 Minghui Wan†
Minghui Wan† Lei Zhang
Lei Zhang Weiguo Song
Weiguo Song- School of Pharmacy, Weifang Medical University, Weifang, China
Based on the structure of sanguinarine, fourteen phenanthridine derivatives were designed and synthesized in the current study. The cytotoxic activities of synthesized compounds were evaluated against five human cancer cell lines (MCF-7, PC3, Hela, A549, and HepG2 cell lines) via MTT assay. Among all the compounds tested, molecule 8a exhibited significant cytotoxic activity against MCF-7 cells with a IC50 value of 0.28 μM. A following up enzymatic assay indicated that compound 8a could inhibit the activity of DNA topoisomerase I/II. Further mechanistic studies performed in the MCF-7 cell line revealed that compound 8a could arrest cell cycle in S phase and induce cell apoptosis via downregulation of Bcl-2 and upregulation of Bax. Collectively, a potent DNA topoisomerase inhibitor (8a) was discovered, which exhibited potential as a candidate chemotherapeutic agent for the management of tumors in the present study.
Introduction
Sanguinarine (SA) belongs to the chrysene-skeleton-based heterocyclic benzo [c] phenanthridine alkaloids family (Figure 1), which are widely distributed in plants, such as Sanguinaria canadensis and Papaveraceae (1–3). Although SA was isolated in the late 1940s (4), extensive research focusing on the molecular mechanism of its anti-tumor effects has commenced only recently (5). SA has attracted extensive attention because of its significant biological activities, including anti-tumor (6, 7), anti-inflammatory, anti-angiogenesis, antiplatelet, antiviral, and anti-fungal effects (8–11). The flat polyaromatic structure of SA enabled it to directly interact with DNA (12). SA-induced cell cycle arrest and apoptosis was found to not only be caused by DNA damage, but also to be a combined result of targeting other cell structures, such as topoisomerases (Top) (13, 14), antiapoptotic protein (6, 15, 16), and mitochondrial membranes (17, 18).
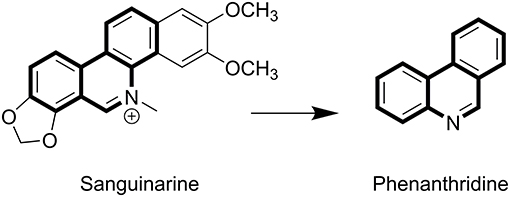
Figure 1. The structures of SA and phenanthridine.
Previous studies reported that SA might interfere with mitochondrial membranes and induce apoptosis in the CEM leukemia cell line HL-60 (18, 19) and KB carcinoma cell line (17). The potential mechanism was associated with nuclear factor (NF-κB) activation (1), mitochondria damage induced caspase activation (20), and increased expression of Bax/Bcl-2 (21, 22). The proapoptotic effects of SA have significant potential in the development of novel antitumor agents with SA as a lead compound. In addition, SA elicited G0/G1 cell cycle arrest (23), which can be associated with the translocation of cyclin D1 and Top II from nucleus to cytoplasm (24, 25). Additionally, NF-κB, AP-1, MMP-9, and STAT3 inhibition were also observed following SA treatment (26–28) and subsequently resulted in suppressed cancer cell metastasis. Moreover, abolishment of VEGF-induced AKT activation was also proposed as another potential mechanism for the antiangiogenic activity of SA (29, 30), which was believed to contribute to its anti-tumor effects in the animal models of melanoma (31) and colorectal cancer (26).
SA exhibited significant potential in the development of new antitumor drugs, as indicated from the results of a wide range of in vitro and in vivo investigations. Due to the structure of multiple aromatic rings, further development of SA as antitumor agent is restricted by its low solubilities and severe side effects. To discover SA analogs with improved solubilities and activities, a series of phenanthridine derivatives with reduced aromaticities were designed and synthesized using phenanthridine as a core scaffold. All the derived compounds were identified with 13C NMR, 1H NMR, HRMS, and biologically evaluated against MCF-7 (human breast cancer), PC3 (human prostatic cancer), Hela (human cervical cancer), A549 (human lung cancer), and HepG2 (human hepatocellular carcinoma) cell lines. During further investigation of the underlying mechanism, molecular techniques such as flow cytometry, hoechst 33258 staining and western blotting were utilized with the representative compounds synthesized in the current study.
Chemistry
The synthetic pathway of phenanthridine derivatives is shown in Scheme 1. As illustrated, amino protection of starting material 1 was performed to afforded compound 2. The following bromine substitution and deprotection of amino group were carried out to generate intermediate 4. Preparation of intermediate 5 was performed by Suzuki coupling of 2-bromoaniline derivatives with corresponding phenylboronic acids. Treatment of intermediate 5 under acidic condition yielded compound 6, and subsequent dehydration of compound 6 afforded 2-isocyanobiphenyls derivatives 7a-t. In the presence of benzoyl peroxide, phenanthridine derivatives 8a-n were derived by reacting of 2-isocyanobiphenyls derivatives with carbon tetrachloride (32).
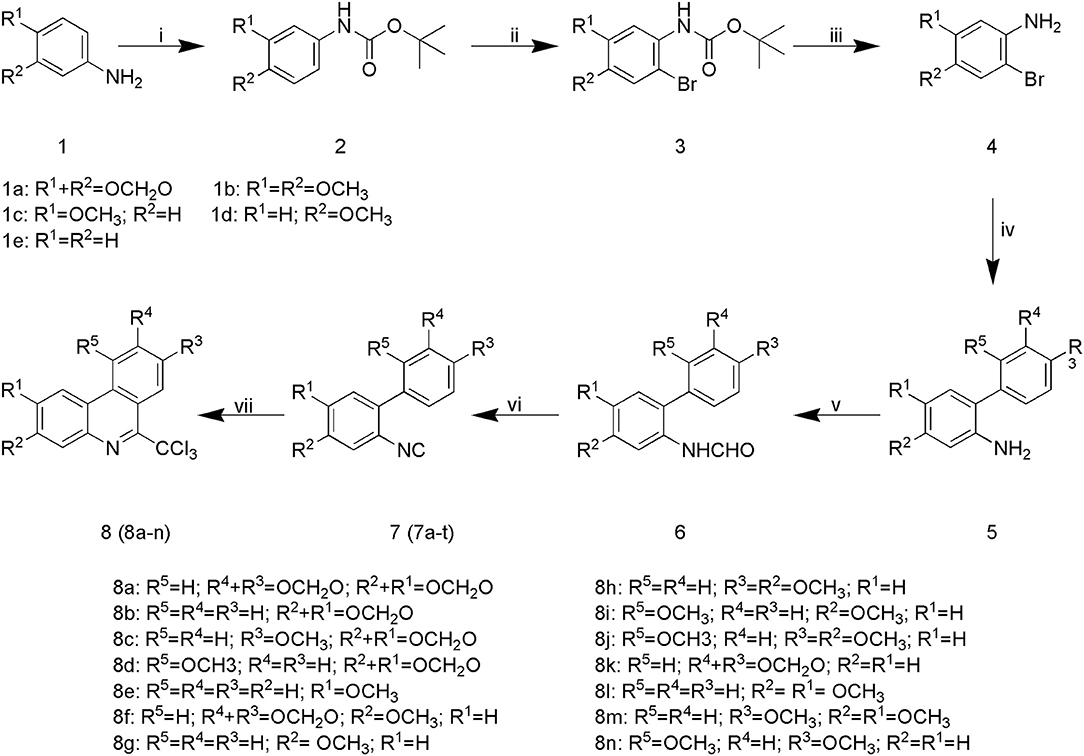
Scheme 1. Synthesis of SA derivatives 8a-n: (i) IPA, Boc2O, ice-bath; (ii) ACN, NBS; (iii) DCM, TFA; (iv) [Pd], K2CO3, DME, 80°C; (v) HCOOH, THF, 60°C; (vi) POCl3, NEt3, THF, 0°C; (vii) BPO, AcONa, reflux.
