2L, and WDR5 interact with the MLL C-terminus to facilitate SET domain-mediated H3K4 methylation. Inhibition of these interactions (MEN1, WDR5i) disrupt the MLL complex and decrease expression of HOXA9 and MEIS1.
In recent years, two tranylcypromine-derivative inhibitors, GSK2879552 (NCT02177812) and iadademstat (ORY-1001; EudraCT 2013-002447-29), have been evaluated in phase I trials in patients with relapsed or refractory AML (Table 1). While the former was terminated due to an unfavorable risk-benefit assessment, preliminary results are available from the latter: iadademstat was well tolerated and induced molecular and morphologic blast cell differentiation in patients harboring MLL gene translocations (37, 38). Preclinical data have suggested the possibility of synergistic effects of LSD1 inhibition with ATRA. As a result, tranylcypromine itself and derivatives, such as IMG-7289 are undergoing evaluation in trials in combination with ATRA (NCT02717884, NCT02273102, NCT02261779, NCT02842827, and EudraCT 2012-002154-23); results are awaited. Following on from phase 1, GSK2879552 and iadademstat are now being evaluated in combination with azacitidine in high risk myelodysplasia (NCT02929498) and AML, respectively (EudraCT 2018-000482-36).
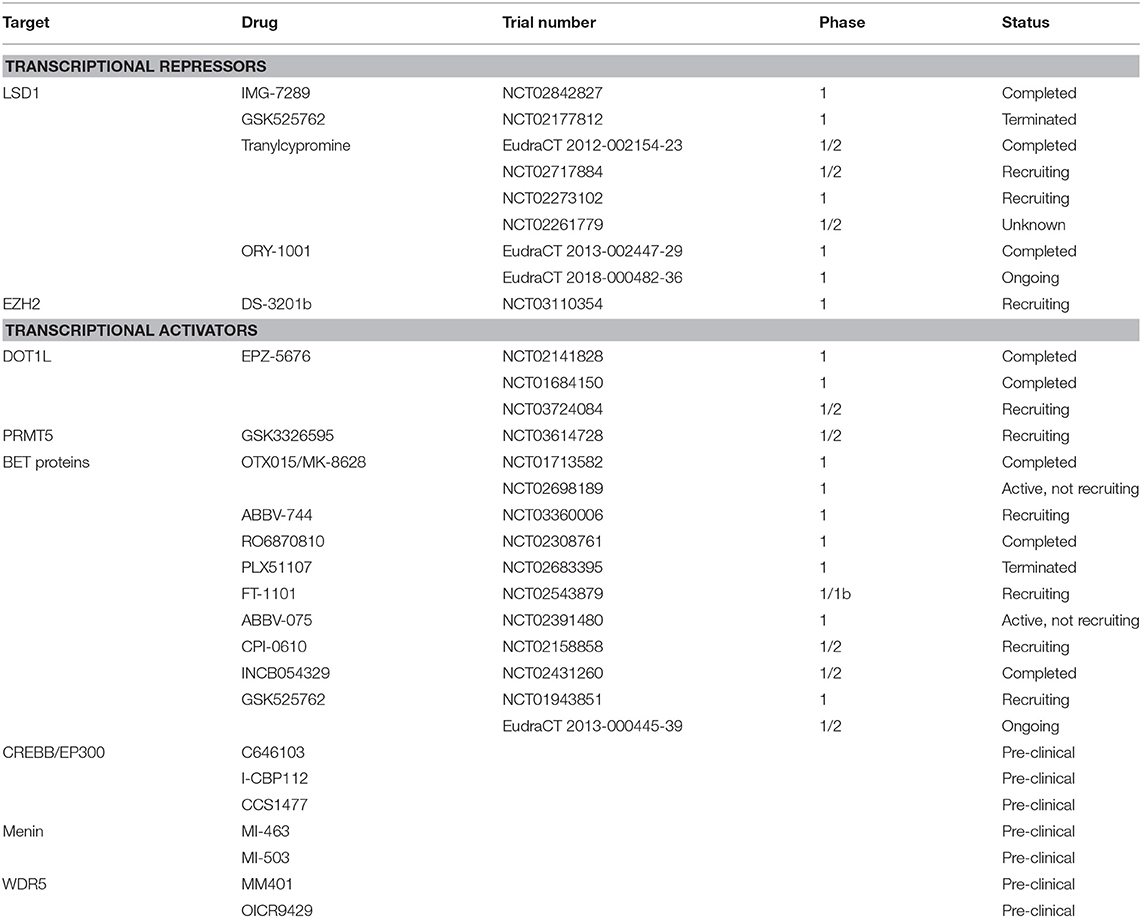
Table 1. Key clinical trials of novel epigenetic therapies in AML (March 2019).
