
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 02 August 2019
Sec. Cancer Genetics
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00673
 Gulnur Zhunussova1,2,3*
Gulnur Zhunussova1,2,3* Georgiy Afonin2,4
Georgiy Afonin2,4 Saltanat Abdikerim1Abai Jumanov2,4Anastassiya Perfilyeva1Dilyara Kaidarova2,4
Saltanat Abdikerim1Abai Jumanov2,4Anastassiya Perfilyeva1Dilyara Kaidarova2,4 Leyla Djansugurova1,3
Leyla Djansugurova1,3Background: Colorectal cancer (CRC) incidence is rising worldwide, as well as in the Republic of Kazakhstan, while its occurrence is also increasing in the younger population. Hereditary forms associated with the development of colon and rectal cancer and early-onset CRC have never been studied in the population of Kazakhstan. The aim of this research was to investigate the spectrum of CRC-related gene mutations to determine which mutations cause early onset of CRC in the Kazakhstan population.
Methods: The study included 125 unrelated patients from Kazakhstan (range 17–50 years in age) with early onset CRC. Genomic DNA was obtained from peripheral blood of the patients. Next-generation sequencing was performed using the TruSightCancer Kit on the MiSeq platform. The Studio Variant was used to annotate and interpret genetic variants.
Results: Bioinformatics analysis of Next-generation sequencing data revealed 11,152 variants from 85 genes, of them, 3,790 missense, 6,254 synonymous variants, 44 3′UTR variants, 10 frameshift variants, five stop-gain variants, four in-frame deletions, two splice donors, one splice acceptor variant, and 1,042 intron or non-coding variants. APC, BRCA2/1, ALK, BRIP1, EGFR, FANCA, FANCD2, FANCI, HNF1A, MEN1, NSD1, PMS2, RECQL4, RET, SLX4, WRN, and XPC genes mutated most often. According to the ACMG guidelines and LOVD/ClinVar databases, 24 variants were pathogenic (10 frameshifts, five missenses, five stop-gain, one in-frame deletion, and three splice-site mutations), and 289 were VUS with population frequency <1%, 131 of them were attributed as deleterious. In the study, 50% of all pathogenic mutations found in Kazakhstani patients with early CRC onset were identified in the subgroups with a family history of CRC and primary multiple tumors. In APC, pathogenic mutations were most often (21%).
Conclusion: Pathogenic and likely pathogenic mutations were found in 20 (16%) out of 125 patients. Eight novel pathogenic mutations detected in FANCI, APC, BMPR1, ATM, and DICER1 genes have not been reported in previous literature. Given the high frequency and wide spectrum of mutations, NGS analysis must be carried out in families with a history of CRC/CRC-related cancers with the purpose to identify cause-effective mutations, clarify the clinical diagnosis, and prevent the development of the disease in other family members.
Currently, colorectal cancer (CRC) is one of the most common types of cancer, with more than 1.8 million new cases and 800,000 deaths registered each year in the world. According to GLOBOCAN, 1,849,518 cases of CRC (10.2% of all cancers) were registered in the world in 2018 (1). According to IARC global data, CRC ranks 3rd in the structure of cancer incidence in men, and 2nd in women (2).
Considering current trends and the dynamics of the demographic situation (population growth and population aging), by 2030, WHO predicts an increase of 77% in global CRC incidence (to 2.2 million new cases per year) and 80% mortality (to 1.1 million deaths from CRC per year). Worldwide, CRC incidence is more influenced by a family history of cancer (FHC) and social (mainly environmental) factors, than by geographical factors (3).
CRC frequency positively and strongly correlates with age. CRC likelihood in the general population increases after age 40, with a dramatic increase after age 50; 90% of CRC cases occur in people over the age of 50 (4).
Colorectal cancer (CRC) has a complex nature which is a sequential interaction of environmental and genetic factors. Sporadic CRC occurs pre-dominantly in the elderly and at a senile age and accounts for nearly 80% of all cases. Early-onset CRC is mostly associated with hereditary syndromes as they pre-dispose the development of CRC as a result of mutations in well-studied genes. Currently, several specific conditions are considered as etiological factors: hereditary non-polyposis colorectal cancer (HNPCC, also known as Lynch syndrome) (5), familial adenomatous polyposis (FAP) syndrome, other rarer syndromes and syndromic associations (Peutz-Jeghers, Gardner-Turner, Bannayan–Riley–Ruvalcaba, Bloom's disease, Tourette's syndrome), inflammatory bowel disease, infection caused by human papillomavirus (squamous cell carcinoma of rectum and anal), and HIV. Lynch syndrome and FAP are the most common known causes of CRC development (6–12).
In the Republic of Kazakhstan, 325,606 patients were registered with malignant tumors from 2008 to 2018. Twenty-eight thousand (8.6%) of them were diagnosed with CRC, including 14,833 (52.9%) cases of colon cancer, and 13,185 (47.1%) cases of rectal cancer. The number of detected CRC cases grew by an average of 845 cases per year, and the CRC incidence rating among other cancers has increased from 5th to 4th place. In the last 10 years, 3,121 (11.1%) of all new CRC patients were younger than 50. In line with a growing CRC incidence among younger adults in Kazakhstan, the number of early-onset CRC patients before age 50 has increased by 14.8%, from 305 cases in 2008 to 350 cases in 2017.
Unfortunately, there are no statistics on hereditary types of CRC in Kazakhstan. For this reason, we are collecting biomaterials and family histories of patients with early-onset CRC.
According to the classic CRC carcinogenesis model, several key genetic changes are required for tumor growth initiation and progression. The earliest genetic trigger is the APC gene inactivation. The mutations in other suppressor genes (SMAD2, SMAD4, DCC, and TP53), oncogenes (KRAS, BRAF), and some other genes contribute to neoplastic transformation in foci of the altered colon mucosa which in turn leads to further degeneration toward malignancy. These mutations may be accompanied by the deregulation of the expression of oncogenes and/or tumor suppressor genes and subsequent epigenetic modifications of their promoter regions. However, other genetic changes are also involved in the development of CRC. Thus, each hereditary syndrome is associated with a specific spectrum of gene mutations (13–15).
The results of genetic analysis of cancer patients indicate an annual increase in the number of genetic changes of prognostic, diagnostic, or clinical significance. Advances in genome and epigenome analysis provide new information on hereditary CRC variants. This creates a need for robust clinical tests able to simultaneously detect multiple genetic changes. Next-generation sequencing (NGS) fully meets this growing demand thanks to the high generating ability of DNA analysis and good clinical throughput. NGS platforms allow for the identification of cause-effect mutations in the early onset of CRC. These mutations have already been described and included in widely used databases such as ClinVar (16), LOVD/InSIGHT (available via the Leiden Open-Source Variation Database (LOVD) http://www.lovd.nl/3.0/home or https://www.insight-group.org/variants/database/), the Catalog of Somatic Mutations in Cancer (COSMIC, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) together with the novel, previously undescribed mutations of key CRC genes.
Therefore, we used multiple gene panels and NGS to screen patients from Kazakhstan with early-onset CRC for potential pathogenic mutations.
The study was conducted on 125 patients aged between 17 and 50 from the Republic of Kazakhstan with early-onset of CRC. All the patients signed the informed consent and filled in the detailed questionnaire which included questions related to the FHC. The consent form and the questionnaire were approved by the IRB/IEC of Kazakh Institute of Oncology and Radiology (KazIOR) and Asfendiyarov Kazakh National Medical University (Almaty, Kazakhstan). In cases observed before 2017, cancer staging was done according to the 7th edition of the AJCC Cancer Staging System; in cases observed after 2017—according to the 8th edition of the AJCC Cancer Staging System (The AJCC Cancer Staging Manual, Eighth Edition, https://cancerstaging.org/references-tools/deskreferences/Pages/8EUpdates.aspx#).
Genomic DNA (gDNA) was isolated from EDTA treated peripheral blood samples using the GeneJET Genomic DNA Purification Kit (Thermo Scientific, USA). Qualitative and quantitative characteristics of the DNA samples were evaluated by the spectrophotometer (Eppendorf Biophotometer plus, Germany) and fluorometer (Quantus™ Promega, USA). At least 50 ng of double-stranded nuclear DNA was obtained per sample.
Next-generation sequencing (NGS) was performed using TruSight Rapid Capture Kit (Illumina, USA) in combination with the TruSight Cancer Sequencing Panel (Illumina, USA) according to the manufacturer's recommendations.
DNA libraries with 4 mM molarities were subjected to clustering using a standard flow cell and were sequenced on the MiSeq platform (Illumina) using the MiSeq Reagent Kit v3 (600 cycles).
Bioinformatics data analysis was carried out using two methodological approaches with different algorithms of analysis. In the first approach, the NGS data were analyzed using MiSeq Reporter v.3.0 software (Illumina). This software allowed mapping and aligning the sequenced sequences and comparing them with the reference sequence of the human genome (GRCH37/hg19) using the Burrows-Wheeler Aligner (BWA) algorithm. Search and detection of variants for specific regions of the genome were carried out using the Genome Analysis Toolkit (GATK, Broad Institute, Cambridge, USA).