Cytotoxicity Assay
The cytotoxicity of synthesized compounds was evaluated against five tumor cell lines (A549, PC3, MCF-7, HepG2, and Hela) via MTT assay. Initially, two doses of each compound (5 and 1 μmol/L) were evaluated. As shown in Table 1, compounds 8a, 8b, 8d, 8e, 8l, 8m, and 8n exhibited significant inhibitory activities against MCF-7, PC3, and Hela cells at the dose of 5 μmol/L. However, when compared with the lead compound SA, molecule 8d, 8l, and 8n exhibited lower inhibitory activity at the dose of 1 μmol/L.
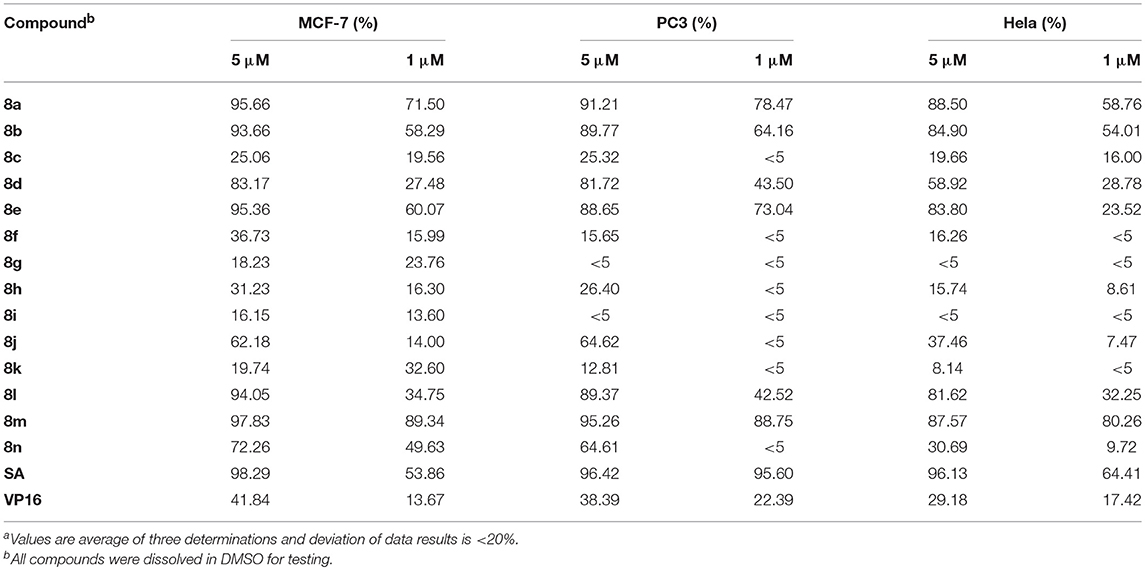
Table 1. The inhibitory activity on tumor cell of phenanthridine derivativesa.
Based on the data mentioned above, compounds 8a, 8b, 8e, and 8m were selected for further test with more doses against the tumor cell lines. The IC50 values of these compounds were summarized in Table 2, all the four compounds exhibited potent cytotoxicity against the five tumor cell lines tested compared with the positive control SA and clinically used antitumor drug Etoposide (VP 16). The results indicated that compounds 8a and 8m exhibited potent activities against all the tested cancer cell lines. Molecule 8a (IC50 = 0.28 ± 0.08) showed potency of over 6 times higher than SA (IC50 = 1.77 ± 0.06) in the inhibition of MCF-7 cells, and molecule 8m (IC50 = 0.39 ± 0.08) exhibited 8.9 times of potency comparing to SA (IC50 = 3.49 ± 0.41) in the inhibition of HepG2 cells. Therefore, 8a, 8b, 8e, and 8m were selected for further mechanistical studies.
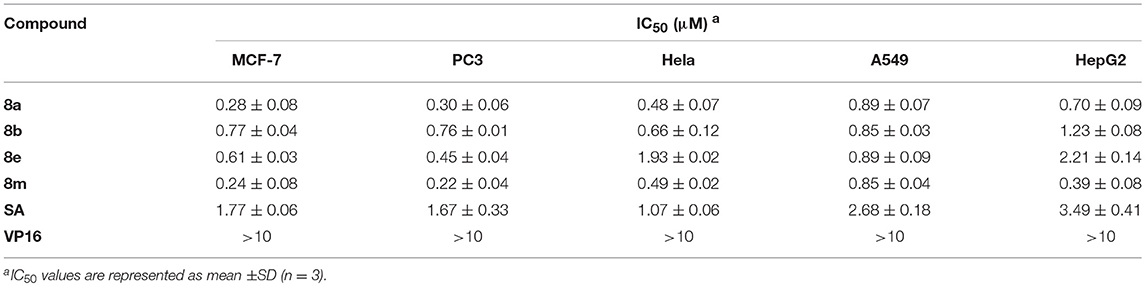
Table 2. The of phenanthridine derivatives.
Topoisomerase Inhibition Assay
To elucidate the target profiles of the cytotoxic compounds (8a, 8b, 8e, and 8m), the inhibitory effects of these compounds were tested against human DNA Top I and IIα by relaxing assay using pBR322 DNA. 10-hydroxy camptothecin (OPT) and VP 16 were used as a positive control for Top I and IIα inhibition, respectively. The Top I/II were able to completely convert the supercoiled DNA to open circular form in the absence of inhibitors (Figure 2, lane B). In contrast, positive control (OPT/VP 16) and active compounds inhibited the activity of Top, which affected the unwinding of the supercoiled DNA, leading to a band pattern similar to the negative control (Figure 2). As shown in Figure 2A, positive control OPT and SA inhibited the activity of both Top I and Top IIα. Compound 8a exhibited weak Top I inhibition, which was similar to OPT. In the Top IIα test, all the tested compounds exhibited potent DNA Top IIα inhibitory activities at the concentration of 100 μM (Figure 2B). Based on the above findings, molecule 8a with most potent cytotoxicity and enzymatic inhibitory activities is chosen as a potential candidate for further investigation.
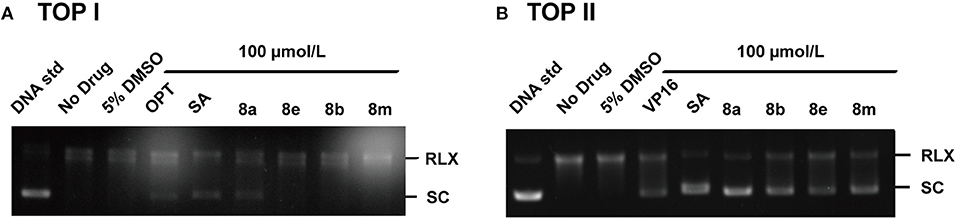
Figure 2. Effects of phenanthridine derivatives and positive control on human Top I (A)/IIa (B). Native superhelix pBR322 was incubated at 37°C for 30 min with 2 units of human Top I/IIα in the absence (lane 2) or presence of compound at concentration 100 μM. One hundred micromolar OPT, VP 16, and SA were used as positive controls, respectively. Negatively supercoiled pBR322 (SC) and relaxed DNA (RLX) were shown. DNA samples were run on agarose gel followed by Genecolour I TM staining.
