 Laura Monaghan1
Laura Monaghan1 Matthew E. Massett1
Matthew E. Massett1 Roderick P. Bunschoten2
Roderick P. Bunschoten2 Alex Hoose2
Alex Hoose2 Petrisor-Alin Pirvan2Robert M. J. Liskamp2
Petrisor-Alin Pirvan2Robert M. J. Liskamp2 Heather G. Jørgensen1
Heather G. Jørgensen1 Xu Huang1*
Xu Huang1*- 1Haemato-Oncology/Systems Medicine Group, Paul O'Gorman Leukemia Research Center, Institute of Cancer Sciences, University of Glasgow, Glasgow, United Kingdom
- 2School of Chemistry, University of Glasgow, Glasgow, United Kingdom
Growing evidence has demonstrated that epigenetic dysregulation is a common pathological feature in human cancer cells. Global alterations in the epigenetic landscape are prevalent in malignant cells across different solid tumors including, prostate cancer, non-small-cell lung cancer, renal cell carcinoma, and in haemopoietic malignancy. In particular, DNA hypomethylation and histone hypoacetylation have been observed in acute myeloid leukemia (AML) patient blasts, with histone methylation being an emerging area of study. Histone 3 lysine 9 trimethylation (H3K9me3) is a post-translational modification known to be involved in the regulation of a broad range of biological processes, including the formation of transcriptionally silent heterochromatin. Following the observation of its aberrant methylation status in hematological malignancy and several other cancer phenotypes, recent studies have associated H3K9me3 levels with patient outcome and highlighted key molecular mechanisms linking H3K9me3 profile with AML etiology in a number of large-scale meta-analysis. Consequently, the development and application of small molecule inhibitors which target the histone methyltransferases or demethylase enzymes known to participate in the oncogenic regulation of H3K9me3 in AML represents an advancing area of ongoing study. Here, we provide a comprehensive review on how this particular epigenetic mark is regulated within cells and its emerging role as a potential therapeutic target in AML, along with an update on the current research into advancing the generation of more potent and selective inhibitors against known H3K9 methyltransferases and demethylases.
Background
The normal process of haematopoietic progression involves the differentiation and self-renewal of haematopoietic stem cells (HSC), as a hierarchical tree the development and progression of blood cells has become a model organism for the study of how the epigenetic landscape changes with cell commitment and ultimately its involvement in cell fate decision making. The precisely controlled expression of lineage specific genes is crucial for the proliferation and differentiation essential for normal development, whereas aberrant transcriptional regulation can result in malignant transformation and oncogenic progression.
The regulation of gene transcription is a tightly controlled process in which DNA associated with a number of transcription factors and histone proteins forms the structure of chromatin, each component playing a role in regulating the gene expression landscape. Epigenetics concerns modification to the marks associates with the DNA and histones which are not mutations but rather alter the structure of the chromatin making genes more or less accessible whilst also recruiting proteins which serve to alter the chromatin structure and promote transcription (1). In 1962, two decades after the coining of the epigenome by Conrad Waddington in 1942 (2), histone methylation was first postulated to be involved with gene expression (3). Over 50 years on and considerable progress has been made in understanding the role of methylated histones. A collection of studies within the first decade of the twenty-first century presented a turning point for our understanding of histone methylation, transforming it from a static histone modification to a dynamic mark whose abundance could be externally altered. Whilst the first methyltransferase SUV39H1 was identified at the turn of the century (4) and hinted that histone methylation was dynamic, it was arguably the subsequent discovery of the first histone demethylase (5) that shifted our perspective of histone methylation. This discovery revealed that the marks were not permanent and paved the way for future studies concerning the regulation of specific methylated histones and their impact on normal tissue development and oncogenic transformation.
Acute Myeloid Leukemia (AML) is an aggressive hematological malignancy characterized by disruption of the normal process of haematopoietic progression involving the differentiation and self-renewal of HSC. These lead to the uncontrolled proliferation of dominant Leukaemic stem cell (LSC) clones and the accumulation of immature myeloid blasts. Originally AML was thought to progress through the two-hit hypothesis in which the acquisition of a type 1 mutation results in an increased rate of proliferation and resistance to apoptosis in collaboration with a type 2 mutation which blocks differentiation (6). With increasing understanding of the epigenome, a third subgroup has been added featuring aberrant epigenetic regulation (7). Genome-wide and candidate gene association studies have highlighted mutations associated with over 70% of de novo AML occurring in genes encoding for epigenetic regulators (8–12), further highlighting the crucial role that epigenetic modifications plays in hematological malignancy.
Moreover, in primary cell sample analysis on comparison of AML samples with normal CD34+ stem and progenitor cells and white blood cells as controls, a preferential decrease in H3K9me3 at core promotor regions is observed in AML blasts, this fundamentally has causal transcription factor changes recruiting varying histone modifying enzymes and altering the chromatin structure (13). Over 2000 loci differed between the sample groups with ~20% of these with a higher than 2-fold change. Fundamentally the changes observed could be significantly centered on the region from −300 to the transcriptional start site and clearly allowed the separation of distinct sample groups (13). By investigating how these enzymes become deregulated in AML and identifying ways to target these, new avenues begin to emerge for possible treatment options to complement the mainstay chemotherapy strategy. In this review, we draw our attention to one particular epigenetic mark, the trimethylation of lysine 9 residues on histone 3 (H3K9me3) which is highly regulated in the normal HSC differentiation and self-renewal alongside the establishment of pre-LSCs (14). A wide range of enzymes are involved in its establishment, recognition, and removal, representing promising therapeutic targets in AML.
Main Text
Role of H3K9me3 in Normal Haematopoiesis and Malignancy
H3K9me3 has a role not only in malignancy but in normal cellular development, acting as a repressor of lineage inappropriate genes and maintaining early cell integrity and genomic stability. In the early 2000's a number of groups provided evidence of its importance in interacting with the evolutionarily conserved amino terminal chromodomain of heterochromatin protein 1 (HP1), a hallmark of heterochromatin, thereby recruiting it to specific chromatin loci (15–17). Heterochromatin is a unique form of chromatin architecture defined as condensed and transcriptionally silent (18). Crucially, HP1 can cause deposition of further H3K9me3 through the recruitment of the methyltransferase SUV39H1 (19) leading to propagation of H3K9me3 across DNA and permitting the establishment of large domains of heterochromatin (20). Fundamental for cell integrity and maintenance, large constitutive heterochromatin facilitated by H3K9me3 maintains repetitive gene clusters and regulatory factors and prevents the unwanted recombination and introduction of mutations. In mouse models, reducing or knocking out the fundamental H3 methyltransferases cause lethality to the embryos at different stages of development (21). By binding with HP1 protein, H3K9me3 recruits further epigenetic modifications which contribute to the maintenance of the heterochromatin (22). However, in facilitative heterochromatin accessing the genes is fundamental for the changing cellular landscape when the cell is forced to adapt, for this reason the mark and subsequent tightly bound heterochromatin must be closely regulated (23). This has been explored through the use of several models which detail the reactions kinetics (24, 25). To date, roles for H3K9me3 have been discovered in regulating apoptosis (26, 27), autophagy (28), development (29, 30), DNA repair (31–35), splicing (36–38), self-renewal (39, 40), transcriptional elongation (41), viral latency (42–44), imprinting (45), aging (46), and cell identity (47).
The role of H3K9me3 within cells is still being explored and has proved to be multi-faceted and intricate in a wide-range of cellular processes, which consequently highlight an extended network of mechanisms which contribute to its accumulation and subsequent genomic stability. H3K9me3 has been implicated in recent years with the progression of pancreatic metastasis, with the somatic mutation burden remaining primarily unchanged within the metastasis a global change is observed in H3K9 methylation status giving a selective advantage and an increased capacity to survive and resist treatment to the metastatic site (48). In AML alterations of H3K9 methylation at promoter regions are associated with an inactivation of tumor suppressor genes and a blockage in differentiation and deregulated proliferation (49, 50). Given the reversible nature of H3K9 trimethylation, this represents an attractive therapeutic target in AML.
The Association of H3K9me3 With Transcriptional Activation
The modifications of histones must not be thought of alone but as a small part of a bigger picture of regulation. Chromatin is not an open and closed book as once was thought but is subject to a number of regulatory factors and enzymes which together influence the outlook. Histone modifications can influence the chromatin structure without direct chromatin access changes (51).An alternative approach to investigating the histone code has identified an opposing role for H3K9me3 in AML. Some evidence postulates a role for H3K9me3 as associated with activation either as a solo mark by selectively interacting with RNA polymerase II to promote mRNA elongation and transcriptional activation or through a specific histone code co-localizing on fundamental genes with activating histone marks H3K9ac and H3K4me2 (52, 53). Similarly, H3K9me3's binding capacity for HP1 can also be questioned as to its mechanism and consequence. HP1 protein exists in three forms α,β, and γ. By binding the α and β subtypes the classical heterochromatin transcriptional repression phenotype is observed but when the mark can be co-localized with the third form HP1γ with 1 kb of the transcriptional start site (TSS) there is RNA polymerase II induced mRNA elongation promoting gene activation which is rapidly lost upon transcriptional termination. This is most clearly seen during the constitutive expression of GAPDH observed with a firm H3K9me3 HP1γ binding phenotype at its TSS (41).
Regulatory Mechanisms of H3K9me3
The dynamic nature of H3K9me3 highlights the number of enzymes which are involved in its regulation. A tightly controlled collection of readers, writers, and erasers of this histone mark establish and maintain transcriptional landscape however small changes in these enzymes can also consequently contribute to disease (Figure 1; Table 1).
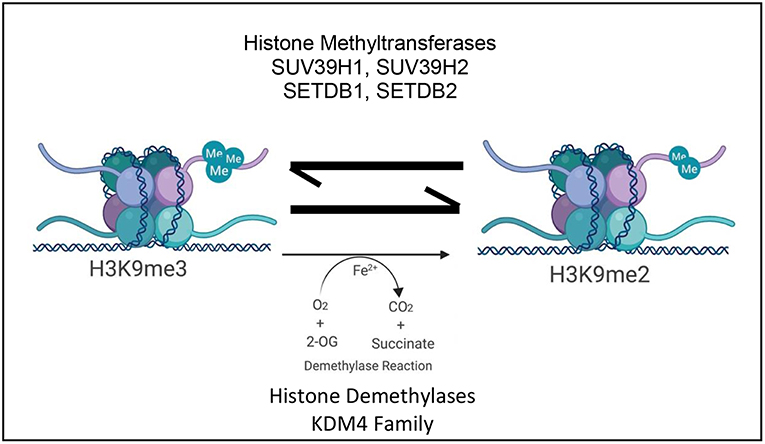
Figure 1. Schematic depicting the current paradigm of H3K9me3 regulation and its influence on transcription by the antagonistic activity of various histone lysine methyltransferases and the KDM4 family of histone lysine specific demethylases ©Created with Biorender.com.