EZH2 is the catalytic subunit of Polycomb Repressive Complex 2 (PRC2) which is responsible for maintaining transcriptional repression of its target genes through tri-methylation of H3 K27 (39, 40) (Figure 1B). This histone mark facilitates recruitment of PRC1 and ubiquitination of H2A K119 to induce a higher repressive state of chromatin (41, 42). EZH2 regulates normal hematopoiesis by maintaining multipotency and self-renewal of hematopietic stem cells (HSCs) (39, 40). However, conditional knockout studies have shown that it is dispensable for HSCs possibly because of redundancy with EZH1 (43). During the last decade, EZH2 has generated much interest as a potential anti-cancer therapeutic strategy. First, several studies have implicated PRC2 complex components including EZH2 in the pathogenesis of diverse cancers including hematopoietic malignancies (44). More recently, distinct cancer-associated mutations in EZH2 have been reported, including gain-of-function mutations in lymphoid malignancies and loss-of-function mutations in myeloid malignancies where they are also associated with a poor prognosis (42). Loss of EZH2 activity in myeloid malignancies also results from differential splicing in the presence of an SRSF2 mutation or consequent upon ASXL1 mutation (45, 46). Interestingly in certain pre-clinical studies using EZH2-deficient mouse models, EZH2 is highlighted as required for the development of myeloid malignancies including MLL-AF9 AML; mutation or deletion leads to a significant loss of LSCs and increased differentiation (47).
The greatest focus for the clinical development of EZH2 inhibitors has been in the setting of lymphoma and some solid tumors (NCT02082977, NCT01897571, NCT02395601) where S-adenosyl methionine-competitive inhibitors, such as GSK2816126, CPI1205, and the most promising, tazemetostat (EPZ-6438), have been evaluated. Another is MAK683 which is an EED-binding complex disrupter under investigation in refractory lymphoma and solid malignancies (NCT02900651). Phase I data for tazemetostat demonstrated an acceptable safety profile and some objective responses (NCT01897571) resulting in initiation of a number of follow on single agent and combination studies (e.g., NCT02875548). On the basis of pre-clinical studies demonstrating the functional importance of PRC2 in MLL-rearranged AML, the dual EZH1–EZH2 inhibitor DS3201b has entered phase 1 as monotherapy in patients with refractory acute leukemia (NCT03110354) (Table 1). However, given a recent report implicating loss of EZH2 and subsequent reduction of histone H3K27 trimethylation in acquired resistance to tyrosine kinase inhibitors (TKIs) and cytotoxic drugs in AML due to derepression of HOX genes (48), cautious selection of specific patient groups is required.
Chromosomal rearrangement of MLL (KMT2A) occurs in around 5% of AML cases, predominantly resulting in an MLL-AF9 fusion gene, although other partner genes occur less frequently (49, 50). The resulting oncoprotein maintains its ability to bind to MLL target genes through N-terminal sequences but recruits additional proteins to MLL target genes through C-terminal sequences. These include members of transcriptional elongation complexes, such as the super elongation complex (SEC) and the H3K79 methyltransferase DOT1L (51).
DOT1L is the only protein methyltransferase responsible for catalyzing methylation of H3K79 (52), a modification generally associated with active transcription (53). Aberrant recruitment of DOT1L results in abnormally high levels of H3K79 methylation on promoters and gene bodies of MLL-fusion target genes, including the HOXA cluster and the homeobox gene MEIS1 (54, 55), which are associated with hematopoietic transformation (Figure 1C). While the precise mechanism by which DOT1L contributes to gene activation is not fully understood, DOT1L inhibits recruitment of a repressive SIRT1 and SUV39H1 complex, thus maintaining an open chromatin state permissive for gene expression (56). Various in vitro and in vivo experimental systems have shown that DOT1L and the interaction between DOT1L and MLL fusion partners is critical for development of leukemia in patients with MLL translocations (57–59).
The S-adenosyl methionine-competitive DOT1L inhibitor pinometostat (EPZ-5676) displays great specificity for DOT1L over other histone methyltransferases (60–63). Preclinical studies revealed that DOT1L inhibition specifically reduces H3K79 methylation and expression of MLL target genes leading to reduction of proliferation and viability as well as increased differentiation of leukemia cells both in vitro and in vivo (61). Phase I clinical studies of single agent pinometostat in adults (NCT01684150) and children (NCT02141828) with advanced or relapsed/refractory MLL-rearranged acute leukemia have recently been completed (64) (Table 1). Despite its limited pharmacokinetics, continuous intravenous administration was sufficient to decrease H3K79 methylation levels and expression of HOXA9 and MEIS1 in individual patients (64). However, only 2 out of 51 adult patients exhibited a clinical response (64) and no objective responses were reported in children (65). These somewhat disappointing results could perhaps be explained by the heterogeneity of MLL fusion proteins which may be differentially sensitive to DOT1L inhibition, uncertainties about optimal dosing, and biological discrepancies between enrolled patients and the preclinical models used to evaluate the effect of pinometostat. Further evaluation of pinometostat in combination with conventional chemotherapy in MLL-rearranged acute leukemia is currently underway (NCT03724084).
