 Yasuhiro Shimizu
Yasuhiro Shimizu Toshihiro Suzuki
Toshihiro Suzuki Toshiaki Yoshikawa1
Toshiaki Yoshikawa1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 10 April 2019
Sec. Cancer Molecular Targets and Therapeutics
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00248
This article is part of the Research Topic Heparan Sulfate Proteoglycans and Their Endogenous Modifying Enzymes: Cancer Players, Biomarkers and Therapeutic Targets View all 13 articles
Glypican-3 (GPC3), a 65 kD protein consisting of 580 amino acids, is a heparan sulfate proteoglycan bound to the cell membrane by glycosylphosphatidylinositol. This protein is expressed in the liver and the kidney of healthy fetuses but is hardly expressed in adults, except in the placenta. Contrarily, GPC3 is specifically expressed in hepatocellular carcinoma (HCC), ovarian clear cell carcinoma, melanoma, squamous cell carcinoma of the lung, hepatoblastoma, nephroblastoma (Wilms tumor), yolk sac tumor, and some pediatric cancers. Although the precise function of GPC3 remains unclear, it has been strongly suggested that it is related to the malignant transformation of HCC. We identified GPC3 as a promising target for cancer immunotherapy and have been working on the development of cancer immunotherapeutic agents targeting it through clinical trials. In some trials, it was revealed that the GPC3 peptide vaccines we developed using human leukocyte antigen-A24- and A2-restricted GPC3-derived peptides could induce GPC3-specific cytotoxic T cells in most vaccinated patients and thereby improve their prognosis. To further improve the clinical efficacy of cancer immunotherapy targeting GPC3, we are also developing next-generation therapeutic strategies using T cells engineered to express antigen-specific T-cell receptor or chimeric antigen receptor. In addition, we have successfully monitored the levels of serum full-length GPC3 protein, which is somehow secreted in the blood. The utility of GPC3 as a biomarker for predicting tumor recurrence and treatment efficacy is now being considered. In this review article, we summarize the results of clinical trials carried out by our team and describe the novel agent targeting the cancer-specific shared antigen, GPC3.
Therapeutic approaches that exploit the immune system are a promising alternative strategy to surgery, radiotherapy, and anticancer drug therapy for cancer treatment. Recent studies have shown that immune checkpoint inhibiters (ICIs) such as antibodies against CTLA-4, programmed cell death (PD)-1, and programmed death ligand 1 have potent and long-term antitumor effects (1, 2); in 2018, Tasuku Honjo and James P Alison won the Nobel Prize for Medicine for their contribution to their development. The extremely high response rates to chimeric antigen receptor-introduced T cell therapy (CAR-T therapy) for cluster of differentiation (CD)19+ hematopoietic tumors and tumor-infiltrative T cell transfer therapy for malignant melanoma have provided further evidence for the efficacy of cancer immunotherapies (3, 4). Additionally, tumor-specific mutant antigens (neoantigen) have attracted attention for their therapeutic potential and clinical trials of personalized cancer vaccines that target neoantigens have been initiated in Europe and the United States (5–7) Meanwhile, peptide vaccines against shared antigens have been developed in Japan but have not yet been approved by the Pharmaceutical Affairs Law.
With the exception of Hodgkin's lymphoma, response rates to ICIs are estimated as no more than 30% in the case of melanoma and 10–20% for other cancers (8, 9). An outstanding challenge is to develop effective therapies for patients who are unresponsive to ICIs. To address this issue, two points must be considered: firstly, the extents to which cancer-responsive T cells are active in patients' bodies; and secondly, how their infiltration into tumors can be enhanced. In cases where cancer-specific effector T cell counts are low, their numbers must be increased by T cell transfer therapy or else the T cells must be induced with cancer vaccines. Inducing inflammation at tumor loci by administration of adjuvants or by chemo- or radiation therapy has been shown to promote T cell infiltration into tumors (10), and ICIs and individualized cancer vaccines derived from neoantigens are thought to be effective in patients with a high frequency of somatic mutations (11). On the other hand, it is difficult to enhance anti-cancer immune responses and even if immunosuppression is overcome (12) in patients with a low, making it necessary to target not only neoantigens but also shared antigens like glypican (GPC)-3. Our clinical trials have shown that cancer peptide vaccines targeting shared antigens can induce peptide-specific cytotoxic T lymphocytes (CTLs) in vaccinated patients without eliciting non-specific autoimmune responses. In addition, gene-based therapy using T cell receptor (TCR) obtained from CTL clones induced by cancer peptide vaccine and isolated from vaccinated patients is expected to have more potent antitumor effects (13). In this review, we summarize the results of cancer immunotherapy targeting GPC3 based on our experience and outline the future prospects.