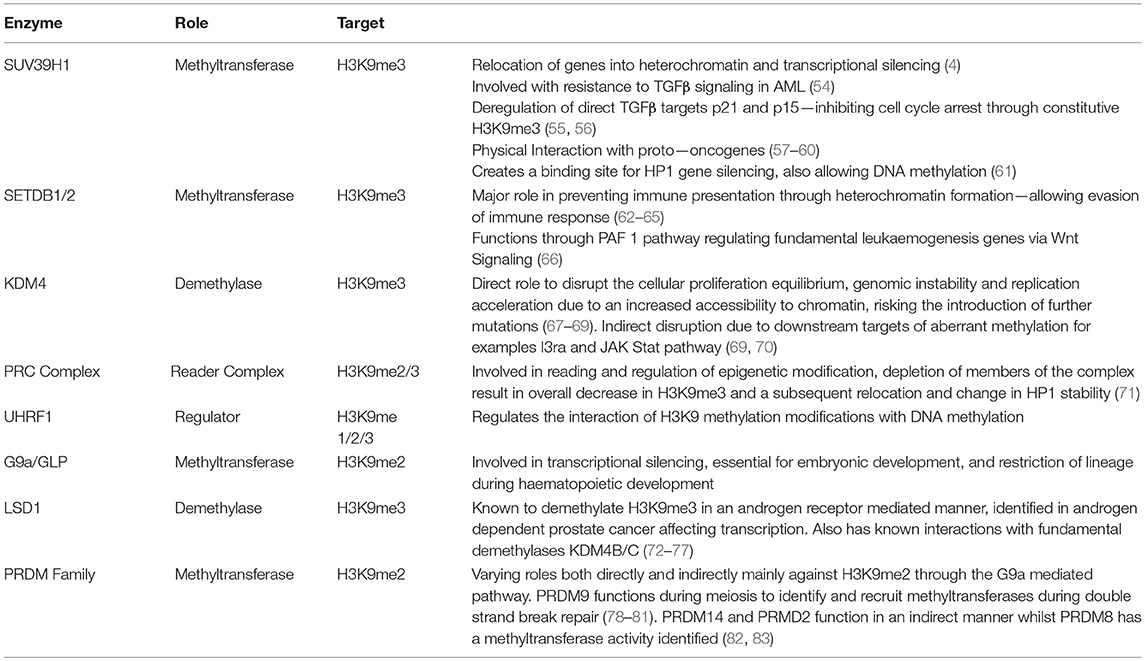
Table 1. Representative summary of enzymes involved in the regulation of H3K9 methylaton—their target state, function, and role in the development of disease.
Writers of H3K9me3 Modifications
SUV39H1
SUV39H1 and SUV39H2 were the earliest methyltransferases identified. Of initial interest due to their respective homologs in model organisms, D. melanogaster and S. pombe, being associated with position effect variegation (PEV) which leads to the relocation of genes into regions of heterochromatin and subsequent transcriptional silencing. Sharing a 59% sequence identity, belonging to the Su (var) gene family and specifically methylating H3K9 (4, 84), these methyltransferases possess two evolutionarily conserved domains, the N-terminal chromo domain (CD) and the C-terminal suppressor of variegation, enhancer of zeste and trithorax (SET) domain. The methyltransferase catalytic activity of the SET domain was elucidated using an in vitro systematic approach, whereby truncated versions of SUV39H1 were tested to observe which conserved regions were required to successfully methylate free histone. This approach also identified H3K9 as the transferases main substrate (4), other studies have shown that by genetically reducing the levels of SUV39H1 there is an observed decrease in global H3K9 methylation (85, 86). Opposed to this but still fundamental for activity the N terminus chromodomain facilitates recruitment of the transferases by binding trimethylated H3K9 and allows the establishment of broad heterochromatin domains due to the successive local spreading of H3K9me3 (25). Interestingly, the spreading mode of H3K9me3 due to SUV39H1 has been explored further, utilizing innovative in vitro synthetic chromatin which closely resembles natural chromatin demonstrating that SUV39H1 first binds its target H3K9 and then engages with the chromatin through a second zinc finger like-domain which acts to enhance its catalytic activity (87). Shirai et al. have also revealed that the CD of SUV39H1 is able to efficiently bind RNA in an H3K9me3 independent manner and that this contributes to efficient recruitment to chromatin (20, 88).
Alongside its role in normal stability SUV39H1 was also the first histone lysine methyltransferase to have its role in AML pathology characterized. One major pathway implicated in the progression of AML and a significant pathway in the regulatory function of SUV39H1 is the acquired resistance to Transforming Growth Factor beta (TGFβ). TGFβ is a signaling pathway frequently deregulated in haematopoietic malignancies with anti-proliferative and differentiation signaling demonstrated to directly arrest growth and inhibit colony formation in the leukaemic stem cell population. Thought to be due to a reduction in receptors on the cell surface as opposed to a mutation, resistance to TGFβ signaling is commonly observed in AML phenotypes (54, 89, 90). MDS1 and EVI1 Complex Locus Protein (MECOM), a potent proto-oncogene known to be involved in stem cell self-renewal and leukaemogenesis physically interacts with the SUV39H1 histone methyltransferase and has been implicated in disease progression of AML in around 5% of cases and its expression negatively correlates with survival in AML. Despite EVI1 not being essential for the methylation functions of the SUV39H1 molecules, this co-localization is necessary for transcriptional repression of the TGFβ pathway associated with the EVI1 AML phenotype. Similarly, the activity of EVI1 requires co-activation of the HOXA9 gene to drive leukaemogenesis, a gene which has been investigated with respect to the demethylase enzyme family and target genes (57–60). Targets of the TGFβ pathways are frequently deregulated in AML. For example, p15 and p21, major tumor suppressors and targets of TGFβ are silenced in AML by pathways involving SUV39H1, giving the leukaemic cells an advantage to proliferate. The gene repression of p15 locus observed in AML is reliant upon several differential epigenetic marks. Aberrant di-methylation of H3K9 on the promoter region of p15 in AML is dependent upon DNA methylation at the promoter region and a specific acetylation pattern on H3K9. A constitutive level of H3K9me3 is required to repress p15 and establish an AML phenotype.
Similarly, an aberrant epigenetic regulation landscape is observed in AML when the expression of p21 is altered, through the action of SUV39H1. Although not carried out on haematopoietic cell lines, Cherrier et al. studied the interaction of SUV39H1 with CTIP2 also known as B-cell lymphoma/leukemia 11B (BCL11B). By increasing H3K9me3 so reducing p21 gene expression, this SUV39H1/CTIP2 complex allows proliferation of leukaemic cells and their failure to undergo apoptosis. BCL11B has been shown to be aberrantly expressed in AML (91, 92). By inhibiting the SUV39H1 induced H3K9me3, p21 facilitated cell cycle arrest can be initiated. p15 and p21 post-translational histone modifications can be restored to normal using an SUV39H1 inhibitor such as chaetocin, which will be discussed in more detail later in this review (55, 56, 61, 93). SUV39H1 dependent methylation of H3K9 creates an enhanced binding site for the HP1 protein, a known gene silencing and enhanced heterochromatin associated protein. This results in an inhibition of gene transcription (61, 94). By creating a complex together, they have been shown to interact with DNA methyltransferases, specifically DMNT3. Originally identified in D. melanogaster (95) loss of specific DNA methylation genes elicit a dual role as H3K9 methylation was also reduced. By investigating the interactions of DNMT and HMT with the HP1 further, a larger network of epigenetic regulation begins to unfold. For example in AML a large crosstalk between epigenetic regulators develops, methylation of H3K9 creates a binding site for HP1 which allows the recruitment of DNMT catalyzing DNA methylation recruiting methyl-CpG-binding proteins which create a location for Histone Deacetylases (HDACs), priming H3K9 sites for methylation (96).
SETDB1/2
SET domain, bifurcated 1 (SETDB1) and SET domain, bifurcated 2 (SETDB2) represent a second family of methyltransferases (97, 98) that specifically target H3K9. They were named after the observation of an intervening 347 amino acid sequence that splits their SET domains (99). These proteins unlike SUV39H family lack a CD, instead possessing methyl-CpG binding domains with SETDB1 additionally possessing a triple Tudor domain (3TD), the function of which has recently been elucidated by crystallographic studies. Revealed to bind a novel bivalent chromatin state defined by H3K9me1/2 and K14ac it subsequently mediates the trimethylation of H3K9 (62). Overexpressed in several cancer phenotypes SETDB1 is necessary for the survival of over 70% of tested AML cell lines. By knocking down SETDB1, AML cells will apoptose via the removal of H3K9me3 on transposable elements, their subsequent expression, and the induction of the interferon immune response identifying it as a fundamental leukaemic cell survival protein (63). One of the major contributing factors of histone methyltransferases in the development of a leukaemic phenotype is their interaction to allow the evasion of the immune system response. Through increasing H3K9me3 at select retrotransposon sites SETDB1 increases the formation of heterochromatin preventing immune presentation allowing leukaemic cells to evade the immune response (63–65). A recently published paper elucidated a role for SETDB1 in the regulation of H3K9me3 through the PAF1 gene known to be associated with histone methylation in Mixed Lineage Leukemia (MLL). By physically interacting with MLL, PAF1 plays a role in regulating fundamental leukaemogenesis genes, for example MEIS1 and the previously described HOXA9. Through critically regulating the Wnt/β-catenin pathway samples with high SETDB1 levels showed a significant reduction in both MEIS1 and HOXA9 by regulating and increasing the silencing associated with the H3K9me3 mark on these specific oncogenic gene promotors (66). Loss of SETDB1 has been identified to cause the differentiation of cells in the embryonic lineage (21).
Erasers of H3K9me3 Modifications: KDM4 Family of Histone Demethylases
At the same time as the methyltransferase enzymes were being investigated, three independent research groups provided insight into the α-ketoglutarate (α-KG), Fe2+, and O2 dependent lysine-specific demethylase 4 (KDM4) JmjC-domain containing demethylase family of proteins, whose catalytic activity reverses the H3K9me3 mark (100–102). Although their function was first characterized by these landmark studies, the KDM4 family was initially described in silico showing the family to be composed of six members (KDM4A-F), four functional members with KDM4E/F hypothesized as pseudo-genes. Each member ubiquitously possesses the JmjC and JmjN domains with KDM4A-C having an additional two Tudor domains (103). KDM4 proteins utilize a distinct hydroxylation chemistry to demethylate H3K9me3 unlike previously identified demethylases such as KDM1A which use a FAD-dependent amine reaction, making their targets mono/di methylated and preventing them from removing the trimethyl marks.