Arginine methylation is increasingly appreciated as an important post-translational modification involved in regulation of transcription and chromatin organization, RNA processing and DNA damage repair (66–68). Arginine methylation is catalyzed by a family of nine protein arginine methyltransferases (PRMTs). However, recent research has mainly focused on the type II protein arginine methyltransferases PRMT1 and PRMT5. PRMT1 promotes H3R4 methylation, which is associated with an active chromatin state at critical promoters during hematopoietic cell differentiation; it is essential for recruitment of the acetyltransferase EP300 (69). PRMT1 can also methylate RUNX1, a key transcription factor required for definitive hematopoiesis, myeloid differentiation, and lymphocyte development (70). PRMT5 modifies H4R3, H2AR3, and H3R8, marks which are associated with transcriptional repression (71–75), and also targets multiple non-histone proteins including components of the spliceosome, PIWI proteins, EGFR, E2F1, TP53, and the NFκB subunit p65 (76–81). Multiple studies have implicated PRMT family members in cancer (82). Importantly, CRISPR-Cas9 screens in MLL-rearranged AML mouse models defined PRMT1 and PRMT5 as essential genes and consequently potential targets in this type of leukemia (83). Although PRMT1 is necessary for leukemic transformation, it is not sufficient for MLL-translocation dependent transformation. PRMT1 needs co-recruitment of KDM4C, an H3K9 demethylase, to regulated expression of MLL-fusion targets, such as HOXA9 (84, 85). Deletion or pharmacologic inhibition of both KDM4C and PRMT1 inhibits transcription and leukemic capacity of MLL fusions in vitro and in vivo (84). In keeping with this, conditional deletion or small molecule inhibition of PRMT5 impaired leukemia development and implicated PRMT5 as an enforcer of the leukemic differentiation block (86).
The first PRMT5 inhibitor to enter clinical trials is GSK3326595, a peptide competitive, S-adenosyl methionine-uncompetitive inhibitor. Although the mechanism of action has not been completely determined, GSK3326595 binds to the substrate recognition site of PRMT5 to inhibit methyltransferase activity and this is associated with decreased proliferation of leukemic cells (87). Results from phase 1 clinical trials in subjects with solid tumors and NHL (NCT02783300) as well as relapsed and refractory myelodysplasia, chronic myelomonocytic leukemia and secondary AML with a low proliferation fraction (NCT03614728) are awaited (Table 1).
BRD4 is a member of the Bromodomain and Extra-Terminal motif (BET) family of proteins, and was identified as a potential cancer therapeutic based on results of a genome-wide shRNA screening in MLL-dependent AML cells (88, 89). BRD4 contains a bromodomain which enables its binding to acetylated lysines in histone H3 and H4 (Figure 1C). As a result, BRD4 is bound to active enhancers genome-wide, but is particularly associated with super-enhancers which are regions characterized by unusually high levels of H3K27 acetylation. BET proteins have been found to maintain aberrant chromatin states in AML and other hematologic malignancies (88, 90–92) in particular through regulation of MYC expression (88). Genetic and shRNA-mediated silencing of BRD4 in MLL-AF9 driven leukemia models not only resulted in the removal of BRD4 from super-enhancers, including the MYC enhancer (93), but also in differentiation of leukemia cells and decrease of leukemogenic potential in vitro and in vivo.
The inhibition of BET proteins with preclinical inhibitors, such as JQ1 showed promising results in several studies in AML cell lines and ex vivo patient samples or mouse models, in particular in specific subtypes with MLL rearrangement, or those with mutations in NPM1, FLT3 or IDH2, or EVI1 overexpression (89, 94–97). Based on these observations, clinical trials of a number of BET inhibitors in AML, lymphoma and solid tumors were initiated including FT1101 (NCT02543879), MK8628 (NCT02698189), RO6870810 (NCT02308761), GSK525762 (NCT01943851, EudraCT 2013-000445-39), ABBV-744 (NCT03360006), ABBV-075 (NCT02391480), CPI-0610 (NCT02158858), and INCB054329 (NCT02431260) (Table 1). Few trial results have been published, but so far their clinical activity as single agents for relapsed or refractory AML appears in the main modest, despite the initial excitement arising from preclinical study data. Alternative combinatorial approaches may still capitalize on the clinical potential of these inhibitors, and studies are underway (98, 99). It is however noteworthy that some complete remissions were seen in a phase 1 study of MK8628 (OTX015), an analog of JQ1 (NCT01713582) (99).
Lysine acetyltransferases (KATs) and histone deacetylases (HDACs) catalyze the dynamic and reversible acetylation of histone and non-histone proteins, and are involved in major epigenetic regulatory mechanism of gene transcription (100) in normal hematopoiesis as well as various malignancies. While HDAC inhibitors have been investigated quite extensively in patients with myeloid malignancies, and without much success, development of KAT inhibitors has been largely neglected. The lysine acetyltransferase paralogs CREBBP (CBP; KAT3A) and EP300 (KAT3B) are transcriptional co-activators regulating a variety of cellular processes. Studies in heterozygous and conditional knockout mice have shown that CREBBP is an essential regulator of HSC differentiation, quiescence, apoptosis and self-renewal (101).
