 Agnès Denys
Agnès Denys Fabrice Allain*
Fabrice Allain*- Univ. Lille, CNRS, UMR 8576 - UGSF - Unité de Glycobiologie Structurale et Fonctionnelle, Lille, France
Alteration in the expression of heparan sulfate (HS)-modifying enzymes has been frequently observed in cancer. Consequently, dysregulation of the HS biosynthetic machinery results in dramatic changes in the HS structure, thereby impacting a range of pivotal cellular processes involved in tumorigenesis and cancer progression including proliferation, migration, apoptosis, and immune escape. HS 3-O-sulfotransferases (HS3STs) catalyse the maturation step of glucosaminyl 3-O-sulfation within HS chains. Although seven HS3ST isozymes have been described in human, 3-O-sulfation is a rare modification and only a few biological processes have been described to be influenced by 3-O-sulfated HS. An aberrant expression of HS3STs has been reported in a variety of cancers. Thus, it was suggested that changes in the expression of these enzymes as a result of tumorigenesis or tumor growth may critically influence cancer cell behavior. In accordance with this assumption, a number of studies have documented the epigenetic repression of HS3ST2 and HS3ST3A in many cancers. However, the situation is not so clear, and there is accumulating evidence that HS3ST2, HS3ST3A, HS3ST3B, and HS3ST4 may also act as tumor-promoting enzymes in a number of cancer cells depending on their phenotypes and molecular signatures. In this mini-review, we focus on the recent insights regarding the abnormal expression of HS3STs in cancer and discuss the functional consequences on tumor cell behavior. In term of clinical outcome, further investigations are needed to explore the potential value of HS3STs and/or their 3-O-sulfated products as targets for therapeutic strategies in cancer treatment.
Introduction
Heparan sulfate (HS) is an anionic and linear polysaccharide, which is covalently attached to core proteins to form HS proteoglycans (HSPG). These molecules are present within the extracellular matrix (ECM) and at the surface of virtually all cells. While the core protein primarily determines the localization of HSPG, HS chains are involved in the binding of a large number of proteins, including growth factors, cytokines, proteases, lipoproteins, and ECM components. HS-protein interactions have multiple effects ranging from simple immobilization to protection against degradation, conformational change, stabilization of receptor-ligand complexes, or protein oligomerization (1–5). Via these interactions, HS does not only regulate physiological processes, such as in embryogenesis, angiogenesis, blood coagulation and inflammation, but are also implicated in many pathologies, including cancer, infectious diseases, and neurodegenerative disorders (6–10).
Structural determinants in HS are derived from enzymatic modifications of the glycan backbone, which is formed by polymerization of the repeat unit consisting of D-glucuronic acid (GlcUA) and N-acetylated D-glucosamine (GlcNAc). In the classical model of biosynthesis, the native polysaccharide is first subject to partial N-deacetylation/N-sulfation of GlcNAc residues. This modification provides the substrate for next modifications, including epimerization of some GlcUA into L-iduronic acid (IdoUA), 2-O-sulfation of uronic acids (mainly IdoUA), and 6-O and/or 3-O-sulfations of GlcN residues (Figure 1). HS-protein interactions are primarily driven by complementarity between positively charged amino acid residues in the ligand and sulfate groups in the HS sequence. However, protein binding to HS does not only rely on the overall degree of sulfation. Instead, a concept has emerged whereby optimal binding depends on the spatial arrangement of sulfate groups in given HS sequences (2, 4, 5, 11, 12).
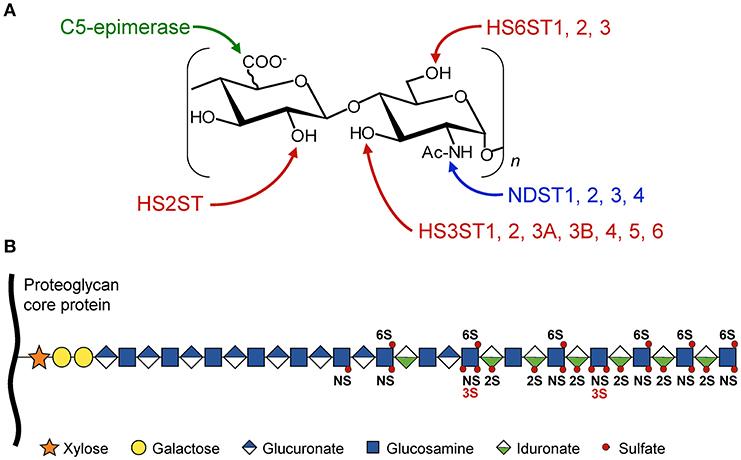
Figure 1. Schematic representation of HS modifications. The HS polysaccharide is linked by the tetrasaccharide linker [Xyl-Gal-Gal-GlcUA] to a specific serine residue within the HSPG core protein. Elongation of the chain is achieved through the alternative addition of GlcUA and GlcNac residues by the polymerases EXT1/EXT2. (A) The disaccharide units [GlcUA-GlcNAc] are modified by the actions of sulfotransferases and epimerase. The sites of modification by N-deacetylases/N-sulfotransferases (NDST), C5-epimerase, HS 2-O-sulfotransferase (HS2ST), HS 6-O-sulfotransferases (HS6STs), and HS 3-O-sulfotransferases (HS3STs) are indicated. (B) HS modifications do not go to completion, resulting in domains with high, intermediate, and low levels of sulfation, enabling the generation of many HS structures and potential ligand-binding sites. NS, N-sulfo group on GlcN residue; 2S, 2-O-sulfo group on uronic acid residue; 3S and 6S, 3-O and 6-O-sulfo groups on GlcN residue.