The structural disparity between the KDM4 members has functional consequences. For example, whilst the KDM4A-C proteins primarily function to demethylate trimethylated H3K9, KDM4D also efficiently demethylates H3K9me2 (104). To discern the essential amino acids and domains of the KDM4 family that are required for their catalytic activity, numerous site-directed mutagenic (105–107) and protein-truncation (102) strategies have been employed. These primarily concern KDM4A and have shown that both the JmjC and JmjN domains are required for demethylation, whereas the plant homeodomain and Tudor domains are not obviously necessary for catalytic activity and are thought to primarily facilitate recruitment and adhesion to specific chromatin loci to remove H3K9me3. For example, crystallographic studies have offered insight concerning the molecular mechanisms of KDM4 proteins and revealed key amino acid residues that determine substrate specificity in the catalytic site of KDM4A which interacts with H3K9me3 (106, 108, 109). Recent work has also shown that the tandem Tudor domain of KDM4C can also bind to H3K4me3 and that this interaction acts to enhance the demethylation of H3K9me3 on the same histone tail, owing to an increased affinity for the mark (110). Another important characteristic recently uncovered is the ability of KDM4A to function as an oxygen detector, restricting H3K9me3 demethylation in hypoxic conditions (111, 112). An unexplored feature of this family is their capability to form homodimers and heterodimers between themselves and the impact this has upon their regulation of H3K9me3. Initial work using a reporter construct suggests that heterodimer formation can synergise or neutralize the transcriptional impact of these proteins at specific gene targets depending on the dimer composition, for example KDM4C and KDM4D heterodimers can synergise to promote higher expression of target genes (104).
Importantly, the KDM4 subfamily is commonly overexpressed in human cancers disrupting the equilibrium of cellular proliferation (67). Given the role of the KDM4 family in demethylating trimethylated H3K9 and the distinct deregulation known for this mark in AML, it is necessary to fully investigate their mechanisms in leukaemogenesis (68). KDM4A has been associated with genomic instability resulting in accelerated replication due to the increase in chromatin accessibility and hence the possibility for the introduction of DNA replication errors to further progress disease (69). It has been investigated and implicated in the disease progression of several cancers via alternative pathways (39, 113–115). Upon knock-out of the KDM4 family (KDM4A-C) a decreased proliferation and colony forming potential of MLL-AF9 leukaemic cells was observed with prolonged survival of murine models (70). ChIP sequencing analysis upon knocking down the KDM4 family members identified thousands of regions which were indicative of KDM4A binding sides, of which 77% were within 1 kb of transcriptional start sites (TSS). A number of these sites show largely increased H3K9 methylation upon deletion of KDM4A-C and localization to sites which are positive for H3K4, demonstrating the crosstalk between modifications. A large amount of transcriptional changes were subsequently observed with 55 genes being up-regulated and 94 genes being down-regulated, with approximately half being associated with binding sides of the KDM4 family (70, 116). The molecular mechanisms elucidated to date in AML suggest KDM4A has roles both directly, dependent on its demethylase activity, and indirectly, through downstream targets of its methylation changes, in oncogenesis. In MLL-AF9 driven leukemia KDM4A has been demonstrated to be involved in the proliferation of leukaemic cells via the aberrant demethylation of the TSS region of l3ra (cd123), the α subunit of the heterodimeric IL-3 receptor that, together with the β subunit (IL3 receptor β), forms a functional high-affinity receptor. Subsequent upregulation of downstream targets and the activation of the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway drive proliferation (69, 70, 117).
Readers of H3K9me3 Modifications
Writers and erasers of histone modifications functions are not exclusive, alongside their dynamic control of the mark they also have a unique and highly controlled feedback system by which they recognize and read the marks to allow for changes in the system to be identified, altered and maintained. H3K9me3 can be read by a number of domains, the PhD and Tudor domains function in the demethylases and the chromo domain which has previously been described in the SUV family of methyltransferases. Others will also contain ankyrin repeats or MBT domains allow them to read and regulate their own function (118). Whilst the core regulatory apparatus governing H3K9me3 levels is composed of methyltransferases and demethylases, another layer of control exists to permit greater flexibility in H3K9me3 regulation. For example, many proteins which belong to the Polycomb Repressive Complexes (PRC) have been implicated in regulating H3K9me3. Depletion of SUZ12, an essential member of the PRC2 complex results in an EZH2-independent simultaneous decrease in overall H3K27me3 and H3K9me3 levels and results in relocation of the HP1α isoform. Whilst the exact mechanism is still elusive, given their localization and demonstrated in vitro (cell line) interactions, SUZ12 is thought to stimulate SUV39H1 activity or be involved with either HP1α recruitment or stability (71). In addition, CBX7, a member of the PRC1 complex has also been shown to play a role in the stability of H3K9me3 at specific loci in a molecular mechanism requiring SUV39H2 recruitment (119).
Complex Regulatory Network
Despite the physical accessibility of the histone modification site being essential, nucleosome occupancy has been identified as a key player in influencing the histone modifications and the complex network of crosstalk they employ to control gene expression and maintain cell identity (51). Transcriptional activation requires both the addition of post-translational marks which promote activation and the removal of post-translational marks which induce repression. A number of enzymes work together to achieve this, each requiring the other to gain the desired effect (120). For example, in AML the protein arginine methyltransferases PRMT4/5 are commonly overexpressed and contribute to the blockage in myeloid differentiation. PRMT1 is a necessary member of the MLL transcriptional complex (116), whilst PRMT6 inhibits the action of MLL to methylate H3K4. Similarly, acetylation of histone complexes (histone acetylates (HATs) have known interactions with the methylation of H3K9 (121). TIP60, which is reduced in AML, has a tumor suppressor role through its recognition of H3K9me3 through the activation of ATM kinase signaling and DNA damage checkpoint activation; it also interacts with p53 protein to initiate apoptosis of damaged cells (122). Similarly competing evidence has been published on the role of H3K9me3 in the establishment and maintenance of specific DNA methylation patterns (123). Originally identified in plant species trimethylation of H3K9 has been shown to directly affect cellular DNA methylation. DNA methylation is governed by two main families of enzymes, Methyl CpG-binding (MBD) family and BR-C, ttk and bab (BTB)/Pox virus and Zinc finger (POZ) family proteins, however these families do not exclusively interact with DNA methylation but also through histone modifications such as histone methylation and acetylation (123). One mechanism of this interaction involves HP1 binding leading to the recruitment of DNA methyltransferases adding a new layer of epigenetic control to the story. HP1 specifically binds during the cell cycle interacting specifically with H3K9me3 recruiting the DNA methyltransferases. During mitosis the binding with HP1 is displaced by the phosphorylation of H3 interrupting the mechanism of DNA methylation (REF 105). A more recent discovery of the ubiquitin-like, PHD, and RING finger containing 1 protein (UHRF1), found a more significant connection between histone methylation and DNA methylation with each reliant on the other for normal function (124). Similarly to the KDM4A family UHRF1 has a Tudor and Plant Homeodomain (PhD) with the former being responsible for binding the histone methyl marks. However, unlike the histone demethylase family UHRF1 has a secondary binding site specific for hypomethylated DNA foci (125). UHRF1 association with methylated H3K9me3 is required for DNA methylation maintenance binding hemimethylated DNA during S phase at the replication fork and recruiting DNA methyltransferases (124). Mutants of UHRF1 which are selectively unable to bind H3K9 methylation or hemi-mCpG, but not both, show a partial defect in heterochromatin, and an ability to bind DNMT1 with a partial rescue of DNA methylation showing that these functions are not mutually exclusive rather that sequence and modification state of DNA can play a part in histone modifications and vice versa (123, 125). These marks must all work together to hold the cell in a homeostatic position; evidence has shown that in AML these mechanisms lose their control and deregulation leads to the phenotype of disease (118).
H3K9me3 is the most prominent and fundamental component to the epigenomic outlook of AML however it is not the only form of histone methylation shown to have influence on the epigenomic and genomic landscapes in AML and other cancer phenotypes. H3K9me3 methyltransferases and demethylases have been shown to form a larger complex and both individually and interdependently influence the epigenetic landscape SETDB1 and SUV39H1 both discussed form a complex with di- and mono-methylase proteins G9A and GLP. In this manner all three forms of H3K9 methylation ultimately rely on the others for maintenance (126). Dimethylation of H3K9 has a major role in transcriptional control and is shown to be erased from tumor suppressors following successful treatment. Specifically in AML large sections of H3K9me2 are hypothesized to promote chromosome instability and mutagenesis changing transcription and silencing tumor suppressors (127). G9a/GLP heteromeric complex is essential for mammalian H3K9 methylation, specifically mono- and di-methylation. With aberrant gene expression following genetic knockdown of both or either of these genes they are hypothesized to be involved in transcriptional silencing (128). The complex is essential for embryonic cell development, lineage commitment, and restriction of reprogramming with spreading H3K9me2 observed during haematopoietic cell lineage development (129, 130). Inhibition of G9a/GLP reduces differentiation, promotes stem cell characteristics delaying disease and reducing LSC frequency via the leukaemic transcription factor HOXA9, implicating a crossover role with the output of H3K9me3 on HOX genes (131, 132).
A related phenomenon within the histone methylation story involves LSD1. LSD1 is a demethylase commonly associated with the demethylation of the activating mark of H3K4 methylation, despite this an alternative role has been postulated for its role in the demethylation of H3K9me3 (72). In the presence of androgen specific receptors LSD1's function alters to selectively control H3K9 methylation resulting in gene repression of androgen specific markers (73, 74).
Originally identified in normal and cancerous prostate cells, LSD1 interacts with androgen nuclear receptor and stimulates androgen receptor dependent transcription via its interaction with a number of factors which together, both as direct effectors or in a combinational manner contribute to the altered transcriptional outlook of disease (75).
Therefore, depending on its associated co-factor LSD1 can either be involved in activation accessibility to chromatin or as a repressive. Similarly this functions in a reciprocal manner, by inhibiting the function of LSD1 in androgen receptor dependent phenotype androgen receptor binding is altered (76). The direct mechanism of this ability to influence the substrate specificity of LSD1 is largely unknown however roles have been postulated for other post translational modifications such as phosphorylation and protein kinases. Similarly a dual interaction has been identified of LSD1 with KDM4C in an androgen receptor mediated system (77).
At a higher level than direct methylation of H3K9, many of the histone modification enzymes are under a form of regulation by the PRDM family of proteins. Themselves with varying known roles in maintaining H3K4 and H3K36 methylation they are thought to recruit intrinsic histone methyltransferases and demethylases to H3K9 influencing methylation status (82). Containing a similar domain to the SET domain conserved within the demethylase family known as PR domain, they interact with H3K9 methylation with varying effects. A few members have been postulated to have roles either directly or indirectly (133). PRDM9 with its main role in the identification and tethering of recombination hotspots during meiosis, binds and recruits other proteins into a multi-protein complex interacting with G9A, previously described as a H3K9me2 methyltransferase and CDYL a reader of the di methyl mark (78–82). PRDM2 is one of few members which have an identified histone methyltransferase activity and is known to work in an estrogen activation dependent manner mediating promotor repression via H3K9 methylation (82). Again PRDM14 functions in a pathway through the G9a protein in early stem cell and embryonic development and has a role not only in H3K9me2 maintenance but also H3K27me3 levels hypothesized to have a role in AML (82). In contrast to this indirect method PRDM8 is thought to have an intrinsic methyltransferase activity in mouse tissue but the function of the human homolog has not been widely explored (82, 83).
