 Tze-Sian Chan
Tze-Sian Chan Yuval Shaked5,6
Yuval Shaked5,6
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 31 July 2019
Sec. Pharmacology of Anti-Cancer Drugs
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00688
This article is part of the Research Topic Therapeutic Targeting of Cancer Stem-Like Cells (CSC) – The Current State of the Art View all 14 articles
Malignant tumors are highly heterogeneous and likely contain a subset of cancer cells termed cancer stem cells (CSCs). CSCs exist in a dynamic equilibrium with their microenvironments and the CSC phenotype is tightly regulated by both cell-intrinsic and cell-extrinsic factors including those derived from their surrounding cells or stroma. Many human solid tumors like breast, lung, colorectal and pancreatic cancers are characterized by a pronounced stromal reaction termed “the desmoplastic response.” Carcinoma-associated fibroblasts (CAFs) derived either from resident fibroblasts or tumor-infiltrating mesenchymal stem cells (MSCs) are a major component of the stroma in desmoplastic cancers. Recent studies identified subpopulations of CAFs proficient in secreting a plethora of factors to foster CSCs, tumor growth, and invasion. In addition, cytotoxic therapy can lead to the enrichment of functionally perturbed CAFs, which are endowed with additional capabilities to enhance cancer stemness, leading to treatment resistance and tumor aggressiveness. When recruited into the tumor stroma, bone-marrow-derived MSCs can promote cancer stemness by secreting a specific set of paracrine factors or converting into pro-stemness CAFs. Thus, blockade of the crosstalk of pro-stemness CAFs and MSCs with CSCs may provide a new avenue to improving the therapeutic outcome of desmoplastic tumors. This up-to-date, in-depth and balanced review describes the recent progress in understanding the pro-stemness roles of CAFs and tumor-infiltrating MSCs and the associated paracrine signaling processes. We emphasize the effects of systemic chemotherapy on the CAF/MSC–CSC interplay. We summarize various promising and novel approaches in mitigating the stimulatory effect of CAFs or MSCs on CSCs that have shown efficacies in preclinical models of desmoplastic tumors and highlight the unique advantages of CAF- or MSC-targeted therapies. We also discuss potential challenges in the clinical development of CSC- or MSC-targeted therapies and propose CAF-related biomarkers that can guide the next-generation clinical studies.
An emerging concept of cancer biology emphasizes the critical role of the hierarchical organization in tumors in the maintenance as well as the progression of the malignant phenotypes. In support of this paradigm, mounting data over recent years, including large-scale genomic analysis and single-cell RNA sequencing analysis, have consistently indicated the existence of a subset of cancer cells termed the tumor-initiating cells (TICs) or CSCs, which are stem-like and have the capability of self-renewing and sustaining tumorigenesis and thereby serve as the driving force of cancer growth, metastasis, and treatment resistance (1–3). CSCs have been found to exist in leukemia and multiple solid tumors, such as glioma, breast cancer, pancreatic ductal adenocarcinoma (PDAC), head and neck squamous cell carcinoma, hepatocellular carcinoma, non-small cell lung cancer (NSCLC), and colorectal cancer (CRC) (4–8).
The recent insights into the complex nature of cancer stemness reveal that CSCs exist in a dynamic equilibrium with their microenvironments and the CSC phenotype is regulated by both cell-intrinsic and cell-extrinsic factors derived by their surrounding cells or stroma cells. The notable examples of the “pro-stemness” or “pro-CSC” factors identified from these studies are inflammatory cytokines, such as interleukin-6 (IL-6), IL-8, and C-C motif chemokine ligand 5 (CCL-5), which play an essential role in CSC regulation as well as invasion and metastasis of tumors (9–11).
Many types of human solid tumors, especially those derived from glandular epithelium, such as breast cancer, NSCLC, PDAC, the scirrhous subtype of gastric adenocarcinoma, and the “stem/serrated/mesenchymal (SSM)” molecular subtype of CRC, are characterized by a pronounced stromal reaction termed “the desmoplastic response” (12–17). CAFs and their collagen matrix products are a major component of the stroma in desmoplastic cancers, comprising a substantial proportion of the tumor mass (18, 19). Instead of being functional inert, there is circumstantial evidence that CAFs are pro-inflammatory due to activation of nuclear factor kappa B (NF-κB), signal transducer and activator of transcription (STAT)-1 and−3, and transforming growth factor (TGF)-β/SMAD signaling and are engaged in active cross-talk with cancer cells (19, 20). Therefore, CAFs can foster tumor cell growth, angiogenesis and invasion (21) by secreting paracrine factors, such as pro-inflammatory cytokines (19), chemokines (14, 19), prostaglandins (PGE) (22), growth factors (23), and proteases (24), and by remodeling the extracellular matrix (ECM) (25–28). CAFs also help foster an immunosuppressive microenvironment in tumors by promoting regulatory T cells (29). Recent studies demonstrated that exosomes derived from CAFs promote cancer progression and treatment resistance (30, 31). Intriguingly, CAFs can even travel with malignant cells to distant sites, where they significantly promote metastasis (32). One of the major mechanisms by which CAFs promote oncogenesis is mediated through their pro-stemness abilities. Recent studies have identified specific subpopulations of CAFs that are proficient in secreting pro-stemness paracrine factors (9–11, 23, 33–35), thereby promoting the conversion of cancer cells into CSCs or supporting the self-renewal and the stemness properties of existing CSCs in tumors. Upon stimulation by cytotoxic stress such as chemotherapy, CAFs can be further induced to secrete pro-stemness cytokines or acquire a senescence-like secretory phenotype and produce large amounts of pro-stemness chemokines to further enhance tumor stemness and aggressiveness following therapy (36, 37).
Although the majority of CAFs in the tumor stroma may be derived from resident stromal fibroblasts, there are now multiple lines of evidence suggesting that a significant proportion of CAFs in tumors are derived from bone marrow-derived mesenchymal stem cells (MSCs). MSCs are pluripotent stem cells that contribute to bone, adipose, cartilage, and muscle tissues and are involved in tissue remodeling, chronic inflammation, immune response, and cancer progression (38). Bone marrow-derived MSCs can be recruited to sites of tissue damages or inflammation by endocrinal signals to exert their tissue repairing functions (39), whereas the tissue-regenerative function of MSCs may go awry in malignant tumors. For instance, in mouse models of breast cancer, PDAC or gastric cancer, bone marrow-derived MSCs are recruited to the tumor microenvironment where they differentiate into CAFs (40–42). Indeed, in a gastric cancer model, approximately 20% of CAFs were found to originate from bone marrow-derived MSCs, which were recruited into the tumors in a transforming growth factor (TGF)-β and C-X-C motif chemokine ligand (CXCL)-12-dependent manner (43). Similarly, MSCs introduced into the tibia trafficked to sites of breast tumor xenografts (44). In an orthotopic murine PDAC model, MSCs were actively recruited into the growing pancreatic tumors (45). Like CAFs, MSCs can significantly influence tumor behaviors and contribute to tumor progression. Most importantly, MSCs promote CSCs by secreting a plethora of pro-stemness cytokines and growth factors or indirectly by differentiating into pro-stemness CAFs (44, 46, 47).
In clinical scenarios, most cancers are treated with certain types of cytotoxic therapies, such as chemotherapy and radiation therapy, which may have profound impacts on the characteristics of tumors including the epithelial and the stromal compartments. Indeed, chemotherapy has been shown to enrich tumor cells for those with mesenchymal and/or CSC features in different types of cancers. CSCs are intrinsically more resistant to therapy and consequently increase disproportionately following systemic chemotherapy and are thought to contribute to tumor relapse and treatment resistance (1, 48, 49). For instance, breast cancers after neoadjuvant chemotherapy are enriched from CD44+CD24− CSCs that also express mesenchymal markers (48, 49). Chronic oxaliplatin or paclitaxel treatment induces an epithelial-mesenchymal transition (EMT) and the enrichment of CSCs in CRC and ovarian cancer (50, 51). Chemotherapy has also been shown to expand CSCs that are dependent on the interleukin (IL)-8–CXCR-1 signaling axis (52). Importantly, CAFs are enriched in chemotherapy-treated human tumor tissues wherein they promote cancer growth, treatment resistance and the self-renewal of CSCs by secreting paracrine factors (36, 53). Moreover, chemotherapy-modulated CAFs secrete a panel of CXCL chemokines to expand CSCs in the treated tumor, leading to paradoxical tumor aggression and treatment failure (37). Thus, adjuvant strategies that target CAFs to temper the chemotherapy-induced enrichment of CSCs may further improve the therapeutic outcome of patients with desmoplastic cancers.
The past two decades of investigations into CSCs and their biology have led to the identification of a number of potentially druggable targets, based on which many CSC-directed therapies have been developed with some of them entering clinical trials (54). Unfortunately, the idea of therapeutic targeting of CSCs has suffered from a series of notable clinical trial failures over recent years, including the focal adhesion kinase (FAK) inhibitor defactinib, the STAT-3 inhibitor napabucasion, the anti-NOTCH-2/3 antibody tarextumab, the anti-delta like canonical notch ligand (DLL)-4 antibody demcizumab, and most recently the multibillion-dollar anti-DLL-3 antibody-drug conjugate rovalpituzumab tesirine (Rova-T). Apparently, there is an urgent need for new and more viable strategies of successfully and safely targeting CSCs. As opposed to the direct targeting of the rare, dynamic and plastic CSC populations, targeting the more abundant, favorably spaced and stable CAFs and MSCs, especially their pro-stemness subsets, presents an attractive strategy to indirectly target cancer stemness to enhance the efficacy of current anti-cancer therapies.