In the second approach, various bioinformatics methods and software packages were used to optimize the workflow. The mapping and alignment of a sequence read against the human reference genome of the GRCH37/hg19 version were conducted using the Bowtie2 algorithm with a very sensitive local parameter. FastQC and MultiQC were used to evaluate the quality of reads of sequences. Picard Tools and SAM Tools/BCF tools were used to convert SAM files into BAM files, sort, and index the mapped reads, remove intermediate files, merge the BAM files, and identify duplicates. Java Runtime Environment and R Bioconductor were used to execute software scripts. GATK was used to re-align the mapped reads around the areas with insertions/deletions. GATK “Haplotype caller” strategy was used to filter and detect genomic variants. Finally, all VCF (variant call format) files containing only alternative variants obtained by GATK were imported into the Variant Studio 3.0 software (Illumina). In the study, only the variants identified by both approaches were considered.
Variant Studio was used to annotate and interpret genetic variants. A Q-score of 30, corresponding to an error rate of 0.1%, was selected as the acceptance threshold value. The following filtration parameters were set to exclude poor quality nucleotide variants: read depth >50×, alternative read depth >20×, quality value >100. Variants that matched the filtering parameters were selected for the study.
Genetic variants were annotated in accordance with the nomenclature of the Human Genome Variation Society (HGVS) (17); the interpretation was done using the Single Nucleotide Polymorphism Database (dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/), ClinVar database, InSIGHT/LOVD database, and COSMIC. Integrative Genomics Viewer was applied to visualize the variants (18, 19).
Identified variants were categorized in accordance with the guidelines of the American College of Medical Genetics and Genomics (ACMG) (20) as pathogenic, likely pathogenic, uncertain significance (VUS), benign, and likely benign. Pathogenic mutations included those resulting in a premature stop codon (frameshift mutations and nonsense), variants with uncorrected splicing and variants affecting protein function according to the results of functional studies. Novel mutations such as frameshift mutations and nonsense mutations leading to protein truncation, as well as mutations without well-established functional studies, were classified as likely pathogenic. Synonymous and intronic variants not affecting splicing were regarded to be benign/probably benign, as well as the variants with a minor allele frequency (MAF) found in 5% or more of the general population according to the 1000 Genomes database (1000G) (21), the Exome Sequencing Project database (ESP6500, https://esp.gs.washington.edu) or the Exome Aggregation Consortium database (ExAC) (20). The remaining variants with no functional data and not fulfilling the criteria of classification as pathogenic/likely pathogenic or benign/likely benign, or with conflicting evidence of their benign and pathogenic nature, were defined as VUS.
In silico, bioinformatics tools such as Sorting Intolerant From Tolerant (SIFT) (22) and Polymorphism Phenotyping-2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/) (23) were used to predict potential pathogenic effects of missense variants on protein structure and function. The variants that matched the following criteria: a SIFT score of <0.05 and a PolyPhen-2 score between 0.95 and 1.0, were considered to be strongly suspected of being deleterious (24).
Demographics, clinical-pathological characteristics, and personal and family history variables were analyzed. Descriptive statistics were summarized as frequency distributions for categorical variables and as means for continuous variables. Wilcoxon rank sum tests were used to compare continuous variables, and the chi-square test was performed to compare categorical variables. The results were considered statistically significant at a p < 0.05. All statistical tests were two-sided at the 5% level of significance. Data were processed and analyzed by using R software, version 3.6.0 (W.N. Venables, D.M. Smith, and the R Core Team).
Sequencing data have been deposited at the NCBI SRA archive with BioProject record PRJNA548114 and SRA accession SUB5747000.
Study cohort included 62 (49.6%) men and 63 (50.4%) women with early onset CRC. Th ethnic composition of the patient group included Kazakhs−61 (48.8%), Russians−36 (28.8%), Uyghurs−7 (5.6%), Azerbajanians−3 (2.4%), Koreans−3 (2.4%), Germans−3 (2.4 %), Ukrainians−2 (1.6%), Tatars−2 (1.6%), Belarusians, Dungans, Turks−1 patient each (0.8% each), patients with mixed ethnic background−5 (4%).
The median age of patients at CRC diagnosis was 40.7 (39.6 in men, 41.7 in women). 25 patients (20.1% of the early CRC onset cohort, of them, 15 men and 10 women) were diagnosed with CRC before the age of 35 (median age 29.08 ± 4.75 for both sexes; 28.4 ± 45.2 in men and 30.1 ± 5.15 in women).
In terms of clinical pathology, patients with early CRC onset differed by TNM staging, tumor localization, and histological types. The pre-dominant histological type was moderately differentiated adenocarcinoma detected in 81 (64.8%) cases. Half of the patients (57.2%) were diagnosed with stage III cancer. The rectum was a more common tumor site in both men and women compared to other sites. No significant statistical difference was found in all clinical characteristics between males and females (Table 1).
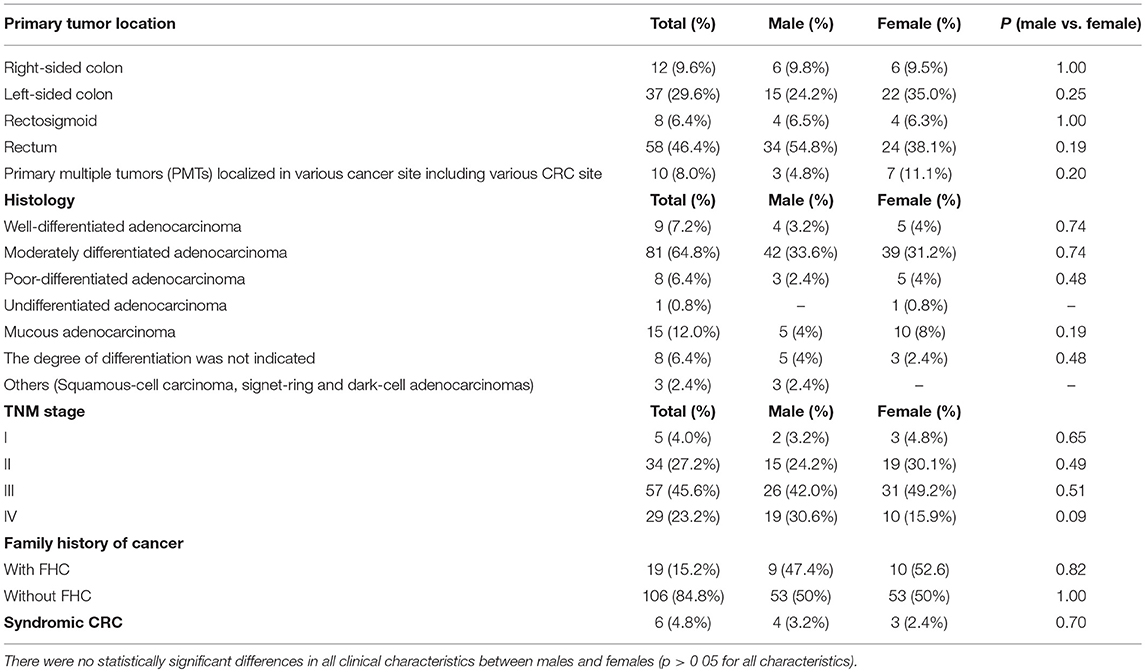
Table 1. Clinicopathological features of patients with early onset of CRC.
Of all the subjects, 19 (15.2%) patients with a family history of cancer (FHC), 6 had the CRC syndrome (Table 2). The characterization of all the patients with early onset CRC is shown in Figure 1.
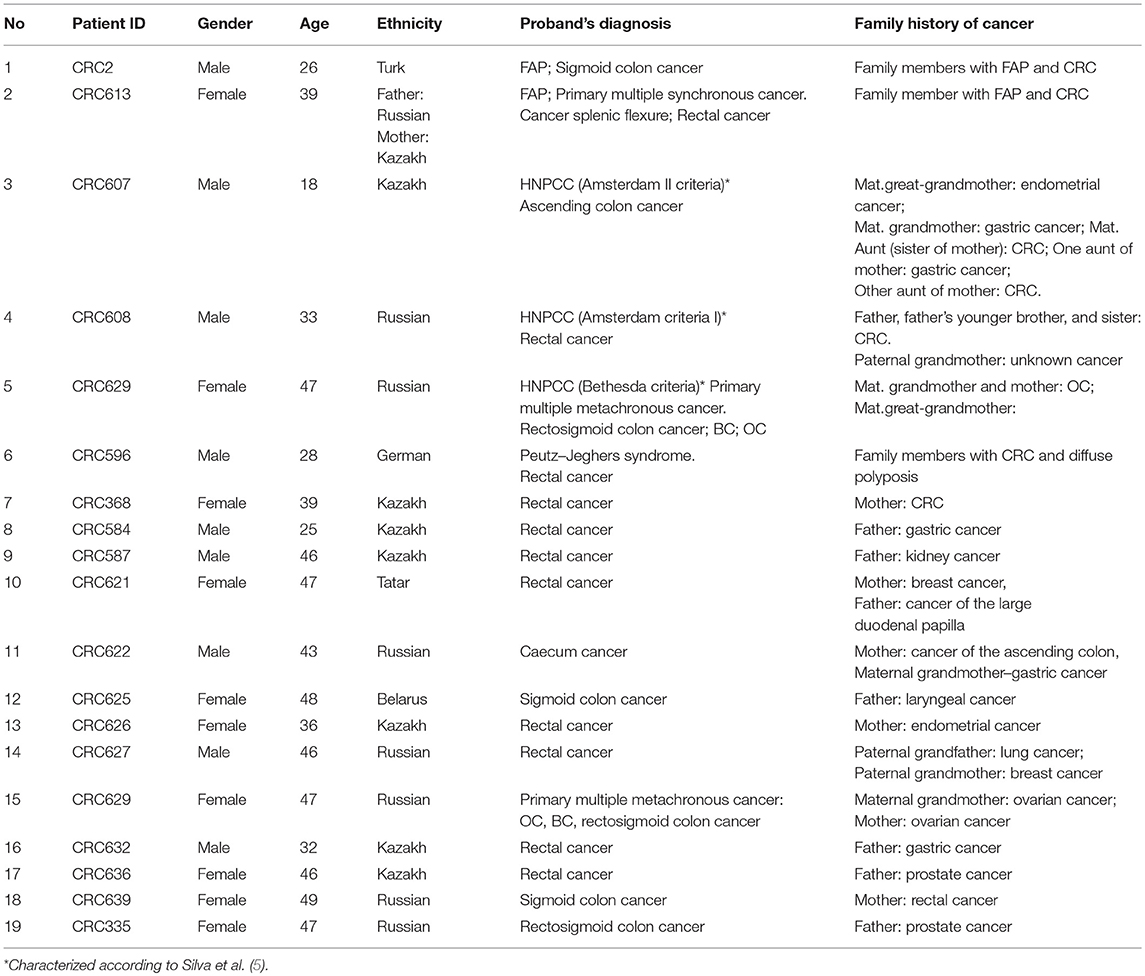
Table 2. Characteristics of the patients with a family history of cancer.