HS3STs represent the largest family of HS-modifying enzymes, and yet the reaction of 3-O-sulfation is the rarest maturation step, when compared to other sulfations. Seven HS3STs have been characterized in human, for which the expression is dependent on cell type and tissue environment (Table 1). HS3ST-mediated 3-O-sulfation leads to at least two distinct forms of 3-O-sulfated motifs. HS3ST1 and HS3ST5 participate in the generation of anticoagulant-active HS/heparin sequences for antithrombin-III, while HS3ST2, HS3ST3A, HS3ST3B, HS3ST4, and HS3ST6 were described to provide the HS-binding motifs for the glycoprotein gD of herpes simplex virus-1 (HSV-1) (13, 14, 33–41). To date, only a few ligands are known to selectively interact with 3-O-sulfated motifs, whereas hundreds of HS-binding proteins have been identified. Consequently, little is known concerning the functions of 3-O-sulfated HS in biological processes, apart from their roles in anticoagulant properties of HS/heparin and entry of HSV-1 into host cells (12, 42, 43).
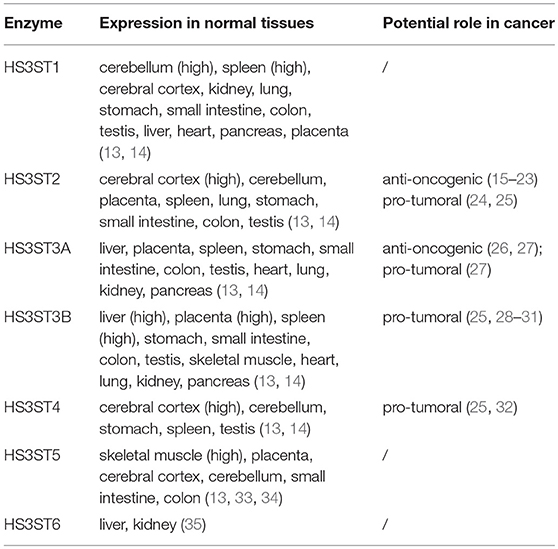
Table 1. Tissue expression of human HS3STs.
Expression of the genes encoding HS-modifying enzymes is frequently dysregulated in cancer and other diseases (42, 44, 45). An aberrant expression of HS3STs has been reported in various cancers, suggesting that these enzymes and their 3-O-sulfated products may be involved in tumorigenesis and cancer progression. However, these reports reveal either anti-oncogenic or tumor-promoting effects (Table 1), and the mechanisms and consequences of HS3ST dysregulation in cancer still remain obscure (15–23, 26–28, 32).
Anti-oncogenic Properties of HS3STs
In cancer cells, hypermethylation of CpG islands in gene promoters has been associated with the loss of expression of some susceptible genes, including tumor suppressor genes, and genes encoding products involved in DNA repair and apoptosis (44, 46, 47). In the attempt to identify novel silenced genes in breast cancer, Miyamoto et al. (22) found that the 5' region of the HS3ST2 gene was hypermethylated in tumor tissue but not in surrounding non-cancerous tissue. As a consequence, the expression level of HS3ST2 was markedly reduced in the cancer sample compared with the matched normal counterpart. Then, they demonstrated that HS3ST2 was not expressed in cell lines representative of the different molecular breast cancer subgroups (48). Reversing methylation restored the expression of the enzyme, confirming the silencing effect of gene methylation. Moreover, HS3ST2 gene hypermethylation was detected in the majority of primary breast cancer samples analysed, and also in human colon, lung and pancreatic cancers (22). Following this work, many clinical studies have been published examining the relationships between aberrant methylation of the HS3ST2 gene and tumorigenesis. Hypermethylation was found at high frequency in gastric, breast, colorectal, prostate and cervix cancers, as well as in hematological neoplasms (15–23). In breast and cervix, hypermethylation of the HS3ST2 gene occurs early during malignant transformation, suggesting a correlation between HS3ST2 silencing and progression of the disease (16, 23). Hwang et al. (18) demonstrated that the exogenous re-expression of HS3ST2 was efficient to inhibit cell migration, invasion and proliferation in various lung cancer cell lines. However, they found that the tumor size was not significantly different between patients with HS3ST2 gene hypermethylation and those without, in spite of the anti-proliferative property of HS3ST2 observed in vitro. Hence, they emphasized the need of further investigations to validate HS3ST2 silencing as a prognostic/predictive biomarker (18).
Besides HS3ST2, an analysis of the methylation status of other genes encoding HS sulfotransferases in chondrosarcoma showed hypermethylation in proximal regions of the HS3ST1 and HS3ST3A1 genes. Exposure to a demethylating agent restored their expression, confirming that aberrant methylation had affected their transcription. Moreover, re-expression of HS3ST3A reduced the proliferative and migratory properties of chondrosarcoma cells, suggesting that silencing of this enzyme may have contributed to tumor cell growth and invasiveness (26). In the following study, Mao et al. (27) demonstrated that the HS3ST3A1 gene is epigenetically repressed in breast cancer cell lines representative of the different molecular subgroups, except in the human epidermal growth factor receptor 2-positive (HER2+) cell lines. Re-expression of the enzyme in luminal A-type MCF-7 and triple negative MDA-MB-231 cell lines reduced cell proliferation in vitro and tumor growth in xenografted mice. Thus, the authors hypothesized that modification in HS structure may have hindered the interactions of growth factors with signalling receptors (27).