In this review, we describe how CAFs and MSCs initiate crosstalk with CSCs and augment cancer stemness in human solid tumors. We emphasize the effects of systemic chemotherapy on CAFs and how these effects can modulate their pro-stemness functions in the treated tumor. We discuss the advantages of targeting CAFs or MSCs over directly targeting CSCs, as well as various promising approaches that aim at disengaging the CAF/MSC–CSC link in preclinical models. This review finally lists potential challenges in the clinical development of pro-stemness-CAF- or MSC-targeted therapies and explores potential biomarkers of pro-stemness CAFs to guide the development of therapeutic strategies to disengage the dangerous interplay between CAFs, MSCs, and CSCs that can be quickly deployed in clinical trials in the treatment of human desmoplastic cancers.
As described above, CAFs are proficient in paracrine signaling and are capable of secreting a plethora of paracrine factors that have been implicated in the maintenance and/or the expansion of CSCs (Figure 1 and Table 1). Among the most extensively studied pro-stemness cytokines secreted by CAFs are IL-6 and IL-8, which have been shown to play an essential role in the regulation of CSCs as well as cancer invasion and metastasis (9–11). Several mechanistic studies have demonstrated that IL-6 participates in the regulation and maintenance of the CSC phenotype mainly through the STAT-3–NF-κB signaling pathway (10, 11, 57). Constitutive IL-6 expression in breast cancer cells maintains their EMT phenotype, which has been implicated in the generation of a CSC phenotype (58, 59). As opposed to the role of the IL-6 inflammatory loop in inducing CSCs with mesenchymal features in breast cancer, IL-8 mainly regulates a subpopulation of epithelial-like CSCs that express high aldehyde dehydrogenase (ALDH) activity and are highly proliferative (52). Consistently, IL-8 was found to profoundly enhance the stemness property of breast cancer and PDAC cells (60–62). A recent proteomic screening identified leukemia inhibitory factor (LIF)-induced STAT-3 activation as the major signaling event in PDAC cells induced by PSCs, leading to activation of stemness programs, including Hippo, Wnt, and STAT-3 (35). Notably, LIF expression is significantly up-regulated in PDAC tissues while the expression of IL-6 does not, underscoring the importance of LIF over IL-6 in PDAC. Aside from interleukins, a multitude of other secretory factors has also been implicated in mediating the pro-stemness capability of CAFs. For instance, in CRC models established using primary carcinoma cells, CAF-derived osteopontin (OPN) has been shown to support the clonogenic capacity of CSCs, which predominantly reside at the tumor edge in close proximity to CAFs (64). Another study also showed that the CAFs freshly isolated from human CRC tumors produced significantly higher levels of CXCL-12, OPN, TGF-β, and hepatocyte growth factor (HGF), which coordinately activated Wnt–β-catenin signaling to induce the expression of the novel CSC marker CD44 variant 6, resulting in an EMT in cancer cells and tumor invasion and metastasis (56). In another CRC model established using freshly isolated carcinoma cells and the paired CAFs, CAFs up-regulated the expression of TGF-β2 and IL-6, which activated the expression of GLI family zinc finger (GLI)-2 in the sonic hedgehog (SHH) pathway, resulting in the transdifferentiation of cancer cells into CSCs and chemotherapy resistance (65). Pancreatic stellate cells (PSCs), a specialized type of CAFs present in the stroma of PDAC, secrete the TGF-β family protein Nodal, which binds to its receptor Activin-like (Alk)-4 and−7 on CSCs to promote their stemness properties (34, 63). In an NSCLC model, insulin-like growth factor (IGF)-II and allied autocrine/paracrine factors secreted by CAFs synergistically activated IGF-1R signaling to induce the expression of the stemness-related gene Nanog, thereby converting cancer cells into CSCs (23). CAFs isolated from human breast cancer secrete abundant levels of PGE-2, which enhances the secretion of IL-6 to expand CSCs (22). Moreover, when co-cultivated with cancer cells, CAFs produced a higher level of CCL-2, which stimulate CSCs by inducing Notch-1 expression and thereby activating the Notch signaling pathway (33).
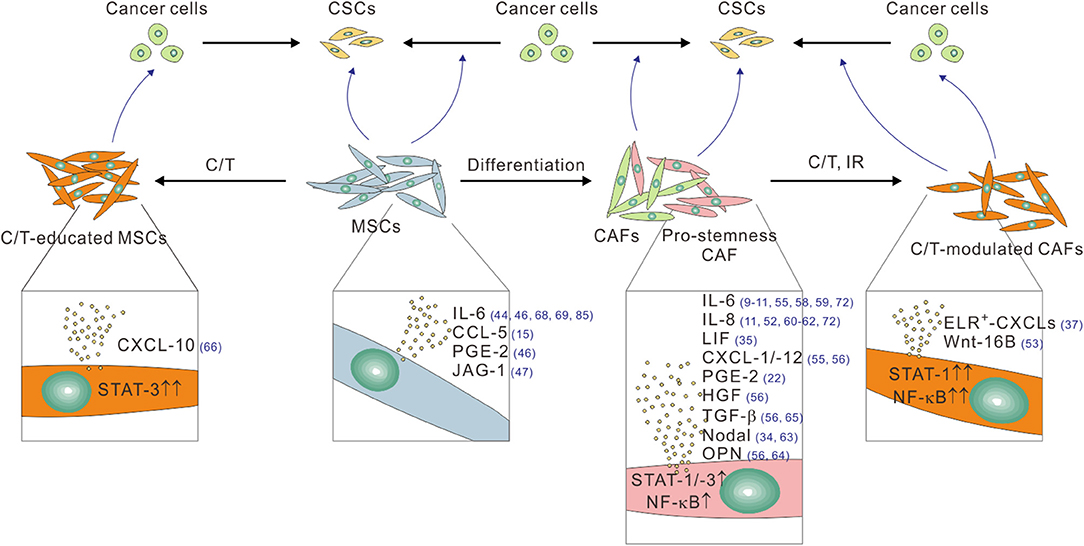
Figure 1. The pro-stemness functions of CAFs and tumor infiltrating MSCs linked to their different functional and treatment status. CAFs, especially their pro-stemness subset, secrete assorted cytokine and chemokines, including IL-6, IL-8, LIF, PGE-2, CXCL-1, CXCL-12, HGF, and TGF-β through heightened STAT, and NF-κB signaling activity to promote the reprogramming of cancer cells into CSCs and/or directly expand the CSC population. CAFs also secret Nodal and osteopontin (OPN) to promote CSCs. Cytotoxic therapies such as chemotherapy (C/T) and ionizing radiation (IR) further potentiate the pro-stemness functions of CAFs by further activating STAT-1 and NF-κB signaling, thereby inducing the secretion of a different panel of pro-stemness factors, including ELR+-CXCL chemokines, and Wnt-16B. Bone marrow-derived and tumor-infiltrating MSCs can promote CSCs by converting into CAFs or by secreting several pro-stemness chemokines such as IL-6, CCL-5, PGE-2, and JAG-1. C/T-educated MSCs further secrete the pro-stemness chemokine CXCL-10 by activating STAT-1 signaling. The reference numbers are shown in blue.
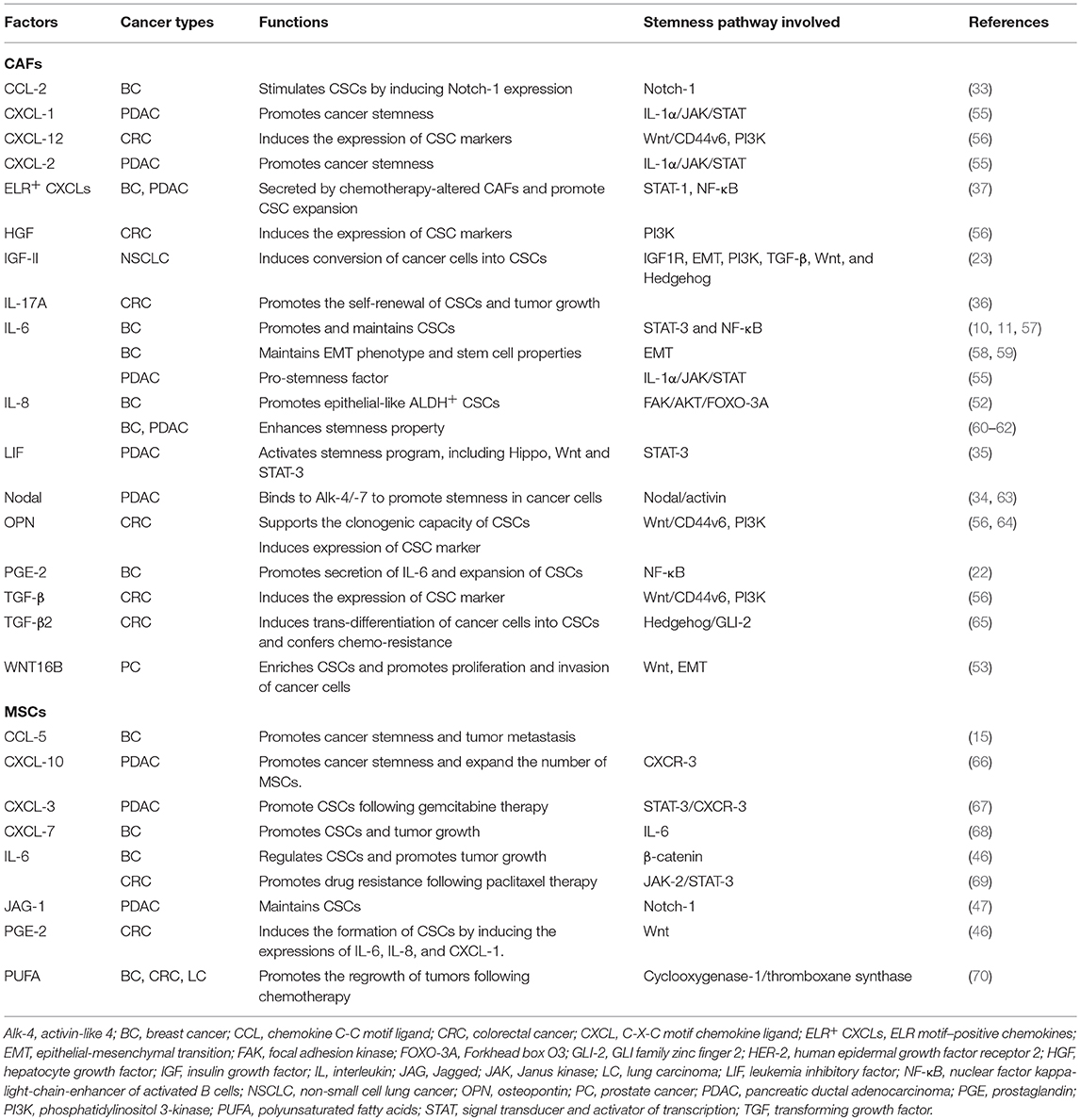
Table 1. Pro-stemness factors secreted by CAFs and MSCs.