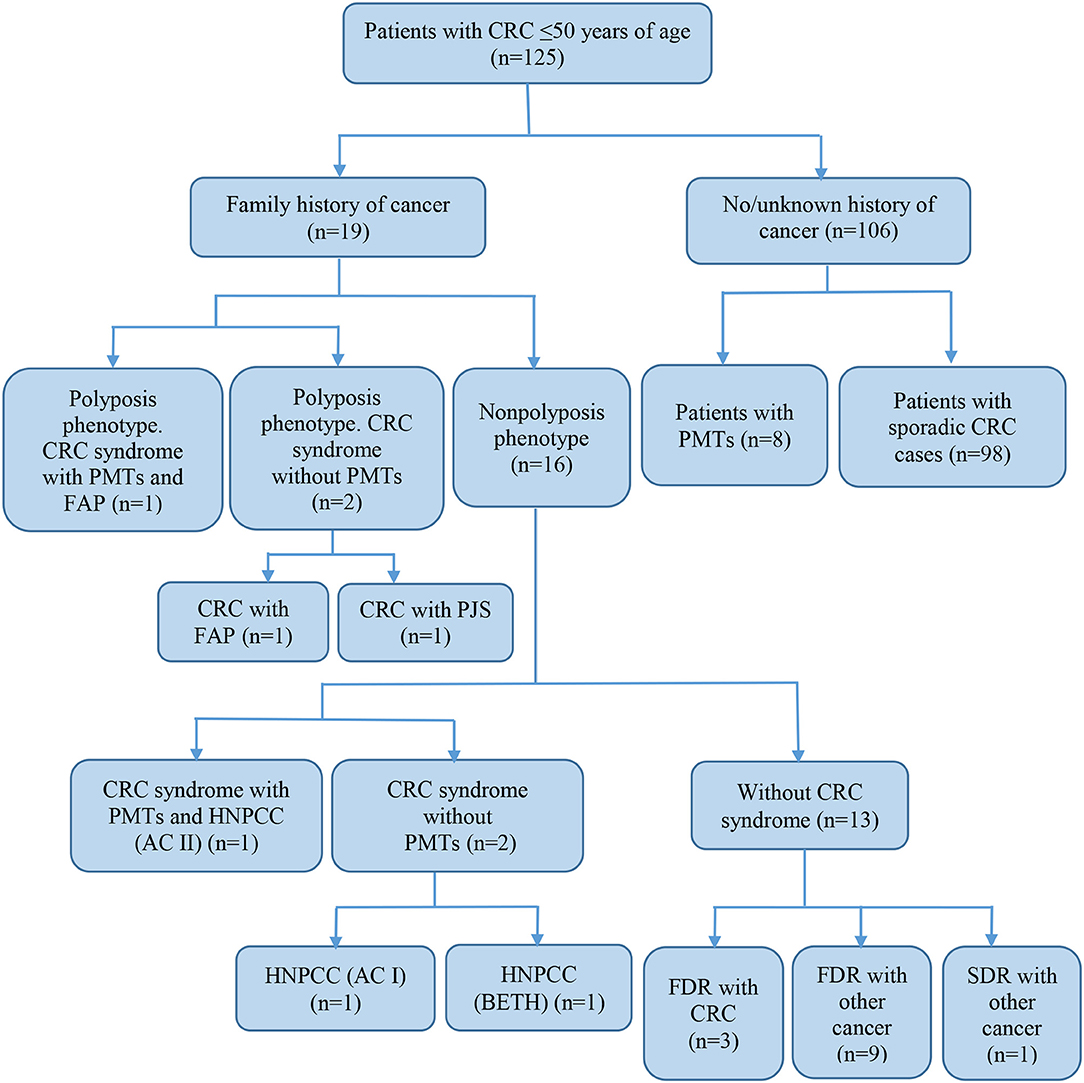
Figure 1. Characteristics of subjects with early-onset CRC. CRC, colorectal cancer; FAP, familial adenomatous polyposis; PJS, Peutz-Jeghers syndrome; HNPCC, hereditary non-polyposis colorectal cancer; AC, Amsterdam I or II criteria; BETH, Bethesda criteria; PMTs, primary multiple tumors; FDR, first-degree relative; SDR, second-degree relative.
A total of 125 unrelated patients with early onset CRC were analyzed by NGS using the TruSight Cancer Sequencing Panel which targeted a set of 94 genes known to play a role in cancer pre-disposition. Bioinformatics analysis of NGS data has revealed 11,152 variants (Supplementary Table 1) in 85 genes, of them, 3,790 missense, 6,254 synonymous variants, 44 3′UTR variants, 10 frameshift variants, five stop-gain variants, four in-frame deletions, two splice donors, one splice acceptor variant, and 1,042 intron or non-coding variants. APC, BRCA2/1, ALK, BRIP1, EGFR, FANCA, FANCD2, FANCI, HNF1A, MEN1, NSD1, PMS2, RECQL4, RET, SLX4, WRN, and XPC genes mutated most often.
The approach to the interpretation of NGS data described in the corresponding databases and guidelines mentioned in the Materials and Methods section has revealed 24 pathogenic/likely pathogenic mutations. 472 variants were classified as VUS; 10,656 variants—as benign/likely benign.
Twenty-four pathogenic and probable pathogenic mutations were detected in 20 patients (16% of the entire group of patients) with cancer of the cecum, ascending colon, hepatic flexure of the colon, splenic flexure of the colon, sigmoid colon, rectosigmoid colon, and rectum. Of them, five patients had a family history of FAP, CRC, gastric cancer, or ovarian cancer. Six patients (four cases of synchronous cancers and two cases of metachronous cancer) had primary multiple tumors (PMTs). Out of those pathogenic and likely pathogenic variants, five were detected in the APC gene, three in FANCI, three in BRCA2, two in BRCA1, two in MLH1, and one mutation each, in MSH6, MUTYH, BLM, NBN, ATM, BMPR1A, CHEK2, AIP, and DICER1 genes. As shown in Table 3, the analysis of mutation type has revealed 10 frameshift mutations, five missense mutations, five stop-gain mutations, one in-frame deletion, and three mutations involving uncorrected splicing. All detected pathogenic and likely pathogenic mutations were in the heterozygous state. In total, 23 mutations were unique, eight of them represented novel variants. Those new mutations have not been previously mentioned in the LOVD and ClinVar databases and have not been described in publications. All mutations were rare, and for 17 of them, frequency data was not available in the 1000G, ESP6500 and ExAC databases (Table 3). 62.5% of the mutations were available in the dbSNP database, 29.1% in the COSMIC catalog, and 38.4% in the ClinVar and/or LOVD databases. Multiple alterations of genes were noticed in three patients. Three frameshift variants including two novel variants (c.907delA in APC gene and c.6743dupA in ATM) and one previously reported mutation (c.657_661delACAAA in NBN) were detected in one patient (male, Kazakh) at the age of 40 with rectum cancer. Two more patients (one male at the age of 26 with FAP and sigmoid colon cancer and one male at the age of 46 with ascending colon cancer and rectum cancer) had one novel frameshift variant each (FANCI c.3340delA and BMPR1A c.152delC, respectively) in combination with other previously reported frameshift variants (APC c.3613delA and CHEK2 c.599T > C, respectively, Table 3).