Tumor-promoting Activities of HS3STs
Albeit that epigenetic repression of the HS3ST2 gene was related to progression of many cancers, Vijaya Kumar et al. (24) reported that its re-expression in MDA-MB-231 cells led to an increase in cell viability and invasion. Likewise, we reported that MDA-MB-231 cells carrying HS3ST2 expression displayed a significant increase in proliferation and survival (25). The pro-invasive phenotype was however not observed in the MCF-7 cell line (24), suggesting that the consequence of HS3ST expression could be different depending on the breast cancer phenotype. In line with this assumption, Mao et al. (27) described that HS3ST3A expression enhanced proliferation and survival of HER2+ SKBR3 cells, but not in MCF-7 cells. However, these authors reported that HS3ST3A was also anti-proliferative in MDA-MB-231 cells, meaning that this isozyme produced opposite effect than the one promoted by HS3ST2 in the same cell line (24, 25). Although intriguing, we also found that overexpression of HS3ST3A did not have any effects on the proliferation of MDA-MB-231 cells. In contrast, forced expression of HS3ST3B and HS3ST4 had the same functional impact as observed in the case of HS3ST2 (25). These results suggest that the impact of HS3ST expression in breast cancer cells could be also dependent on the type of isozyme. One explanation may be that each HS3ST exhibits subtle differences in their substrate requirement. On this assumption, HS3ST3A may have a restricted substrate specificity, making it incapable of synthesizing 3-O-sulfated HS with a tumor-promoting property in MDA-MB-231 cells. Conversely, HS3ST2, HS3ST3B, and HS3ST4 may exhibit a broader selectivity or share, at least in part, some common acceptors.
In pancreatic cancer cells, high level expression of HS3ST3B was reported to induce epithelial-mesenchymal transition (EMT) and to enhance cell invasiveness in vitro. Moreover, HS3ST3B overexpression was associated with an increased angiogenesis in graft-bearing mice, supporting the idea that HS3ST3B could favor pancreatic cancer progression (29). In the continuity of this study, Zhang et al. (30) reported that high expression of HS3ST3B in U937 leukemia cells enhanced cell proliferation and survival, while its silencing had opposite effects. The advantage given by HS3ST3B was related to an increase in the production of vascular endothelial growth factor (VEGF) and activation of downstream signalling pathways. Next, the authors demonstrated that conditioned medium of HS3ST3B-expressing U937 cells had a promoting effect on angiogenesis, which was dependent on the secretion of VEGF. Finally, they confirmed that HS3ST3B effectively promoted leukemia cell proliferation and VEGF-dependent angiogenesis in xenografted mice (30). Most recently, a clinical study conducted in a cohort of lung cancer patients uncovered that HS3ST3B expression was upregulated in tumor biopsies compared to that in matched normal tissues (28). A high level expression of the enzyme was also observed in NSCLC cell lines. Silencing its expression reversed the mesenchymal phenotype, meaning that HS3ST3B is involved in the regulation of EMT in lung cancer cells in the same way as in pancreatic cells (28, 29). High expression level of TRF2 (telomere repeat binding factor 2), a protein normally involved in telomere protection, has been observed in various human cancers. Interestingly, the HS3ST4 gene was identified as a transcriptional target of TRF2, and increasing TRF2 level led to an up-regulation of HS3ST4 gene expression. Moreover, exogenous expression of either TRF2 or HS3ST4 in various tumor cell lines similarly resulted in increased tumor growth in xenografted mice, which suggests that the expression of this enzyme may be part of a pro-oncogenic pathway (32).
HS3STs and Modulation of Signalling Pathways
Consistent with a pro-invasive phenotype, Erk1/2 and β-catenin signalling was upregulated in HS3ST2-expressing cells in an HS-dependent manner (24). As a consequence, the expression of several target genes involved in cancer cell invasiveness and survival was increased. High level expression of HS3ST3B in U937 leukemia cells was associated with activation of Notch-1, Erk1/2 and Akt signalling (30), and more recently, the tumor-promoting effects of HS3ST2, HS3ST3B, and HS3ST4 were related to sustained activation of Src, Akt, and NF-κB, and up-regulation of the anti-apoptotic proteins survivin and XIAP (25). Importantly, all these signalling molecules have been well described to play a critical role in promoting tumor growth and resistance to apoptosis (49, 50).
Most of the studies conducted with cancer cell lines reported that HS3ST overexpression resulted in an increase in the level of 3-O-sulfated motifs (24, 25, 27). Consequently, 3-O-sulfation may have influenced ligand binding to cell surface HS, leading to an alteration of diverse signalling processes. Whether 3-O-sulfation can modulate ligand-receptor interactions was however unknown, until neuropilin-1 (Nrp1) was described as a preferential ligand for 3-O-sulfated HS (43). Initially described as a co-receptor for VEGFs and class 3 semaphorins in endothelial cells and neurons, there is now evidence that Nrp1 is also expressed in a number of cancer cells, wherein it regulates cell growth, migration, invasion, and immune escape, by interacting with a broad spectrum of growth factors (51–53). Importantly, HS was reported to contribute to formation of a high-affinity complex incorporating Nrp1, VEGF, and cognate signalling receptors (54, 55). Zhang et al. (30) described that the tumor-promoting effect of HS3ST3B in leukemia cells was dependent on an autocrine activation of VEGF-dependent signalling pathways. Thus, it may be suggested that 3-O-sulfation of HS has improved interplay between Nrp1, VEGF, and its receptors. Besides VEGF, transforming growth factor (TGF)-β has been also identified as a ligand of Nrp1. Interestingly, HS3ST3B was described as a regulator of TGF-β-mediated EMT in NSCLC cells (28). Though not mentioned in the study, these findings suggest a possible participation of Nrp1 and 3-O-sulfated HS in the response induced by TGF-β. Along the same lines, we demonstrated that silencing of Nrp1 in MDA-MB-231 cells reversed the advantage given by HS3ST3B (31). Hence, these findings raise the possibility that the tumor-promoting properties of HS3ST3B could be dependent on the formation of signalling complexes containing Nrp1.