Mounting data accumulated over recent years have suggested that CAFs in desmoplastic cancers are phenotypically, functionally and genetically heterogeneous and are likely dynamically controlled by their environments and origins (29, 71–76). Indeed, CAFs have been classified into various functional subtypes according to a panel of surface markers, including such as α-SMA, fibroblast activation protein (FAP), fibroblast specific protein (FSP)-1, and platelet-derived growth factor receptor (PDGFR)-α/β (19, 77, 78). At the functional level, FAP+ CAFs are enriched in low stiffness and fibronectin-rich ECMs, whereas α-SMA+ CAFs are found in stiffer ECM contexts (76). In a transgenic model of PDAC, depletion of α-SMA+ CAFs did not affect the number of FAP+ CAFs, indicating that they represent different CAF subpopulations (79). Interestingly, FAP+ CAFs are predominantly involved in the synthesis and the turnover of ECM while α-SMA+ CAFs mediate contraction. Importantly, the recently identified CAF heterogeneity relates to the pro-stemness and pro-oncogenic capabilities of CAFs. For instance, in oral squamous cell carcinoma, a subgroup of CAFs termed “CAF-D” has been shown to induce EMT of malignant keratinocytes through secreting TGF-β (74). Since the EMT program in cancer cells imparts them with CSC features (59, 80, 81), it is likely that this specific subpopulation of CAFs might have induced the phenotypic conversion of keratinocytes into CSCs. In PDAC, two distinct subgroups of PSCs have been identified in mouse and human PDAC tissues (71). Remarkably, only those PSCs located away from tumor cells, denoted as “inflammatory CAFs (iCAFs),” were proficient in secreting pro-stemness factors, including IL-6, CXCL-1, and CXCL-2, through activation of IL-1α-Janus kina (JAK)–STAT signaling (55). By contrast, PSCs located adjacent to tumor cells have the propensity of differentiating into collagen-producing and α-smooth muscle actin (α-SMA)-positive myofibroblasts. In analogous to this emerging paradigm of the functional heterogeneity of CAFs, in human breast cancer and NSCLC tissues, a distinct subpopulation of CAFs was found to express CD10 as well as the complement 5 a receptor G-protein coupled receptor 77 (GPR-77) and are proficient in promoting CSCs and their stemness properties and inducing chemoresistance of tumor cells through persistent NF-κB activation along with the resultant IL-6 and IL-8 secretion (72). Of note, these pro-stemness subset of CAFs were either defined by surface markers (e.g., CD10 and GPR-77), their transcriptome and secretome (e.g., CAF-D), or a specific set of secretory factors (e.g., IL-6 and LIF in iCAFs). It remains to be established if the pro-stemness subset of CAFs indeed vary among different types of cancers or can be molecularly defined in a more precise manner.
In chemotherapy-treated desmoplastic cancers, CAFs are endowed with additional pro-stemness and pro-oncogenic capabilities as a result of the stress-induced chronic phenotypic and functional alterations. For instance, in prostate cancer, the genotoxic agent mitoxantrone stimulated Wnt-16B secretion by stromal fibroblasts, which promoted the proliferation and invasion of carcinoma cells, which likely contained the enriched CSCs (53). In human CRC, chemotherapy led to the enrichment of IL-17A-producing CAFs within the tumor stroma, which in turn promoted the self-renewal of CSCs and tumor growth (36). In addition, following systemic chemotherapy, breast and pancreatic CAFs secreted large amounts of the “ELR-motif-positive” (ELR+) CXCL chemokines through chronic activation of the STAT-1 and NF-κB transcriptional activities, which stimulated CXCR-2 signaling in cancer cells to elicit their transdifferentiation into CSCs and thereby promoted post-treatment tumor aggression and treatment failure (37).
Despite the multiple lines of evidence supporting the pro-stemness functions of CAFs and therapy-modulated CAFs, it is worth noting that, as highlighted above, CAFs are capable of promoting tumor progression and treatment resistance through a multitude of mechanisms. Therefore, the tumor-promoting effects of CAFs summarized herein may likely be mediated by the concerted actions of a plurality of mechanisms and should not be attributed only to the pro-stemness functions of CAFs. Moreover, since most of these studies were conducted in immuno-compromised or immune-deficient animal models, caution should be exercised while interpreting the results related to the CAF-derived pro-stemness cytokines and chemokines that are actively involved in inflammation and immune regulation. Whether or not CAFs exert similar positive regulatory effects on CSCs in immunocompetent backgrounds and how these effects work in concert with other tumor-promoting mechanisms of CAFs await further in-depth investigation.
MSCs were initially considered to be cells promoting the regenerative properties of wounds and damaged tissues. A growing body of evidence indicated that the regenerative function of MSCs are hijacked by malignant tumors such that a significant number of bone marrow-derived MSCs are recruited to the tumor microenvironment, where a considerable proportion of them differentiate into CAFs (40, 41). Like CAFs, MSCs can secrete a plethora of cytokines and growth factors, which make them proficient in paracrine and heterotypic signaling processes. For instance, a recent comprehensive cytokine secretion profile of human MSCs identified IL-6, IL-8, TIMP metallopeptidase inhibitor 2 (TIMP-2), CCL-2 (MCP-1), and vascular endothelial growth factor (VEGF) as the most abundantly secreted factors (82). Other studies reported that MSCs promote cancer metastasis by secreting CCL-5 (15), CXCL-12, and IGF-1 (83). MSCs also contribute to tumor angiogenesis by secreting vascular endothelial growth factor (VEGF), and β-fibroblast growth factor (FGF) (67). Furthermore, MSCs promote immunomodulation by upregulating cytokines such as IL-6, IL-8, and TGF-β (68, 69, 84). In breast cancer, the cancer cells stimulate the secretion of CCL-5 from MSCs, which acts in a paracrine fashion on the cancer cells to enhance their motility, invasion, and metastasis (15). Importantly, several studies have directly implicated MSCs in the regulation of CSCs (Figure 1). MSCs in breast cancer regulate CSCs through cytokine loops involving IL-6 and CXCL-7, thereby accelerating tumor growth (44). In CRC, MSCs secrete prostaglandin E2 (PGE-2) in response to IL-1 released by carcinoma cells, which act in an autocrine fashion to induce the expression of IL-6, IL-8 and CXCL-1, which together induce the formation of CSCs (46). Once differentiated into CAFs, MSCs can maintain CSCs through secreting the Notch ligand Jagged-1 (47). In analogous to the effect of chemotherapy on the number and the pro-stemness property of CAFs, the number of bone marrow-derived MSCs significantly increased following gemcitabine treatment in the tumor stroma in a mouse xenograft model of PDAC (66). Importantly, these gemcitabine-educated MSCs were found to have a positive regulatory effect on CSCs through the STAT-3–CXCL-10–CXCR-3 paracrine signaling axis. Similarly, following paclitaxel treatment or hyperthermia therapy, MSCs secreted IL-6, IL-7, IL-8, EGF, and IGF, which supported drug resistance (85, 86). In another study, cisplatin-activated MSCs produced specific polyunsaturated fatty acids which in turn promoted the regrowth of tumors following therapy (70).
Collectively, the ample evidence underscore the important role of CAFs and tumor-infiltrating MSCs in the maintenance and the expansion of CSCs and suggest that targeting this component of the tumor stroma may provide a new avenue to improving the therapeutic outcome of human desmoplastic cancers.