CREBBP and EP300 have been implicated in the development of various malignancies, including solid tumors and hematologic diseases (102). Indeed they are among the most frequently mutated KATs in blood cancers, in particular in lymphoma, with inactivating mutations mainly affecting the acetyltransferase domain. Importantly, CREBBP and EP300 are also found as oncogenic fusion partners of the histone acetyltransferase gene MOZ or MLL in leukemia (103, 104) (Figure 1D). In the MLL-CREBBP fusion, the bromo- and acetyltransferase domains of CREBBP are retained and are required for transformation. Additionally, downstream of fusion oncoproteins, recruitment of CREBBP and EP300 to chromatin binding sites for the transcription factor MYB is essential for the differentiation block in leukemias initiated by a range of fusions including AML1-ETO and MLL-AF9 (105, 106). In AML1-ETO AML, EP300 interacts directly with the AML1-ETO protein to regulate transcription of AML1-ETO target genes that are important for leukemic stem cells proliferation and self-renewal (107). CREBBP and EP300 have also been associated with transcriptional activation in collaboration with other leukemogenic proteins, such as NUP98-HOXA9 (108). Thus, there is quite some evidence that CREBBP and EP300 serve important roles in leukemic hematopoiesis and that their therapeutic targeting might be beneficial.
The multidomain organization of CREBBP and EP300 paralogs has prompted several inhibitor development projects. The most potent KAT inhibitors developed so far have been C646 (109) and I-CBP112 (110), an acetyl-lysine competitive protein–protein interaction inhibitor. Both induced differentiation and impaired leukemia-initiating potential in AML1-ETO+ or MLL-AF9+ AML cells in vitro and in vivo. More recently, a CREBBP and EP300 bromodomain inhibitor (CCS1477) has been demonstrated to have potent anti-proliferative and pro-differentiation activity in AML cell lines and primary patient samples (111); a first-in-human phase 1 study has commenced in castration-resistant prostate cancer (NCT03568656) and a related study will shortly commence in patients with multiple hematologic malignancies, including AML.
Recent reports have demonstrated that MLL-driven gene expression is dependent on the interaction of MLL with menin (MEN1) (112). MEN1 serves as an adaptor for the interaction of MLL with LEDGF, a protein that tethers the MLL complex to chromatin (112). This interaction is also crucial for leukemic transformation, proliferation, and expression of leukemia associated genes including the HOXA-cluster and MEIS1 (113) (Figure 1E). Interestingly, while the interaction of MEN1 with MLL1 is not essential for normal hematopoiesis (114), genetic disruption of the MEN1-MLL fusion protein interaction abrogates the oncogenic properties of MLL fusion proteins and blocks the development of AML in vivo (113), highlighting this interaction as an attractive therapeutic target to develop targeted drugs for MLL leukemia patients. Another potentially interesting approach to disrupt the MLL-fusion complex is inhibition of the interaction between WD repeat domain 5 (WDR5) and MLL1. WDR5 directly interacts with SETD1A, SETD1B or one of four homologous MLL methyltransferases (115, 116), which are components of the MLL methyltransferase complex. This interaction is required for the catalytic activity of the enzymes and is responsible for H3K4-specific methylation, a histone mark generally associated with transcriptional activation.
Several small molecules and tool compounds targeting the interactions of components of the MLL complex have been recently developed. Inhibitors targeting MEN1-MLL have been shown to reverse HOXA and MEIS gene expression, thereby releasing the differentiation block associated with MLL-rearranged leukemia (117–121). Similarly MM401 and OICR9429, two compounds which disrupt the MLL-WDR5 interaction, inhibit the proliferation of AMLs harboring MLL translocations (122) or CEBPA mutations (123). However, limited bioavailability and efficacy in vivo is still an important hurdle to overcome.
Epigenetic regulators are central players in the initiation and maintenance of hematopoietic malignancies, an observation which has resulted in myriad opportunities for development of targeted therapies. In particular, where epigenetic mechanisms are specifically disordered in malignant but not normal blood cells, there exists the potential for a significant therapeutic window. Along these lines, there has been significant progress in understanding the role of epigenetic modifications and their modifiers in cancer in general and in AML in particular. The discovery and development of small-molecule inhibitors targeting certain epigenetic regulators has already led to opportunities for clinical trial evaluation and potential patient benefit; impending trial results will inform on efficacy and safety. While the evaluation of many of these compounds for their single agent activity is an essential first step, it will be critical to test efficacy in combination with either standard-of-care chemotherapies or novel therapeutics, to determine their optimal role in the treatment of leukemia. In time, personalization of therapeutic regimens according to patient cytogenetics and molecular mutations may become of essential importance. A final point is that the interdependence of cancer epigenetics and immunological responses has to be taken in consideration. In the setting of leukemia, this includes the therapeutic modality of allogeneic stem cell transplantation. It is clear that epigenetic therapies can induce cellular responses in tumor cells that interact with the immune system and which may contribute to their efficacy; for example, effects of adoptive immunotherapies and immune checkpoint inhibitors can be potentiated by epigenetic therapies (124). As such, the combination of immunotherapies and epigenetic therapies also holds potential promise for the development of additional therapeutic options in AML.
BW and TS conceived and wrote the manuscript.