Besides the roles attributed to HS moieties, HSPG core proteins have binding properties that engage them in specific interactions with proteins involved in signalling and cytoskeleton organization (1, 3). A number of studies have reported that the expression of HSPG is dysregulated in many cancers, thus altering key biological processes involved in cell proliferation and survival (9). However, there is no evidence that changes in the expression of a core protein can alter the sulfation patterns within HS chains (56). Recently, Corti et al. (57) reported that HS chains of syndecan-2 contained higher levels of 6-O and 3-O sulfations, which was related to an increase in formation of a signalling complex between syndecan-2, VEGF and its receptor in endothelial cells. These last findings demonstrate the existence of a regulatory mechanism wherein a core protein determines the sulfation pattern of its own HS chains. High level expression of syndecan-2 has been observed in many cancers (9). This suggests that such a regulatory mechanism of HS sulfation may also occur in cancer cells. This could lead to the appearance of HSPG with specific HS chains, thereby enhancing the binding and functions of certain HS ligands. This assumption deserves additional works to identify the HSPG and their relevant ligands that interact with 3-O-sulfated HS in cancer cells.
HS3STs and Escape to Immune Surveillance
A body of evidence has accumulated over the past two decades indicating that HS3ST2 is epigenetically silenced in a wide range of cancers and tumor cell lines (15–23). However, the authors did not address the possibility that another HS3ST could be expressed in place of HS3ST2. This assumption is supported by clinical studies showing that HS3ST3A and HS3ST3B were highly expressed in biopsies from patients with HER2+ breast cancer (27) and lung cancer biopsies (28), respectively. These observations suggest that HS3STs can compensate each other for loss of their expression depending on the molecular signature and tissue environment of cancer cells.
During cancer progression, developing tumor cells are exposed to pro-inflammatory mediators that enhance immune anti-tumoral response. In order to evade this immune pressure, tumor cells can change their intrinsic features, thereby resulting in the emergence of cellular variants with increased activation of pro-oncogenic pathways, and less immunogenic phenotype (58). It is of note that upregulation of the expression of HS3ST3B has been observed in many cell types exposed to inflammatory stimuli (28, 59–62). On the other hand, a progressive upregulation of TRF2 was observed during progression of colon cancer. Increased expression of TRF2 was associated with an abnormal expression of HS3ST4, which in turn led to inhibition of NK cell activation and recruitment. The same effects were observed with cancer cells carrying an exogenous expression of HS3ST4, suggesting that the isozyme may be involved in a mechanism of immune escape (32). In line with these findings, we reported that HS3ST-transfected MDA-MB-231 cells were more resistant to apoptosis induced by death receptors ligands or NK cells in vitro (25). The functions of certain NK cell receptors can be modulated through interactions in cis with HS on NK cells themselves or in trans with HS on target cells. Disruption of cis-interactions releases NK receptors and enhances NK cell functional response. Interestingly, silencing of HS3ST3B in NK cells was found to down-regulate the cis-interactions between HS and the NK receptors KIR2DL4 and NKp46, meaning that the functions of these receptors can be regulated through interactions with 3-O-sulfated HS (63, 64). It is thus tempting to speculate that 3-O-sulfation in cancer cells may allow cell surface HS to engage in trans interactions with NK cell receptors. Accordingly, upregulation of the expression of certain HS3STs, such as HS3ST3B or HS3ST4, may be a mechanism that permits cancer cells to impact NK cell activation and to escape their elimination. On that assumption, the tumor-promoting properties of HS3STs may rely not only on alteration of intrinsic processes in cancer cells but also on a non-cell autonomous mechanism bypassing immune surveillance.
Conclusions and Prospects
The regulation of HS biosynthesis is still poorly understood, and whether other factors can influence specific HS sulfation in a given cell type remains largely unknown. In this respect, we demonstrated that HS3ST3B is a Golgi-resident enzyme, while HS3ST2 is specifically addressed to the plasma membrane. This suggests that different subcellular location of HS3STs may be a regulatory mechanism to produce distinct 3-O-sulfated motifs (65). Initially, HS3STs have been divided into two groups, based on their contribution to the synthesis of anticoagulant-active sequences and binding motifs for HSV-1 gD protein. However, there is recent evidence that the situation is not so simple, and a better characterization of the catalytic activity of HS3STs and its regulation is required to define more precisely the biological functions of each isozyme (40, 42, 66, 67). It is also of note that most of the effects attributed to HS3STs in cancer are arising from in vitro experiments. These findings need to be regarded with caution, because interference with other metabolic processes can have a dramatic impact on cell behavior, without being linked necessarily to changes in HS sulfation (68, 69). All of this suggests that alteration in the expression of HS3STs in cancer cells may have diverse functional impacts, which could explain the different action of a particular isoform in a given cell type. Accordingly, the roles of HS3STs in cancer need to be further explored to evaluate the potential of these enzymes as targets for therapeutic strategies in cancer treatment.