Given that CAFs positively regulate CSCs through the secretion of pro-stemness paracrine factors, a number of preclinical studies have exploited the therapeutic potential of the functional blockade of the CAF-to-CSC paracrine signaling process to improve the treatment of desmoplastic cancers (Figure 2). For instance, loss of PTEN in a HER2-overexpression genetic background or the trastuzumab resistance in breast cancer cells has been linked to activation of the IL-6/STAT-3/NF-κB inflammatory loop, which induced an EMT phenotype and expansion of the CSC population. Therefore, blocking this loop by a function-blocking anti-IL-6 receptor antibody could effectively revert these phenotypes (10). In another study, functional inhibition of the subgroup of IL-6- and IL-8-secreting CD10+GPR-77+ CAFs with anti-IL-6 and anti-IL-8 antibodies, together with docetaxel chemotherapy, has led to a near complete remission of tumors in a patient-derived xenograft (PDX) model of breast cancer (72). Interestingly, this study also demonstrated that an anti-GPR-77 antibody in combination with docetaxel therapy exerted anti-tumor efficacy comparable to that induced by the combination anti-IL-6/anti-IL-8 therapy, which significantly reduced the number of CD10+GPR-77+ CAFs and the proportion of CSCs in the treated tumors (72). In keeping with the critical role of IL-8 in cancer stemness, a function-blocking antibody against its receptor, CXCR-1, or a small-molecule inhibitor of CXCR-1 and CXCR-2, repertaxin, could deplete CSCs and inhibit tumor aggressiveness in human breast cancer xenografts (5). Of particular clinical relevance was the recent finding that repertaxin in combination with paclitaxel demonstrated a 30% response rate in a phase 1b study of metastatic breast cancer (87). Other small-molecule inhibitors of CXCR-2, including AZ13381758 and SB225002, have also shown preclinical efficacy in transgenic or PDX models of PDAC (37, 88). Of note, since that CXCR-2 is also expressed by myeloid-derived immunosuppressive cells in PDAC (88), its inhibitor may exert anti-tumor efficacy through multiple mechanisms of action. Aside from the molecular targeting of IL-6, IL-8 and their receptors, targeting their downstream signaling components in cancer cells and/or CSCs offer other viable opportunities for disabling the CAF–CSC crosstalk. For instance, a small-molecule inhibitor of STAT-3, BBI608, has been reported to significantly inhibit cancer stemness in a variety of cancer types (89), whereas the results from some of the recent clinical trials were discouraging. Furthermore, several novel therapeutics targeting the CAF-to-CSC IL-6–STAT-3 signaling axis are under development. These include a high-affinity anti-IL-6 antibody, MEDI5117, which has been shown to enhance the anti-tumor efficacy of chemotherapy or gefitinib in several types of tumors that are known to be driven by the IL-6–STAT-3 signaling and especially target the CD44+CD24− CSCs in trastuzumab-resistant and HER-2+ breast cancer cells (90). Another example is a cyclic oligonucleotide decoy that corresponds to the STAT-3 response element of STAT-3-targeted genes, which showed promising anti-tumor efficacy in NSCLC models (91). Recently, another interleukin family protein, LIF, was found to be the major PSC-derived factors that promote CSCs in PDAC cells and tissues (35). Accordingly, systemic administration of a LIF-neutralizing antibody in combination with chemotherapy reduced the percentage of CSCs and mesenchymal-transited cancer cells and extended the survival of tumor-bearing mice in a transgenic model of PDAC.
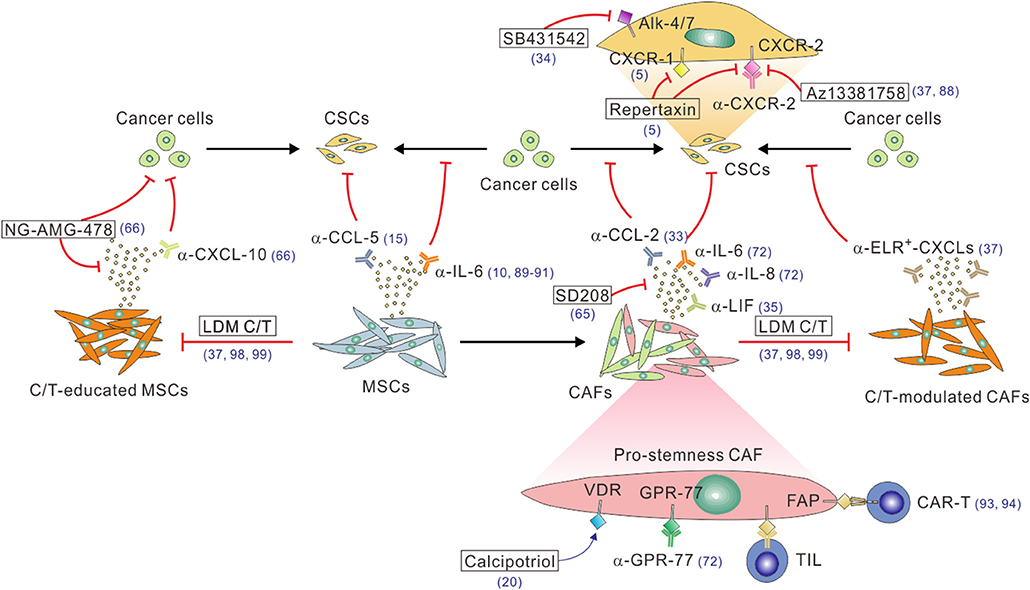
Figure 2. A multitude of approaches to block the CAF/MSC–CSC crosstalk. Function-blocking antibodies, including α-IL-6, α-IL-8, α-LIF, α-CCL-2, and α-CCL5, or small molecular inhibitors, such as the TGF-β inhibitor SD208, can be used to block the stimulatory effect of these pro-stemness factors secreted by CAFs or C/T-modulated CAFs. On the other hand, the CAF-CSC paracrine signaling can be blocked by function-blocking antibodies (e.g., α-CXCR-1, α-CXCR-2) or small molecule inhibitors (e.g., repertaxin, AZ13381758, SB431542) of the receptors on CSCs and/or cancer cells. Likewise, the pro-stemness functions of tumor-infiltrating MSCs can be antagonized by function-blocking antibodies against IL-6 or CCL-5. The enhanced pro-stemness functions of C/T-modulated CAFs or MSCs can be potentially blocked by function-blocking anti-ELR+-CXCL-chemokine antibodies, anti-CXCL-10 antibody, the CXCL-10 inhibitor AMG-478 (encapsulated by MSC-derived nano-ghost, NG), or adopting low-dose metronomic (LDM) C/T regimens. Function-blocking α-GRP-77 antibodies can be used to reduce the tumor infiltration of CD10+GPR-77+ pro-stemness CAFs. Calcipotriol can activate VDR signaling to inhibit IL-6, CCL-2, and CXCL-1 production by CAFs. Finally, FAP+ or GPR-77+ CAFs can be depleted by using DNA vaccines to induce CAF-specific tumor-infiltrating T cells (TILs) or administrating CAF-specific CAR-T cell or other types of engineered immune cells. The reference numbers are shown in blue.
Aside from inhibiting interleukin paracrine signaling, a number of studies have explored the therapeutic potential of inhibiting other CAF-derived pro-stemness factors. For instance, the CCL-2 neutralizing antibody has been shown to significantly suppress tumorigenesis and inhibit pro-stemness Notch signaling in an orthotopic breast cancer model involving the co-implantation of cancer cells and CAFs (33). The TGF-β inhibitor SD208 has been shown to reduce the CAF-induced expression of stemness markers and simultaneously induced the expression of differentiation markers in CAF-cocultivated CRC cells (65). Consequently, SD208 in combination with the small molecule inhibitor of GLI-2, a transcriptional factor in the SHH pathway, restored the sensitivity of the tumors to chemotherapy in mouse PDX models of CRC. SB431542, an inhibitor of the Nodal receptor Alk-4/7 expressed on pancreatic CSCs, could block the stemness and invasive capacities of CSCs and thereby inhibited PDAC progression especially when used in conjunction with a Smoothened (a SHH pathway receptor) inhibitor that depleted the tumor stroma (34). Nevertheless, it is worthy of note that these promising results should be interpreted with caution since most of the studies were conducted in immuno-deficient mice without the potential influence from the immune system, which is especially relevant as most of the pro-stemness mediators studied also have pro-inflammatory and immune-related functions.