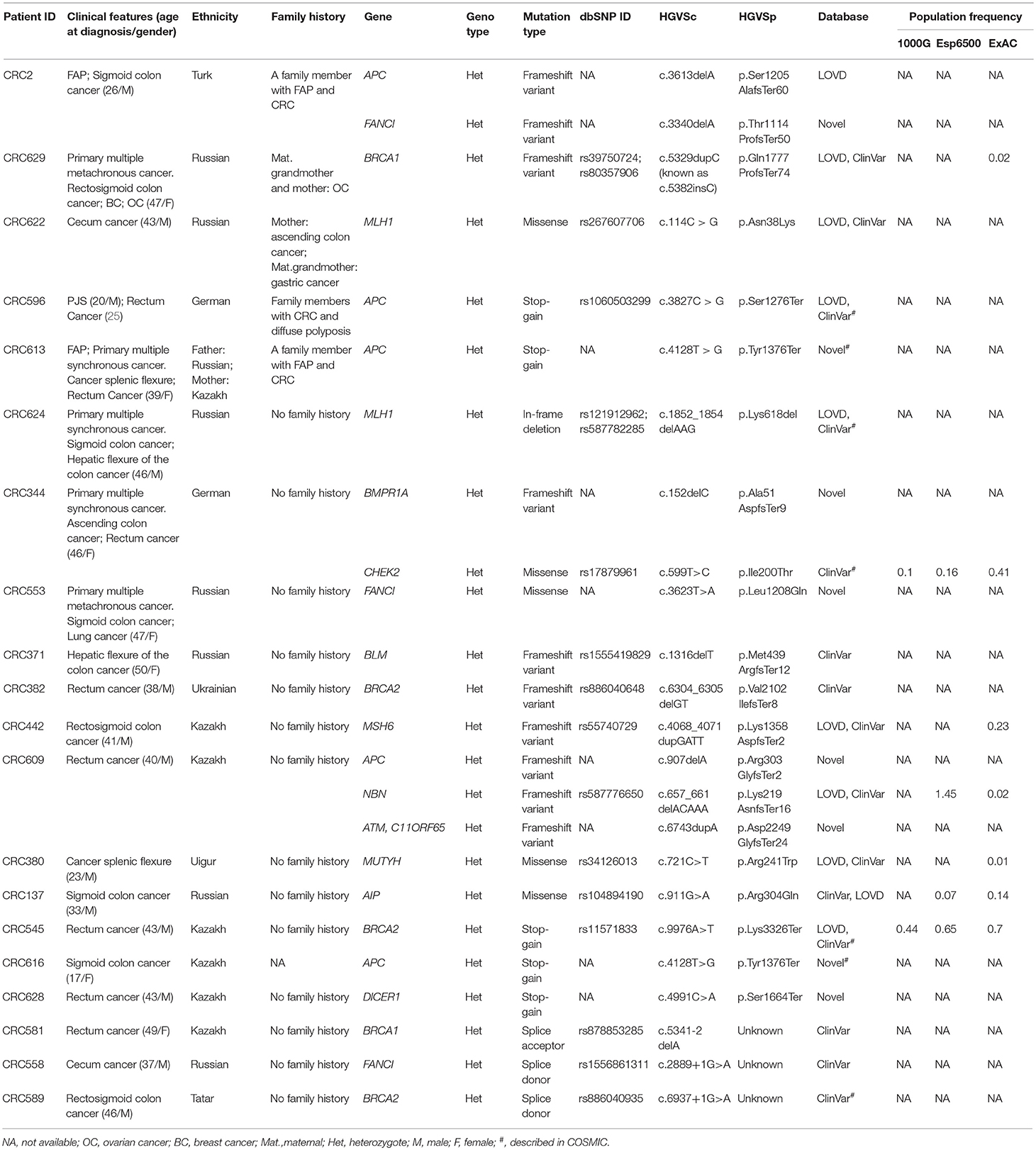
Table 3. Pathogenic and likely pathogenic variants identified by TruSight cancer sequencing panel in 20 patients with early-onset CRC.
Pathogenic mutations were most common in the APC gene that encoded tumor suppressor protein acting as an antagonist of the Wnt signaling pathway. APC gene mutations were strongly associated with CRC carcinogenesis (12, 13). Most of them were found in CRC patients with polyposis, FAP, and PJS that could be considered as disease-causing mutations. Pathogenic mutations were also common in Fanconi Anemia associated gene (FANCI, 12.5%) known to play a significant role in repairing double-stranded DNA breaks by homologous recombination and restoring inter-chain DNA crosslinks (26). Fanconi Anemia genes have been reported to play an important role in CRC inherited pre-disposition (27). The mutations of one of the mismatch repair (MMR) genes, MLH1 associated with Lynch syndrome (28, 29) were also quite frequent (8.3%).
Mutations were also found in non-CRC susceptibility genes such as BRCA1 (8.3%) and BRCA2 (12.5%). Those tumor suppressor genes controlling genome integrity were strongly associated with breast and ovarian cancer (30, 31). Pearlman et al. have shown a high prevalence of mutations in the non-CRC genes (BRCA1/2−8.3%) among patients with early onset of CRC (32). However, some studies have demonstrated a modest link between CRC risk and mutations in BRCA1/2 (33, 34). Notably, 1 female (Patient ID: CRC629) with a BRCA1 mutation (c.5329dupC (also known as c.5382insC)/p.Gln1777ProfsTer74, 0.8%) with primary multiple cancer (CRC, ovarian, and breast cancer) had a family history of ovarian cancer. In the studies of CRC patients from different populations, BRCA1 c.5382insC mutation was found in unselected Jewish Ashkenazi patients (0.44%) (25, 35), in unselected Polish patients (0.29%) and in patients with a reported family history of CRC (0.93%) (34).
Of all the 472 identified VUS, 131 were potentially deleterious according to at least one in silico prediction tool (PolyPhen-2 and SIFT) (Supplementary Table 2). Forty-five of all VUS were selected according to the classification shown in Figure 2 and they were categorized as being strongly deleterious (Supplementary Table 3). Of these 45 VUS (42 missense variants in 35 patients and three in-frame deletions in three patients) were identified. Three patients (Patient ID: CRC530, CRC597, and CRC625) had in-frame deletions in the APC, FANCE, and TSC2 gene, respectively. The largest number of VUSs were found in MSH2 involved in mismatch repair and BRCA2 which is a tumor suppressor gene involved in the maintenance of genome stability, specifically the homologous recombination pathway for double-strand DNA repair (Figure 3).
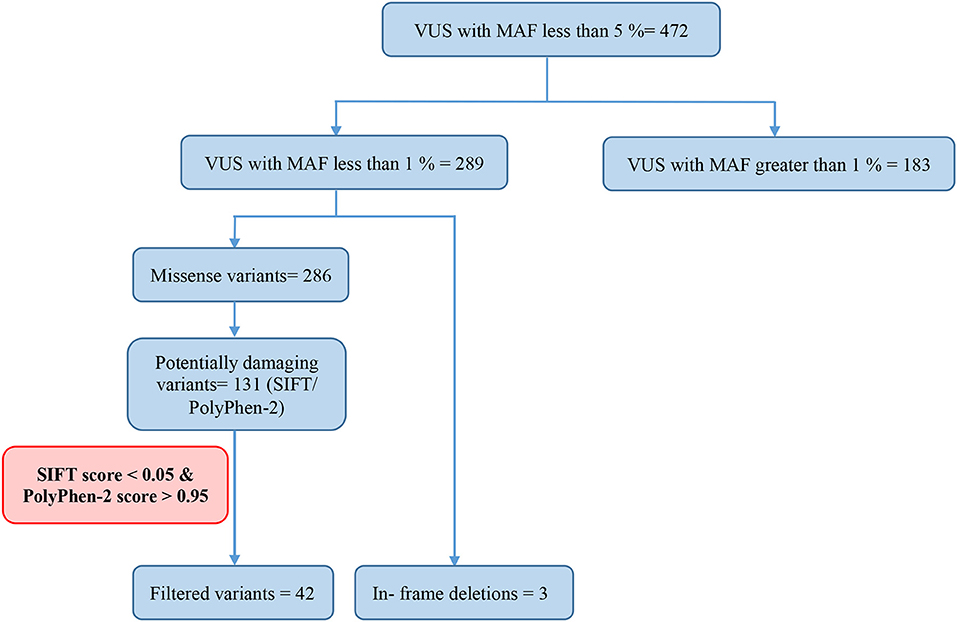
Figure 2. Classification of variant of uncertain significance (VUS).
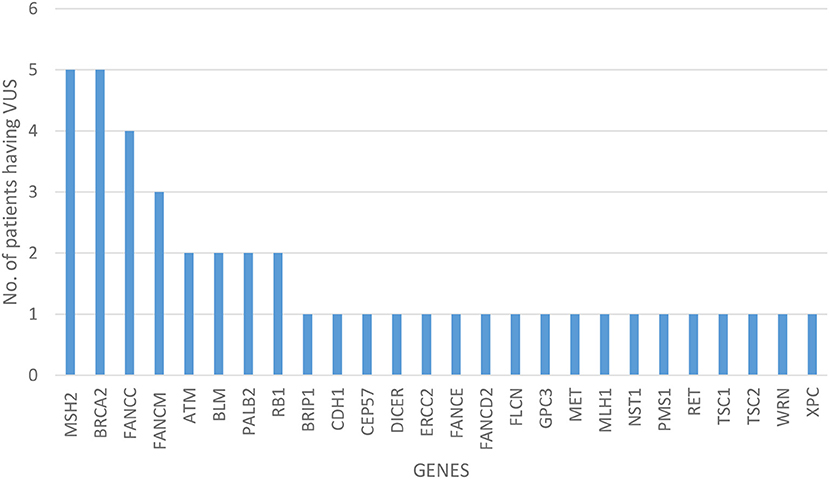
Figure 3. Distribution of variant of uncertain significance.