Up to now, a lot of attention has been focused on HS mimetics (6, 70). A typical example is the synthesis of the pentasaccharide that binds antithrombin III (71). However, it is still difficult to synthesize oligosaccharides with complex sulfation patterns. A seducing alternative is the use of a chemo-enzymatic approach, in which controlled sulfation could be achieved by recombinant enzymes (42, 72, 73). To date, most of the HS3STs have been cloned and used to prepare 3-O-sulfated oligosaccharides (13, 40, 41, 43). Some of them have proven to be effective as anticoagulant agents (74) and inhibitors of HSV-1 entry (75). On the other hand, targeting HS3STs directly to hinder the reaction of 3-O-sulfation may be a challenging endeavor. Byrne et al. (76) reported that HS2ST was a target for a variety of cell-permeable small molecules, including kinase inhibitors. These findings suggest that such molecules could be redesigned for specific inhibition of HS sulfotransferases. On this assumption, designing specific HS3ST inhibitors via high-throughput screening of bio-active agents might be a future strategy to control HS 3-O-sulfation in cancer cells. In conclusion, a better understanding of the functions of HS3STs in cancer cells may provide opportunities to use these HS-modifying enzymes as molecular targets to improve therapeutic strategies.
Author Contributions
AD and FA: conception, design, writing, and review of the manuscript.
Funding
This work was supported by the University of Lille, France and by the Centre National de la Recherche Scientifique (CNRS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors would like to thank Kenji Uchimura for critical reading of the manuscript. This work was conducted in the framework of the GDR GAG (GDR 3739 CNRS).
References
1. Afratis NA, Nikitovic D, Multhaupt HA, Theocharis AD, Couchman JR, Karamanos NK. Syndecans - key regulators of cell signaling and biological functions. FEBS J. (2017) 284:27–41. doi: 10.1111/febs.13940
2. Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulphate. Annu Rev Biochem. (2002) 71:435–71. doi: 10.1146/annurev.biochem.71.110601.135458
3. Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. (2011) 3:a004952. doi: 10.1101/cshperspect.a004952
4. Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. (2001) 11:75–82. doi: 10.1016/S0962-8924(00)01897-3
5. Zhang L. Glycosaminoglycan (GAG) biosynthesis and GAG-binding proteins. Prog Mol Biol Transl Sci. (2010) 93:1–17. doi: 10.1016/S1877-1173(10)93001-9
6. Lanzi C, Cassinelli G. Heparan sulfate mimetics in cancer therapy: the challenge to define structural determinants and the relevance of targets for optimal activity. Molecules. (2018) 23:E2915. doi: 10.3390/molecules23112915
7. Maïza A, Chantepie S, Vera C, Fifre A, Huynh MB, Stettler O, et al. The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. (2018) 592:3806–18. doi: 10.1002/1873-3468.13082
8. Mohamed S, Coombe DR. Heparin mimetics: their therapeutic potential. Pharmaceuticals. (2017) 10:E78. doi: 10.3390/ph10040078
9. Nagarajan A, Malvi P, Wajapeyee N. Heparan sulfate and heparan sulfate proteoglycans in cancer initiation and progression. Front Endocrinol. (2018) 9:483. doi: 10.3389/fendo.2018.00483
10. Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan sulphate glycosaminoglycans in cancer. Nat Rev Cancer. (2002) 2:521–8. doi: 10.1038/nrc842
11. Carlsson P, Presto J, Spillmann D, Lindahl U, Kjellén L. Heparin/heparan sulfate biosynthesis. J Biol Chem. (2008) 283:20008–14. doi: 10.1074/jbc.M801652200
12. Thacker BE, Xu D, Lawrence R, Esko JD. Heparan sulfate 3-O-sulfation: a rare modification in search of a function. Matrix Biol. (2014) 35:60–72. doi: 10.1016/j.matbio.2013.12.001
13. Mochizuki H, Yoshida K, Shibata Y, Kimata K. Tetrasulfated disaccharide unit in heparan sulfate: enzymatic formation and tissue distribution. J Biol Chem. (2008) 283:31237–45. doi: 10.1074/jbc.M801586200
14. Shworak NW, Liu JA, Petros LM, Zhang LJ, Kobayashi M, Copeland NG, et al. Multiple isoforms of heparan sulfate D-glucosaminyl 3-O-sulfotransferase - isolation, characterization, and expression of human cDNAs and identification of distinct genomic loci. J Biol Chem. (1999) 274:5170–84.