Cell Cycle Analysis
To elucidate the effects of molecule 8a on cell cycle distributions, MCF-7 cells were treated with various doses of molecule 8a (0, 0.15, 0.3, and 0.6 μM) for 24 h. As shown in Figure 3, compound 8a treatment led to significant accumulation of MCF-7 cells at S phase (from 18.86 to 42.99%) dose-dependently. While reduced cells at the G2/M phase was detected from 23.46 to 10.45% (0.15 μM), 8.69% (0.3 μM), and 5.62% (0.6 μM) following treatment with compound 8a dose-dependently. These results suggest that compound 8a exhibited a significant antitumor effect and led to MCF-7 cell cycle arrest at the S phase in a dose-dependent manner.
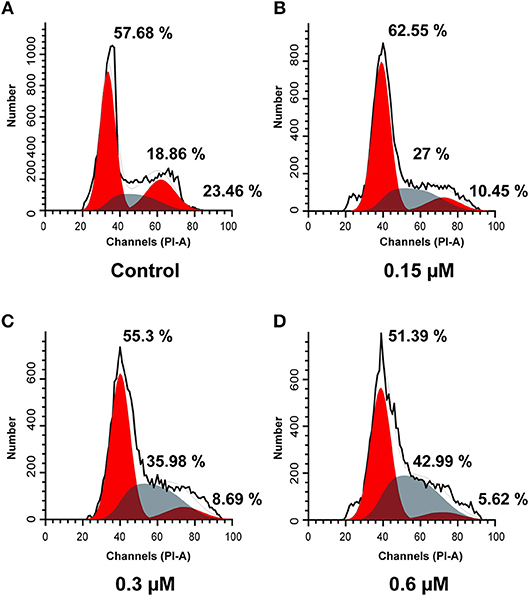
Figure 3. Cell cycle analysis using PI staining of compound 8a on MCF-7 cells. Cells were treated with compound 8a at 0.15 (B), 0.3 (C), and 0.6 (D) μM for 24 h, compared with the control (A). Cell cycle were detected by flow cytometry.
Cell Apoptosis Assay
To further investigate the role of apoptosis in the antitumor effect of compound 8a, Hoechst 33258 staining was performed to investigate the nuclear morphological changes following molecule 8a treatment on MCF-7 cells. Hoechst 33258 is a fluorescent stain used to label DNA; live cells nuclei will be stained with uniformly light blue and apoptotic cells nuclei will be stained with bright blue because of chromatin condensation. As shown in Figure 4A, higher levers of apoptotic cells with nuclear condensation, nuclear fragmentation and enhanced brightness were detected in the cells following treatment with various doses of molecule 8a (0.15, 0.3, and 0.6 μM). To quantify the number of apoptotic cells and to distinguish early apoptosis and secondary necrosis, MCF-7 cells were stained with annexin V-FITC/PI. As shown in Figure 4B, after treatment with difference doses of compound 8a (0, 0.15, 0.3, and 0.6 μM), the percentage of apoptotic cells were significantly increased from 11.16% of the control to 14.35, 22.79, and 28.98%, respectively, indicating that induction of cell apoptosis contributes to the antitumor effect of compound 8a.
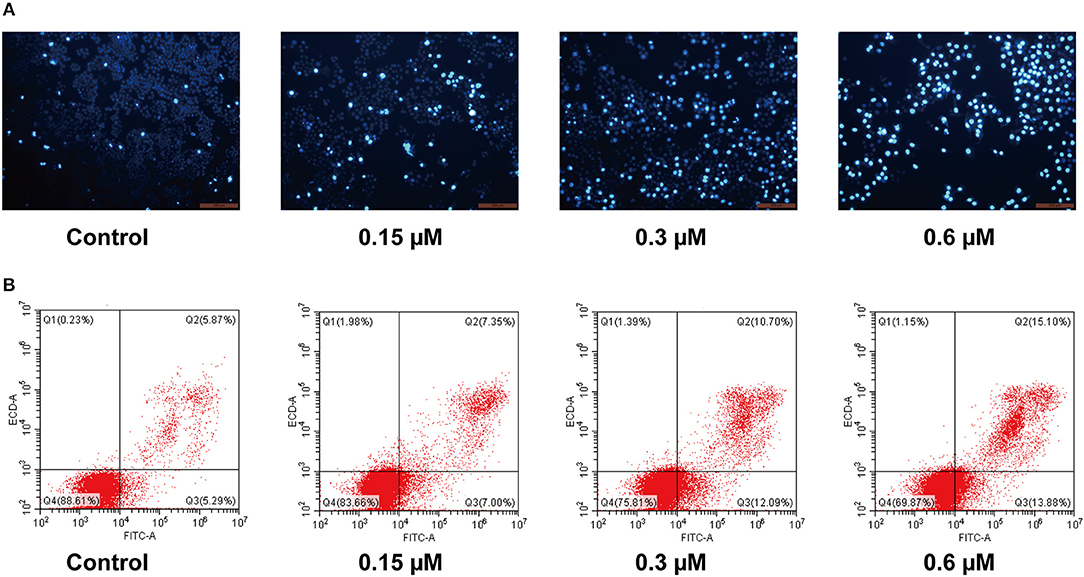
Figure 4. Pro-apoptotic effect of compound 8a on MCF-7 cells. (A) Apoptotic assay by Hoechst 33258. MCF-7 cells were treated with compound 8a at 0.15, 0.3, and 0.6 μM for 24 h, and then cells were stained with Hoechst 33258 and visualized under a fluorescent microscope. (B) Apoptotic assay by flow cytometry. MCF-7 cells were treated with compound 8a at 0.15, 0.3, and 0.6 μM for 24 h. Then cells were stained with Annexin V-FITC/PI and were detected by flow cytometry analysis.
Protein Expressions of Bcl-2 and Bax
Apoptosis is a heavily regulated cell death process influenced by a series of regulatory molecules (33). The mitochondria-dependent pathway has been described as an important signaling pathway of cell apoptosis regulated by the Bcl-2 family including the pro- and anti-apoptotic proteins such as Bax (pro-apoptotic protein) and Bcl-2 (anti-apoptotic protein) (34–36). Moreover, the ratio of Bax/Bcl-2 is important for apoptosis induced by the mitochondrial pathway. Therefore, the effect of compound 8a on the levels of Bax and Bcl-2 was evaluated in MCF-7 cells. The results indicated that compound 8a could significantly downregulate Bcl-2 levels and upregulate Bax levels in MCF-7 cells, increasing the ratio of Bax/Bcl-2 in a dose-dependent manner (Figure 5). Collectively, these results suggest that compound 8a induced apoptosis by regulating the expression of apoptosis-related proteins.
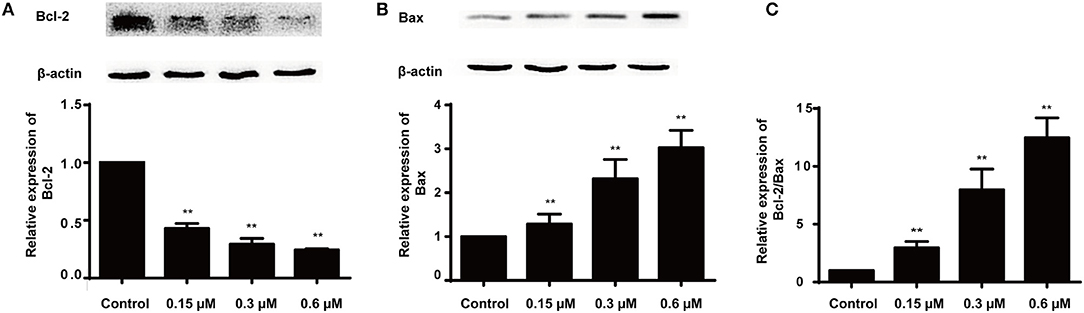
Figure 5. Effects of 8a on the expressions of Bcl-2 (A), Bax (B) and the expression ratio (C) in MCF-7 cells. The cells were treated with different concentrations (0.15, 0.3, and 0.6 μM) for 24 h; β-actin served as an internal control. All date were represented as mean ± SD (n = 3). **p < 0.01, compared with control group.