Eight novel alterations were identified. Two of the novel alterations, c.7429G>A/p.Gly2477Arg in the ATM gene and c.1306C > A/p.Leu436Met in the FANCD2 gene were identified in patients with FHC (Patient ID: CRC335 and CRC587). Other six novel variants were detected in patients with no FHC, c.1135G > A/p.Ala379Thr in NSD1 gene, c.2777A > G/p.Glu926Gly in RB1 gene, c.2693G > A/p.Arg898Lys in the BLM gene, c.1031A > C/p.Gln344Pro in MSH2 gene, c.1493T > G p.Phe498Cys in DICER1 gene and c.278G > A/p.Arg93His in PMS1 gene in a patient (Patient ID: CRC380, CRC442, CRC599, CRC600, and CRC635).
The three novel variants in patients (Patient ID: CRC599, CRC600, CRC635) were found in high and moderate penetrance genes (BLM, MSH2, and PMS1) which are known to be associated with CRC so these mutations can be regarded as one of the risk factors for developing CRC.
The patient cohort under study was conditionally divided into three subgroups by their clinical and molecular characteristics: patients with FHC, patients with PMTs, and patients with sporadic cases (SC) of the disease had the average age 38.9 ± 9.1, 44.6 ± 3.3, and 40.7 ± 7.3, respectively. Detailed information about the most significant mutations with deleterious effects found in patients with FHC (n = 19), PMTs (n = 10), and SCs (n = 98) are represented in Supplementary Tables 4–6, respectively. For patients with FHC, PMTs and SC, 32, 9, and 93 variants were classified as VUS with potentially damaging effects according to the criteria of PolyPhen and SIFT databases and six, five, and eleven variants identified were considered as pathogenic in accordance with the ACMG guidelines, respectively. Early-onset CRC patients with pathogenic variants and VUS were evaluated according to the level of cancer pre-disposition genes (high and moderate penetrance genes listed in the National Comprehensive Cancer Network (NCCN) (36, 37), which are shown in Figures 4, 5, respectively. Pathogenic mutations in high penetrance CRC genes were higher in patients with FHC (21.1 vs. 3.1%; P = 0.0002) and in patients with PMTs (20.0 vs. 3.1%; P = 0.0004) compared with patients without/unknown FHC (3.1%) (Figure 4).
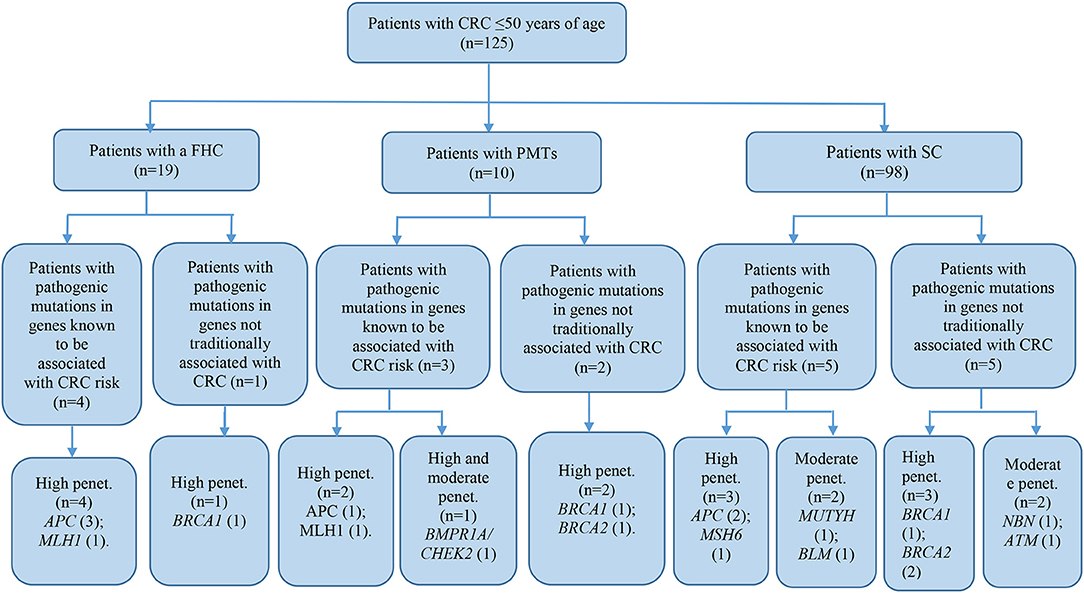
Figure 4. Risk assessment of pathogenic variants by the level of cancer pre-disposition genes (high and moderate) in patients with early onset of colorectal cancer. We described only patients with pathogenic mutations in genes recommended by NCCN (36, 37). CRC, colorectal cancer; FHC, family history of cancer; PMTs, primary multiple tumors; SC, sporadic cases; Penet., penetrance genes.
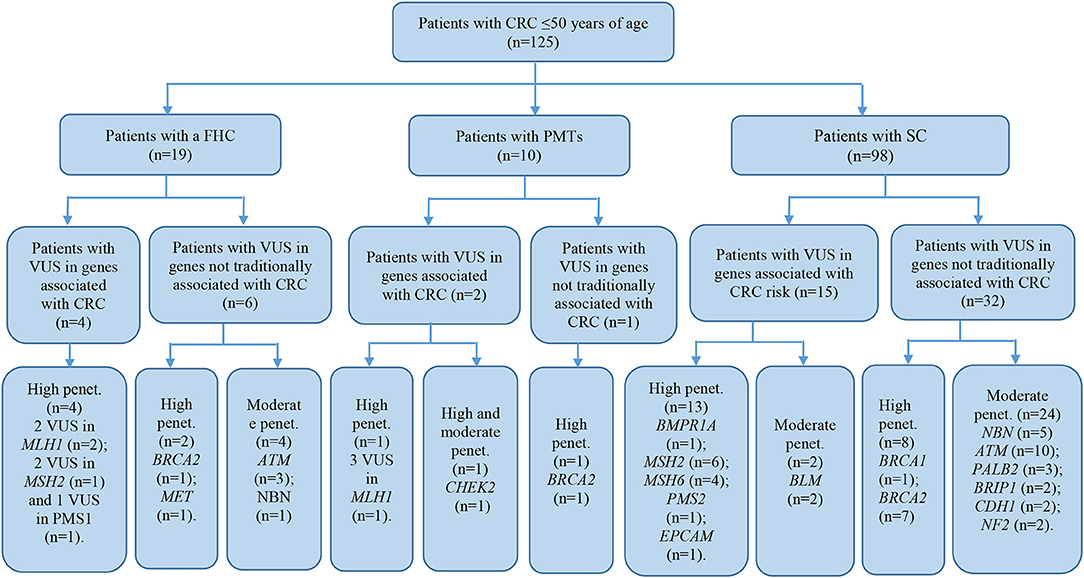
Figure 5. Risk assessment of VUS by the level of cancer pre-disposition genes (high and moderate) in patients with early onset of colorectal cancer. We described patients with VUS in genes recommended by NCCN (36, 37). CRC, colorectal cancer; FHC, family history of cancer; PMTs, primary multiple tumors; SC, sporadic cases; Penet., penetrance genes.
Segregation analysis was done only for two patients with FAP and CRC with the information of relatives (Patient ID: CRC613: sister and daughter, Patient ID: CRC2 brother) but not for the whole cohort because of the unavailability of data (Figure 6). These patients had three frameshifts and two stop codon mutations in APC, MLH1, and FANCI genes. One of those mutations in the APC gene c.3613delA (p.Ser1205AlafsTer60) was previously described in the Polish population among family members with FAP (38). Other mutations [one in FANCI c.3340delA (p.Thr1114ProfsTer50), two in APC c.4128T > G (p.Tyr1376Ter) and c.2835delG (p.Thr946HisfsTer9), and one in MLH1gene c.436C > T (p.Gln146Ter)] have not been previously described in the literature. However, APC c.4128T > G somatic mutation has been reported in the COSMIC database.