Given the profound impacts of CAFs on the tumor stemness and aggressiveness in desmoplastic cancers, depleting CAFs in the tumor stroma provided another viable option in attenuating the CAF–CSC interplay (Figure 2). Along this line, an oral DNA vaccine targeting FAP, a CAF-specific marker, has been demonstrated to suppress tumor growth and metastasis and confer a survival benefit in murine models of CRC and breast cancer (92). Moreover, adoptive transfer of FAP-targeted chimeric antigen receptor (CAR) T cells could specifically kill FAP+ CAFs and induce multiple beneficial stroma alterations, leading to delayed tumor growth and survival extension in mouse models of NSCLC and PDAC (93, 94). Interestingly, a combined targeting of FAP+ CAFs and EPH receptor A2 (EphA2)+ cancer cells led to a nearly complete remission of the tumors (93), suggesting that CAF-targeted approaches have the potential to supplement and synergize with conventional cancer-cell-targeted therapies. Notwithstanding these promising results, caution must be exercised with the application of CAF-depleting strategy as the genetic depletion of CAFs using the conditional knockout of SHH signaling or the ganciclovir-induced depletion of CAFs in PDAC unexpectedly led to invasive and undifferentiated tumors along with unfavorable immunosuppression (79, 95). Furthermore, depletion of FAP+ stromal cells in a transgenic mouse model has been associated with muscle wasting and impaired erythropoiesis (96), implicating the potential adverse effects of CAF-depleting therapies. In this regard, the functional inhibition of CAFs instead of their depletion may be a safer and more desirable therapeutic approach than the direct depletion of CAFs. Several recent studies toward this direction have shown promises. For instance, vitamin D receptor (VDR) signaling has been shown to antagonize TGF-β/SMAD signaling-induced activation of PSCs in PDAC tissues, which was mediated by the pro-stemness factors IL-6, CCL-2, and CXCL-1 (20). As such, calcipotriol, a potent vitamin D analog that controls VDR induction, inhibited inflammatory signaling in CAFs and reduced the expressions of IL-6, CCL-2, and CXCL-1. When combined with gemcitabine treatment, calcipotriol synergized with chemotherapy to control tumor growth and extend survival in transgenic mouse models of PDAC. Another approach involved transducing CAFs with a nanocarrier-formulated plasmid encoding a secretable form of the death ligand TNF-related apoptosis-inducing ligand (TRAIL) termed sTRAIL. Since CAFs are intrinsically resistant the killing effect of TRAIL, once transduced with the sTRAIL-expressing plasmid, they were converted into sTRAIL-producing cells and thereby triggered apoptosis of neighboring cancer cells (97). Surprisingly, the expression of sTRAIL also reprogrammed CAFs into a quiescent state. This approach demonstrated strong anti-tumor efficacy in a PDAC model.
Recently, low-dose metronomic (LDM) chemotherapy has emerged as a highly clinical applicable strategy to enhance the tumoral treatment response by tempering the therapy-induced stromal alterations in desmoplastic cancers (37, 98, 99). Compelling evidence from laboratory-based and clinical correlative studies have demonstrated that conventional chemotherapy administered at a maximum tolerated dose (MTD) induces myriad alterations in stromal cells, including endothelial cells and their progenitor cells, immune cells, and CAFs (53, 100). In keeping with this paradigm, our group recently demonstrated that systemic MTD chemotherapy of assorted agents, including paclitaxel, gemcitabine, doxorubicin, and cyclophosphamide, had profound impacts on CAFs in human breast cancer and PDAC tissues, which acquired the ability to secret large amounts of pro-stemness ELR+ CXCL chemokines through the chronic activation of STAT-1 and NF-κB signaling (37). The CSC niche microenvironment generated by therapy-modulated CAFs could be attenuated by pretreating the tumors with a CXCR-2 inhibitor or by switching the dosing schedule to LDM regimens, which had a much less stimulatory effect on CAFs. We envisage that this approach has multiple benefits. First, it obviates the lengthy and costly process of developing new CAF- and/or CSC-targeted agents, which has an especially high attrition rate according to past experiences. Second, an increasing number of oral chemotherapeutic agents are available for clinical use, making the concept of LDM chemotherapy immediately clinical applicable. Third, as mentioned earlier, LDM chemotherapy not only may prevent the CAF–CSC interplay but may also exert multiple favorable effects on other cells in the tumor stroma, including tumor-associated macrophages, myeloid-derived suppressor cells, and blood vessel cells (98, 99, 101, 102). We thus foresee that LDM chemotherapy will become the treatment of choice in many types of desmoplastic cancers.
Due to their pro-tumorigenic activities, a number of studies had been carried out to try and target MSCs as a therapeutic approach in cancer (103). Specifically, given that tumor-infiltrating MSCs can directly support CSCs through multiple paracrine signaling pathways, including IL-6, IL-7, CXCL-1, PGE-2, Jagged-1, and CXCL-10 (44, 46, 47, 66), blockade of the molecular crosstalk between MSCs and CSCs may be potentially useful in inhibiting cancer stemness in desmoplastic cancers. Indeed, a recently study exemplified the potential utility of this approach (66). In a mouse model of PDAC, MSCs were found in close proximity to CSCs following gemcitabine chemotherapy, implicating MSCs as the CSC niche. Mechanistically, gemcitabine-exposed MSCs secrete high levels of CXCL-10 that activate its receptor CXCR-3 on CSCs, activating STAT-3 signaling and promoting the survival of CSCs. Consistently, systemic administration of the CXCL-10 inhibitor AMG487 formulated with MSC-derived membrane-based nanoparticles termed “nano-ghost (NG)” led to its intratumoral accumulation in close proximity to CSCs, thereby reducing the percentage of CSCs and augmenting the therapeutic efficacy of gemcitabine. In analogous to the directly depletion of CAFs, the direct depletion of MSCs might provide an alternative approach to nullify their crosstalk with CSCs. However, whether MSC-deprived host for a limited time may cause toxicity on its own remains an open question. Alternatively, given that MSCs secrete CXCL-10 in response to gemcitabine treatments, and that gemcitabine given at an LDM regimen could attenuate therapy-induced production of pro-stemness chemokines from CAFs in PDAC (37), it is highly likely that LDM chemotherapy may also prevent or at least attenuate chemotherapy-induced activation of MSCs and their secretion of pro-stemness chemokines. This possibility merits further investigations. On the other hand, since MSCs secrete the pro-stemness cytokine IL-6 (46), the various anti-IL-6 antibodies and/or STAT-3 inhibitors developed to inhibit the CAF–CSC crosstalk can also be adopted to block the interaction between MSCs and CSCs (10, 89–91). This raises the possibility that MSC- and CAF-targeted therapeutics may synergize with each other. We thus envisage that the CAFs and MSCs dual targeting approach may provide an opportunity to more thoroughly block the stroma-derived pro-stemness signals to maximize the anti-tumor efficacy in the treatment of desmoplastic cancers.
As opposed to the direct targeting of CSCs, which poses significant challenges, targeting CAFs or MSCs along with the pro-stemness niches they generate may have several advantages in the treatment of desmoplastic cancers (Table 2). First and foremost, a growing body of evidence now suggests that CSCs are highly heterogeneous and plastic and the conversion between different CSC populations plays an important role in tumor progression and treatment response (104). For instance, breast cancer CSCs exist in alternative mesenchymal-like and epithelial-like states which can transition between each other (105–107). CSCs can also be derived from differentiated cancer cells through cellular reprogramming or transdifferentiation (11), which can be particularly facilitated by cytotoxic stresses such as chemotherapy and ionizing radiation (37, 108). The highly dynamic nature of CSCs makes them moving targets in cancer therapy, which presents a daunting challenge to therapeutic efforts aiming at completely eradicating them. Echoing this notion, two recent studies in the organoid models of CRC highlighted the difficulty of eradicating CSCs. Specifically, ablation of LGR-5+ CSCs halted tumor growth, whereas the tumors resumed growth following the removal of the cell death inducers due to the re-emergence of CSCs from differentiated tumor cells (109, 110). As a comparison, CAFs are both genetically and phenotypically stable; therefore, CAF-directed therapies may lead to a more stable and sustainable anti-CSC effect compared with that results from the direct targeting of CSCs. Second, the recent discoveries of specific subpopulations of pro-stemness CAFs have rendered CAF-directed therapy more feasible as they not only provide novel therapeutic targets, such as GPR-77 (72), but also rendered the related therapies more specific and safer than the non-specific targeting of CAFs (79). Another unique advantage of targeting pro-stemness CAFs relates to their spatial distributions within desmoplastic cancers. Specifically, CAFs and tumor-infiltrating MSCs exist in large numbers in the tumor stroma, which contrasts sharply with CSCs that comprise only a small or even a rare subpopulation of cancer cells and exist within cancer cell nests or as individually dispersed cells or small cell clusters at the tumor periphery or the invasive front (111, 112). In desmoplastic cancers such as PDAC, there are abundant CAFs in the stroma, which can account for more than 90% of the total tumor volume (113, 114). Thus, there are a far larger number of CAFs or MSCs that can be exposed to the therapeutics administrated at a given tissue concentration than that of CSCs. Accordingly, CAF- or MSC-targeted therapeutics may have better pharmacodynamic effects than CSC-targeted agents in the treatment of desmoplastic cancers. Moreover, CAFs are often localized to the periphery of the tumor cell nests or glands and close to blood vessels, rendering them directly accessible to the therapeutics diffused from the blood circulation (115). By contrast, carcinoma cells, including the small population of CSCs, are frequently distantly spaced from blood vessels in desmoplastic tumors. In fact, CAFs per se constitute a significant barrier for the therapeutic delivery of drugs and even nanoparticles to cancer cells (97, 116). Echoing the importance of the spatial distribution of cells in the treatment of poorly perfused desmoplastic tumors, clinical data has confirmed that the majority of therapeutics, such as gemcitabine, can only reach the stroma of human PDAC tissues (117). Collectively, these factors make targeting the link between CAFs or MSCs with CSCs more justified, feasible and clinically promising than the direct targeting of CSCs in the treatment of desmoplastic cancers.
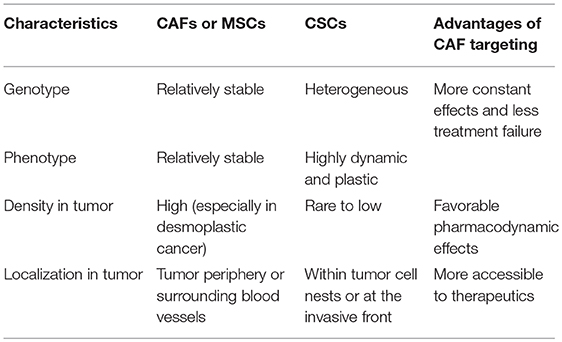
Table 2. The potential advantages of targeting pro-stemness CAFs and MSCs.