Conclusions
Based on the structure of sanguinarine, fourteen phenanthridine derivatives 8a-m were synthesized and evaluated for their cytotoxic activity against five different human cancer cell lines. Among the evaluated compounds, 8a exhibited a broad spectrum of anti-proliferative activities against all the tested cancer cell lines. Further mechanistic assay revealed that compound 8a could inhibit the activity of both DNA Top I and Top II, as well as preventing cell transition from S to G2 phase dose-dependently. Apoptosis studies against MCF-7 cells indicated that downregulation of Bcl-2 and upregulation of Bax expression may contribute to the anti-proliferative activities. In summary, these findings suggest that molecule 8a is a potent lead compound in the derived phenanthridine derivatives. Further molecule 8a based structural modification may be beneficial in the discovery of novel anticancer agents with improved antitumor activity and reduced side effects.
Materials and Methods
Chemistry
All chemicals were obtained from commercial suppliers and used without further purification. Reactions progress was detected by thin layer chromatography (TLC) and visualized under UV light. Two hundred to three hundred mesh silica gel was used for column chromatography. All compounds were characterized by 13C NMR, 1H NMR, and HRMS. 1H and 13C NMR spectra were recorded on Mercury Plus-400 with internal standard used TMS and recorded in parts per million (ppm). Date were reported as s (singlet), br (broad), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and coupling constant (J) in hertz (Hz). Melting point was determined by MP 100 Automatic Melting Point Apparatus.
Representative Procedure for the Synthesis of Compounds 7a-t
To dissolve compound 6, THF and NEt3 was added, the solution was added to POCl3 (11 mmol) until the solution was cooled to 0°C. The reaction was quenched by saturated Na2CO3 until complete consumption of starting material, monitored by TLC. The solution of the crude product was extracted with ethyl acetate, and organic layer was dried over Na2SO4 and evaporated to dryness. The residue was purified by column chromatography with silica gel (200–300 mesh).
2-isocyano-3′,4′-Methylenedioxy-4,5-methylenedioxy-1,1′-biphenyl (7a)
Yellowish -white solid, Yield 78%; Mp (154.4–156.1°C); 1H NMR (400 MHz, CDCl3) δ 6.90 (d, J = 8.9 Hz, 4H), 6.78 (s, 1H), 6.05 (s, 2H), 6.01 (s, 2H).
2-isocyano-4,5-methylenedioxy-1,1′-biphenyl (7b)
Brown solid, Yield 80%; Mp (90.1–90.3°C); 1H NMR (400 MHz, CDCl3) δ 7.54–7.30 (m, 5H), 6.91 (s, 1H), 6.82 (s, 1H), 6.05 (s, 2H).
2-isocyano-4,5-methylenedioxy-4′-methoxy-1,1′-biphenyl (7c)
White solid, Yield 81%; Mp (132–133.1°C);1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.7 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 6.90 (s, 1H), 6.80 (s, 1H), 6.05 (s, 2H), 3.85 (s, 3H).
2-isocyano-4,5-methylenedioxy-2′-methoxy-1,1′-biphenyl (7d)
Yellowish-white solid, Yield 82%; Mp (139.4–140.7°C); 1H NMR (400 MHz, CDCl3) δ 7.43–7.34 (m, 1H), 7.20 (dd, J = 7.5, 1.8 Hz, 1H), 7.07–6.96 (m, 2H), 6.90 (s, 1H), 6.79 (s, 1H), 6.05 (s, 2H), 3.83 (s, 3H).
2-isocyano-4,5-methylenedioxy-2′,4′-dimethoxy-1,1′-biphenyl (7e)
Brown solid, Yield 79%; Mp (161.4–161.9°C); 1H NMR (400 MHz, CDCl3) δ 7.10 (s, 1H), 6.88 (s, 1H), 6.77 (s, 1H), 6.56 (dt, J = 5.2, 2.5 Hz, 2H), 6.04 (s, 2H), 3.85 (s, 3H), 3.81 (s, 3H).
2-isocyano-3′,4′-methylenedioxy-5-methoxy-1,1′-biphenyl (7f)
Yellowish-white solid, Yield 81%; Mp (119.6–120.1°C); 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.6 Hz, 1H), 7.12 (d, J = 1.7 Hz, 1H), 7.09–6.95 (m, 4H), 6.10 (s, 2H), 3.84 (s, 3H).
2-isocyano-5-methoxy-1,1′-biphenyl (7g)
Black oil, Yield 83%; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 8.7 Hz, 1H), 7.58–7.42 (m, 5H), 7.08–6.99 (m, 2H), 3.85 (s, 3H).
2'-isocyano-3,4-methylenedioxy-1,1′-biphenyl (7h)
Green solid, Yield 85%; Mp (71.6–73.9°C); 1H NMR (400 MHz, CDCl3) δ 6.02 (s, 2H), 7.02–6.94 (m, 2H), 6.94–6.87 (m, 1H), 7.46 (d, J = 9.3 Hz, 1H), 7.43–7.30 (m, 3H).
2'-isocyano-2,4-dimethoxy-1,1′-biphenyl (7i)
Yellowish-white solid, Yield 79%; Mp (90.1–90.5°C); 1H NMR (400 MHz, CDCl3) δ 7.41 (ddd, J = 8.9, 7.4, 1.8 Hz, 2H), 7.38–7.29 (m, 2H), 7.18–7.10 (m, 1H), 6.58 (dd, J = 5.7, 2.2 Hz, 2H), 3.86 (s, 3H), 3.81 (s, 3H).
2′-isocyano-2,4,5′-trimethoxy-1,1′-biphenyl (7j)
Yellow solid, Yield 80%; Mp (104.6–104.9°C); 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 8.9 Hz, 1H), 6.88–6.79 (m, 2H), 6.58 (dd, J = 5.4, 2.3 Hz, 2H), 3.86 (s, 3H), 3.82 (s, 6H).
2-isocyano-3′,4′-methylenedioxy-4-methoxy-1,1′-biphenyl (7k)
Yellowish-white solid, Yield 75%; Mp (120.6–120.9°C); 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.4 Hz, 1H), 7.02–6.86 (m, 5H), 6.02 (s, 2H), 3.85 (s, 3H).
2-isocyano-4-methoxy-1,1′-biphenyl (7l)
Yellow solid, Yield 78%; Mp (117.3–117.6°C); 1H NMR (400 MHz, CDCl3) δ 7.53–7.43 (m, 4H), 7.43–7.36 (m, 1H), 7.33 (d, J = 9.0 Hz, 1H), 7.05–6.97 (m, 2H), 3.86 (s, 3H).
2-isocyano-4,4′-dimethoxy-1,1′-biphenyl (7m)
Yellowish brown solid, Yield 83%; Mp (102.4–102.8°C); 1H NMR (400 MHz, CDCl3) δ 7.44–7.37 (m, 2H), 7.30 (d, J = 9.0 Hz, 1H), 6.99 (d, J = 9.0 Hz, 4H), 3.85 (d, J = 4.0 Hz, 6H).