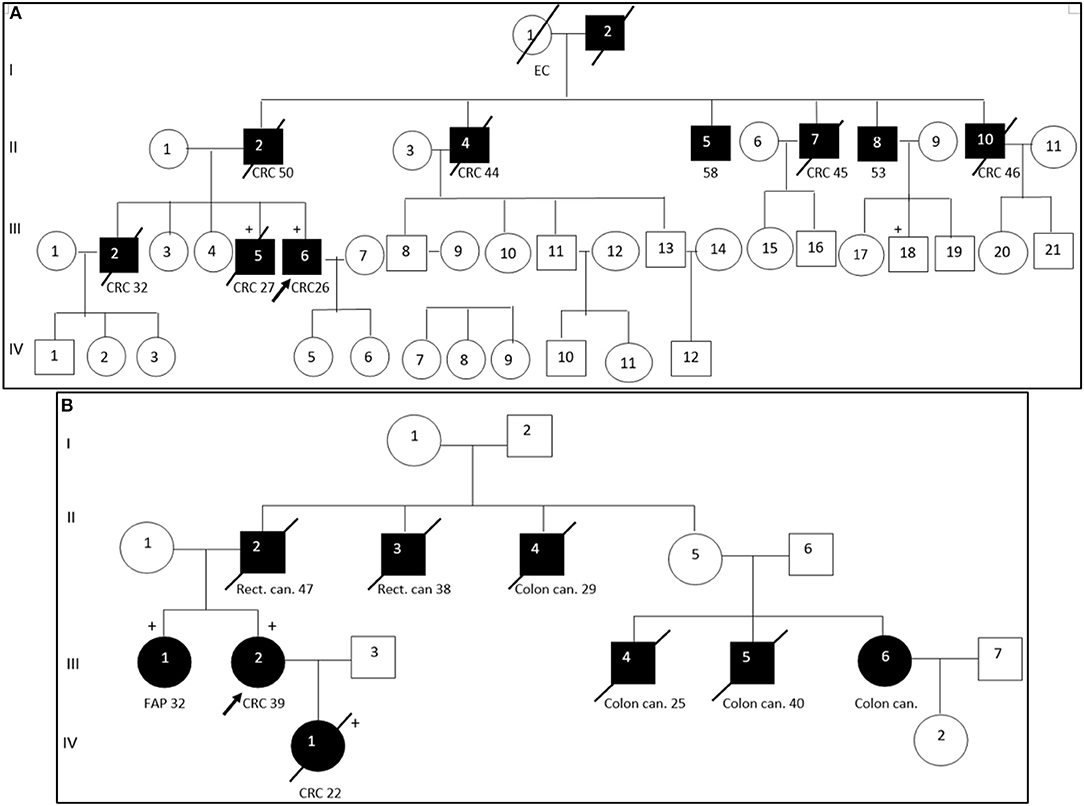
Figure 6. Pedigree of two patients with familial adenomatous polyposis. Arrow points to the proband. Shading indicates family members that were diagnosed with FAP. Squares and circles denote males and females, respectively. Roman number indicates generations. NGS tested patients, depicted by a plus. (A). APC c. c.3613delA (pathogenic mutation) and MLH1 c. c.2146G > A (VUS) were identified in III-6 (proband CRC2) and III-5 (proband's brother). FANCI c. c.3340delA (pathogenic mutation) were identified in III-6 (proband). (B). APC c.4128T > G (pathogenic mutation) and FANCM c.4881T > G (VUS) were identified in III-2 (proband CRC613) and IV-1 (proband's daughter). APC c.2835delG and MLH1 c.436C > T pathogenic mutations were identified in III-1 (proband's sister). CRC, colorectal cancer; Rect. can., rectal cancer; Colon can., colon cancer; FAP, familial adenomatous polyposis; EC, endometrial cancer.
DNA sequencing has revealed identical genetic defects in the proband's brother aged 28: germline hereditary mutations [one missense (c.2146G > A) in the MLH1 gene and one pathogenic frameshift mutation (c.3613delA) in the APC gene] and daughter of a FAP (CRC613) proband with primary multiple CRC tumors had identical APC 4128T > G/p.Tyr1376Ter pathogenic stop-gain and FANCM c.4881T > G missense mutations, while the same proband's sister had another APC c.2835delG/p.Thr946HisfsTer9 pathogenic frameshift mutation (novel) and a MLH1 c.436C > T/p.Gln146Ter pathogenic stop-gain mutation (described in ClinVar and LOVD) (Table 3).
The discovery of the molecular genetic causes of many of the hereditary syndromes associated with CRC (FAP, Lynch syndrome, Peutz-Jeghers, Gardner-Turner, Bannayan–Riley–Ruvalcaba, Bloom's disease, Tourette's syndrome, and others) has increased the importance of genetic testing (39). The limited capacity of old genetic screening methods, due to their low sensitivity and a small number of studied genes, has led to the introduction of multigenic testing in oncology (40, 41). The advantages of multigenic analysis contribute to its widespread use in clinical practice (42, 43).
The study of hereditary forms of the colon, rectal cancer and early-onset CRC in the Kazakhstani population has not been conducted previously due to the lack of statistical data. Most patients were observed at late CRC stages, and FHC was not considered. NGS-based analysis of Kazakhstani patients with early-onset CRC has been conducted for the first time. The data on the cohort of 125 patients with early CRC onset was collected in the last 5 years. According to the patient-filled questionnaire, only 19 (15.2%) patients had CRC, endometrial cancer, ovarian, breast, prostate, lung, gastric, kidney, or laryngeal cancer in their family history. Such frequency of FHC corresponds to the published data on early-onset CRC without known genetic background (44). CRC is known to be a multifactorial disease, the development of which can be influenced by lifestyle and dietary patterns (5–11). According to most of the questionnaires filled by patients with early-onset CRC, its early development can be caused by a sedentary lifestyle, smoking, as well as a diet high in red meat and low in fiber, a characteristic of traditional Kazakh cuisine.
Considering the clinical requirements in genetic testing, conducting NGS we supposed that understanding of molecular genetic variants presented in our cohort with early-onset CRC would allow segregating its hereditary forms from sporadic cases.
Using the TruSight Cancer Sequencing Panel (Illumina, USA), we have identified in total 11,152 variants in 85 genes (Supplementary Table 1). The most frequently mutated genes included: known tumor suppressor genes controlling genetic integrity (APC, BRCA2/1, and ATM), oncogenes encoding tyrosine-protein kinase transmembrane receptor activity (RET and ALK), helicase gene family (BRIP1, WRN, and RECQL4), Fanconi anemia (FA) genes (FANCA, FANCD2, and FANCI), mismatch repair endonuclease (PMS2) and nucleotide excision repair genes (XPC), nuclear tumor suppressor MEN1 that regulates gene transcription by coordinating chromatin remodeling, NSD1 gene that controlled the androgen receptor transactivation, SLX4 gene that encoded an assembly component of multiple structure-specific endonucleases, and HNF1A gene that encoded the transcriptional factor for some specific liver enzymes responsible for detoxification. We did not find the modified variants of nine genes that were used from the panel (CDC73, CDK4, ERCC5, MAX, SDHAF2, SDHC, SDHD, SMAD4, and XPA).
Among the 289 variants of uncertain significance, 131 were attributed to potential deleterious variants. Potential deleterious VUS were obtained by mutational frequency analysis (<1% in the general population) using 1000G, ESP6500, and ExAC databases and using in silico predictive tools (SIFT and Polyphen2).
According to ACMG guidelines and LOVD/ClinVar databases, 24 of the altered variants were found to be pathogenic. Eight mutations were novel and were attributed to likely be pathogenic. The panel allowed the detection of the pathogenic mutations in well-established CRC susceptibility genes, such as APC, MLH1, and MSH6 in high-penetrance genes, and in moderate-penetrance genes like MUTYH (monoallelic), BMPR1A, BLM, and CHEK2 in 11 patients (8.8%) of our cohort. These findings were lower than those reported by Pearlman et al. (16%) (32). Patients with pathogenic mutations identified in CRC related and non-CRC related genes were slightly lower (16%) than the results reached by Martin-Morales et al. (18.4%) (44). In our study half of the patients with pathogenic mutations had high or moderate penetrance genes not traditionally associated with CRC (2, BRCA1; 3, BRCA2; 1, ATM/NBN; 3, FANCI; 1, DICER1; and 1, AIP). Similar results were reported in some recent studies conducted on early-onset CRC patients and unselected CRC patients of all ages (43, 45–48). The obtained data in this study cannot confirm a causal link between mutations that were detected in genes, whose significance regarding CRC pre-disposition is unknown such as breast cancer genes, Fanconi Anemia genes, etc. and developing CRC in patients. With the discovery of the germline heterozygous mutations in the study, we believe that further studies on the somatic level, such as the analysis of loss of heterozygosity, should be conducted to confirm the causative effects of mutations. Despite the fact that some studies have shown a link between CRC risk and non-CRC susceptibility gene mutations, these findings are probably incidental. However, the cancer spectrum and penetrance for many well-defined and recently discovered genes will likely redefine present multigene panel testing as using NGS is becoming more routine (32).
Dividing our study cohort into a subgroup according to the clinical features of patients with PMTs, patients with FHC, and patients with no/unknown FHC, we noticed that 50% of the pathogenic and likely pathogenic mutations were found in subgroups with FHC and PMTs. Patients with FHC and PMTs were more likely to report pathogenic mutations in high penetrance CRC genes. We assumed that those circumstances might have contributed to the active participation of those mutations in the carcinogenesis of CRC.
The syndromic nature of CRC was clinically noted in six cases (4.8%): in two cases of FAP, in three cases of LS and in one case of PJS. Mutation spectra were considered to confirm the clinical diagnosis. Thus, the patients with FAP syndrome had deleterious mutations of APC, MLH1, FANCM, and FANC1 genes, while Lynch syndrome patients from our cohort had causative mutations in MSH2, TSC1, ERCC2, and KIT genes.
No pathogenic mutations were found in two patients with Lynch syndrome. However, one proband had VUS in DNA repair system genes (MSH2 c.2078G > A (p.Cys693Tyr) and c.2072T > C (p.Ile691Thr), TSC1 c.1460C > G (p.Ser487Cys) and ERCC2c.691G > A (p.Val231Met) which were considered as potentially pathogenic by two predictive analyzes in silico. Missense variant c.2078G > A in mismatch repair gene MSH2 was previously described in Brazilian patients with LS (49, 50). Another mutation MSH2 c.2072T > C/p.Ile691Thr was also reported in individuals with LS from Swiss families and in individuals from Utah and California (51, 52). This missense variant could affect the protein structure and function, but such predictions have not been verified by any functional study, and their clinical significance is still unknown. Second LS proband had no pathogenic or likely pathogenic mutations in MMR genes, but polymorphisms (PRF1 c.272C > T/p.Ala91Val, FANCI c.164C > T/p.Pro55Leu, and BRCA1 c.3113A > G/p.Glu1038Gly described in ClinVar as VUS) have been detected. The 1000G, ESP6500, and ExAC databases reported that the population frequency of those polymorphisms exceeded 1%, and two of the in silico predictive tools considered them as deleterious.