We identified GPC3 in a cDNA microarray screen of several tens of thousands of genes for novel cancer antigens (14). GPC3, 65-kDa protein consisting of 580 amino acids is a heparan sulfate chain proteoglycan bound to the cell membrane by a glycosylphosphatidylinositol (GPI) anchor (Figure 1A) (15). GPC3 regulates cell proliferation signals by binding growth factors such as Wnt, fibroblast growth factor, and insulin-like growth factor and plays an important role in the proliferation and differentiation of embryonic cells (16–18). In addition, the gene is present on the X chromosome (Xq26) and shows high homology between humans and mice. Gene mutations and deletions cause gigantism with various malformations and Simpson-Golabi-Behmel syndrome in humans, with similar phenotypic manifestations in mice (16, 19). GPC3 is expressed in various fetal tissues (liver, lung, kidney, and placenta) but is not detected in normal postnatal tissue due to DNA methylation-induced epigenetic silencing (20, 21). On the other hand, GPC3 is expressed in hepatocellular carcinoma (HCC), melanomas, ovarian clear cell carcinoma (OCCC), lung squamous cell carcinomas, and some childhood cancers (hepatoblastomas, nephroblastomas, and yolk sac tumors) (14, 21). Particularly, GPC3 is detected in ≥80% of patients with HCC caused by hepatitis B or C (Figure 1B) (14, 22, 23). The function of membrane-anchored GPC3 in these cancers is unknown, but it is likely involved in the neoplastic transformation of HCC (23). Additionally, immunohistochemical analysis of HCC tissues have revealed at least three GPC3 expression patterns, which we have classified as diffuse, membrane-localized, and granular (Figure 1C) (Shimizu, manuscript in preparation).
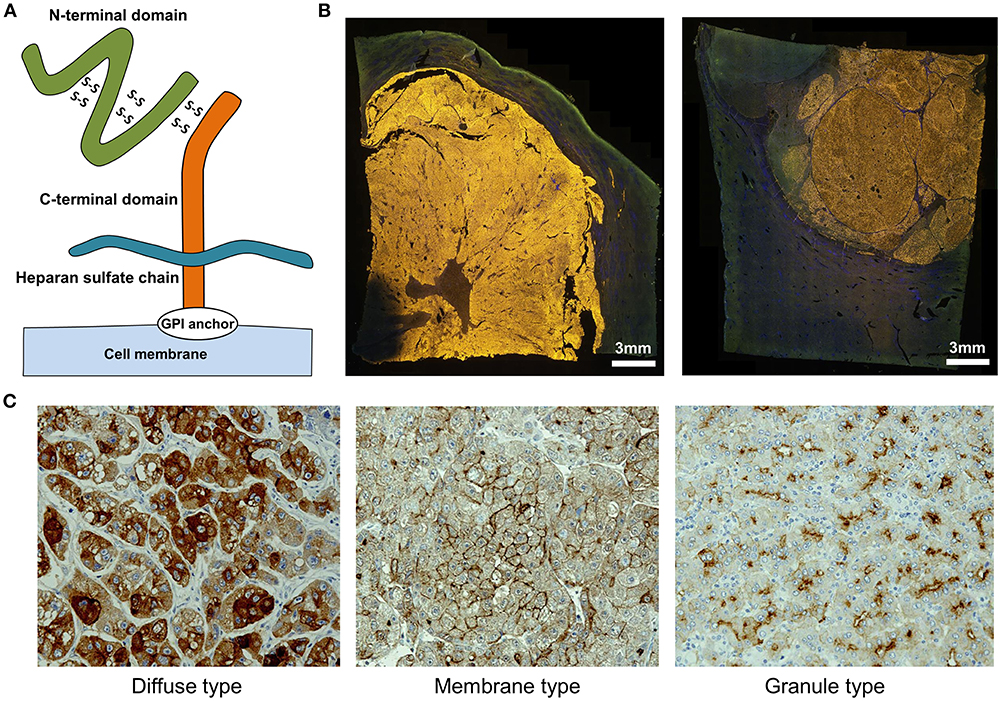
Figure 1. Characteristics of GPC3. (A) Schema of GPC3. (B) Fluorescence micrographs of GPC3-positive HCC. GPC3 labeling appears as a yellow color. (C) GPC3 expression patterns can be classified into diffuse, membrane, and granule types by immunohistochemistry.