Thus, elucidating the regulatory mechanisms contributing to the multifaceted role of H3K9me3 in hematological malignancies will provide insights in aid of novel anti-leukaemic therapy development.
Therapeutic Relevance of H3K9 Methylation and Its Regulators in AML
H3K9 Methylation Status Can Determine Survival in AML
In AML, specific histone methylome patterns owing to the deregulation of histone methyltransferases and demethylases bring with them a phenotype of disease, this could prove to be key in assessing patient's outcome and treatment regime (69, 134). Recent work has been carried out with the aim of improving the current prognostic diagnosis of AML via the incorporation of both epigenetic factors and genetic markers (135). In terms of histone methylation, it has been determined that global epigenome changes can refine the disease classification system in a large cohort of AML patients. Over 2000 genomic loci, around 453 promoters were found to have differential expression owing to changes in H3K9me3. It was then further investigated if these changes could distinguish the cell's origin and if a signature of histone modifications could effectively predict the outcome of the disease. By comparing AML to Acute Lymphoblastic Leukemia (ALL) and normal white blood cell (WBC) populations it was found that independent of karyotype, age or mutation status, promoter H3K9me3 levels correlated with the outcome of the disease in >70% of cases (13).
H3K9 Methylation Regulators as Potential Therapeutic Targets in AML
A better understanding of the regulatory pathways which govern this dynamic mark has continuously proven to be key in the precise selection of fundamental functional targets. The increasing importance of post translational regulatory proteins highlight them as potential therapeutically targets. Lysine-methylation and other modifications are complex networks which will take significant time to completely reveal its mechanisms (136). However, a number of studies and groups both industrial and academic are currently focused on collaborating to develop synthetic, organic and peptidomimetric chemistry to produce a wide variety of inhibitors that are capable of targeting epigenetic regulating enzymes. While great progress is being made, selectivity remains a significant challenge.
Inhibitors of Writers of H3K9 Methylation as Novel Therapeutic Agents1
Originally, Chaetocin 1 (Figure 2; Table 2) was reported as the first specific inhibitor for SUV39H1/H2, with an IC50 of 0.8 uM (61, 138). However, follow-up studies showed that this inhibition was non-specific and time-dependent, therefore, Chaetocin was unsuitable to act as a selective chemical probe for histone lysine methyl transferases (139, 140, 143). Most current drug developments involve targeting G9a including competition with the S-adenosylmethionine (SAM) cofactor for example inhibitor BRD4770 2 (144). It is postulated that G9a/GLP, SUV39H1, and SETDB1 coexist in the same multi-protein complex together with other post-translational modifying proteins. Having an interdependency on one another knockdown or inhibition of SUV39H1 has the ability to not only also destabilize the G9a,GLP, and SETDB1 proteins but also affect the downstream targets of these proteins independently with a causal effect in histone methylase levels and activity (126). High throughput screening of over 125,000 compounds identified BIX01294 3, one of only seven compounds which showed any efficacy via inhibition of G9a/GLP against H3K9me2 (145). It was selected for further analysis due to its alternative method of binding in comparison with non-specific generic analogs which inhibit via cofactor S-adenosyl-methionine (SAM), such as methylthio-adenosine (MTA), sinefungin, and S-adenosyl-homocysteine (SAH) binding (141). However, it was found to have a high degree of toxicity against human cells restraining its potential (141). Using the 2,4-diamino-6,7-dimethoxyquinazoline template, Kim et al. developed UNC0632 4, a selective G9a and GLP inhibitor that was less toxic but also less potent than BIX01294 because of poor cell permeability (146).
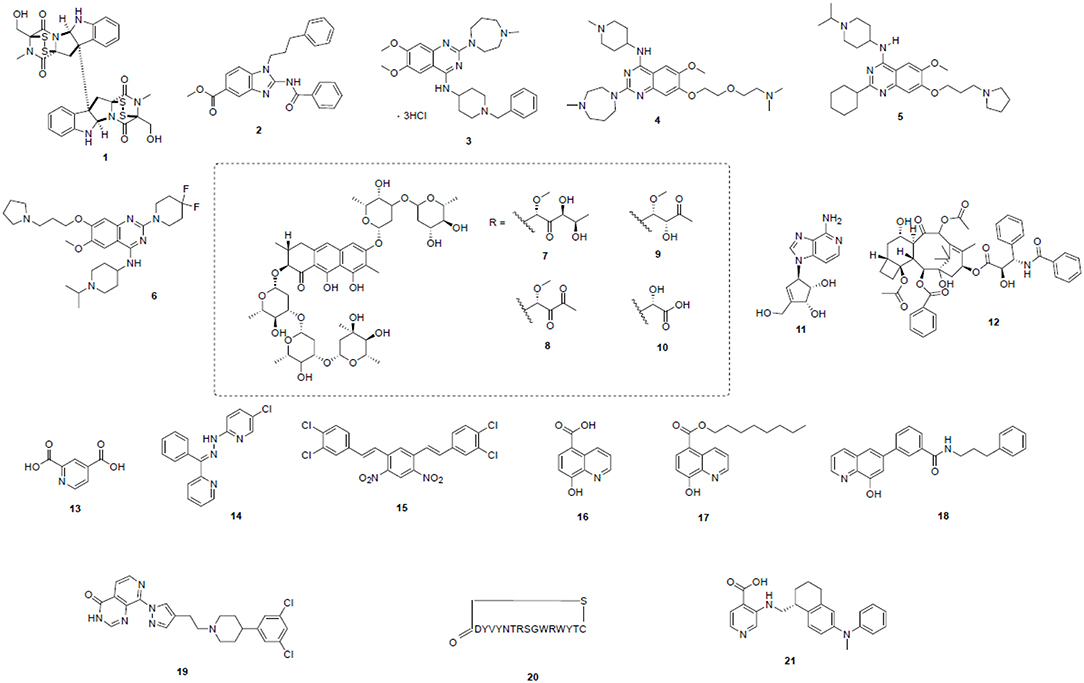
Figure 2. Potent epigenetic inhibitors targeting H3K9 modifying enzymes—including SUV39H1/2 (1), G9a (2–6), SETDB1 (7–12), and KDMs (13–21). Compound structures are numbered according to Table 2 and throughout the text.
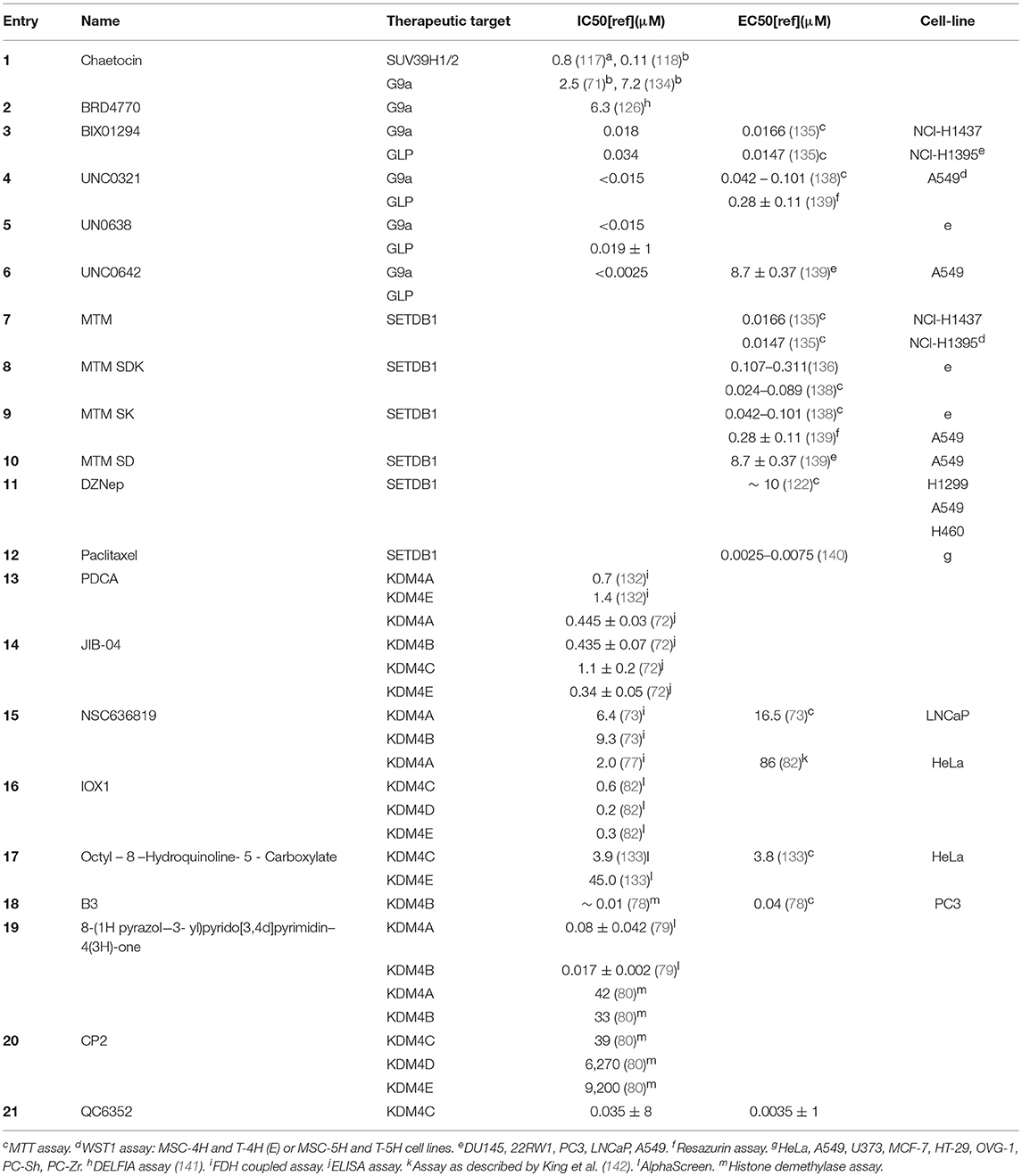
Table 2. Overview of aAssay as described by Eskeland et al. (137) bAlphaLISA assay detecting H3K9 methylation.
To improve the cell permeability they utilized a previously established structure activity relationship of the quinazoline scaffold to design a new generation of inhibitors with better cell permeability while maintaining the same potency. Among these compounds UNC0638 5 had the best potency and physiochemical potency and subsequently UNC0642 6 a selective inhibitors of EHMT2 (G9a) inducing a selective H3K9me2 decrease (146).
Although the importance of SETDB1 as a potential therapeutic target was recently demonstrated, selective inhibitors for SETDB1 have not been extensively described (147). A number of potential nonspecific inhibitors including mithramycin (MTM, 7) and its analogs eg. MTM SDK 8, MTM SK 9, and MTM SD 10, together with the cyclopentenyl analog of 3-deazaadenosine, DZNep 11 have been postulated. The latter non-specifically inhibits histone methylases (148), and a decreased methylation of H3K9 was observed in the nucleus of DZNep-treated cells. Transcription of SETDB1 can be regulated by DZNep and its expression was decreased in lung cancer (149). Similarly Paclitaxel 12 is a, widely used anti-cancer drug and downregulation of SETDB1 at the transcriptional level by 12 resulted in the death of human lung cancer cells (150). However, little is known about the effectiveness of these drugs against hematological malignancies.