15. Bernal C, Aguayo F, Villarroel C, Vargas M, Díaz I, Ossandon FJ, et al. Reprimo as a potential biomarker for early detection in gastric cancer. Clin Cancer Res. (2008) 14 6264–9. doi: 10.1158/1078-0432.CCR-07-4522
16. Dietrich D, Lesche R, Tetzner R, Krispin M, Dietrich J, Haedicke W, et al. Analysis of DNA methylation of multiple genes in microdissected cells from formalin-fixed and paraffin-embedded tissue. J Histochem Cytochem. (2009) 57:477–89. doi: 10.1369/jhc.2009.953026
17. Gonzalo V, Lozano JJ, Muñoz J, Balaguer F, Pellisé M, Rodríguez de Miguel C, et al. Aberrant gene promoter methylation associated with sporadic multiple colorectal cancer. PLoS ONE. (2010) 5:e8777. doi: 10.1371/journal.pone.0008777
18. Hwang JA, Kim Y, Hong SH, Lee J, Cho YG, Han JY, et al. Epigenetic inactivation of heparan sulfate (glucosamine) 3-O-sulfotransferase 2 in lung cancer and its role in tumorigenesis. PLoS ONE. (2013) 8:e79634. doi: 10.1371/journal.pone.0079634
19. Lim EH, Ng SL, Li JL, Chang AR, Ng J, Ilancheran A, et al. Cervical dysplasia: assessing methylation status (Methylight) of CCNA1, DAPK1, HS3ST2, PAX1 and TFPI2 to improve diagnostic accuracy. Gynecol Oncol. (2010) 119:225–31. doi: 10.1016/j.ygyno.2010.07.028
20. Mahapatra S, Klee EW, Young CY, Sun Z, Jimenez RE, Klee GG, et al. Global methylation profiling for risk prediction of prostate cancer. Clin Cancer Res. (2012) 18:2882–95. doi: 10.1158/1078-0432.CCR-11-2090
21. Martin-Subero JI, Ammerpohl O, Bibikova M, Wickham-Garcia E, Agirre X, Alvarez S, et al. A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PLoS ONE. (2009) 4:e6986. doi: 10.1371/journal.pone.0006986
22. Miyamoto K, Asada K, Fukutomi T, Okochi E, Yagi Y, Hasegawa T, et al. Methylation-associated silencing of heparan sulfate D-glucosaminyl 3-O-sulfotransferase-2 (3-OST-2) in human breast, colon, lung and pancreatic cancers. Oncogene. (2003) 22:274–80. doi: 10.1038/sj.onc.1206146
23. Shivapurkar N, Sherman ME, Stastny V, Echebiri C, Rader JS, Nayar R, et al. Evaluation of candidate methylation markers to detect cervical neoplasia. Gynecol Oncol. (2007) 107:549–53. doi: 10.1016/j.ygyno.2007.08.057
24. Vijaya-Kumar A, Salem-Gassar E, Spillmann D, Stock C, Sen YP, Zhang T, et al. HS3ST2 modulates breast cancer cell invasiveness via MAP kinase- and Tcf4 (Tcf7l2)-dependent regulation of protease and cadherin expression. Int J Cancer. (2014) 135:2579–92. doi: 10.1002/ijc.28921
25. Hellec C, Delos M, Carpentier M, Denys A, Allain F. The heparan sulfate 3-O-sulfotransferases (HS3ST) 2, 3B and 4 enhance proliferation and survival in breast cancer MDA-MB-231 cells. PLoS ONE. (2018) 13:e0194676. doi: 10.1371/journal.pone.0194676
26. Bui C, Ouzzine M, Talhaoui I, Sharp S, Prydz K, Coughtrie MW, et al. Epigenetics: methylation-associated repression of heparan sulfate 3-O-sulfotransferase gene expression contributes to the invasive phenotype of H-EMCSS chondrosarcoma cells. FASEB J. (2010) 24:436–50. doi: 10.1096/fj.09-136291
27. Mao X, Gauche C, Coughtrie MW, Bui C, Gulberti S, Merhi-Soussi F, et al. The heparan sulfate sulfotransferase 3-OST3A (HS3ST3A) is a novel tumor regulator and a prognostic marker in breast cancer. Oncogene. (2016) 35:5043–55. doi: 10.1038/onc.2016.44
28. Zhang Z, Jiang H, Wang Y, Shi M. Heparan sulfate D-glucosamine 3-O-sulfotransferase 3B1 is a novel regulator of transforming growth factor-beta-mediated epithelial-to-mesenchymal transition and regulated by miR-218 in non-small cell lung cancer. J Can Res Ther. (2018) 14:24–9. doi: 10.4103/jcrt.JCRT_659_17
29. Song K, Li Q, Jiang ZZ, Guo CW, Li P. Heparan sulfate D-glucosaminyl 3-O sulfotransferase-3B1, a novel epithelial-mesenchymal transition inducer in pancreatic cancer. Cancer Biol Ther. (2011) 12:88–398. doi: 10.4161/cbt.12.5.15957
30. Zhang L, Song K, Zhou L, Xie Z, Zhou P, Zhao Y, et al. Heparan sulfate D-glucosaminyl 3-O-sulfotransferase-3B1 (HS3ST3B1) promotes angiogenesis and proliferation by induction of VEGF in acute myeloid leukemia cells. J Cell Biochem. (2015) 116:1101–12. doi: 10.1002/jcb.25066
31. Hellec C, Diawara M, Carpentier M, Denys A, Allain F. The pro-tumoral activity of heparan sulfate 3-O-sulfotransferase 3B (HS3ST3B) in breast cancer MDA-MB-231 cells is dependent on the expression of neuropilin-1. Molecules. (2018) 23:E2718. doi: 10.3390/molecules23102718
32. Biroccio A, Cherfils-Vicini J, Augereau A, Pinte S, Bauwens S, Ye J, et al. TRF2 inhibits a cell-extrinsic pathway through which natural killer cells eliminate cancer cells. Nat Cell Biol. (2013) 15:818–28. doi: 10.1038/ncb2774
33. Mochizuki H, Yoshida K, Gotoh M, Sugioka S, Kikuchi N, Kwon YD, et al. Characterization of a heparan sulfate 3-O-sulfotransferase-5, an enzyme synthesizing a tetrasulfated disaccharide. J Biol Chem. (2003) 278:26780–87. doi: 10.1074/jbc.M301861200
34. Xia G, Chen J, Tiwari V, Ju W, Li JP, Malmstrom A, et al. Heparan sulfate 3-O-sulfotransferase isoform 5 generates both an antithrombin-binding site and an entry receptor for herpes simplex virus type 1. J Biol Chem. (2002) 277:37912–19. doi: 10.1074/jbc.M204209200
35. Xu D, Tiwari V, Xia G, Clement C, Shukla D, Liu J. Characterization of heparan sulphate 3-O-sulphotransferase isoform 6 and its role in assisting the entry of herpes simplex virus type 1. Biochem J. (2005) 385:451–9. doi: 10.1042/BJ20040908
36. Liu J, Shworak NW, Fritze LM, Edelberg JM, Rosenberg RD. Purification of heparan sulfate D-glucosaminyl 3-O-sulfotransferase. J Biol Chem. (1996) 271:27072–82.