Membrane-bound GPC3 can be cleaved and secreted into the blood (Figure 1A). Mammalian GPC family members are cleaved at the GPI anchor level by endogenous GPI phospholipase D (24). It was previously proposed that Notum (a conserved secretory feedback inhibitory protein of the Wnt signaling pathway) targets the GPI anchor in a manner similar to phospholipase and draws the GPC/Wnt complex away from the cell surface to inhibit Wnt signaling; however, it is now thought to control the signal without acting on the anchor (25, 26). It is presumed that GPC3 is cleaved between Arg358 and Ser359, which releases the N-terminal region as a soluble protein from cancer cells into the circulation (27). Thus, various forms of GPC3 protein are present in blood, although their functions remain unclear.
Given these features, GPC3 is an ideal target for cancer immunotherapy. We identified each peptide that can bind to human leukocyte antigen (HLA)-A24 or -A2 and induce GPC3 peptide-specific CTLs (28, 29). Furthermore, we conducted clinical trials of vaccines based on these peptides (30–34). In the future, we envision an array of GPC3-based strategies, not only as cancer vaccines, but also in antibody therapy, adoptive immunotherapy with TCR- or CAR-transduced T cells, and others. We also anticipate that plasma GPC3 will be validated as a biomarker for HCC or for evaluating the efficacy of cancer immunotherapy against GPC3.
We have identified HLA-A24- and HLA-A2-restricted GPC3-derived peptides; approximately 60% of Japanese are positive for HLA-A24 and 40% for HLA-A2, which is the major type in Europeans and North Americans (28, 29). Peptide-specific CTLs were induced in mice immunized with these GPC3 peptides, which exerted antitumor effects without eliciting an autoimmune response (28, 29). In preparation for clinical trials using these HLA-restricted peptides, we investigated whether differences in peptide dosage would affect the efficacy of vaccination, and performed studies in mice to determine the optimal adjuvant (35). We compared five groups: peptide-only and peptide combined with incomplete Freund's adjuvant (IFA), CpG, α-GalCel, or aluminum and found that the GPC3-specific CTLs were only induced in the IFA combination group, demonstrating that the peptide alone was ineffective. We therefore used the peptides plus IFA as cancer vaccines in clinical trials and observed that stronger immune responses were induced by varying peptide dosage.
At the National Cancer Center Hospital East (Kashiwa, Japan), we conducted a phase I clinical trial of GPC3 peptide vaccines in a cohort comprising 33 cases of advanced HCC from February 2007 to November 2009 (UMIN Clinical Trials Registry: 000001395) (Table 1) (30, 38). The primary endpoints were safety and immune response. In a dose escalation study of 0.3, 1, 3, 10, and 30 mg there was no dose-limiting toxicity (DLT), making it difficult to determine the maximum tolerated dose. Although the partial clinical response in a patient treated with 30 mg as well as dose-dependent immunological reactions suggested a greater efficacy of high dosages, a dose of 30 mg required a total vaccine volume of 6 ml, which was difficult to administer and caused pain along with reddening and induration at the site of administration. Based on these observations, we determined that a dosage of 3 mg would be appropriate for the next-phase trial.