2-isocyano-2′,4-dimethoxy-1,1′-biphenyl (7n)
White solid, Yield 82%; Mp (125.5–126°C); 1H NMR (400 MHz, CDCl3) δ 7.48–7.34 (m, 1H), 7.33–7.17 (m, 2H), 7.08–6.94 (m, 4H), 3.84 (d, J = 5.4 Hz, 6H).
2-isocyano-2′,4,4′-trimethoxy-1,1′-biphenyl (7o)
Yellowish-white solid, Yield 80%; Mp (105.9–107.3°C); 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 1.7 Hz, 1H), 7.12 (d, J = 8.9 Hz, 1H), 7.01–6.93 (m, 2H), 6.61–6.53 (m, 2H), 3.90–3.79 (m, 9H).
2-isocyano-3′,4′-methylenedioxy-4,5-dimethoxy-1,1′-biphenyl (7p)
Brown solid, Yield 84%; Mp (171.7–172.3°C); 1H NMR (400 MHz, CDCl3) δ 6.99–6.86 (m, 4H), 6.80 (s, 1H), 6.02 (s, 2H), 3.91 (d, J = 2.5 Hz, 6H).
2-isocyano-4,5-dimethoxy-1,1′-biphenyl (7q)
Yellowish-white solid, Yield 82%; Mp (139.4–139.9°C); 1H NMR (400 MHz, CDCl3) δ 7.27 (s, 1H), 7.01 (s, 1H), 3.85 (d, J = 3.7 Hz, 5H), 7.58–7.47 (m, 3H), 7.47–7.39 (m, 1H).
2-isocyano-4,4′,5-trimethoxy-1,1′-biphenyl (7r)
Yellowish brown solid, Yield 84%; Mp (102.7–103.7°C); 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 6.93 (s, 1H), 6.82 (s, 1H), 3.92 (d, J = 1.5 Hz, 6H), 3.86 (s, 3H).
2-isocyano-2′,4,5-trimethoxy-1,1′-biphenyl (7s)
Yellow solid, Yield 83%; Mp (103–103.6°C); 1H NMR (400 MHz, CDCl3) δ 7.44–7.35 (m, 1H), 7.28–7.20 (m, 1H), 7.09–6.98 (m, 2H), 6.94 (s, 1H), 6.82 (s, 1H), 3.94–3.82 (m, 9H).
2-isocyano-2′,4,4′,5-tetramethoxy-1,1′-biphenyl (7t)
Yellowish-white solid, Yield 82%; Mp (123.4–123.9°C); 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 8.9 Hz, 1H), 6.92 (s, 1H), 6.79 (s, 1H), 6.57 (dq, J = 4.2, 2.4 Hz, 2H), 3.91 (s, 3H), 3.88 (s, 3H), 3.86 (s, 3H), 3.82 (s, 3H).
Representative Procedure for the Synthesis of Compounds 8a-8n
A mixture was produced of 2-isocyanobiphenyls derivatives (0.5 mmol), benzoyl peroxide (0.6 mmol), AcONa (1.0 mmol) in CCl4 (2 mL) under an atmosphere of N2. The reaction was stirred under reflux until complete consumption of starting material, monitored by TLC (about 16h). The solution of the crude product was extracted with ethyl acetate. The organic layers were washed with a saturated solution of NaHCO3 and dried over Na2SO4 and evaporated to dryness. The residue was purified by column chromatography with silica gel (200–300 mesh) to afford the product 6-trichloromethylphenanthridine.
2,3-methylenedioxy-8,9-methylenedioxy-6-(trichloromethyl)phenanthridine (8a)
Yellow solid, Yield 40%; Mp (198.7–199.6°C); 1H NMR (400 MHz, CDCl3): δ 8.23 (s, 1H), 7.77 (s, 1H), 7.67 (s, 1H), 7.53 (s, 1H), 6.18 (d, J = 8.5 Hz, 4H); 13C NMR(101 MHz, DMSO): δ 151.43, 150.27, 149.97, 149.09, 148.77, 147.57, 133.32, 129.68, 128.98, 115.89, 107.21, 104.01, 103.18, 102.90, 101.57, 100.33; HRMS (ESI) m/z 383.9592 (M+H).
2,3-methylenedioxy-6-(trichloromethyl)phenanthridine (8b)
Yellowish solid, Yield 39%; Mp (175.4–176.5°C); 1H NMR(400 MHz, CDCl3): δ 8.92 (d, J = 8.4 Hz, 1H), 8.49 (d, J = 8.2 Hz, 1H), 7.91–7.78 (m, 2H), 7.68 (t, J = 7.6 Hz, 1H), 7.60 (s, 1H), 6.19 (s, 2H); 13C NMR(101 MHz, DMSO): δ 150.59, 150.37, 150.29, 137.46, 134.81, 131.31, 127.60, 127.04, 124.18, 121.88, 119.36, 107.77, 103.08, 100.50; HRMS (ESI) m/z 339.9696 (M+H).
2,3-methylenedioxy-8-methoxy-6-(trichloromethyl)phenanthridine (8c)
Brown solid, Yield 41%; Mp (93.8–95.0°C); 1H NMR(400 MHz, CDCl3) δ 8.12–8.05 (m, 8H), 7.80 (s, 1H), 7.67 (t, J = 7.5 Hz, 4H), 6.17 (s, 2H), 4.01 (s, 3H); 13C NMR(101 MHz, DMSO) δ 167.67, 163.08, 162.77, 135.60, 134.08, 133.34, 131.07, 130.82, 129.93, 129.87, 129.78, 129.70, 129.31, 129.01, 128.45, 124.97; HRMS (ESI) m/z 369.9804 (M+H).
2,3-methylenedioxy-10-methoxy-6-(trichloromethyl)phenanthridine(8d)
Yellow solid; Yield 38%; Mp (219.7–222.3°C); 1H NMR (400 MHz, DMSO) δ 7.95 (d, J = 6.3 Hz, 1H), 7.81 (t, J = 8.3 Hz, 1H), 7.59 (d, J = 3.6 Hz, 2H), 7.31 (t, J = 7.8 Hz, 1H), 6.32 (s, 2H), 3.69 (s, 3H); HRMS (ESI) m/z 369.9804 (M+H).
2-methoxy-6-(trichloromethyl)phenanthridine (8e)
Yellowish-white solid; Yield 37%; Mp (119–120.9°C); 1H NMR (400 MHz, CDCl3): δ 8.96 (d, J = 8.2 Hz, 1H), 8.65 (d, J = 8.4 Hz, 1H), 8.19 (d, J = 9.0 Hz, 1H), 7.94–7.83 (m, 2H), 7.80–7.71 (m, 1H), 7.42 (dd, J = 9.0, 2.7 Hz, 1H), 4.05 (s, 3H); 13C NMR (101 MHz, DMSO): δ 160.62, 149.93, 135.31, 134.27, 132.51, 131.38, 128.05, 127.82, 126.70, 124.59, 120.53, 120.27, 104.00, 98.71, 56.46; HRMS (ESI) m/z 325.9901 (M+H).
8,9-methylenedioxy-3-methoxy-6-(trichloromethyl)phenanthridine (8f)
Brown solid; Yield 32%; Mp (197.5–197.8°C); 1H NMR (400 MHz, CDCl3) δ 8.36–8.19 (m, 2H), 7.91 (s, 1H), 7.59 (d, J = 2.7 Hz, 1H), 7.35 (dd, J = 9.1, 2.7 Hz, 1H), 6.20 (s, 2H), 4.00 (s, 3H); 13C NMR (101 MHz, DMSO): δ 160.32, 151.87, 141.76, 133.80, 124.77, 120.86, 119.65, 115.39, 109.96, 108.77, 104.45, 103.88, 103.25, 101.29, 100.64, 56.08; HRMS (ESI) m/z 369.9798 (M+H).