37. O'Donnell CD, Tiwari V, Oh MJ, Shukla D. A role for heparan sulfate 3-O sulfotransferase isoform 2 in herpes simplex virus type 1 entry and spread. Virology. (2006) 346:452–9. doi: 10.1016/j.virol.2005.11.003
38. Shukla D, Liu J, Blaiklock P, Shworak NW, Bai XM, Esko JD, et al. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. (1999) 99:13–22.
39. Tiwari V, O'Donnell CD, Oh MJ, Valyi-Nagy T, Shukla D. A role for 3-O sulfotransferase isoform-4 in assisting HSV-1 entry and spread. Biochem Biophys Res Commun. (2005) 338:930–7. doi: 10.1016/j.bbrc.2005.10.056
40. Wang Z, Hsieh PH, Xu Y, Thieker D, Chai EJ, Xie S, et al. Synthesis of 3-O-sulfated oligosaccharides to understand the relationship between structures and functions of heparan sulfate. J Am Chem Soc. (2017) 139:5249–56. doi: 10.1021/jacs.7b01923
41. Zhang LJ, Lawrence R, Schwartz JJ, Bai XM, Wei G, Esko JD, et al. The effect of precursor structures on the action of glucosaminyl 3-O-sulfotransferase-1 and the biosynthesis of anticoagulant heparan sulfate. J Biol Chem. (2001) 276:28806–13. doi: 10.1074/jbc.M100204200
42. Kaltenbach DD, Jaishankar D, Hao M, Beer JC, Volin MV, Desai UR, et al. Sulfotransferase and heparanase: remodeling engines in promoting virus infection and disease development. Front Pharmacol. (2018) 9:1315. doi: 10.3389/fphar.2018.01315
43. Thacker BE, Seamen E, Lawrence R, Parker MW, Xu Y, Liu J, et al. Expanding the 3-O-sulfate proteome - Enhanced binding of neuropilin-1 to 3-O-sulfated heparan sulfate modulates its activity. ACS Chem Biol. (2016) 11:971–80. doi: 10.1021/acschembio.5b00897
44. Ropero S, Setien F, Espada J, Fraga MF, Herranz M, Asp J, et al. Epigenetic loss of the familial tumor-suppressor gene exostosin-1 (EXT1) disrupts heparan sulfate synthesis in cancer cells. Hum Mol Genet. (2004) 13:2753–65. doi: 10.1093/hmg/ddh298
45. Sanderson RD, Yang Y, Kelly T, MacLeod V, Dai Y, Theus A. Enzymatic remodeling of heparan sulfate proteoglycans within the tumor microenvironment: growth regulation and the prospect of new cancer therapies. J Cell Biochem. (2005) 96:897–905. doi: 10.1002/jcb.20602
46. Gronbaek K, Hother C, Jones PA. Epigenetic changes in cancer. APMIS. (2007) 115:1039–59. doi: 10.1111/j.1600-0463.2007.apm_636.xml.x
47. Vucic EA, Brown CJ, Lam WL. Epigenetics of cancer progression. Pharmacogenomics. (2008) 9:215–34. doi: 10.2217/14622416.9.2.215
48. Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer. (2010) 4:35–41. doi: 10.1177/117822341000400004
49. Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem J. (2010) 430:199–205. doi: 10.1042/BJ20100814
50. Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harb Perspect Med. (2015) 5:a006098. doi: 10.1101/cshperspect.a006098
51. Prud'homme GJ, Glinka Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget. (2012) 3:921–39. doi: 10.18632/oncotarget.626
52. Raimondi C, Ruhrberg C. Neuropilin signalling in vessels, neurons and tumours. Semin Cell Dev Biol. (2013) 24:172–8. doi: 10.1016/j.semcdb.2013.01.001
53. West DC, Rees CG, Duchesne L, Patey SJ, Terry CJ, Turnbull JE, et al. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J Biol Chem. (2005) 280:13457–64. doi: 10.1074/jbc.M410924200
54. Sarabipour S, Mac Gabhann F. VEGF-A121a binding to neuropilins - A concept revisited. Cell Adh Migr. (2018) 12:204–14. doi: 10.1080/19336918.2017.1372878
55. Teran M, Nugent MA. Synergistic binding of vascular endothelial growth factor-A and its receptors to heparin selectively modulates complex affinity. J Biol Chem. (2015) 290:16451–62. doi: 10.1074/jbc.M114.627372
56. Zako M, Dong J, Goldberger O, Bernfield M, Gallagher JT, Deakin JA. Syndecan-1 and−4 synthesized simultaneously by mouse mammary gland epithelial cells bear heparan sulfate chains that are apparently structurally indistinguishable. J Biol Chem. (2003) 278:13561–9. doi: 10.1074/jbc.M209658200
57. Corti F, Wang Y, Rhodes JM, Atri D, Archer-Hartmann S, Zhang J, et al. N-terminal syndecan-2 domain selectively enhances 6-O heparan sulfate chains sulfation and promotes VEGFA165-dependent neovascularization. Nat Commun. (2019) 10:2124. doi: 10.1038/s41467-019-10205-0
58. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. (2002) 3:991–8. doi: 10.1038/ni1102-991
59. Krenn EC, Wille I, Gesslbauer B, Poteser M, van Kuppevelt TH, Kungl AJ. Glycanogenomics: a qPCR-approach to investigate biological glycan function. Biochem Biophys Res Commun. (2008) 375:297–302. doi: 10.1016/j.bbrc.2008.07.144