BW and TS were supported by Cancer Research UK grant number C5759/A20971.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, et al. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. (2013) 31:1133–6. doi: 10.1038/nbt.2701
2. Gallipoli P, Giotopoulos G, Huntly BJ. Epigenetic regulators as promising therapeutic targets in acute myeloid leukemia. Therap Adv Hematol. (2015) 6:103–19. doi: 10.1177/2040620715577614
3. Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. (2016) 127:42–52. doi: 10.1182/blood-2015-07-604512
4. Glass JL, Hassane D, Wouters BJ, Kunimoto H, Avellino R, Garrett-Bakelman FE, et al. Epigenetic identity in AML depends on disruption of nonpromoter regulatory elements and is affected by antagonistic effects of mutations in epigenetic modifiers. Cancer Discov. (2017) 7:868–83. doi: 10.1158/2159-8290.CD-16-1032
5. Cai SF, Chen CW, Armstrong SA. Drugging chromatin in cancer: recent advances and novel approaches. Mol Cell. (2015) 60:561–70. doi: 10.1016/j.molcel.2015.10.042
6. Brien GL, Valerio DG, Armstrong SA. Exploiting the epigenome to control cancer-promoting gene-expression programs. Cancer Cell. (2016) 29:464–76. doi: 10.1016/j.ccell.2016.03.007
7. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
8. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
9. Kuchenbauer F, Buske C. Revisiting thrombocytopenia in acute promyelocytic leukemia. Leukemia. (2018) 32:1477–8. doi: 10.1038/s41375-018-0105-1
10. Lubbert M, Suciu S, Hagemeijer A, Ruter B, Platzbecker U, Giagounidis A, et al. Decitabine improves progression-free survival in older high-risk MDS patients with multiple autosomal monosomies: results of a subgroup analysis of the randomized phase III study 06011 of the EORTC Leukemia Cooperative Group and German MDS Study Group. Ann Hematol. (2016) 95:191–9. doi: 10.1007/s00277-015-2547-0
11. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. (2010) 28:562–9. doi: 10.1200/JCO.2009.23.8329
12. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. (2017) 130:722–31. doi: 10.1182/blood-2017-04-779405
13. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. (2017) 377:454–64. doi: 10.1056/NEJMoa1614359
14. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. (2018) 378:2386–98. doi: 10.1056/NEJMoa1716984
15. DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. (2018) 19:216–28. doi: 10.1016/S1470-2045(18)30010-X
16. Wei A, Strickland SA, Roboz GJ, Hou J-Z, Fiedler W, Lin TL, et al. Phase 1/2 study of venetoclax with low-dose cytarabine in treatment-naive, elderly patients with acute myeloid leukemia unfit for intensive chemotherapy: 1-year outcomes. Blood. (2017) 130:890.
17. Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. (2019) 33:379–89. doi: 10.1038/s41375-018-0312-9
18. Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. (2017) 130:2373–6. doi: 10.1182/blood-2017-09-797712
19. Perl AE, Cortes JE, Strickland SA, Ritchie EK, Neubauer A, Martinelli G, et al. An open-label, randomized phase III study of gilteritinib versus salvage chemotherapy in relapsed or refractory FLT3 mutation-positive acute myeloid leukemia. J Clin Oncol. (2017) 35(15_Suppl.):TPS7067. doi: 10.1200/JCO.2017.35.15_suppl.TPS7067
20. Gallipoli P, Huntly BJP. Novel epigenetic therapies in hematological malignancies: current status and beyond. Semin Cancer Biol. (2018) 51:198–210. doi: 10.1016/j.semcancer.2017.07.005
21. Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. (2019) 25:403–18. doi: 10.1038/s41591-019-0376-8
22. Agrawal K, Das V, Vyas P, Hajduch M. Nucleosidic DNA demethylating epigenetic drugs–a comprehensive review from discovery to clinic. Pharmacol Therap. (2018) 188:45–79. doi: 10.1016/j.pharmthera.2018.02.006
23. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012
24. Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. (2005) 437:436–9. doi: 10.1038/nature04020
25. Maiques-Diaz A, Somervaille TC. LSD1: biologic roles and therapeutic targeting. Epigenomics. (2016) 8:1103–16. doi: 10.2217/epi-2016-0009
26. Humphrey GW, Wang Y, Russanova VR, Hirai T, Qin J, Nakatani Y, et al. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J Biol Chem. (2001) 276:6817–24. doi: 10.1074/jbc.M007372200
27. You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc Natl Acad Sci USA. (2001) 98:1454–8. doi: 10.1073/pnas.98.4.1454
28. Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. (2002) 10:1119–28. doi: 10.1016/S1097-2765(02)00740-2
29. Hakimi MA, Dong Y, Lane WS, Speicher DW, Shiekhattar R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J Biol Chem. (2003) 278:7234–9. doi: 10.1074/jbc.M208992200
30. Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. (2009) 138:660–72. doi: 10.1016/j.cell.2009.05.050
31. Baron R, Binda C, Tortorici M, McCammon JA, Mattevi A. Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure. (2011) 19:212–20. doi: 10.1016/j.str.2011.01.001
32. Maiques-Diaz A, Spencer GJ, Lynch JT, Ciceri F, Williams EL, Amaral FMR, et al. Enhancer Activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. (2018) 22:3641–59. doi: 10.1016/j.celrep.2018.03.012
33. Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. (2010) 29:1803–16. doi: 10.1038/emboj.2010.63
34. Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell. (2007) 27:562–72. doi: 10.1016/j.molcel.2007.06.039
35. Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. (2012) 18:605–11. doi: 10.1038/nm.2661
36. Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. (2012) 21:473–87. doi: 10.1016/j.ccr.2012.03.014
37. Somervaille T, Salamero O, Montesinos P, Willekens C, Perez Simon JA, Pigneux A, et al. Safety, phamacokinetics (PK), pharmacodynamics (PD) and preliminary activity in acute leukemia of Ory-1001, a first-in-class inhibitor of lysine-specific histone demethylase 1A (LSD1/KDM1A): initial results from a first-in-human phase 1 study. Blood. (2016) 128:4060.
38. Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. (2018) 33:495–511 e412. doi: 10.1016/j.ccell.2018.02.002
39. Di Croce L, Helin K. Transcriptional regulation by polycomb group proteins. Nat Struct Mol Biol. (2013) 20:1147–55. doi: 10.1038/nsmb.2669
40. Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. (2011) 469:343–9. doi: 10.1038/nature09784
41. Haladyna JN, Yamauchi T, Neff T, Bernt KM. Epigenetic modifiers in normal and malignant hematopoiesis. Epigenomics. (2015) 7:301–20. doi: 10.2217/epi.14.88
42. Lund K, Adams PD, Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia. (2014) 28:44–9. doi: 10.1038/leu.2013.288
43. Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. (2014) 14:68–80. doi: 10.1016/j.stem.2013.10.001
45. Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. (2016) 22:672–8. doi: 10.1038/nm.4097
46. Micol JB, Pastore A, Inoue D, Duployez N, Kim E, Lee SC, et al. ASXL2 is essential for haematopoiesis and acts as a haploinsufficient tumour suppressor in leukemia. Nat Commun. (2017) 8:15429. doi: 10.1038/ncomms15429
47. Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci USA. (2012) 109:5028–33. doi: 10.1073/pnas.1202258109
48. Gollner S, Oellerich T, Agrawal-Singh S, Schenk T, Klein HU, Rohde C, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med. (2017) 23:69–78. doi: 10.1038/nm.4247
49. Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. (2013) 27:2165–76. doi: 10.1038/leu.2013.135
50. Neff T, Armstrong SA. Recent progress toward epigenetic therapies: the example of mixed lineage leukemia. Blood. (2013) 121:4847–53. doi: 10.1182/blood-2013-02-474833
51. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. (2011) 20:66–78. doi: 10.1016/j.ccr.2011.06.010
52. Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. (2008) 28:2825–39. doi: 10.1128/MCB.02076-07
53. McLean CM, Karemaker ID, van Leeuwen F. The emerging roles of DOT1L in leukemia and normal development. Leukemia. (2014) 28:2131–8. doi: 10.1038/leu.2014.169
54. Deshpande AJ, Deshpande A, Sinha AU, Chen L, Chang J, Cihan A, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell. (2014) 26:896–908. doi: 10.1016/j.ccell.2014.10.009
55. Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, et al. hDOT1L links histone methylation to leukemogenesis. Cell. (2005) 121:167–78. doi: 10.1016/j.cell.2005.02.020
56. Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med. (2015) 21:335–43. doi: 10.1038/nm.3832
57. Chang MJ, Wu H, Achille NJ, Reisenauer MR, Chou CW, Zeleznik-Le NJ, et al. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. (2010) 70:10234–42. doi: 10.1158/0008-5472.CAN-10-3294
58. Jo SY, Granowicz EM, Maillard I, Thomas D, Hess JL. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. (2011) 117:4759–68. doi: 10.1182/blood-2010-12-327668
59. Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. (2011) 117:6912–22. doi: 10.1182/blood-2011-02-334359
60. Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. (2013) 122:1017–25. doi: 10.1182/blood-2013-04-497644
61. Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. (2011) 20:53–65. doi: 10.1016/j.ccr.2011.06.009
62. Cao K, Collings CK, Morgan MA, Marshall SA, Rendleman EJ, Ozark PA, et al. An Mll4/COMPASS-Lsd1 epigenetic axis governs enhancer function and pluripotency transition in embryonic stem cells. Sci Adv. (2018) 4:eaap8747. doi: 10.1126/sciadv.aap8747
63. Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. (2012) 3:1288. doi: 10.1038/ncomms2304
64. Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. (2018) 131:2661–9. doi: 10.1182/blood-2017-12-818948
65. Shukla N, Wetmore C, O'Brien MM, Silverman LB, Brown P, Cooper TM, et al. Final report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with relapsed or refractory MLL-r acute leukemia. Blood. (2016) 128:2780.
66. Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. (2005) 18:263–72. doi: 10.1016/j.molcel.2005.04.003
67. Wang L, Pal S, Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol Cell Biol. (2008) 28:6262–77. doi: 10.1128/MCB.00923-08
68. Yang Y, McBride KM, Hensley S, Lu Y, Chedin F, Bedford MT. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol Cell. (2014) 53:484–97. doi: 10.1016/j.molcel.2014.01.011
69. An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. (2004) 117:735–48. doi: 10.1016/j.cell.2004.05.009
70. Shia WJ, Okumura AJ, Yan M, Sarkeshik A, Lo MC, Matsuura S, et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood. (2012) 119:4953–62. doi: 10.1182/blood-2011-04-347476
71. Aggarwal P, Vaites LP, Kim JK, Mellert H, Gurung B, Nakagawa H, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. (2010) 18:329–40. doi: 10.1016/j.ccr.2010.08.012
72. Chen H, Lorton B, Gupta V, Shechter D. A TGFbeta-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene. (2017) 36:373–86. doi: 10.1038/onc.2016.205
73. Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. (2007) 26:3558–69. doi: 10.1038/sj.emboj.7601794
74. Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol. (2004) 24:9630–45. doi: 10.1128/MCB.24.21.9630-9645.2004
75. Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. (2009) 16:304–11. doi: 10.1038/nsmb.1568
76. Kirino Y, Kim N, de Planell-Saguer M, Khandros E, Chiorean S, Klein PS, et al. Arginine methylation of Piwi proteins catalysed by dPRMT5 is required for Ago3 and Aub stability. Nat Cell Biol. (2009) 11:652–8. doi: 10.1038/ncb1872
77. Vagin VV, Wohlschlegel J, Qu J, Jonsson Z, Huang X, Chuma S, et al. Proteomic analysis of murine Piwi proteins reveals a role for arginine methylation in specifying interaction with Tudor family members. Genes Dev. (2009) 23:1749–62. doi: 10.1101/gad.1814809
78. Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang DB, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc Natl Acad Sci USA. (2013) 110:13516–21. doi: 10.1073/pnas.1311784110
79. Hsu JM, Chen CT, Chou CK, Kuo HP, Li LY, Lin CY, et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol. (2011) 13:174–81. doi: 10.1038/ncb2158
80. Zheng S, Moehlenbrink J, Lu YC, Zalmas LP, Sagum CA, Carr S, et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol Cell. (2013) 52:37–51. doi: 10.1016/j.molcel.2013.08.039
81. Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, et al. Arginine methylation regulates the p53 response. Nat Cell Biol. (2008) 10:1431–9. doi: 10.1038/ncb1802
82. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. (2013) 13:37–50. doi: 10.1038/nrc3409
83. Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. (2015) 33:661–7. doi: 10.1038/nbt.3235
84. Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, et al. Targeting aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute myeloid leukemia. Cancer Cell. (2016) 29:32–48. doi: 10.1016/j.ccell.2015.12.007
85. Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. (2007) 9:1208–15. doi: 10.1038/ncb1642
86. Kaushik S, Liu F, Veazey KJ, Gao G, Das P, Neves LF, et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia. (2018) 32:499–509. doi: 10.1038/leu.2017.206
87. Rasco D, Tolcher A, Siu LL, Heinhuis K, Postel-Vinay S, Barbash O, et al. Abstract CT038: a phase I, open-label, dose-escalation study to investigate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of GSK3326595 in subjects with solid tumors and non-Hodgkin's lymphoma. Cancer Res. (2017) 77:CT038. doi: 10.1158/1538-7445.AM2017-CT038
88. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. (2011) 478:524–8. doi: 10.1038/nature10334
89. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. (2011) 478:529–33. doi: 10.1038/nature10509
90. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. (2011) 146:904–17. doi: 10.1016/j.cell.2011.08.017
91. Emadali A, Rousseaux S, Bruder-Costa J, Rome C, Duley S, Hamaidia S, et al. Identification of a novel BET bromodomain inhibitor-sensitive, gene regulatory circuit that controls Rituximab response and tumour growth in aggressive lymphoid cancers. EMBO Mol Med. (2013) 5:1180–95. doi: 10.1002/emmm.201202034
92. Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. (2012) 120:2843–52. doi: 10.1182/blood-2012-02-413021
93. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. (2013) 153:320–34. doi: 10.1016/j.cell.2013.03.036
94. Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BAM, Erpelinck C, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. (2014) 157:369–81. doi: 10.1016/j.cell.2014.02.019
95. Fiskus W, Sharma S, Qi J, Shah B, Devaraj SG, Leveque C, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Therap. (2014) 13:2315–27. doi: 10.1158/1535-7163.MCT-14-0258
96. Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. (2014) 28:311–20. doi: 10.1038/leu.2013.338
97. Chen C, Liu Y, Lu C, Cross JR, Morris JPt, Shroff AS, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. (2013) 27:1974–85. doi: 10.1101/gad.226613.113
98. Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. (2016) 3:e196–204. doi: 10.1016/S2352-3026(16)00021-1
99. Berthon C, Raffoux E, Thomas X, Vey N, Gomez-Roca C, Yee K, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. (2016) 3:e186–95. doi: 10.1016/S2352-3026(15)00247-1
100. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. (2014) 15:703–8. doi: 10.1038/nrm3890
101. Chan WI, Hannah RL, Dawson MA, Pridans C, Foster D, Joshi A, et al. The transcriptional coactivator Cbp regulates self-renewal and differentiation in adult hematopoietic stem cells. Mol Cell Biol. (2011) 31:5046–60. doi: 10.1128/MCB.05830-11
102. Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. (2004) 23:4225–31. doi: 10.1038/sj.onc.1207118
103. Katsumoto T, Yoshida N, Kitabayashi I. Roles of the histone acetyltransferase monocytic leukemia zinc finger protein in normal and malignant hematopoiesis. Cancer Sci. (2008) 99:1523–7. doi: 10.1111/j.1349-7006.2008.00865.x
104. Zhang Y, Zeleznik-Le N, Emmanuel N, Jayathilaka N, Chen J, Strissel P, et al. Characterization of genomic breakpoints in MLL and CBP in leukemia patients with t(11;16). Genes Chromosomes Cancer. (2004) 41:257–65. doi: 10.1002/gcc.20077
105. Zhao L, Glazov EA, Pattabiraman DR, Al-Owaidi F, Zhang P, Brown MA, et al. Integrated genome-wide chromatin occupancy and expression analyses identify key myeloid pro-differentiation transcription factors repressed by Myb. Nucleic Acids Res. (2011) 39:4664–79. doi: 10.1093/nar/gkr024
106. Pattabiraman DR, McGirr C, Shakhbazov K, Barbier V, Krishnan K, Mukhopadhyay P, et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood. (2014) 123:2682–90. doi: 10.1182/blood-2012-02-413187
107. Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol Cell Biol. (2006) 26:2782–90. doi: 10.1128/MCB.26.7.2782-2790.2006
108. Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. (1999) 19:764–76. doi: 10.1128/MCB.19.1.764
109. Santer FR, Hoschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Therap. (2011) 10:1644–55. doi: 10.1158/1535-7163.MCT-11-0182
110. Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. (2015) 75:5106–19. doi: 10.1158/0008-5472.CAN-15-0236
111. Pegg N, Worthington J, Young B, Prosser A, Gaughan L, Spencer G, et al. Abstract 3991: Novel small molecule inhibitors of p300/CBP down-regulate androgen receptor (AR) and c-Myc for the treatment of prostate cancer and beyond. Cancer Res. (2018) 78:3991. doi: 10.1158/1538-7445.AM2018-3991
112. Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. (2008) 14:36–46. doi: 10.1016/j.ccr.2008.05.003
113. Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. (2005) 123:207–18. doi: 10.1016/j.cell.2005.09.025
114. Li BE, Gan T, Meyerson M, Rabbitts TH, Ernst P. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood. (2013) 122:2039–46. doi: 10.1182/blood-2013-03-486647
115. Dharmarajan V, Lee JH, Patel A, Skalnik DG, Cosgrove MS. Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J Biol Chem. (2012) 287:27275–89. doi: 10.1074/jbc.M112.364125
116. Zhang P, Lee H, Brunzelle JS, Couture JF. The plasticity of WDR5 peptide-binding cleft enables the binding of the SET1 family of histone methyltransferases. Nucleic Acids Res. (2012) 40:4237–46. doi: 10.1093/nar/gkr1235
117. Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. (2015) 27:589–602. doi: 10.1016/j.ccell.2015.02.016
118. Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. (2012) 8:277–84. doi: 10.1038/nchembio.773
119. He S, Senter TJ, Pollock J, Han C, Upadhyay SK, Purohit T, et al. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J Med Chem. (2014) 57:1543–56. doi: 10.1021/jm401868d
120. Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, et al. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. (2012) 120:4461–9. doi: 10.1182/blood-2012-05-429274
121. Xu Y, Yue L, Wang Y, Xing J, Chen Z, Shi Z, et al. Discovery of novel inhibitors targeting the menin-mixed lineage leukemia interface using pharmacophore- and docking-based virtual screening. J Chem Inform Model. (2016) 56:1847–55. doi: 10.1021/acs.jcim.6b00185
122. Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. (2014) 53:247–61. doi: 10.1016/j.molcel.2013.12.001
123. Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R, et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat Chem Biol. (2015) 11:571–8. doi: 10.1038/nchembio.1859
Keywords: AML, LSD1, EZH2, BET bromodomain, DOT1L, PRMT5, EP300, MLL
Citation: Wingelhofer B and Somervaille TCP (2019) Emerging Epigenetic Therapeutic Targets in Acute Myeloid Leukemia. Front. Oncol. 9:850. doi: 10.3389/fonc.2019.00850
Received: 05 April 2019; Accepted: 19 August 2019;
Published: 06 September 2019.
Edited by:
Cyrus Khandanpour, University Hospital Münster, GermanyReviewed by:
Maximilian Christopeit, Universität Hamburg, GermanyCopyright © 2019 Wingelhofer and Somervaille. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim C. P. Somervaille, dGltLnNvbWVydmFpbGxlQGNydWsubWFuY2hlc3Rlci5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.