3-methoxy-6-(trichloromethyl)phenanthridine (8g)
Yellow solid; Yield 26%; Mp (175.1–175.3°C); 1H NMR (400 MHz, CDCl3): δ 8.93 (d, J = 8.7 Hz, 1H), 8.62 (d, J = 8.4 Hz, 1H), 8.48 (d, J = 9.1 Hz, 1H), 7.90–7.81 (m, 1H), 7.72–7.63 (m, 2H), 7.44–7.36 (m, 1H), 4.02 (s, 3H). 13C NMR (101 MHz, DMSO): δ 160.81, 152.92, 142.01, 135.11, 131.94, 127.91, 126.82, 124.58, 123.65, 120.95, 119.09,119.06, 110.67, 98.60, 56.13. HRMS (ESI) m/z 325.9899 (M+H).
3,8-dimethoxy-6-(trichloromethyl)phenanthridine (8h)
Yellow solid; Yield 40%; Mp (146.7–147.2°C); 1H NMR (400 MHz, CDCl3): δ 8.52 (d, J = 9.2 Hz, 1H), 8.39 (d, J = 9.1 Hz, 1H), 8.28 (d, J = 2.6 Hz, 1H), 7.63 (d, J = 2.7 Hz, 1H), 7.50 (dd, J = 9.2, 2.6 Hz, 1H), 7.38 (dd, J = 9.1, 2.7 Hz, 1H), 4.01 (d, J = 2.4 Hz, 6H); 13C NMR (101 MHz, DMSO): δ 160.06, 157.07, 151.84, 141.12, 129.60, 125.46, 124.08, 122.20, 121.18, 120.32, 119.35, 110.40, 108.81, 56.07, 55.91; HRMS (ESI) m/z 356.0009 (M+H).
3,10-dimethoxy-6-(trichloromethyl)phenanthridine (8i)
Yellow solid; Yield 37%; Mp (149.9–150.9°C). 1H NMR (400 MHz, CDCl3): δ 9.46 (d, J = 9.5 Hz, 1H), 8.60 (d, J = 8.5 Hz, 1H), 7.72–7.56 (m, 2H), 7.41–7.29 (m, 2H), 4.16 (s, 3H), 4.02 (s, 3H); 13C NMR (101 MHz, DMSO) δ 159.78, 157.90, 152.59, 142.64, 129.33, 127.12, 125.28, 120.88, 120.27, 120.03, 118.54, 113.19, 110.92, 56.63, 55.97; HRMS (ESI) m/z 356.0009 (M+H).
3,8,10-trimethoxy-6-(trichloromethyl)phenanthridine (8j)
Yellow solid; Yield 35%; Mp (97–97.3°C); 1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 2.3 Hz, 1H), 7.70–7.60 (m, 3H), 7.35 (dd, J = 9.5, 2.9 Hz, 1H), 4.12 (s, 3H), 4.01 (d, J = 2.9 Hz, 6H); 13C NMR (101 MHz, DMSO): δ 162.77, 159.25, 159.09, 157.44, 135.58, 130.82, 129.78, 129.00, 128.38, 120.61, 120.42, 110.71, 103.67, 101.47, 56.84, 55.95, 55.92; HRMS (ESI) m/z 386.0112 (M+H).
8,9-methylenedioxy-6-(trichloromethyl)phenanthridine (8k)
Yellowish solid; Yield 32%; Mp (164.4–165°C); 1H NMR (400 MHz, CDCl3) δ 8.43–8.36 (m, 1H), 8.30 (s, 1H), 8.27–8.20 (m, 1H), 8.02 (s, 1H), 7.73 (tt, J = 7.1, 5.3 Hz, 2H), 6.24 (d, J = 16.4 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 151.82, 151.22, 148.15, 140.03, 133.36, 130.68, 129.57, 125.30, 123.39, 118.56, 116.49, 114.73, 104.69, 103.41, 101.84; HRMS (ESI) m/z 339.9697 (M+H).
2,3-dimethoxy-6-(trichloromethyl)phenanthridine (8l)
Yellow solid; Yield 43%; Mp (174.5–176.1°C); 1H NMR (400 MHz, CDCl3): δ 8.95 (d, J = 8.6 Hz, 1H), 8.58 (d, J = 8.4 Hz, 1H), 8.16–8.08 (m, 4H), 7.86 (s, 2H), 7.74–7.58 (m, 4H), 7.49 (t, J = 7.8 Hz, 4H), 4.16 (s, 3H), 4.11 (s, 3H); 13C NMR (101 MHz, DMSO): δ 167.75, 151.93, 151.86, 133.29, 131.16, 129.69, 128.99, 127.70, 126.77, 124.17, 119.98, 119.33, 110.56, 102.99, 56.68, 56.30; HRMS (ESI) m/z 356.0010 (M+H).
2,3,8-trimethoxy-6-(trichloromethyl)phenanthridine (8m)
Yellow solid; Yield 39%; Mp (125.5–126.9°C); 1H NMR (400 MHz, CDCl3): δ 8.48 (d, J = 9.1 Hz, 1H), 8.29 (d, J = 2.5 Hz, 1H), 7.77 (s, 1H), 7.62 (s, 1H), 7.50 (dd, J = 9.2, 2.6 Hz, 1H), 4.14 (s, 3H), 4.09 (s, 3H), 4.02 (s, 3H); 13C NMR (101 MHz, DMSO): δ 157.06, 152.00, 151.26, 149.05, 135.36, 129.09, 128.95, 126.01, 121.63, 120.63, 120.29, 110.36, 108.35, 102.47, 56.64, 56.23, 55.89; HRMS (ESI) m/z 386.0115 (M+H).
8,10-dimethoxy-6-(trichloromethyl) phenanthridine (8n)
Yellow solid; Yield 39%; Mp (161.3–162°C); 1H NMR (400 MHz, CDCl3): δ 9.45–9.38 (m, 1H), 8.28–8.21 (m, 1H), 8.01 (d, J = 2.3 Hz, 1H), 7.71 (dd, J = 6.5, 3.5 Hz, 2H), 7.00 (d, J = 2.4 Hz, 1H), 4.13 (s, 3H), 4.02 (s, 3H); 13C NMR (101 MHz, DMSO): δ 160.02, 158.37, 151.25, 139.93, 130.95, 130.05, 128.51, 127.15, 124.74, 122.76, 119.71, 103.62, 101.98, 99.01, 56.93, 56.04; HRMS (ESI) m/z 356.0007 (M+H).
Pharmacology
Cell Culture
A549, PC3, MCF-7, HepG2 and Hela cell lines were obtained from the Chinese Academy of Sciences Cell Bank. A549, Hela and PC3 were cultured in RPMI-1640 medium supplemented with 10% FBS, MCF-7 cells were maintained in MEM medium supplemented with 10% FBS, HepG2 cells were cultured in DMEM medium supplemented with 10% FBS. All the cell lines were cultured at humidified atmosphere containing 5% CO2 at 37°C. The stock solutions (20 mM) of phenanthridine derivatives were prepared in DMSO and added at desired concentrations to the cell culture. DMSO concentration did not exceed 1:1,000 in the final culture.