60. Martinez P, Denys A, Delos M, Sikora AS, Carpentier M, Julien S, et al. Macrophage polarization alters the expression and sulfation pattern of glycosaminoglycans. Glycobiology. (2015) 25:502–13. doi: 10.1093/glycob/cwu137
61. Sikora AS, Delos M, Martinez P, Carpentier M, Allain F, Denys A. Regulation of the expression of heparan sulfate 3-O-sulfotransferase 3B (HS3ST3B) by inflammatory stimuli in human monocytes. J Cell Biochem. (2016) 117:1529–42. doi: 10.1002/jcb.25444
62. Sikora AS, Hellec C, Carpentier M, Martinez P, Delos M, Denys A, et al. Tumour-necrosis factor-α induces heparan sulfate 6-O-endosulfatase 1 (Sulf-1) expression in fibroblasts. Int J Biochem Cell Biol. (2016) 80:57–65. doi: 10.1016/j.biocel.2016.09.021
63. Brusilovsky M, Cordoba M, Rosental B, Hershkovitz O, Andrake MD, Pecherskaya A, et al. Genome-wide siRNA screen reveals a new cellular partner of NK cell receptor KIR2DL4: heparan sulfate directly modulates KIR2DL4-mediated responses. J Immunol. (2013) 191:5256–67. doi: 10.4049/jimmunol.1302079
64. Brusilovsky M, Radinsky O, Cohen L, Yossef R, Shemesh A, Braiman A, et al. Regulation of natural cytotoxicity receptors by heparan sulfate proteoglycans in -cis: A lesson from NKp44. Eur J Immunol. (2015) 45:1180–91. doi: 10.1002/eji.201445177
65. Delos M, Foulquier F, Hellec C, Vicogne D, Fifre A, Carpentier M, et al. Heparan sulfate 3-O-sulfotransferase 2 (HS3ST2) displays an unexpected subcellular localization in the plasma membrane. Biochim Biophys Acta Gen Subj. (2018) 1862:1644–55. doi: 10.1016/j.bbagen.2018.04.013
66. Gesteira TF, Coulson-Thomas VJ. Structural basis of oligosaccharide processing by glycosaminoglycan sulfotransferases. Glycobiology. (2018) 28:885–97. doi: 10.1093/glycob/cwy055
67. Meneghetti MCZ, Gesteira Ferreira T, Tashima AK, Chavante SF, Yates EA, Liu J, et al. Insights into the role of 3-O-sulfotransferase in heparan sulfate biosynthesis. Org Biomol Chem. (2017) 15:6792–9. doi: 10.1039/c7ob01533j
68. Guo Y, Feng Y, Li Z, Lin X. Drosophila heparan sulfate 3-O sulfotransferase B null mutant is viable and exhibits no defects in Notch signaling. J Genet Genomics. (2014) 41:369–78. doi: 10.1016/j.jgg.2014.04.006
69. Viola M, Brüggemann K, Karousou E, Caon I, Caravà E, Vigetti D, et al. MDA-MB-231 breast cancer cell viability, motility and matrix adhesion are regulated by a complex interplay of heparan sulfate, chondroitin-/dermatan sulfate and hyaluronan biosynthesis. Glycoconj J. (2017) 34:411–20. doi: 10.1007/s10719-016-9735-6
70. Lindahl U. Heparan sulfate-protein interactions: a concept for drug design? Thromb Haemost. (2007) 98:109–15. doi: 10.1160/TH07-04-0310
71. Petitou MC, van Boeckel AA. A synthetic antithrombin III binding pentasaccharide is now a drug! what comes next? Angew Chem Int Ed. (2004) 43:3118–33. doi: 10.1002/anie.200300640
72. Chen J, Avci FY, Muñoz EM, McDowell LM, Chen M, Pedersen LC, et al. Enzymatic redesigning of biologically active heparan sulfate. J Biol Chem. (2005) 280:42817–25. doi: 10.1074/jbc.M504338200
73. Kuberan B, Lech MZ, Beeler DL, Wu ZL, Rosenberg RD. Enzymatic synthesis of antithrombin III-binding heparan sulfate pentasaccharide. Nat Biotechnol. (2003) 21:1343–6. doi: 10.1038/nbt885
74. Jin W, Li S, Chen J, Liu B, Li J, Li X, et al. Increased soluble heterologous expression of a rat brain 3-O-sulfotransferase 1 - A key enzyme for heparin biosynthesis. Protein Expr Purif . (2018) 151:23–9. doi: 10.1016/j.pep.2018.06.007
75. Copeland R, Balasubramaniam A, Tiwari V, Zhang F, Bridges A, Linhardt RJ, et al. Using a 3-O-sulfated heparin octasaccharide to inhibit the entry of herpes simplex virus type 1. Biochemistry. (2008) 47:5774–83. doi: 10.1021/bi800205t
Keywords: heparan sulfate, sulfotransferase, cancer, epigenetic regulation, immune escape
Citation: Denys A and Allain F (2019) The Emerging Roles of Heparan Sulfate 3-O-Sulfotransferases in Cancer. Front. Oncol. 9:507. doi: 10.3389/fonc.2019.00507
Received: 05 April 2019; Accepted: 28 May 2019;
Published: 12 June 2019.
Edited by:
Cinzia Lanzi, National Tumor Institute (Italy), ItalyReviewed by:
Helena Bonciani Nadeer, Federal University of São Paulo, BrazilDavide Vigetti, University of Insubria, Italy
Copyright © 2019 Denys and Allain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrice Allain, ZmFicmljZS5hbGxhaW5AdW5pdi1saWxsZS5mcg==