Tumor cells are highly heterogeneous in terms of their phenotypes, genotypes, and functions. As aforementioned, it is increasingly recognized that the intra-tumoral heterogeneity not only exists in the epithelial compartment but also the stromal compartment of the tumors, including CAFs (29, 71, 72). As such, human desmoplastic cancers may vary considerably with respect to the number as well as the composition of CAFs, including those with pro-stemness properties. Clinical trials investigating therapies targeting the CAF-to-CSC crosstalk should be ideally conducted in a patient- and tumor-tailored manner based on surrogate markers of CAF activation and/or their pro-stemness functions. We list a number of CAF-related biomarkers that may potentially fulfill this purpose (Table 3). First, a high density of α-SMA+ CAFs in tumors has been linked to the resistance to neoadjuvant chemotherapy in breast cancer (72). Therefore, the density of CAFs may serve as a simple and immediately clinically applicable biomarker based on which CAF-targeted therapies can be implemented. Likewise, the density of CAFs also significantly increased following systemic chemotherapy in human CRC tissues (36). A plausible corollary is that the density of CAFs positively correlates with the likelihood of treatment resistance in most desmoplastic cancer and thus can serve as a universal biomarker to guide CAF-targeted therapies. Notably, since different CAF markers, including such as α-SMA, FAP, and FSP-1, may identify functionally distinct CAF populations that vary among different cancer types of subtypes (76, 77, 79), it remains to be established which CAF marker or any of their combinations can serve as a clinically informed biomarker. Beyond simply measuring the density of CAFs, the staining intensity of phosphorylated STAT-1 in CAFs, which reflects their ability to produce pro-stemness chemokines following chemotherapy (37), may also aid the clinical decision-making regarding when CAF-directed therapies should be implemented. On the other hand, in untreated tumors, the density of pro-stemness CAFs, such as α-SMA−PDGF-Rα+IL-6+ iCAFs in PDAC and CD10+GPR-77+ CAFs in breast cancer and NSCLC (71, 72), can serve as a companion diagnostic to guide the selection of patients for anti-CAF/CSC therapies, especially those targeting the IL-6 and/or the IL-8 paracrine signaling pathways. In theory, these CAF-related biomarkers can be further combined with widely used surrogate markers of CSCs, such as ALDH, CD133, CD44, CD24, CD90, and EpCAM (118, 119), to increase their predictive power and clinical utility. We predict that the application of these CAF-related stemness markers may increase the success rate of the related clinical trials and pave the road for the next-generation patient-tailored anti-cancer therapies.
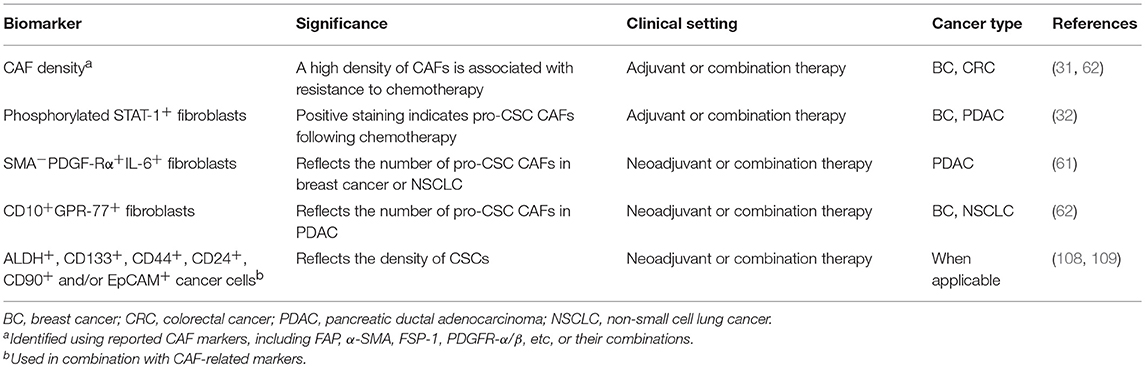
Table 3. Biomarkers linked to pro-stemness CAFs that can guide clinical studies.
Whilst targeting pro-stemness CAFs and MSCs have multiple theoretical advantages over the direct targeting of CSCs, several potential challenges remain and require careful considerations at the various developmental stages of the therapies. First, since CAFs or MSCs maintain their crosstalk with CSCs mainly through pro-stemness cytokines and chemokines, the majority of CAF- or MSC-targeted therapeutics are function-blocking antibodies (Figure 2). It is widely accepted that large-molecule therapeutics like antibodies have very limited penetration into desmoplastic tissues and may only be able to reach CAFs or MSCs spaced at the outer rim of tumors or those located surrounding or near blood vessels. If so, their anti-CSC and anti-tumor efficacy will be severely compromised (120). One potential solution for this problem is pre-treating desmoplastic tumors with agents that can reduce the number of CAFs and/or the desmoplastic reaction they produce, which can be exemplified by the stroma-reducing effect of nab-paclitaxel and SHH inhibitors in human PDAC (34, 117). Another solution is by using small-molecule inhibitors or nanoparticles designed to block pro-stemness factors or their receptors, such as repertaxin, SD208, BBI608, calcipotriol, and NG-AMG487, which have the ability to diffuse deeply into the desmoplastic stroma and reach their intended target cells compared with antibodies. Second, CSC-directed therapies, which target only a small subpopulation of cancer cells, would not be expected to produce measurable changes in tumor burden according to conventional Response Evaluation Criteria in Solid Tumors (RECIST) criteria. Therefore, more pertinent, “stemness-informed” surrogate markers of response that are applicable to anti-CSC agents should be developed to guide the conduction of clinical trials, especially at the phase II stage (121). This concern should be also taken into consideration when conducting clinical trial testing CAF- or MSC-targeted therapies designed to specifically target CSCs. We propose that this problem can be at least partially tackled by introducing stemness-informed CAF- or MSC-related biomarkers as described in Table 2. Third, as described above, CAFs or MSCs are spaced in the tumor stroma, whereas CSCs exist mainly within tumor nests or as individually dispersed cells or small cell clusters at the tumor periphery or the invasive front. Therefore, a plausible corollary is that the CAF/MSC–CSC crosstalk through paracrine signaling will predominantly take place at the tumor periphery. If so, pathological biomarkers and criteria that reflect the distance between CAFs or MSCs and CSCs should be developed to select those tumors that most likely respond to therapies directed at disrupting the CAF/MSC–CSC interplay. Finally, the timing of implementing pro-stemness-CAF- or MSC-targeted therapies will be another important consideration in the design of the related clinical trials. For the agents designed to target treatment-naïve CAFs or MSCs, it is critical to dose patients in early phases of cancer treatment before or concurrently with neoadjuvant chemotherapy as CSCs are less frequent and may be more susceptible to CSC-directed agents (54). By contrast, for the therapeutics targeting chemotherapy-modulated CAFs or MSCs, they should be administered following the initiation or during the course of chemotherapy, depending on when and the extent to which the pro-stemness functions of CAFs or MSCs are activated. Another timing of CAF/MSC-CSC-directed therapy is at the adjuvant setting following the removal of primary tumors, at which the therapy is designed to target micro-metastatic and circulating tumor cells that are known to contain enriched CSC populations (122–124). In this scenario, the blockade of CAF- or MSC-derived pro-stemness factors is expected to prevent the formation of CSC niches in primary or distant sites to reduce tumor recurrence and/or metastasis following surgery. Again, appropriate stemness- and/or stroma-informed biomarkers will be required to guide patient selection as well as the prediction of response in this type of trial.
The first generation of therapeutic strategies aiming at blocking the CAF-derived pro-stemness factors has remained largely in preclinical stages or been tested in early-phase clinical trials. Further optimization and improvements in the potency of antibody and small-molecule therapeutics or the introduction of novel therapeutic entities, such as the STAT-3-targeted oligonucleotide (91), may hold promises to overcoming current developmental hurdles. Alternatively, functional targeting or the specific depletion of the pro-stemness subpopulation of CAFs using such as FAP- or GPR-77-targeted antibodies, DNA vaccine, and immune cell therapeutics, provides promising next-generation approaches to preventing the cross-talk between CAFs and CSCs. Suppressing the pro-stemness factors secreted by MSCs or the direct depletion of MSCs also represents an interesting and promising opportunity of antagonizing their pro-oncogenic effects. The pro-stemness-CAF- or MSC-targeted therapies offer a novel opportunity of enhancing the treatment response of cytotoxic therapies such as chemotherapy and IR to prevent treatment-triggered expansion and activation of CSCs. Moreover, pro-stemness-CAF- and MSC-targeted therapies may synergize with CSC-targeted agents to reduce cancer stemness and aggressiveness, ultimately improving the therapeutic outcome of patients with desmoplastic cancers. Pro-stemness-CAF-related biomarkers are expected to aid the design of clinical trials and guide patient selection in CAF-/MSC-targeted therapies. Whilst these novel stroma-targeted approaches may potentially renew the interest in CSC-directed therapies in solid tumors, whether or not they can indeed fulfill their promise remains to be validated by more meticulously designed clinical trials.
T-SC: conceptualization, drafting of the manuscript, and critical revision of the manuscript. YS: drafting of the manuscript. KT: conceptualization, drafting of the manuscript, critical revision of the manuscript, obtained funding, final supervision, review, and editing.