MTT Assay
Cytotoxic activities of the phenanthridine derivatives was evaluated by MTT assay. The stock solutions of phenanthridine derivatives were diluted with culture medium. The cells were seeded in 96-well plates at a density 5 × 103 cells per well and incubated until confluency 90–95%, then each well was treated with 100 μL medium containing the desired concentrations of phenanthridine derivatives and incubated for 48 h. 20 μL MTT working solution (5 mg/mL) was then added to each well and incubated for another 4 h. At the end of incubation, the medium was carefully removed, and 200 μL DMSO was added. The optical density at 490 nm and 630 nm were then measured with a microplate reader (MODEL). The percentage of cell growth inhibition was calculated with the following equation: % inhibition = [1–(Sample group OD490 - Sample group OD630)/(Control group OD490-Control group OD630)] × 100%. The IC50 values were calculated with Origin 7.5 software, and standard deviations of the IC50 values were obtained from at least 3 independent experiments.
DNA Top I and IIα Relaxation Assay In vitro
The human Top I and IIα inhibitory activity was determined by agarose gel electrophoresis. Reaction mixture was prepared with 0.5 μg pBR322 supercoiled DNA (TaKaRa) and human Top I (TaKaRa) or IIα (TopoGEN) enzyme in the absence or presence of compound in the Top reaction buffer (Top I: DNA Top I buffer 2 μL, DNA Top I 1U, 0.1% BSA 2 μL and sterile water up to 20 μL; Top IIα: DNA Top IIα buffer A 2 μL, DNA Top IIα buffer B 2 μL, DNA Top IIα 1U and sterile water up to 20 μL). After 30 min of incubation at 37°C, the reaction mixture was electrophoresed on 0.8% agarose gel at 80 V for 50 min with TAE running buffer. The gel was then immersed in the Genecolour I TM staining solution for 45 min and photographed under UV light.
Cell Cycle Assay
MCF-7 cells in logarithmic growth phase were seeded in 6-well plates (6 × 105 cells/well) and incubated with different doses of compound 8a (0, 0.15, 0.3, and 0.6 μM) for 24 h. Cells were then washed twice with cold PBS and fixed in 70% precooled ethanol at 4°C for 12 h. After the fixation, cells were washed again with PBS and stained with PI/RNase A for 30 min at room temperature, and eventually subjected to flow cytometry (CytoFLEX, Beckman Coulter). for cell cycle distribution determination.
Hoechst 33258 Staining
MCF-7 cells in logarithmic growth phase were seeded in 6-well plates (4 × 105 cells/well) and incubated with different doses of compound 8a (0, 0.15, 0.3, and 0.6 μM) for 24 h. Cells were then washed twice with PBS and stained with Hoechst 33258 working solution for 30 min at 37°C under 5% CO2. The morphological changes of apoptotic cells were observed with a fluorescence microscope (Leica DMI 4000B) with blue filter.
Annexin V/PI Detection
MCF-7 cells in logarithmic growth phase were seeded in 6-well plates (4 × 105 cells/well) and incubated with different doses of compound 8a (0, 0.15, 0.3, and 0.6 μM) for 24 h. After the incubation, cells were washed with PBS, collected, resuspended with binding buffer from the Annexin V-FITC kit (Thermo fisher Co., USA), and then added with 5 μl annexin V-FITC and mixed gently. After 10 min of incubation, 1 μl PI was added to each sample and mixed gently. After incubation at room temperature for another 20 min in the dark, cells were subjected to flow cytometer (CytoFLEX, Beckman Coulter).
Western Blotting
MCF-7 cells were incubated with different doses of compound 8a (0, 0.15, 0.3, and 0.6 μM) for 24 h, and then total cell proteins were extracted with RIPA buffer supplemented with 1:100 protease inhibitor (info) and phosphatase inhibitor (info). Sample protein concentrations were determined with BCA assay (ComWin Biotech Co., Beijing, China), then equal amounts of protein (30 μg) were mixed with sampling buffer and denatured for 5 min at 100°C. Resulting samples were then subjected to Sodium dodecyl sulfate-polyacrylamide electrophoresis (SDS-PAGE). After electrophoresis, proteins were transferred to polyvinylidene difluoride (PVDF) membrane (Millipore) and blocked with 5% fat-free dry milk in 1 × Tris-buffered saline (TBST) for 2 h at room temperature. Membranes were then probed with Bcl-2 (rabbit, 1:1,000, Santa Cruz, CA), Bax (rabbit, 1:1,000, Santa Cruz, CA) and β-actin antibodies at 4°C overnight. The membranes were then washed with TBST three times and incubated with anti-rabbit secondary antibody (Santa Cruz, CA) and visualized with ECL-detecting reagents (ComWin Biotech Co., Beijing, China). The images were obtained from 6000 pro (Clinx Science Instruments Co., Ltd., Shanghai, China) and analyzed with Image Studio Lite software.
Statistical Analysis
Results were expressed as mean ± standard deviation (SD) of three independent experiments performed in triplicates (n = 3). SPSS 19.0 software were used for statistical analysis and the means between two groups were compared by one way analysis of variance (ANOVA) with Dunnett's test, P < 0.05 was considered significant.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
WS and FY designed the project. MW, YC, QL, and QX performed the experiments. MW and LZ analyzed the data and wrote the manuscript. All authors discussed the results and contributed to the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (Youth Found, Grant No. 81803343).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chaturvedi MM, Kumar A, Darnay BG, Chainy GB, Agarwal S, Aggarwal BB. Sanguinarine (pseudochelerythrine) is a potent inhibitor of NF-kappaB activation, IkappaBalpha phosphorylation, and degradation. J Biol Chem. (1997) 272:30129–34. doi: 10.1074/jbc.272.48.30129
2. Croaker A, King GJ, Pyne JH, Anoopkumar-Dukie S, Liu L. Sanguinaria canadensis: traditional medicine, phytochemical composition, biological activities and current uses. Int J Mol Sci. (2016) 17:17091414. doi: 10.3390/ijms17091414
3. Achkar IW, Mraiche F, Mohammad RM, Uddin S. Anticancer potential of sanguinarine for various human malignancies. Future Med Chem. (2017) 9:933–50. doi: 10.4155/fmc-2017-0041
4. Sarkar SM. Isolation from argemone oil of dihydrosanguinarine and sanguinarine; toxicity of sanguinarine. Nature. (1948) 162:265. doi: 10.1038/162265a0
5. Kalogris C, Garulli C, Pietrella L, Gambini V, Pucciarelli S, Lucci C, et al. Sanguinarine suppresses basal-like breast cancer growth through dihydrofolate reductase inhibition. Biochem Pharmacol. (2014) 90:226–34. doi: 10.1016/j.bcp.2014.05.014
6. Slaninová I, Pěnčíková K, Urbanová J, Slanina J, Táborská E. Antitumour activities of sanguinarine and related alkaloids. Phytochemistry Rev. (2014) 13:51–68. doi: 10.1007/s11101-013-9290-8
7. Zhang R, Wang G, Zhang PF, Zhang J, Huang YX, Lu YM, et al. Sanguinarine inhibits growth and invasion of gastric cancer cells via regulation of the DUSP4/ERK pathway. J Cell Mol Med. (2017) 21:1117–27. doi: 10.1111/jcmm.13043
8. Eun JP, Koh GY. Suppression of angiogenesis by the plant alkaloid, sanguinarine. Biochem Biophys Res Commun. (2004) 317:618–24. doi: 10.1016/j.bbrc.2004.03.077
9. Miao F, Yang XJ, Zhou L, Hu HJ, Zheng F, Ding XD, et al. Structural modification of sanguinarine and chelerythrine and their antibacterial activity. Nat Prod Res. (2011) 25:863–75. doi: 10.1080/14786419.2010.482055
10. Yang XJ, Miao F, Yao Y, Cao FJ, Yang R, Ma YN, et al. In vitro antifungal activity of sanguinarine and chelerythrine derivatives against phytopathogenic fungi. Molecules. (2012) 17:13026–35. doi: 10.3390/molecules171113026
11. Cee VJ, Chavez F Jr, Herberich B, Lanman BA, Pettus LH, Reed AB, et al. Discovery and optimization of macrocyclic quinoxaline-pyrrolo-dihydropiperidinones as potent pim-1/2 kinase inhibitors. ACS Med Chem Lett. (2016) 7:408–12. doi: 10.1021/acsmedchemlett.5b00403