Inhibitors of Erasers of H3K9 Methylation as Novel Therapeutic Agents
As previously described lysine specific demethylation in humans is catalyzed by two subfamilies of demethylases, namely, 2-oxoglutarate (2OG)—and flavin-dependent lysine demethylases. Their active sites consist of the JmjC- and amine oxidase-like (AOL) domains, respectively. Structural similarities and the conservation of active sites within the KDM families, makes design of specific inhibitors challenging. The concepts of KDM inhibition have been well-covered by Thinnes, Kaniskan and Lohse et al., who have described a wide variety of JmjC inhibitors (114, 136, 151). Specific inhibitors for KDM4 have been described in detail (67, 152). In AML with a major focus on H3K9me3 and specifically the KDM4A-D subfamily which catalyze its demethylation inhibitors of this specific family will be discussed here in more detail.
The KDM subfamily containing the JmjC domain has a Fe (II) containing catalytic site (153). Unlike the AOL domain, the JmjC domain is able to demethylate trimethyl lysine. The catalytic cycle requires the co-factors molecular oxygen and 2OG (154). Most inhibitors are small molecules and will, either act as 2OG mimics competing with 2OG, or act by chelating Fe2+ in the active site. Early promising metal binding inhibitors included pyridine-2, 4-dicarboxylic acid (PDCA, 13), capable of selective inhibition of KDM4A and KDM4E (IC50 = 0.7 and 1.4 μM, respectively) over the other JmjC containing KDMs (155). In the search for potential inhibitors for 2OG-oxygenases, Wang et al. uncovered JIB-04 14, a hydrazone derivative. Although 14 was a potent inhibitor, it was not selective and a range of KDM family members were inhibited including KDM4A (IC50: 0.445 ± 0.03 μM) and KDM4C (IC50:1.1 ± 0.2 μM) (156). NSC636819 15, was uncovered via a structure guided compound screen with available docking and domain knowledge. Although it was capable of inhibiting KDM4's with an association for KDM4A (IC50 = 6.4 μM) and KDM4B (IC50 = 9.3 μM) it was not as potent as other inhibitors published at the time (namely JIB-04) (157) and in a prostate cancer model has a reduced inhibitory concentration of 16.5 μM showing a reduced cell permeability and specificity. 15 functions as a competitive inhibitor and despite have a relatively low efficacy against KDM4 family members may provide an alternative framework base for drug development. Similar to 14, 15 is also capable of inhibition of Prolyl-hydroxylase and HIF1α in the micromolar range (158). A clear disadvantage in inhibitor 15 is the presence of nitro aromatic moieties, which are potentially carcinogenic and therefore undesirable (159, 160). Interestingly downstream gene targets of prostate cancer cell lines treated with 11 showed a number of pathways alongside 27% androgen response gene targets identifying a possible crossover function of LSD1 and KDM4A/B.
High throughput screening of potential inhibitors for 2OG-oxygenases resulted in uncovering 8-hydroxyquinoline-5-carboxylic acid (IOX1, 16), with an IC50 of 0.2 μM against KDM4A specifically (142). in addition to an EC50 value of in HeLa cells of 86 μM. Clearly, the polar character of IOX1 caused by the carboxylic acid moiety, hydroxyl, and amino groups hinder membrane permeability, explaining the >400-fold decrease in potency biochemical assays as compared to that in cells (161). To improve the membrane permeability, Schiller et al. (161) synthesized ester derivatives of 16 with varying lengths of alkyl groups. Evaluation of activity against KDM4A as a representative histone demethylase by immunofluorescence identified by transiently overexpressing KDM4A in a HeLa cell line model and then subsequently treating with each compound individually, H3K9me3 levels following treatment significantly increased on comparison with both control and overexpression of a mutant KDM4A protein, as a negative. They found that an n-octyl ester derivative, octyl 8-hydroxyquinoline-5-carboxylate 17 was superior to its parent compound likely due to increased membrane permeability (162). A more extended ALPHA screen assay was then used to compare the selectivity of 17 against other 2OG oxygenase families in comparison to their KDM4 function. The results support the classification of 16 as a broad-spectrum 2OG-oxygenases inhibitor (161). Compound 17 restricted the observed inhibitory activity to the KDM4 subfamily. Specifically, KDM4C was inhibited most potently with an IC50 value of 0.6 μM (161). 17 was also the least cytotoxic compound and the most potent inhibitor of H3K9me3 demethylation with an IC50 of 3.8 μM (162).
Additionally, an 8-hydroxyquinoline containing compound was utilized to inhibit KDM4B. 3-(8-Hydroxyquinolin-6-yl)-N-(3-phenylpropyl) benzamide (B3, 18) inhibited KDM4B with an IC50 of ca. 10 nM in enzymatic studies and also significantly inhibited the enzymatic activity of other KDM4 isoforms (163). Another very potent KDM4A/B inhibitor class was developed by Bavetsias et al. (139) and comprised the N-substituted4-(pyridin-2-yl) thiazole-2-amine derivatives which were optimized to give 8-(1H-pyrazol-3-yl)pyrido[3,4-d]pyrimidin-4(3H)-ones 19. Having a 4- (3,5-dichlorophenyl) piperidine linker this compound was the most potent in the series, capable of inhibition of KDM4A with an IC50 value of 80 nM and KDM4B with an IC50 value of 17 nM (164). Kawamura et al. reported cyclic peptide inhibitors of KDM4A-C demethylase enzymes with selectivity over other KDMs, including closely related KDM4D/E isoforms. CP2 20 was uncovered as the most promising cyclic peptide. 20 selectively inhibited KDM4A-C with IC50 values of 42, 33 and 39 nM, respectively (165).
The KDM4 family are deregulated in many cancer phenotypes, one which has been the focus of drug discovery in recent times is breast cancer. Breast cancer stem cells as they have been termed have been postulated to be responsible for metastasis and resistance to current therapies. This stem cell population is poorly characterized but thought to involve KDM4 family members for maintenance. Using a 3D tissue model and cell free assays Metzger et al. (166) showed that knockdown of KDM4A had effect in breast cancer tumors in mouse models through its control of cell proliferation (166). They went on to utilize a compound identified by Chen et al. (167) known as QC6532 to chemically inhibit the action of KDM4 family members. QC6532 (21) was developed through a complex strategy of compound modification. Through screening of an established database they selected an appropriate base structure with known efficacy by substituting the carboxylic acid position they selectively increased hydrophobic interactions and improved cellular permeability, alongside optimizing the position of tetrahydronaphthalene to increase potency (167). Intermediate compounds showed favorable interactions with the KDM4 family and through using crystal structural analysis the group designed compounds substituting on the active site aromatic ring. QC6352 showed cellular activity with a relatively reduced potency however perceived activity against KDM5 family remained significantly high to an extent that it may influence the phenotype observed (167). It has promising efficacy against breast cancer cell lines and 3D culture models and encouragingly is readily orally available in mouse models with low clearance and a high volume of distribution (166). Other inhibitors such as A366 have been identified however as their main target so far has only been identified to be KDM4 family action on H3K4 methylation they are outwith the scope of this review (168).
Conclusion
H3K9me3 is a dynamic covalent post-translational histone modification which is known to be pathologically deregulated in a number of diseases including haematopoietic malignancies. It is tightly controlled by a number of enzymes which place, maintain and remove H3K9me3 such as the KDM4 family which are rapidly emerging as factors which demonstrate a strong regulatory influence on oncogenic gene transcription in AML. Whilst progress has been made in targeting and developing of inhibitors for the specific H3K9me3 methyltransferases or demethylases, selectivity remains an unmet challenge. The worrying statistics which accompany AML with a 5 year survival rate of <40%, highlight the necessity to continually drive to identify new mechanisms for the development of de-novo AML both causative and as consequences of driver mutations. Alongside this identifying and developing the ability to target these functions specifically. A comprehensive understanding of the oncogenic function of H3K9me3 within AML cells and the respective epigenetic regulators that fine-tune its abundance will permit the development of more efficient therapeutic strategies in the future.
Author Contributions
LM, MM, RB, AH, and P-AP drafted the manuscript and prepared figures. RL, HJ, and XH critically analyzed and revised the manuscript to its final form. All authors read and approved the final manuscript.
Funding
This work was supported by the Wellcome Trust [105614/Z/14/Z]; Leuka [2016/JGF/0005]; the Howat Foundation and Friends of Paul O'Gorman; and SULSA-PECRE; XH is a John Goldman Fellow; LM is a recipient of Adam Renwick Martin-Friends of Paul O'Gorman Ph.D. Studentship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AML, Acute myeloid leukemia; H3K9me3, Histone 3 lysine 9 trimethylation; HSC, Haematopoietic stem cells; LSC, Leukaemic stem cell; HP1, Heterochromatin protein 1; TSS, Transcriptional start site; HAT, Histone acetylates; PEV, Position effect variegation; CD, Chromo domain; SET, Suppressor of variegation, enhancer of zeste and trithorax; TGFβ, Transforming Growth Factor beta; MECOM, MDS1 and EVI1 Complex Locus Protein; BCL11B, B-cell lymphoma/leukemia 11B; HDAC, Histone Deacetylases; SETDB1, SET domain, bifurcated 1; SETDB2, SET domain, bifurcated 2; MLL, Mixed Lineage Leukemia; α-KG, α-ketoglutarate; KDM4, Lysine-specific demethylase 4; JAK/STAT, Janus Kinase/Signal Transducer and Activator of Transcription; PRC, Polycomb Repressive Complexes; ALL, Acute Lymphoblastic Leukemia; WBC, Normal white blood cell; SAM, S-adenosylmethionine; 2OG, 2-oxoglutarate; AOL, JmjC- and amine oxidase-like.