This work was supported in part by Ministry of Science and Technology, Taiwan (MOST 102-2628-B-400-MY3, 104-2314-B-400-022, MOST 105-2314-B-400-018 to KT; MOST 107-2314-B-038-103 to T-SC), Taipei Medical University (DP2-107-21121-C-04 to KT), and Wan Fang Hospital, Chi-Mei Medical Center, and Hualien Tzu-Chi Hospital Joint Cancer Center Grant, Ministry of Health and Welfare (MOHW108-TDU-B-212-124020 to KT), Taipei Medical University – Wan Fang Hospital (107TMU-WFH-16 to T-SC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. (2008) 8:755–68. doi: 10.1038/nrc2499
2. Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. (2018) 173:338–54.e315. doi: 10.1016/j.cell.2018.03.034
3. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. (2014) 344:1396–401. doi: 10.1126/science.1254257
4. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. (2004) 432:396–401. doi: 10.1038/nature03128
5. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
6. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. (2007) 67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030
7. van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzman-Ramirez N, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. (2010) 70:5163–73. doi: 10.1158/0008-5472.CAN-09-3806
8. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. (2007) 445:106–10. doi: 10.1038/nature05372
9. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. (2011) 121:3804–9. doi: 10.1172/JCI57099
10. Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. (2012) 47:570–84. doi: 10.1016/j.molcel.2012.06.014
11. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. (2011) 108:1397–402. doi: 10.1073/pnas.1018898108
12. Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. (2001) 411:375–9. doi: 10.1038/35077241
13. Shiao SL, Coussens LM. The tumor-immune microenvironment and response to radiation therapy. J Mammary Gland Biol Neoplasia. (2010) 15:411–21. doi: 10.1007/s10911-010-9194-9
14. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
15. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. (2007) 449:557–63. doi: 10.1038/nature06188
16. Minamoto T, Ooi A, Okada Y, Mai M, Nagai Y, Nakanishi I. Desmoplastic reaction of gastric carcinoma: a light- and electron-microscopic immunohistochemical analysis using collagen type-specific antibodies. Hum Pathol. (1988) 19:815–21. doi: 10.1016/S0046-8177(88)80265-X
17. Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. (2015) 47:312–9. doi: 10.1038/ng.3224
18. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. (2006) 6:392–401. doi: 10.1038/nrc1877
19. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. (2010) 17:135–47. doi: 10.1016/j.ccr.2009.12.041
20. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. (2014) 159:80–93. doi: 10.1016/j.cell.2014.08.007
21. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. (2016) 16:582–98. doi: 10.1038/nrc.2016.73
22. Rudnick JA, Arendt LM, Klebba I, Hinds JW, Iyer V, Gupta PB, et al. Functional heterogeneity of breast fibroblasts is defined by a prostaglandin secretory phenotype that promotes expansion of cancer-stem like cells. PLoS ONE. (2011) 6:e24605. doi: 10.1371/journal.pone.0024605
23. Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun. (2014) 5:3472. doi: 10.1038/ncomms4472
24. Cheng JD, Dunbrack RL Jr, Valianou M, Rogatko A, Alpaugh RK, Weiner LM. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. (2002) 62:4767–72.
25. Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. (2011) 146:148–63. doi: 10.1016/j.cell.2011.05.040
26. Serebriiskii I, Castello-Cros R, Lamb A, Golemis EA, Cukierman E. Fibroblast-derived 3D matrix differentially regulates the growth and drug-responsiveness of human cancer cells. Matrix Biol. (2008) 27:573–85. doi: 10.1016/j.matbio.2008.02.008
27. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. (2009) 139:891–906. doi: 10.1016/j.cell.2009.10.027
28. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. (2019) 18:70. doi: 10.1186/s12943-019-0994-2
29. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. (2018) 33:463–79.e410. doi: 10.1016/j.ccell.2018.01.011
30. Luga V, Wrana JL. Tumor-stroma interaction: revealing fibroblast-secreted exosomes as potent regulators of Wnt-planar cell polarity signaling in cancer metastasis. Cancer Res. (2013) 73:6843–7. doi: 10.1158/0008-5472.CAN-13-1791
31. Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE. (2015) 10:e0125625. doi: 10.1371/journal.pone.0125625
32. Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci USA. (2010) 107:21677–82. doi: 10.1073/pnas.1016234107
33. Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. (2012) 72:2768–79. doi: 10.1158/0008-5472.CAN-11-3567
34. Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. (2011) 9:433–46. doi: 10.1158/1538-7445.FBCR11-B45
35. Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature. (2019) 569:131–5. doi: 10.1038/s41586-019-1130-6
36. Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J Exp Med. (2013) 210:2851–72. doi: 10.1084/jem.20131195
37. Chan TS, Hsu CC, Pai VC, Liao WY, Huang SS, Tan KT, et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J Exp Med. (2016) 213:2967–88. doi: 10.1084/jem.20151665
38. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. (1999) 284:143–7. doi: 10.1126/science.284.5411.143
39. Fox JM, Chamberlain G, Ashton BA, Middleton J. Recent advances into the understanding of mesenchymal stem cell trafficking. Br J Haematol. (2007) 137:491–502. doi: 10.1111/j.1365-2141.2007.06610.x
40. Guo X, Oshima H, Kitmura T, Taketo MM, Oshima M. Stromal fibroblasts activated by tumor cells promote angiogenesis in mouse gastric cancer. J Biol Chem. (2008) 283:19864–71. doi: 10.1074/jbc.M800798200
41. Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. (2008) 68:4331–9. doi: 10.1158/0008-5472.CAN-08-0943
42. Beckermann BM, Kallifatidis G, Groth A, Frommhold D, Apel A, Mattern J, et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer. (2008) 99:622–31. doi: 10.1038/sj.bjc.6604508
43. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. (2011) 19:257–72. doi: 10.1016/j.ccr.2011.01.020
44. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. (2011) 17:211–5. doi: 10.1038/nm.2284
45. Zischek C, Niess H, Ischenko I, Conrad C, Huss R, Jauch KW, et al. Targeting tumor stroma using engineered mesenchymal stem cells reduces the growth of pancreatic carcinoma. Ann Surg. (2009) 250:747–53. doi: 10.1097/SLA.0b013e3181bd62d0
46. Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. (2012) 2:840–55. doi: 10.1158/2159-8290.CD-12-0101
47. Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y, et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci. (2013) 104:157–64. doi: 10.1111/cas.12059
48. Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. (2008) 100:672–9. doi: 10.1093/jnci/djn123
49. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. (2009) 106:13820–5. doi: 10.1073/pnas.0905718106
50. Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. (2006) 12:4147–53. doi: 10.1158/1078-0432.CCR-06-0038
51. Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, et al. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. (2007) 31:277–83. doi: 10.3892/ijo.31.2.277
52. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. (2010) 120:485–97. doi: 10.1172/JCI39397
53. Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. (2012) 18:1359–68. doi: 10.1038/nm.2890
54. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. (2019) 24:25–40. doi: 10.1016/j.stem.2018.11.017
55. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. (2019) 9:282–301. doi: 10.1158/2159-8290.CD-18-0710
56. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. (2014) 14:342–56. doi: 10.1016/j.stem.2014.01.009
57. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. (2009) 139:693–706. doi: 10.1016/j.cell.2009.10.014
58. Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. (2009) 28:2940–7. doi: 10.1038/onc.2009.180
59. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
60. Kim MP, Fleming JB, Wang H, Abbruzzese JL, Choi W, Kopetz S, et al. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS ONE. (2011) 6:e20636. doi: 10.1371/journal.pone.0020636
61. Chen L, Fan J, Chen H, Meng Z, Chen Z, Wang P, et al. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci Rep. (2014) 4:5911. doi: 10.1038/srep05911
62. Singh JK, Farnie G, Bundred NJ, Simoes BM, Shergill A, Landberg G, et al. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin Cancer Res. (2013) 19:643–56. doi: 10.1158/1078-0432.CCR-12-1063
63. Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. (2012) 11:1282–90. doi: 10.4161/cc.19679
64. Lenos KJ, Miedema DM, Lodestijn SC, Nijman LE, van den Bosch T, Romero Ros X, et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat Cell Biol. (2018) 20:1193–202. doi: 10.1038/s41556-018-0179-z
65. Tang YA, Chen YF, Bao Y, Mahara S, Yatim S, Oguz G, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1alpha and TGF-beta2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci USA. (2018) 115:E5990–9. doi: 10.1073/pnas.1801348115
66. Timaner M, Letko-Khait N, Kotsofruk R, Benguigui M, Beyar-Katz O, Rachman-Tzemah C, et al. Therapy-educated mesenchymal stem cells enrich for tumor-initiating cells. Cancer Res. (2018) 78:1253–65. doi: 10.1158/0008-5472.CAN-17-1547