12. Maiti M, Nandi R, Chaudhuri K. Sanguinarine: a monofunctional intercalating alkaloid FEBS Lett. (1982) 142:280–4.
13. Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. (2001) 70:369–413. doi: 10.1146/annurev.biochem.70.1.369
14. Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. (2002) 3:430–40. doi: 10.1038/nrm831
15. Chen XM, Zhang M, Fan PL, Qin YH, Zhao HW. Chelerythrine chloride induces apoptosis in renal cancer HEK-293 and SW-839 cell lines. Oncol Lett. (2016) 11:3917–24. doi: 10.3892/ol.2016.4520
16. Wang X, Tanaka M, Krstin S, Peixoto HS, Wink M. The interference of selected cytotoxic alkaloids with the cytoskeleton: an insight into their modes of action. Molecules. (2016) 21:906. doi: 10.3390/molecules21070906
17. Chang MC, Chan CP, Wang YJ, Lee PH, Chen LI, Tsai YL, et al. Induction of necrosis and apoptosis to KB cancer cells by sanguinarine is associated with reactive oxygen species production and mitochondrial membrane depolarization. Toxicol Appl Pharmacol. (2007) 218:143–51. doi: 10.1016/j.taap.2006.10.025
18. Kaminskyy V, Kulachkovskyy O, Stoika R. A decisive role of mitochondria in defining rate and intensity of apoptosis induction by different alkaloids. Toxicol Lett. (2008) 177:168–81. doi: 10.1016/j.toxlet.2008.01.009
19. Vrba J, Dolezel P, Vicar J, Ulrichova J. Cytotoxic activity of sanguinarine and dihydrosanguinarine in human promyelocytic leukemia HL-60 cells. Toxicol In Vitro. (2009) 23:580–8. doi: 10.1016/j.tiv.2009.01.016
20. Adhami VM, Aziz MH, Mukhtar H, Ahmad N. Activation of prodeath Bcl-2 family proteins and mitochondrial apoptosis pathway by sanguinarine in immortalized human HaCaT keratinocytes. Clin Cancer Res. (2003) 9:3176–82.
21. Ahsan H, Reagan-Shaw S, Breur J, Ahmad N. Sanguinarine induces apoptosis of human pancreatic carcinoma AsPC-1 and BxPC-3 cells via modulations in Bcl-2 family proteins. Cancer Lett. (2007) 249:198–208. doi: 10.1016/j.canlet.2006.08.018
22. Liu Y, Jiao R, Ma ZG, Liu W, Wu QQ, Yang Z, et al. Sanguinarine inhibits angiotensin II-induced apoptosis in H9c2 cardiac cells via restoring reactive oxygen species-mediated decreases in the mitochondrial membrane potential. Mol Med Rep. (2015) 12:3400–8. doi: 10.3892/mmr.2015.3841
23. Lee B, Lee SJ, Park SS, Kim SK, Kim SR, Jung JH, et al. Sanguinarine-induced G1-phase arrest of the cell cycle results from increased p27KIP1 expression mediated via activation of the Ras/ERK signaling pathway in vascular smooth muscle cells. Arch Biochem Biophys. (2008) 471:224–31. doi: 10.1016/j.abb.2008.01.008
24. Holy J, Lamont G, Perkins E. Disruption of nucleocytoplasmic trafficking of cyclin D1 and topoisomerase II by sanguinarine. BMC Cell Biol. (2006) 7:13. doi: 10.1186/1471-2121-7-13
25. Xu JY, Meng QH, Chong Y, Jiao Y, Zhao L, Rosen EM, et al. Sanguinarine inhibits growth of human cervical cancer cells through the induction of apoptosis. Oncol Rep. (2012) 28:2264–70. doi: 10.3892/or.2012.2024
26. Pica F, Balestrieri E, Serafino A, Sorrentino R, Gaziano R, Moroni G, et al. Antitumor effects of the benzophenanthridine alkaloid sanguinarine in a rat syngeneic model of colorectal cancer. Anticancer Drugs. (2012) 23:32–42. doi: 10.1097/CAD.0b013e32834a0c8e
27. Sun M, Liu C, Nadiminty N, Lou W, Zhu Y, Yang J, et al. Inhibition of Stat3 activation by sanguinarine suppresses prostate cancer cell growth and invasion. Prostate. (2012) 72:82–9. doi: 10.1002/pros.21409
28. Park SY, Jin ML, Kim YH, Lee SJ, Park G. Sanguinarine inhibits invasiveness and the MMP-9 and COX-2 expression in TPA-induced breast cancer cells by inducing HO-1 expression. Oncol Rep. (2014) 31:497–504. doi: 10.3892/or.2013.2843
29. Basini G, Bussolati S, Santini SE, Grasselli F. Sanguinarine inhibits VEGF-induced angiogenesis in a fibrin gel matrix. Biofactors. (2007) 29:11–8. doi: 10.1002/biof.5520290102
30. Dong XZ, Zhang M, Wang K, Liu P, Guo DH, Zheng XL, et al. Sanguinarine inhibits vascular endothelial growth factor release by generation of reactive oxygen species in MCF-7 human mammary adenocarcinoma cells. Biomed Res Int. (2013) 2013:517698. doi: 10.1155/2013/517698
31. De Stefano I, Raspaglio G, Zannoni GF, Travaglia D, Prisco MG, Mosca M, et al. Antiproliferative and antiangiogenic effects of the benzophenanthridine alkaloid sanguinarine in melanoma. Biochem Pharmacol. (2009) 78:1374–81. doi: 10.1016/j.bcp.2009.07.011
32. Zhou Y, Wu C, Dong X, Qu J. Synthesis of 6-trichloromethylphenanthridines by transition metal-free radical cyclization of 2-isocyanobiphenyls. J Org Chem. (2016) 81:5202–8. doi: 10.1021/acs.joc.6b00885
33. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. (2011) 147:742–58. doi: 10.1016/j.cell.2011.10.033
34. Nunez G, Clarke MF. The Bcl-2 family of proteins: regulators of cell death and survival. Trends Cell Biol. (1994) 4:399–403. doi: 10.1016/0962-8924(94)90053-1
35. Boise LH, Gottschalk AR, Quintans J, Thompson CB. Bcl-2 and Bcl-2-related proteins in apoptosis regulation. Curr Top Microbiol Immunol. (1995) 200:107–21. doi: 10.1007/978-3-642-79437-7_8
Keywords: phenanthridine, anticancer, topoisomerase, apoptosis, cell cycle arrest
Citation: Wan M, Zhang L, Chen Y, Li Q, Fan W, Xue Q, Yan F and Song W (2019) Synthesis and Anticancer Activity Evaluation of Novel Phenanthridine Derivatives. Front. Oncol. 9:274. doi: 10.3389/fonc.2019.00274
Received: 24 February 2019; Accepted: 25 March 2019;
Published: 16 April 2019.
Edited by:
Zhe-Sheng Chen, St. John's University, United StatesReviewed by:
Xiangbao Meng, Peking University, ChinaPranav Gupta, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2019 Wan, Zhang, Chen, Li, Fan, Xue, Yan and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Yan, eWFuZmFuZzMwM0AxNjMuY29t
Weiguo Song, c29uZ3dnQDEzOS5jb20=
†These authors have contributed equally to this work