References
1. Felsenfeld G. A brief history of epigenetics. Cold Spring Harb Pers pect Biol. (2014) 6:a018200. doi: 10.1101/cshperspect.a018200
3. Allfrey VG, Mirsky AE. Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science. (1964) 144:559. doi: 10.1126/science.144.3618.559
4. Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. (2000) 406:593–9. doi: 10.1038/35020506
5. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012
6. Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. (2002) 3:179–98. doi: 10.1146/annurev.genom.3.032802.115046
7. Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. (2012) 12:599–612. doi: 10.1038/nrc3343
8. Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood. (2013) 121:3563–72. doi: 10.1182/blood-2013-01-451781
9. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. (2009) 361:1058–66. doi: 10.1056/NEJMoa0903840
10. Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. (2011) 43:309–15. doi: 10.1038/ng.788
11. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
12. Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. (2012) 26:934–42. doi: 10.1038/leu.2011.326
13. Müller-Tidow C, Klein HU, Hascher A, Isken F, Tickenbrock L, Thoennissen N, et al. Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood. (2010) 116:3564–71. doi: 10.1182/blood-2009-09-240978
14. Sharma S, Gurudutta G. Epigenetic regulation of hematopoietic stem cells. Int J Stem Cells. (2016) 9:36–43. doi: 10.15283/ijsc.2016.9.1.36
15. Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. (2001) 410:116–20. doi: 10.1038/35065132
16. Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. (2001) 292:110–3. doi: 10.1126/science.1060118
17. Nielsen AL, Oulad-Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol Cell. (2001) 7:729–39. doi: 10.1016/S1097-2765(01)00218-0
18. Allshire RC, Madhani HD. Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol. (2018) 19:229–44. doi: 10.1038/nrm.2017.119
19. Muramatsu D, Kimura H, Kotoshiba K, Tachibana M, Shinkai Y. Pericentric H3K9me3 formation by HP1 interaction-defective histone methyltransferase Suv39h1. Cell Struct Funct. (2016) 41:145–52. doi: 10.1247/csf.16013
20. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. (2001) 410:120–4. doi: 10.1038/35065138
21. Kim J, Kim H. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J. (2012) 53:232–9. doi: 10.1093/ilar.53.3-4.232
22. Becker JS, Nicetto D, Zaret KS. H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends Genet. (2016) 32:29–41. doi: 10.1016/j.tig.2015.11.001
23. Foret MR, Sandstrom RS, Rhodes CT, Wang Y, Berger MS, Lin CH. Molecular targets of chromatin repressive mark H3K9me3 in primate progenitor cells within adult neurogenic niches. Front Genet. (2014) 5:252. doi: 10.3389/fgene.2014.00252
24. Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. Dynamics and memory of heterochromatin in living cells. Cell. (2012) 149:1447–60. doi: 10.1016/j.cell.2012.03.052
25. Zhang K, Mosch K, Fischle W, Grewal SI. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol. (2008) 15:381–8. doi: 10.1038/nsmb.1406
26. Olcina MM, Leszczynska KB, Senra JM, Isa NF, Harada H, Hammond EM. H3K9me3 facilitates hypoxia-induced p53-dependent apoptosis through repression of APAK. Oncogene. (2016) 35:793–9. doi: 10.1038/onc.2015.134
27. Lu C, Yang D, Sabbatini ME, Colby AH, Grinstaff MW, Oberlies NH, et al. Contrasting roles of H3K4me3 and H3K9me3 in regulation of apoptosis and gemcitabine resistance in human pancreatic cancer cells. BMC Cancer. (2018) 18:149. doi: 10.1186/s12885-018-4061-y
28. Biga PR, Latimer MN, Froehlich JM, Gabillard JC, Seiliez I. Distribution of H3K27me3, H3K9me3, and H3K4me3 along autophagy-related genes highly expressed in starved zebrafish myotubes. Biol Open. (2017) 6:1720–5. doi: 10.1242/bio.029090
29. Fujiwara K, Fujita Y, Kasai A, Onaka Y, Hashimoto H, Okada H, et al. Deletion of JMJD2B in neurons leads to defective spine maturation, hyperactive behavior and memory deficits in mouse. Transl Psychiatry. (2016) 6:e766. doi: 10.1038/tp.2016.31
30. Magaraki A, van der Heijden G, Sleddens-Linkels E, Magarakis L, van Cappellen WA, Peters A, et al. Silencing markers are retained on pericentric heterochromatin during murine primordial germ cell development. Epigenetics Chromatin. (2017) 10:11. doi: 10.1186/s13072-017-0119-3
31. Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. (2009) 11:1376–82. doi: 10.1038/ncb1982
32. Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci USA. (2014) 111:9169–74. doi: 10.1073/pnas.1403565111
33. Khoury-Haddad H, Nadar-Ponniah PT, Awwad S, Ayoub N. The emerging role of lysine demethylases in DNA damage response: dissecting the recruitment mode of KDM4D/JMJD2D to DNA damage sites. Cell Cycle. (2015) 14:950–8. doi: 10.1080/15384101.2015.1014147
34. Wu R, Wang Z, Zhang H, Gan H, Zhang Z. H3K9me3 demethylase Kdm4d facilitates the formation of pre-initiative complex and regulates DNA replication. Nucleic Acids Res. (2017) 45:169–80. doi: 10.1093/nar/gkw848
35. Hausmann M, Wagner E, Lee JH, Schrock G, Schaufler W, Krufczik M, et al. Super-resolution localization microscopy of radiation-induced histone H2AX-phosphorylation in relation to H3K9-trimethylation in HeLa cells. Nanoscale. (2018) 10:4320–31. doi: 10.1039/C7NR08145F
36. Saint-Andre V, Batsche E, Rachez C, Muchardt C. Histone H3 lysine 9 trimethylation and HP1gamma favor inclusion of alternative exons. Nat Struct Mol Biol. (2011) 18:337–44. doi: 10.1038/nsmb.1995
37. Bieberstein NI, Kozakova E, Huranova M, Thakur PK, Krchnakova Z, Krausova M, et al. TALE-directed local modulation of H3K9 methylation shapes exon recognition. Sci Rep. (2016) 6:29961. doi: 10.1038/srep29961
38. Barrand S, Andersen IS, Collas P. Promoter-exon relationship of H3 lysine 9, 27, 36 and 79 methylation on pluripotency-associated genes. Biochem Biophys Res Commun. (2010) 401:611–7. doi: 10.1016/j.bbrc.2010.09.116
39. Pedersen MT, Kooistra SM, Radzisheuskaya A, Laugesen A, Johansen JV, Hayward DG, et al. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. (2016) 35:1550–64. doi: 10.15252/embj.201593317
40. Loh YH, Zhang W, Chen X, George J, Ng HH. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. (2007) 21:2545–57. doi: 10.1101/gad.1588207
41. Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell. (2005) 19:381–91. doi: 10.1016/j.molcel.2005.06.011
42. Maksakova IA, Goyal P, Bullwinkel J, Brown JP, Bilenky M, Mager DL, et al. H3K9me3-binding proteins are dispensable for SETDB1/H3K9me3-dependent retroviral silencing. Epigenetics Chromatin. (2011) 4:12. doi: 10.1186/1756-8935-4-12
43. Imai K, Kamio N, Cueno ME, Saito Y, Inoue H, Saito I, et al. Role of the histone H3 lysine 9 methyltransferase Suv39 h1 in maintaining Epsteinn-Barr virus latency in B95–8 cells. FEBS J. (2014) 281:2148–58. doi: 10.1111/febs.12768
44. Lang F, Li X, Vladimirova O, Hu B, Chen G, Xiao Y, et al. CTCF interacts with the lytic HSV-1 genome to promote viral transcription. Sci Rep. (2017) 7:39861. doi: 10.1038/srep39861
45. Fukuda A, Tomikawa J, Miura T, Hata K, Nakabayashi K, Eggan K, et al. The role of maternal-specific H3K9me3 modification in establishing imprinted X-chromosome inactivation and embryogenesis in mice. Nat Commun. (2014) 5:5464. doi: 10.1038/ncomms6464
46. Mendelsohn AR, Larrick JW. Stem cell depletion by global disorganization of the H3K9me3 epigenetic marker in aging. Rejuvenation Res. (2015) 18:371–5. doi: 10.1089/rej.2015.1742
47. Koide S, Oshima M, Takubo K, Yamazaki S, Nitta E, Saraya A, et al. Setdb1 maintains hematopoietic stem and progenitor cells by restricting the ectopic activation of nonhematopoietic genes. Blood. (2016) 128:638–49. doi: 10.1182/blood-2016-01-694810
48. McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. (2017) 49:367–76. doi: 10.1038/ng.3753
49. Chung YR, Schatoff E, Abdel-Wahab O. Epigenetic alterations in hematopoietic malignancies. Int J Hematol. (2012) 96:413–27. doi: 10.1007/s12185-012-1181-z
50. Lehmann U, Brakensiek K, Kreipe H. Role of epigenetic changes in hematological malignancies. Ann Hematol. (2004) 83:137–52. doi: 10.1007/s00277-003-0798-7
51. Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. (2011) 27:389–96. doi: 10.1016/j.tig.2011.06.006
52. Wiencke JK, Zheng S, Morrison Z, Yeh RF. Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene. (2008) 27:2412–21. doi: 10.1038/sj.onc.1210895
53. Xu J, Kidder BL. H4K20me3 co-localizes with activating histone modifications at transcriptionally dynamic regions in embryonic stem cells. BMC Genomics. (2018) 19:514. doi: 10.1186/s12864-018-4886-4
54. Ruscetti FW, Akel S, Bartelmez SH. Autocrine transforming growth factor-beta regulation of hematopoiesis: many outcomes that depend on the context. Oncogene. (2005) 24:5751–63. doi: 10.1038/sj.onc.1208921
55. Geyer CR. Strategies to re-express epigenetically silenced p15(INK4b) and p21(WAF1) genes in acute myeloid leukemia. Epigenetics. (2010) 5:696–703. doi: 10.4161/epi.5.8.13276
56. Cherrier T, Suzanne S, Redel L, Calao M, Marban C, Samah B, et al. p21(WAF1) gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene. (2009) 28:3380–9. doi: 10.1038/onc.2009.193
57. Hinai AA, Valk PJ. Review: aberrant EVI1 expression in acute myeloid leukaemia. Br J Haematol. (2016) 172:870–8. doi: 10.1111/bjh.13898
58. Lugthart S, van Drunen E, van Norden Y, van Hoven A, Erpelinck CA, Valk PJ, et al. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. (2008) 111:4329–37. doi: 10.1182/blood-2007-10-119230
59. Glass C, Wuertzer C, Cui X, Bi Y, Davuluri R, Xiao YY, et al. Global identification of EVI1 target genes in acute myeloid leukemia. PLoS ONE. (2013) 8:e67134. doi: 10.1371/journal.pone.0067134
60. Goyama S, Nitta E, Yoshino T, Kako S, Watanabe-Okochi N, Shimabe M, et al. EVI-1 interacts with histone methyltransferases SUV39H1 and G9a for transcriptional repression and bone marrow immortalization. Leukemia. (2010) 24:81–8. doi: 10.1038/leu.2009.202
61. Lai YS, Chen JY, Tsai HJ, Chen TY, Hung WC. The SUV39H1 inhibitor chaetocin induces differentiation and shows synergistic cytotoxicity with other epigenetic drugs in acute myeloid leukemia cells. Blood Cancer J. (2015) 5:e313. doi: 10.1038/bcj.2015.37