The pathogenic effects of two mutations (c.4128T > G and c.3613delA) in the APC gene were confirmed in patients with FAP (Patient ID: CRC613 and CRC2) by segregation analysis and defined as disease-causing mutations. Interestingly, the same stop-gain mutation in nucleotide position c.4128T > G (p.Tyr1376Ter) in the APC gene was detected in a patient with sigmoid colon cancer at the age of 17 (Patient ID: CRC616). As the patient (CRC616) had no evidence of polyposis but had the same pathogenic mutation as that of the patient aged 39 with FAP (Patient ID: CRC613), this patient was allocated to a group with a high risk of polyposis.
The patient with clinically diagnosed PJS had no mutations in the STK11 gene which is associated with PJS (53). However, a stop-gain mutation in the APC gene c.3827C > G/p.Ser1276Ter was identified in that patient. Unfortunately, a segregated study was not possible because of the unavailability of the patient's relative data. We supposed possible errors in clinical diagnostics of this case. This mutation of the APC gene has previously been reported in Danish, Italian and Irish FAP families (54–56). Multiple pathogenic frameshift mutations in a set of genes like APC, ATM, NBN, BMPR1A, and CHEK2 can be crucial in the diseases' development. Moreover, we have found multiple gene alterations with pathogenic mutations and VUS in APC and other MMR related genes in some cases. We assume that such alterations detected in key genes with a role in tumor suppression and mismatch repair can be the risk factor for developing polyposis and Lynch syndrome.
It should be noted, that 18 mismatch repair variants of uncertain significance (MLH1, MSH2, MSH6, PMS1, and PMS2 with MAF <1% of general populations) found in 15 early-onset CRC patients could be very relevant, in particular if the tumors have high-level microsatellite instability (MSI-H) (14). However, there were no available data of the MSI instability levels for these patients.
Eleven pathogenic mutations in 11 cases were identified among 98 patients with early-onset sporadic CRC. Four of those pathogenic variants were novel. The mutation spectrum was wide and covered the genes of general replication/transcription/recombination (BLM, DICER1), common control of genome stability/proliferation/cell division (BRCA1/2, APC, ATM), DNA repair systems (MSH6, NBN, MUTYH, FANCI), and transcription of xenobiotic metabolizing enzymes (AIP). Pathogenic mutations (stop-gain, frameshift, and splice donor) were most frequent in BRCA2, APC, and FANCI genes. That subgroup of patients had many cases of VUS and polymorphisms, but a case-control study was required to assess the involvement of certain mutations in specific CRC carcinogenesis. Our previous case-control study of a panel of candidate polymorphisms has shown a significant association of the following genotypes with increased CRC risk: DCC (32008376 G/G(A), MLH1 (-93G/G(A), TP53 (Pro72Pro), GSTT1 deletions, and GSTM1 deletions (57). In the current study, control samples collected for NGS analysis were matched to the enrolled CRC patients by age, gender, and ethnicity to determine the role of identified variants in patients with early onset sporadic CRC.
In conclusion, we believe that despite the high cost, NGS analysis is necessary for families with a family history of CRC or CRC-related cancers to identify cause-effect mutations, clarify the clinical diagnosis and to prevent the development of disease in close relatives. The novel pathogenic mutations in high-penetrance genes obtained in our study can contribute to the explanation of CRC heritability not only in Kazakhstani families. They can supplement the list of CRC-related mutations and promote the development of cheaper specific tests for the detection of hereditary forms of CRC.
All patients gave written informed consent in accordance with the Declaration of Helsinki. The study protocol was approved by the Ethics Committee of the Asfendiyarov Kazakh National Medical University (Almaty, Kazakhstan).
DK: supervision, funding acquisition, and project administration. SA, AP, and GZ: performed the NGS library preparation, sequencing, and bioinformatics analyses. GZ: supervised all bioinformatics analyses. GA and AJ: collected the patients' samples and clinic data. GZ and GA: writing-original draft manuscript preparation. LD and DK designed the study and contributed with the valuable discussion and revision of the manuscript. All authors read and approved the final manuscript.
This work was supported by grant 49019/PTF-MHSD-ot-18 of the Ministry of Healthcare of the Republic of Kazakhstan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are sincerely grateful to all patients who participated in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00673/full#supplementary-material
AC, Amsterdam I or II criteria; ACMG, American College of Medical Genetics and Genomics; BC, breast cancer; BG, Bethesda guidelines/criteria; BWA, Burrows-Wheeler Aligner; COSMIC, Catalog of Somatic Mutations in Cancer; CRC, colorectal cancer; ExAC, Exome Aggregation Consortium database; FAP, familial adenomatous polyposis; FDR, first-degree relative; FHC, family history of cancer; GATK, Genome Analysis Toolkit; gDNA, Genomic DNA; GWAS, genome-wide association studies; HGVS, Human Genome Variation Society; HNPCC, hereditary non-polyposis colorectal cancer; InSIGHT, International Society of Gastrointestinal Hereditary Tumors; LOVD, Leiden Open Variation Database; MAF, minor allele frequency; MMR, mismatch repair; NGS, Next-generation sequencing; OC, ovarian cancer; PJS, Peutz-Jeghers syndrome; PMTs, primary multiple tumors; SC, sporadic cases; SDR, second-degree relative; VUS, variant of uncertain significance.
1. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. (2019) 144:1941–53. doi: 10.1002/ijc.31937
2. Global Cancer Observatory. International Agency for Research on Cancer 2019. Cancer Today. Colorectum. Estimated age-standardized incidence rates (World) in 2018 worldwide, females, all ages (2018) Available online at: http://gco.iarc.fr/today/online-analysis-multi-bars?v=2018&mode=cancer&mode_population (accessed March, 2019).
3. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. (2017) 66:683–91. doi: 10.1136/gutjnl-2015-310912
4. Haggar FA, Boushey RP. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. (2009) 22:191–7. doi: 10.1055/s-0029-1242458
5. DA Silva FC, Wernhoff P, Dominguez-Barrera C, Dominguez-Valentin M. Update on Hereditary colorectal cancer. Anticancer Res. (2016) 36:4399–405. doi: 10.21873/anticanres.10983
6. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. (2010) 138:2044–58. doi: 10.1053/j.gastro.2010.01.054
7. Al-Sukhni W, Aronson M, Gallinger S. Hereditary colorectal cancer syndromes: familial adenomatous polyposis and lynch syndrome. Surg Clin North Am. (2008) 88:819–44. doi: 10.1016/j.suc.2008.04.012
8. Radice P, Cama A, Mariani-Costantini R. Molecular genetics of polyposis and hereditary colorectal cancer. FORUM–Trends Exp Clin Med. (1996) 6:75–291.
9. Ekbom A, Helmick C, Zack M, Adami H-O. Ulcerative-colitis and colorectal-cancer–a population-based study. N Engl J Med. (1990) 323:1228–33. doi: 10.1056/NEJM199011013231802
10. Ryan DP, Compton CC, Mayer RJ. Carcinoma of the anal canal. N Engl J Med. (2000) 342:792–800. doi: 10.1056/NEJM200003163421107
11. Goedert JJ, Coté TR, Virgo P, Scoppa SM, Kingma DW, Gail MH, et al. Spectrum of AIDS-associated malignant disorders. Lancet. (1998) 351:1833–9. doi: 10.1016/S0140-6736(97)09028-4
12. Ponz De Leon M, Benatti P, Borghi F, Pedroni M, Scarselli A, Di Gregorio C, et al. Aetiology of colorectal cancer and relevance of monogenic inheritance. Gut. (2004) 53:115–22. doi: 10.1136/gut.53.1.115
13. Migliore L, Migheli F, Spisni R, Copped F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J Biomed Biotechnol. (2011) 2011:792362. doi: 10.1155/2011/792362
14. Tariq K, Ghias K. Colorectal cancer carcinogenesis: a review of mechanisms. Cancer Biol Med. (2016) 13:120–35. doi: 10.28092/j.issn.2095-3941.2015.0103
15. Vaiopoulos AG, Athanasoula KC, Papavassiliou AG. Epigenetic modifications in colorectal cancer: Molecular insights and therapeutic challenges. Biochim Biophys Acta–Mol Basis Dis. (2014) 1842:971–80. doi: 10.1016/j.bbadis.2014.02.006
16. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. (2014) 42:D980–5. doi: 10.1093/nar/gkt1113
17. den Dunnen J. Nomenclature for the Description of Sequence Variants. Nijmegen: Human Genome Variation Society (2013). Available online at: https://www.hgvs.org/mutnomen (accessed March, 2019).
18. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. (2011) 29:24–6. doi: 10.1038/nbt.1754
19. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Geno mics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. (2013) 14:178–92. doi: 10.1093/bib/bbs017
20. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
21. Altshuler DL, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. A map of human genome variation from population-scale sequencing. Nature. (2010) 467:1061–73. doi: 10.1038/nature09534
22. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
23. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. (2013). 76:7–20. doi: 10.1002/0471142905.hg0720s76
24. Lin P-H, Kuo W-H, Huang A-C, Lu Y-S, Lin C-H, Kuo S-H, et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget. (2016) 7:8310–20. doi: 10.18632/oncotarget.7027
25. Chen-Shtoyerman R, Figer A, Fidder HH, Rath P, Yeremin L, Meir SB, et al. The frequency of the predominant Jewish mutations in BRCA1 and BRCA2 in unselected Ashkenazi colorectal cancer patients. Br J Cancer. (2001) 84:475–7. doi: 10.1054/bjoc.2000.1598
26. Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. (2016) 35:909–23. doi: 10.15252/embj.201693860
27. Esteban-Jurado C, Franch-Expósito S, Muñoz J, Ocaña T, Carballal S, López-Cerón M, et al. The Fanconi anemia DNA damage repair pathway in the spotlight for germline predisposition to colorectal cancer. Eur J Hum Genet. (2016) 24:1501–5. doi: 10.1038/ejhg.2016.44
28. Peltomaki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet. (2002) 10:735–40. doi: 10.1093/hmg/10.7.735
29. Pino MS, Mino-Kenudson M, Wildemore BM, Ganguly A, Batten J, Sperduti I, et al. Deficient DNA mismatch repair is common in Lynch syndrome-associated colorectal adenomas. J Mol Diagnostics. (2009) 11:238–47. doi: 10.2353/jmoldx.2009.080142
30. Teng LS, Zheng Y, Wang HH. BRCA1/2 associated hereditary breast cancer. J Zhejiang Univ Sci B. (2008) 9:85–9. doi: 10.1631/jzus.B0710617
31. Girolimetti G, Perrone AM, Santini D, Barbieri E, Guerra F, Ferrari S, et al. BRCA-Associated Ovarian Cancer: from molecular genetics to risk management. Biomed Res Int. (2014) 2014:787143. doi: 10.1155/2014/787143
32. Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. (2017) 3:464–71. doi: 10.1001/jamaoncol.2016.5194
33. Phelan CM, Iqbal J, Lynch HT, Lubinski J, Gronwald J, Moller P, et al: Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: Results from a follow-up study. Br J Cancer. (2014) 110:530–4. doi: 10.1038/bjc.2013.741
34. Suchy J, Cybulski C, Górski B, Huzarski T, Byrski T, D?bniak T, et al. BRCA1 mutations and colorectal cancer in Poland. Fam Cancer. (2010) 9:541–4. doi: 10.1007/s10689-010-9378-x
35. Kirchhoff T, Satagopan JM, Kauff ND, Huang H, Kolachana P, Palmer C, et al. Frequency of BRCA1 and BRCA2 mutation in unselected Ashkenazi Jewish patients with colorectal cancer. J Natl Cancer Inst. (2004) 96:68–70. doi: 10.1093/jnci/djh006
36. National Cancer Care Network. Referenced With Permission From the NCCN Guidelines for Genetic/Familial High-RiskAssessment: Colorectal v.1.2016. National Comprehensive Cancer Network IArrAJ. Available online at: https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#detection (accessed March, 2019).
37. National Cancer Care Network. Genetic/Familial High Risk Assessment: Breast and/or Ovarian and Colorectal. NCCN Clinical Practice Guidelines in Oncology. (2016). Available online at: https://www.nccn.org/ (accessed March, 2019).
38. Plawski A, Slomski R. APC gene mutations causing familial adenomatous polyposis in Polish patients. J Appl Genet. (2008) 49:407–14. doi: 10.1007/BF03195640
39. Lynch HT, Lynch JF, Lynch PM, Attard T. Hereditary colorectal cancer syndromes: Molecular genetics, genetic counseling, diagnosis, and management. Fam Cancer. (2008) 7:27–39. doi: 10.1007/s10689-007-9165-5
40. Slavin TP, Niell-Swiller M, Solomon I, Nehoray B, Rybak C, Blazer KR, et al. Clinical application of multigene panels: challenges of next-generation counseling and cancer risk management. Front Oncol. (2015) 5:208. doi: 10.3389/fonc.2015.00208
41. Desmond A, Kurian AW, Gabree M, Mills MA, Anderson MJ, Kobayashi Y, et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol. (2015) 1:943–51. doi: 10.1001/jamaoncol.2015.2690
42. Espenschied CR, LaDuca H, Li S, McFarland R, Gau CL, Hampel H. Multigene panel testing provides a new perspective on Lynch syndrome. J Clin Oncol. (2017) 35:2568–75. doi: 10.1200/JCO.2016.71.9260
43. Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P, et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology. (2015) 149:604–13.e20. doi: 10.1053/j.gastro.2015.05.006
44. Martin-Morales L, Rofes P, Diaz-Rubio E, Llovet P, Lorca V, Bando I, et al. Novel genetic mutations detected by multigene panel are associated with hereditary colorectal cancer predisposition. PLoS ONE. (2018) 13:e0203885. doi: 10.1371/journal.pone.0203885
45. Stoffel EM, Koeppe E, Everett J, Ulintz P, Kiel M, Osborne J, et al. Germline genetic features of young individuals with colorectal cancer. Gastroenterology. (2018) 154:897–905. doi: 10.1053/j.gastro.2017.11.004
46. Yurgelun MB, Kulke MH, Fuchs CS, Allen BA, Uno H, Hornick JL, et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J Clin Oncol. (2017) 35:1086–95. doi: 10.1200/JCO.2016.71.0012
47. Cragun D, Radford C, Dolinsky JS, Caldwell M, Chao E, Pal T. Panel-based testing for inherited colorectal cancer: a descriptive study of clinical testing performed by a US laboratory. Clin Genet. (2014) 86:510–20. doi: 10.1111/cge.12359
48. Yurgelun MB, Masciari S, Joshi VA, Mercado RC, Lindor NM, Gallinger S, et al. Germline TP53 mutations in patients with early-onset colorectal cancer in the colon cancer family registry. JAMA Oncol. (2015) 1:214–21. doi: 10.1001/jamaoncol.2015.0197
49. Rossi BM, Palmero EI, López-Kostner F, Sarroca C, Vaccaro CA, Spirandelli F, et al. A survey of the clinicopathological and molecular characteristics of patients with suspected Lynch syndrome in Latin America. BMC Cancer. (2017) 17:623. doi: 10.1186/s12885-017-3599-4
50. Schneider NB, Pastor T, de Paula AE, Achatz MI, Santos ÂR dos, Vianna FSL, et al. Germline MLH1, MSH2 and MSH6 variants in Brazilian patients with colorectal cancer and clinical features suggestive of Lynch Syndrome. Cancer Med. (2018) 7:2078–88. doi: 10.1002/cam4.1316
51. Kovac M, Laczko E, Haider R, Jiricny J, Mueller H, Heinimann K, et al. Familial colorectal cancer: eleven years of data from a registry program in Switzerland. Fam Cancer. (2011) 10:605–16. doi: 10.1007/s10689-011-9458-6
52. Samowitz WS, Curtin K, Lin HH, Robertson MA, Schaffer D, Nichols M, et al. The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer. Gastroenterology. (2001) 121:830–8. doi: 10.1053/gast.2001.27996
53. Chae HD, Jeon CH. Peutz-Jeghers syndrome with germline mutation of STK11. Ann Surg Treat Res. (2014) 86:325–30. doi: 10.4174/astr.2014.86.6.325
54. Bisgaard ML, Ripa RS, Bülow S. Mutation analysis of the adenomatous polyposis coli (APC) gene in Danish patients with familial adenomatous polyposis (FAP). Hum Mutat. (2004) 23:522. doi: 10.1002/humu.9234
55. Giarola M, Stagi L, Presciuttini S, Mondini P, Radice MT, Sala P, et al. Screening for mutations of the APC gene in 66 Italian familial adenomatous polyposis patients: evidence for phenotypic differences in cases with and without identified mutation. Hum Mutat. (1999) 13:116–23.
56. O'Sullivan MJ, Mulcahy TM, Cambell J, O'Suilleabhain CB, Kirwan WO, Doyle CT, et al. Detection of five novel germline mutations of the APC gene in Irish familial adenomatous polyposis families. Hum Mutat. (1998) 11(Suppl 1):S251–3.
Keywords: early-onset CRC, next-generation sequencing, pathogenic mutation, primary multiple tumors, family history of cancer
Citation: Zhunussova G, Afonin G, Abdikerim S, Jumanov A, Perfilyeva A, Kaidarova D and Djansugurova L (2019) Mutation Spectrum of Cancer-Associated Genes in Patients With Early Onset of Colorectal Cancer. Front. Oncol. 9:673. doi: 10.3389/fonc.2019.00673
Received: 23 April 2019; Accepted: 10 July 2019;
Published: 02 August 2019.
Edited by:
Steven M. Lipkin, Weill Cornell Medicine, Cornell University, United StatesReviewed by:
Felipe Cavalcanti Carneiro Silva, Federal University of Piauí, BrazilCopyright © 2019 Zhunussova, Afonin, Abdikerim, Jumanov, Perfilyeva, Kaidarova and Djansugurova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gulnur Zhunussova, Z3VsbnVyX2pAbWFpbC5ydQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.