67. Schmidt A, Ladage D, Schinkothe T, Klausmann U, Ulrichs C, Klinz FJ, et al. Basic fibroblast growth factor controls migration in human mesenchymal stem cells. Stem Cells. (2006) 24:1750–8. doi: 10.1634/stemcells.2005-0191
68. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. (2011) 71:614–24. doi: 10.1158/0008-5472.CAN-10-0538
69. Tsai KS, Yang SH, Lei YP, Tsai CC, Chen HW, Hsu CY, et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology. (2011) 141:1046–56. doi: 10.1053/j.gastro.2011.05.045
70. Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. (2011) 20:370–83. doi: 10.1016/j.ccr.2011.08.010
71. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
72. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. (2018) 172:841–56.e816. doi: 10.1016/j.cell.2018.01.009
73. Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res. (2017) 23:1710–21. doi: 10.1158/1078-0432.CCR-15-2851
74. Costea DE, Hills A, Osman AH, Thurlow J, Kalna G, Huang X, et al. Identification of two distinct carcinoma-associated fibroblast subtypes with differential tumor-promoting abilities in oral squamous cell carcinoma. Cancer Res. (2013) 73:3888–901. doi: 10.1158/0008-5472.CAN-12-4150
75. Bartoschek M, Oskolkov N, Bocci M, Lovrot J, Larsson C, Sommarin M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun. (2018) 9:5150. doi: 10.1038/s41467-018-07582-3
76. Avery D, Govindaraju P, Jacob M, Todd L, Monslow J, Pure E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. (2018) 67:90–106. doi: 10.1016/j.matbio.2017.12.003
77. Park CK, Jung WH, Koo JS. Expression of cancer-associated fibroblast-related proteins differs between invasive lobular carcinoma and invasive ductal carcinoma. Breast Cancer Res Treat. (2016) 159:55–69. doi: 10.1007/s10549-016-3929-2
78. O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci USA. (2011) 108:16002–7. doi: 10.1073/pnas.1109493108
79. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
80. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. (2012) 148:349–61. doi: 10.1016/j.cell.2011.11.025
81. Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. (2011) 145:926–40. doi: 10.1016/j.cell.2011.04.029
82. Park CW, Kim KS, Bae S, Son HK, Myung PK, Hong HJ, et al. Cytokine secretion profiling of human mesenchymal stem cells by antibody array. Int J Stem Cells. (2009) 2:59–68. doi: 10.15283/ijsc.2009.2.1.59
83. Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. (2013) 154:1060–73. doi: 10.1016/j.cell.2013.07.036
84. Tang RJ, Shen SN, Zhao XY, Nie YZ, Xu YJ, Ren J, et al. Mesenchymal stem cells-regulated Treg cells suppress colitis-associated colorectal cancer. Stem Cell Res Ther. (2015) 6:71. doi: 10.1186/s13287-015-0055-8
85. Scherzed A, Hackenberg S, Froelich K, Kessler M, Koehler C, Hagen R, et al. BMSC enhance the survival of paclitaxel treated squamous cell carcinoma cells in vitro. Cancer Biol Ther. (2011) 11:349–57. doi: 10.4161/cbt.11.3.14179
86. Lis R, Touboul C, Mirshahi P, Ali F, Mathew S, Nolan DJ, et al. Tumor associated mesenchymal stem cells protects ovarian cancer cells from hyperthermia through CXCL12. Int J Cancer. (2011) 128:715–25. doi: 10.1002/ijc.25619
87. Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin Cancer Res. (2017) 23:5358–65. doi: 10.1158/1078-0432.CCR-16-2748
88. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. (2016) 29:832–45. doi: 10.1016/j.ccell.2016.04.014
89. Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci USA. (2015) 112:1839–44. doi: 10.1073/pnas.1424171112
90. Zhong H, Davis A, Ouzounova M, Carrasco RA, Chen C, Breen S, et al. A novel IL6 antibody sensitizes multiple tumor types to chemotherapy including trastuzumab-resistant tumors. Cancer Res (2016) 76:480–90. doi: 10.1158/0008-5472.CAN-15-0883
91. Njatcha C, Farooqui M, Kornberg A, Johnson DE, Grandis JR, Siegfried JM. STAT3 cyclic decoy demonstrates robust antitumor effects in non-small cell lung cancer. Mol Cancer Ther. (2018) 17:1917–26. doi: 10.1158/1535-7163.MCT-17-1194
92. Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. (2006) 116:1955–62. doi: 10.1172/JCI26532
93. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. (2013) 21:1611–20. doi: 10.1038/mt.2013.110
94. Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. (2015) 75:2800–10. doi: 10.1158/0008-5472.CAN-14-3041
95. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
96. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. (2013) 210:1137–51. doi: 10.1084/jem.20122344
97. Miao L, Liu Q, Lin CM, Luo C, Wang Y, Liu L, et al. Targeting tumor-associated fibroblasts for therapeutic delivery in desmoplastic tumors. Cancer Res. (2017) 77:719–31. doi: 10.1158/0008-5472.CAN-16-0866
98. Shaked Y, Pham E, Hariharan S, Magidey K, Beyar-Katz O, Xu P, et al. Evidence implicating immunological host effects in the efficacy of metronomic low-dose chemotherapy. Cancer Res. (2016) 76:5983–93. doi: 10.1158/0008-5472.CAN-16-0136
99. Hasnis E, Alishekevitz D, Gingis-Veltski S, Bril R, Fremder E, Voloshin T, et al. Anti-Bv8 antibody and metronomic gemcitabine improve pancreatic adenocarcinoma treatment outcome following weekly gemcitabine therapy. Neoplasia. (2014) 16:501–10. doi: 10.1016/j.neo.2014.05.011
100. Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. (2012) 21:488–503. doi: 10.1016/j.ccr.2012.02.017
101. Kerbel RS, Shaked Y. Therapy-activated stromal cells can dictate tumor fate. J Exp Med. (2016) 213:2831–3. doi: 10.1084/jem.20161845
102. Andre N, Tsai K, Carre M, Pasquier E. Metronomic chemotherapy: direct targeting of cancer cells after all? Trends Cancer. (2017) 3:319–25. doi: 10.1016/j.trecan.2017.03.011
103. Poggi A, Varesano S, Zocchi MR. How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. Front Immunol. (2018) 9:262. doi: 10.3389/fimmu.2018.01342
104. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. (2009) 9:265–73. doi: 10.1038/nrc2620
105. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. (2014) 2:78–91. doi: 10.1016/j.stemcr.2013.11.009
106. Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, et al. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. (2013) 5:1228–42. doi: 10.1016/j.celrep.2013.11.014
107. Colacino JA, Azizi E, Brooks MD, Harouaka R, Fouladdel S, McDermott SP, et al. Heterogeneity of human breast stem and progenitor cells as revealed by transcriptional profiling. Stem Cell Rep. (2018) 10:1596–609. doi: 10.1016/j.stemcr.2018.03.001
108. Lau J, Ilkhanizadeh S, Wang S, Miroshnikova YA, Salvatierra NA, Wong RA, et al. STAT3 blockade inhibits radiation-induced malignant progression in glioma. Cancer Res. (2015) 75:4302–11. doi: 10.1158/0008-5472.CAN-14-3331
109. Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature. (2017) 545:187–92. doi: 10.1038/nature22081
110. de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature. (2017) 543:676–80. doi: 10.1038/nature21713
111. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov. (2014) 13:497–512. doi: 10.1038/nrd4253
112. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
113. Yen TW, Aardal NP, Bronner MP, Thorning DR, Savard CE, Lee SP, et al. Myofibroblasts are responsible for the desmoplastic reaction surrounding human pancreatic carcinomas. Surgery. (2002) 131:129–34. doi: 10.1067/msy.2002.119192
114. Waghray M, Yalamanchili M, di Magliano MP, Simeone DM. Deciphering the role of stroma in pancreatic cancer. Curr Opin Gastroenterol. (2013) 29:537–43. doi: 10.1097/MOG.0b013e328363affe
115. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. (2009) 324:1457–61. doi: 10.1126/science.1171362
116. Miao L, Lin CM, Huang L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J Control Release. (2015) 219:192–204. doi: 10.1016/j.jconrel.2015.08.017
117. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. (2011) 29:4548–54. doi: 10.1200/JCO.2011.36.5742
118. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. (2011) 17:313–9. doi: 10.1038/nm.2304
119. Islam F, Gopalan V, Smith RA, Lam AK. Translational potential of cancer stem cells: a review of the detection of cancer stem cells and their roles in cancer recurrence and cancer treatment. Exp Cell Res. (2015) 335:135–47. doi: 10.1016/j.yexcr.2015.04.018
120. Tsai KK, Chan TS, Shaked Y. Next viable routes to targeting pancreatic cancer stemness: learning from clinical setbacks. J Clin Med. (2019) 8:702. doi: 10.3390/jcm8050702
121. Vidal SJ, Rodriguez-Bravo V, Galsky M, Cordon-Cardo C, Domingo-Domenech J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene. (2014) 33:4451–63. doi: 10.1038/onc.2013.411
122. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. (2015) 526:131–5. doi: 10.1038/nature15260
123. Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. (2018) 18:669–80. doi: 10.1038/s41568-018-0056-x
Keywords: cancer-associated fibroblasts, mesenchymal stem cells, cancer stem cells, paracrine signaling, desmoplasia
Citation: Chan T-S, Shaked Y and Tsai KK (2019) Targeting the Interplay Between Cancer Fibroblasts, Mesenchymal Stem Cells, and Cancer Stem Cells in Desmoplastic Cancers. Front. Oncol. 9:688. doi: 10.3389/fonc.2019.00688
Received: 27 April 2019; Accepted: 12 July 2019;
Published: 31 July 2019.
Edited by:
Alexandre Prieur, ECS-Screening SA, SwitzerlandReviewed by:
Aamir Ahmad, Mitchell Cancer Institute, United StatesCopyright © 2019 Chan, Shaked and Tsai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelvin K. Tsai, dHNhaWtAdG11LmVkdS50dw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.