62. Jurkowska RZ, Qin S, Kungulovski G, Tempel W, Liu Y, Bashtrykov P, et al. H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat Commun. (2017) 8:2057. doi: 10.1038/s41467-017-02259-9
63. Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol. (2017) 216:3535–49. doi: 10.1083/jcb.201612160
64. Robbez-Masson L, Tie CHC, Rowe HM. Cancer cells, on your histone marks, get SETDB1, silence retrotransposons, and go! J Cell Biol. (2017) 216:3429–31. doi: 10.1083/jcb.201710068
65. Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. (2002) 16:919–32. doi: 10.1101/gad.973302
66. Ropa J, Saha N, Chen Z, Serio J, Chen W, Mellacheruvu D, et al. PAF1 complex interactions with SETDB1 mediate promoter H3K9 methylation and transcriptional repression of. Oncotarget. (2018) 9:22123–36. doi: 10.18632/oncotarget.25204
67. Labbé RM, Holowatyj A, Yang Z-Q. Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. Am J Transl Res. (2014) 6:1–15. Available online at: http://www.ajtr.org/
68. Boila LD, Chatterjee SS, Banerjee D, Sengupta A. KDM6 and KDM4 histone lysine demethylases emerge as molecular therapeutic targets in human acute myeloid leukemia. Exp Hematol. (2018) 58:44–51.e7. doi: 10.1016/j.exphem.2017.10.002
69. D'Oto A, Tian Q-W, Davidoff AM, Yang J. Histone demethylases and their roles in cancer epigenetics. J Med Oncol Ther. (2016) 1:34–40. Available online at: http://www.alliedacademies.org/medical-oncology-therapeutics/
70. Agger K, Miyagi S, Pedersen MT, Kooistra SM, Johansen JV, Helin K. Jmjd2/Kdm4 demethylases are required for expression of Il3ra and survival of acute myeloid leukemia cells. Genes Dev. (2016) 30:1278–88. doi: 10.1101/gad.280495.116
71. de la Cruz CC, Kirmizis A, Simon MD, Isono K, Koseki H, Panning B. The polycomb group protein SUZ12 regulates histone H3 lysine 9 methylation and HP1 alpha distribution. Chromosome Res. (2007) 15:299–314. doi: 10.1007/s10577-007-1126-1
72. Cucchiara V, Yang JC, Mirone V, Gao AC, Rosenfeld MG, Evans CP. Epigenomic regulation of androgen receptor signaling: potential role in prostate cancer therapy. Cancers. (2017) 9:9. doi: 10.3390/cancers9010009
73. Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. (2005) 437:436–9. doi: 10.1038/nature04020
74. Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, et al. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep. (2014) 9:1618–27. doi: 10.1016/j.celrep.2014.11.008
75. Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. (2011) 20:457–71. doi: 10.1016/j.ccr.2011.09.001
76. Sun H, Liang L, Li Y, Feng C, Li L, Zhang Y, et al. Lysine-specific histone demethylase 1 inhibition promotes reprogramming by facilitating the expression of exogenous transcriptional factors and metabolic switch. Sci Rep. (2016) 6:30903. doi: 10.1038/srep30903
77. Wissmann M, Yin N, Müller JM, Greschik H, Fodor BD, Jenuwein T, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. (2007) 9:347–53. doi: 10.1038/ncb1546
78. Walker M, Billings T, Baker CL, Powers N, Tian H, Saxl RL, et al. Affinity-seq detects genome-wide PRDM9 binding sites and reveals the impact of prior chromatin modifications on mammalian recombination hotspot usage. Epigenetics Chromatin. (2015) 8:31. doi: 10.1186/s13072-015-0024-6
79. Houle AA, Gibling H, Lamaze FC, Edgington HA, Soave D, Fave MJ, et al. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res. (2018) 28:1611–20. doi: 10.1101/gr.231696.117
80. Parvanov ED, Tian H, Billings T, Saxl RL, Spruce C, Aithal R, et al. PRDM9 interactions with other proteins provide a link between recombination hotspots and the chromosomal axis in meiosis. Mol Biol Cell. (2017) 28:488–99. doi: 10.1091/mbc.e16-09-0686
81. Powers NR, Parvanov ED, Baker CL, Walker M, Petkov PM, Paigen K. The Meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots in vivo. PLoS Genet. (2016) 12:e1006146. doi: 10.1371/journal.pgen.1006146
82. Hohenauer T, Moore AW. The Prdm family: expanding roles in stem cells and development. Development. (2012) 139:2267–82. doi: 10.1242/dev.070110
83. Eom GH, Kim K, Kim SM, Kee HJ, Kim JY, Jin HM, et al. Histone methyltransferase PRDM8 regulates mouse testis steroidogenesis. Biochem Biophys Res Commun. (2009) 388:131–6. doi: 10.1016/j.bbrc.2009.07.134
84. O'Carroll D, Scherthan H, Peters AH, Opravil S, Haynes AR, Laible G, et al. Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol. (2000) 20:9423–33. doi: 10.1128/MCB.20.24.9423-9433.2000
85. Czvitkovich S, Sauer S, Peters AH, Deiner E, Wolf A, Laible G, et al. Over-expression of the SUV39H1 histone methyltransferase induces altered proliferation and differentiation in transgenic mice. Mech Dev. (2001) 107:141–53. doi: 10.1016/S0925-4773(01)00464-6
86. Patnaik D, Chin HG, Estève PO, Benner J, Jacobsen SE, Pradhan S. Substrate specificity and kinetic mechanism of mammalian G9a histone H3 methyltransferase. J Biol Chem. (2004) 279:53248–58. doi: 10.1074/jbc.M409604200
87. Muller MM, Fierz B, Bittova L, Liszczak G, Muir TW. A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat Chem Biol. (2016) 12:188–93. doi: 10.1038/nchembio.2008
88. Shirai A, Kawaguchi T, Shimojo H, Muramatsu D, Ishida-Yonetani M, Nishimura Y, et al. Impact of nucleic acid and methylated H3K9 binding activities of Suv39h1 on its heterochromatin assembly. Elife. (2017) 6. doi: 10.7554/eLife.25317
89. Fortunel NO, Hatzfeld A, Hatzfeld JA. Transforming growth factor-beta: pleiotropic role in the regulation of hematopoiesis. Blood. (2000) 96:2022–36. Available online at: http://www.bloodjournal.org/
90. Dong M, Blobe GC. Role of transforming growth factor-beta in hematologic malignancies. Blood. (2006) 107:4589–96. doi: 10.1182/blood-2005-10-4169
91. Abbas S, Sanders MA, Zeilemaker A, Geertsma-Kleinekoort WM, Koenders JE, Kavelaars FG, et al. Integrated genome-wide genotyping and gene expression profiling reveals BCL11B as a putative oncogene in acute myeloid leukemia with 14q32 aberrations. Haematologica. (2014) 99:848–57. doi: 10.3324/haematol.2013.095604
92. Bezrookove V, van Zelderen-Bhola SL, Brink A, Szuhai K, Raap AK, Barge R, et al. A novel t((6) 14)(q25-q(27) q32) in acute myelocytic leukemia involves the BCL11B gene. Cancer Genet Cytogenet. (2004) 149:72–6. doi: 10.1016/S0165-4608(03)00302-9
93. Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene. (2010) 29:576–88. doi: 10.1038/onc.2009.361
94. Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. (2013) 73:2936–42. doi: 10.1158/0008-5472.CAN-12-4300
95. Lyko F, Ramsahoye BH, Jaenisch R. DNA methylation in Drosophila melanogaster. Nature. (2000) 408:538–40. doi: 10.1038/35046205
96. Fuks F. DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev. (2005) 15:490–5. doi: 10.1016/j.gde.2005.08.002
97. Falandry C, Fourel G, Galy V, Ristriani T, Horard B, Bensimon E, et al. CLLD8/KMT1F is a lysine methyltransferase that is important for chromosome segregation. J Biol Chem. (2010) 285:20234–41. doi: 10.1074/jbc.M109.052399
98. Yang L, Xia L, Wu DY, Wang HB, Chansky HA, Schubach WH, et al. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene. (2002) 21:148–52. doi: 10.1038/sj.onc.1204998
99. Harte PJ, Wu W, Carrasquillo MM, Matera AG. Assignment of a novel bifurcated SET domain gene, SETDB1, to human chromosome band 1q21 by in situ hybridization and radiation hybrids. Cytogenet Cell Genet. (1999) 84:83–6. doi: 10.1159/000015220
100. Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. (2006) 125:467–81. doi: 10.1016/j.cell.2006.03.028
101. Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. (2006) 442:307–11. doi: 10.1038/nature04837
102. Fodor BD, Kubicek S, Yonezawa M, O'Sullivan RJ, Sengupta R, Perez-Burgos L, et al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. (2006) 20:1557–62. doi: 10.1101/gad.388206
103. Katoh M. Identification and characterization of JMJD2 family genes in silico. Int J Oncol. (2004) 24:1623–8. doi: 10.3892/ijo.25.3.759
104. Shin S, Janknecht R. Diversity within the JMJD2 histone demethylase family. Biochem Biophys Res Commun. (2007) 353:973–7. doi: 10.1016/j.bbrc.2006.12.147
105. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. (2006) 439:811–6. doi: 10.1038/nature04433
106. Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol. (2007) 14:689–95. doi: 10.1038/nsmb1273
107. Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. (2006) 312:748–51. doi: 10.1126/science.1125162
108. Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BM, et al. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. (2007) 448:87–91. doi: 10.1038/nature05971
109. Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. (2006) 125:691–702. doi: 10.1016/j.cell.2006.04.024
110. Pack LR, Yamamoto KR, Fujimori DG. Opposing chromatin signals direct and regulate the activity of lysine demethylase 4C (KDM4C). J Biol Chem. (2016) 291:6060–70. doi: 10.1074/jbc.M115.696864
111. Hancock RL, Masson N, Dunne K, Flashman E, Kawamura A. The activity of JmjC histone lysine demethylase KDM4A is highly sensitive to oxygen concentrations. ACS Chem Biol. (2017) 12:1011–9. doi: 10.1021/acschembio.6b00958
112. Tausendschon M, Dehne N, Brune B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine. (2011) 53:256–62. doi: 10.1016/j.cyto.2010.11.002
113. Greenblatt SM, Nimer SD. Chromatin modifiers and the promise of epigenetic therapy in acute leukemia. Leukemia. (2014) 28:1396–406. doi: 10.1038/leu.2014.94
114. Lohse B, Kristensen JL, Kristensen LH, Agger K, Helin K, Gajhede M, et al. Inhibitors of histone demethylases. Bioorg Med Chem. (2011) 19:3625–36. doi: 10.1016/j.bmc.2011.01.046
115. Højfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. (2013) 12:917–30. doi: 10.1038/nrd4154
116. Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, et al. Targeting aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute myeloid leukemia. Cancer Cell. (2016) 29:32–48. doi: 10.1016/j.ccell.2015.12.007
117. Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. (2002) 100:2980–8. doi: 10.1182/blood-2002-03-0852
118. Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. (2015) 16:1467–81. doi: 10.15252/embr.201540945
119. Li Q, Wang X, Lu Z, Zhang B, Guan Z, Liu Z, et al. Polycomb CBX7 directly controls trimethylation of histone H3 at lysine 9 at the p16 locus. PLoS ONE. (2010) 5:e13732. doi: 10.1371/journal.pone.0013732
120. Linggi BE, Brandt SJ, Sun ZW, Hiebert SW. Translating the histone code into leukemia. J Cell Biochem. (2005) 96:938–50. doi: 10.1002/jcb.20604
121. Godley LA, Le Beau MM. The histone code and treatments for acute myeloid leukemia. N Engl J Med. (2012) 366:960–1. doi: 10.1056/NEJMcibr1113401
122. Kaidi A, Jackson SP. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. (2013) 498:70–4. doi: 10.1038/nature12201
123. Rose NR, Klose RJ. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta. (2014) 1839:1362–72. doi: 10.1016/j.bbagrm.2014.02.007
124. Rothbart SB, Krajewski K, Nady N, Tempel W, Xue S, Badeaux AI, et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol. (2012) 19:1155–60. doi: 10.1038/nsmb.2391
125. Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J, et al. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat Commun. (2013) 4:1563. doi: 10.1038/ncomms2562
126. Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell. (2010) 37:46–56. doi: 10.1016/j.molcel.2009.12.017
127. Salzberg AC, Harris-Becker A, Popova EY, Keasey N, Loughran TP, Claxton DF, et al. Genome-wide mapping of histone H3K9me2 in acute myeloid leukemia reveals large chromosomal domains associated with massive gene silencing and sites of genome instability. PLoS ONE. (2017) 12:e0173723. doi: 10.1371/journal.pone.0173723
128. Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. (2008) 27:2681–90. doi: 10.1038/emboj.2008.192
129. Liu N, Zhang Z, Wu H, Jiang Y, Meng L, Xiong J, et al. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev. (2015) 29:379–93. doi: 10.1101/gad.254425.114
130. Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C, et al. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell. (2014) 53:277–89. doi: 10.1016/j.molcel.2013.12.005
131. Kondengaden SM, Luo LF, Huang K, Zhu M, Zang L, Bataba E, et al. Discovery of novel small molecule inhibitors of lysine methyltransferase G9a and their mechanism in leukemia cell lines. Eur J Med Chem. (2016) 122:382–93. doi: 10.1016/j.ejmech.2016.06.028
132. Lehnertz B, Pabst C, Su L, Miller M, Liu F, Yi L, et al. The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev. (2014) 28:317–27. doi: 10.1101/gad.236794.113
133. Fog CK, Galli GG, Lund AH. PRDM proteins: important players in differentiation and disease. Bioessays. (2012) 34:50–60. doi: 10.1002/bies.201100107
134. Chopra M, Bohlander SK. Disturbing the histone code in leukemia: translocations and mutations affecting histone methyl transferases. Cancer Genet. (2015) 208:192–205. doi: 10.1016/j.cancergen.2014.10.005
135. Marcucci G, Yan P, Maharry K, Frankhouser D, Nicolet D, Metzeler KH, et al. Epigenetics meets genetics in acute myeloid leukemia: clinical impact of a novel seven-gene score. J Clin Oncol. (2014) 32:548–56. doi: 10.1200/JCO.2013.50.6337
136. Thinnes CC, England KS, Kawamura A, Chowdhury R, Schofield CJ, Hopkinson RJ. Targeting histone lysine demethylases - progress, challenges, and the future. Biochim Biophys Acta. (2014) 1839:1416–32. doi: 10.1016/j.bbagrm.2014.05.009
137. Eskeland R, Czermin B, Boeke J, Bonaldi T, Regula JT, Imhof A. The N-terminus of drosophila SU(VAR)3–9 mediates dimerization and regulates its methyltransferase activity. Biochemistry. (2004) 43:3740–9. doi: 10.1021/bi035964s
138. Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat Chem Biol. (2005) 1:143–5. doi: 10.1038/nchembio721
139. Cherblanc FL, Chapman KL, Brown R, Fuchter MJ. Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases. Nat Chem Biol. (2013) 9:136–7. doi: 10.1038/nchembio.1187
140. Cherblanc FL, Chapman KL, Reid J, Borg AJ, Sundriyal S, Alcazar-Fuoli L, et al. On the histone lysine methyltransferase activity of fungal metabolite chaetocin. J Med Chem. (2013) 56:8616–25. doi: 10.1021/jm401063r
141. Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. (2007) 25:473–81. doi: 10.1016/j.molcel.2007.01.017
142. King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, et al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS ONE. (2010) 5:e15535. doi: 10.1371/journal.pone.0015535
143. Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Reply to “Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases.” Nat Chem Biol. (2013) 9:137. doi: 10.1038/nchembio.1188
144. Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, et al. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol. (2012) 7:1152–7. doi: 10.1021/cb300139y
145. Upadhyay AK, Rotili D, Han JW, Hu R, Chang Y, Labella D, et al. An analog of BIX-01294 selectively inhibits a family of histone H3 lysine 9 Jumonji demethylases. J Mol Biol. (2012) 416:319–27. doi: 10.1016/j.jmb.2011.12.036
146. Kim Y, Lee HM, Xiong Y, Sciaky N, Hulbert SW, Cao X, et al. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat Med. (2017) 23:213–22. doi: 10.1038/nm.4257
147. Karanth AV, Maniswami RR, Prashanth S, Govindaraj H, Padmavathy R, Jegatheesan SK, et al. Emerging role of SETDB1 as a therapeutic target. Expert Opin Ther Targets. (2017) 21:319–31. doi: 10.1080/14728222.2017.1279604
148. Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. (2009) 8:1579–88. doi: 10.1158/1535-7163.MCT-09-0013
149. Lee JK, Kim KC. DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem Biophys Res Commun. (2013) 438:647–52. doi: 10.1016/j.bbrc.2013.07.128
150. Bocci G, Di Paolo A, Danesi R. The pharmacological bases of the antiangiogenic activity of paclitaxel. Angiogenesis. (2013) 16:481–92. doi: 10.1007/s10456-013-9334-0
151. Kaniskan HU, Martini ML, Jin J. Inhibitors of Protein Methyltransferases and Demethylases. Chem Rev. (2018) 118:989–1068. doi: 10.1021/acs.chemrev.6b00801
152. Chin YW, Han SY. KDM4 histone demethylase inhibitors for anti-cancer agents: a patent review. Expert Opin Ther Pat. (2015) 25:135–44. doi: 10.1517/13543776.2014.991310
153. Bollinger JM, Price JC, Hoffart LM, Barr EW, Krebs C. Mechanism of Taurine: α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Eur J Inorg Chem. (2005) 2005:4245–54. doi: 10.1002/ejic.200500476
154. Williams ST, Walport LJ, Hopkinson RJ, Madden SK, Chowdhury R, Schofield CJ, et al. Studies on the catalytic domains of multiple JmjC oxygenases using peptide substrates. Epigenetics. (2014) 9:1596–603. doi: 10.4161/15592294.2014.983381
155. Rose NR, Ng SS, Mecinovic J, Lienard BM, Bello SH, Sun Z, et al. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J Med Chem. (2008) 51:7053–6. doi: 10.1021/jm800936s
156. Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. (2013) 4:2035. doi: 10.1038/ncomms3035
157. Chu CH, Wang LY, Hsu KC, Chen CC, Cheng HH, Wang SM, et al. KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor. J Med Chem. (2014) 57:5975–85. doi: 10.1021/jm500249n
158. Wang J, Wang H, Wang LY, Cai D, Duan Z, Zhang Y, et al. Silencing the epigenetic silencer KDM4A for TRAIL and DR5 simultaneous induction and antitumor therapy. Cell Death Differ. (2016) 23:1886–96. doi: 10.1038/cdd.2016.92
159. Strauss MJ. The nitroaromatic group in drug design. pharmacology and toxicology (for Nonpharmacologists). Ind Eng Chem Product Res Dev. (1979) 18:158–66. doi: 10.1021/i360071a002
160. Walsh JS, Miwa GT. Bioactivation of drugs: risk and drug design. Annu Rev Pharmacol Toxicol. (2011) 51:145–67. doi: 10.1146/annurev-pharmtox-010510-100514
161. Hopkinson RJ, Tumber A, Yapp C, Chowdhury R, Aik W, Che KH, et al. 5-Carboxy-8-hydroxyquinoline is a broad spectrum 2-oxoglutarate oxygenase inhibitor which causes iron translocation. Chem Sci. (2013) 4:3110–7. doi: 10.1039/c3sc51122g
162. Schiller R, Scozzafava G, Tumber A, Wickens JR, Bush JT, Rai G, et al. A cell-permeable ester derivative of the JmjC histone demethylase inhibitor IOX1. ChemMedChem. (2014) 9:566–71. doi: 10.1002/cmdc.201300428
163. Duan L, Rai G, Roggero C, Zhang QJ, Wei Q, Ma SH, et al. KDM4/JMJD2 histone demethylase inhibitors block prostate tumor growth by suppressing the expression of AR and BMYB-regulated genes. Chem Biol. (2015) 22:1185–96. doi: 10.1016/j.chembiol.2015.08.007
164. Bavetsias V, Lanigan RM, Ruda GF, Atrash B, McLaughlin MG, Tumber A, et al. 8-Substituted Pyrido[3,4-d]pyrimidin-4(3H)-one derivatives as potent, cell permeable, KDM4 (JMJD2) and KDM5 (JARID1) histone lysine demethylase inhibitors. J Med Chem. (2016) 59:1388–409. doi: 10.1021/acs.jmedchem.5b01635
165. Kawamura A, Munzel M, Kojima T, Yapp C, Bhushan B, Goto Y, et al. Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat Commun. (2017) 8:14773. doi: 10.1038/ncomms14773
166. Metzger E, Stepputtis SS, Strietz J, Preca B-T, Urban S, Willmann D, et al. KDM4 inhibition targets breast cancer stem-like cells. Cancer Res. (2017) 77:5900–12. doi: 10.1158/0008-5472.CAN-17-1754
167. Chen YK, Bonaldi T, Cuomo A, Del Rosario JR, Hosfield DJ, Kanouni T, et al. Design of KDM4 inhibitors with antiproliferative effects in cancer models. ACS Med Chem Lett. (2017) 8:869–74. doi: 10.1021/acsmedchemlett.7b00220
Keywords: acute myeloid leukemia (AML), Leukaemic stem cell (LSC), Histone 3 lysine 9 trimethylation (H3K9me3), lysine specific demethylase (KDM), heterochromatin, gene suppression
Citation: Monaghan L, Massett ME, Bunschoten RP, Hoose A, Pirvan P-A, Liskamp RMJ, Jørgensen HG and Huang X (2019) The Emerging Role of H3K9me3 as a Potential Therapeutic Target in Acute Myeloid Leukemia. Front. Oncol. 9:705. doi: 10.3389/fonc.2019.00705
Received: 16 April 2019; Accepted: 16 July 2019;
Published: 02 August 2019.
Edited by:
Cyrus Khandanpour, University Hospital Münster, GermanyReviewed by:
Zeng-quan Yang, Wayne State University, United StatesShilpa Dhar, University of Texas MD Anderson Cancer Center, United States
Manfred Jung, University of Freiburg, Germany
Copyright © 2019 Monaghan, Massett, Bunschoten, Hoose, Pirvan, Liskamp, Jørgensen and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Huang, eHUuaHVhbmdAZ2xhc2dvdy5hYy51aw==