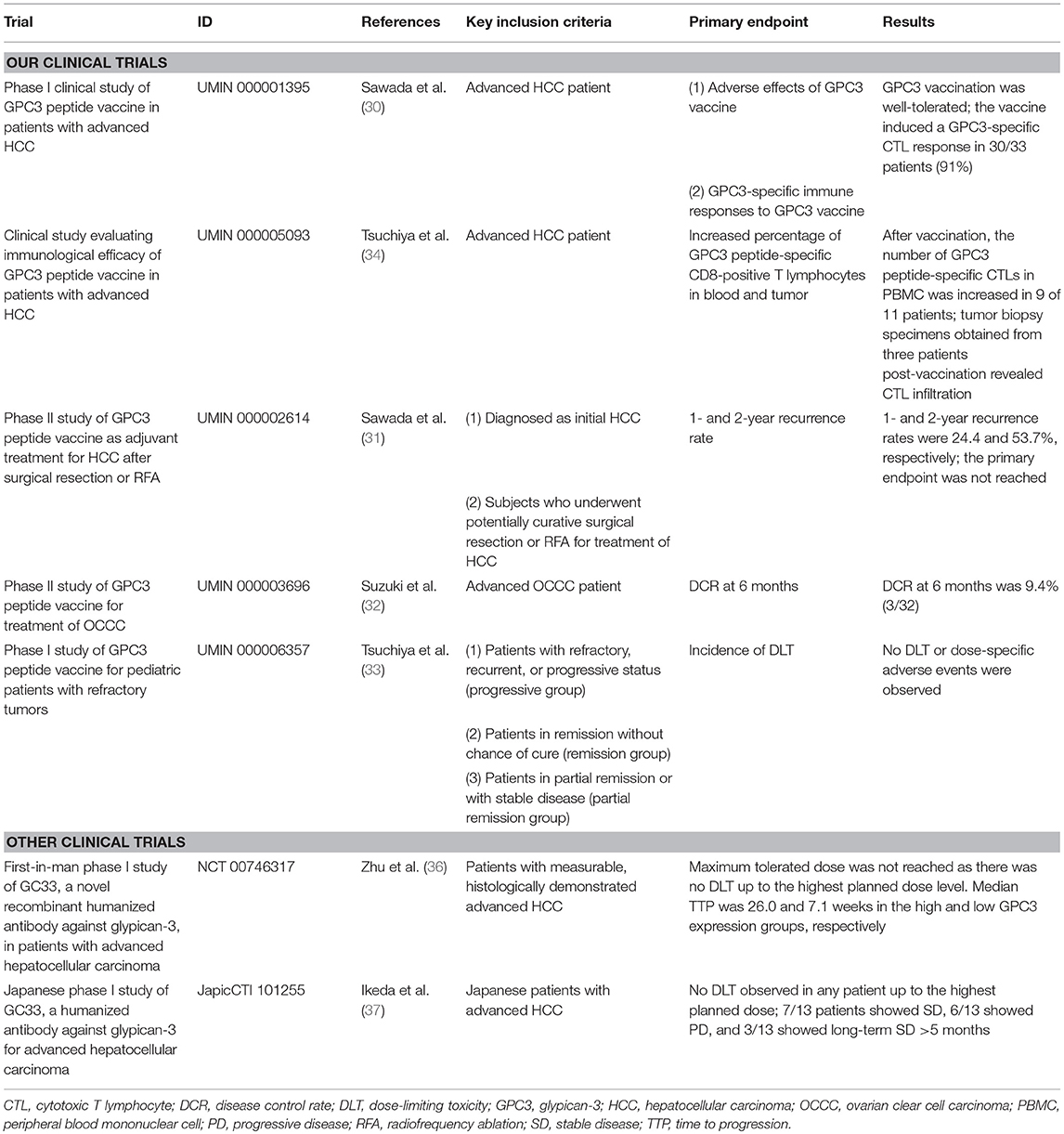
Table 1. Summary of clinical trials for cancer immunotherapy targeting GPC3.
In this first-in-human study of GPC3 peptide vaccines, we confirmed the safety of GPC3 peptide vaccines and obtained promising clinical results. We also detected an elevation in peptide-specific CTL counts in peripheral blood by interferon-γ enzyme-linked immunosorbent spot assay, demonstrating that these vaccines can induce immune responses even in humans. By analyzing tumor biopsies we identified cases where infiltrating CD8+ CTLs were present after but not before vaccination, confirming the immunological effects of our vaccines (30, 38).
A subsequent phase I trial was initiated to investigate the extent of CTL infiltration into tumor tissues (UMIN Clinical Trials Registry: 000005093) by analyzing tumor biopsies obtained before and after vaccination (Table 1) (33). The primary endpoint was GPC3 peptide-specific immune response induced by vaccination. However, this trial was conducted after approval of sorafenib treatment, and patients with extremely late-stage HCC were registered only after sorafenib had ceased to be effective; indeed, most of the patients showed negligible response to the vaccine, probably because of their endogenous immunosuppressive states. Additionally, post-vaccination biopsies were completed in just 11 cases. Nevertheless, we were able to glean useful data from this trial: in one case the HCC tissue became inflamed and then necrotic after two injections of the vaccine, despite ongoing liver dysfunction (39); and we established GPC3 peptide-specific CTL clones from a tumor biopsy specimen (33).
We performed a single-arm phase II clinical trial to evaluate 1- and 2-year relapse rates in 41 cases following radical treatment of HCC using GPC3 peptide vaccine as adjuvant therapy (Table 1) (31). GPC3 peptide-specific CTL responses were detected in 35 of the 41 patients (85.4%) after vaccination. Since the absence of GPC3 expression is correlated with good prognosis, we compared patients with GPC3-positive HCC and control subjects and found that post-surgical administration of GPC3 peptide vaccine can extend the recurrence-free survival period; moreover, overall survival was prolonged in cases where CTL induction was observed. This is likely due to the suppression of GPC3-positive HCC with highly malignant features. We plan to report these results in the future (Miura, manuscript in preparation). Meanwhile, there were two cases of relapse despite the presence of numerous induced peptide-specific CTLs in peripheral blood due to vaccine administration. In these cases, GPC3 was expressed in the primary tumor, but in recurrent cancer, the expression was undetectable (31). These observations suggest that peptide vaccine targeting one type of shared antigen—which could eliminate tumor cells expressing the antigen—may not completely prevent tumor growth due to the increased heterogeneity of the cancer. In such instances cancer peptide vaccines that target multiple shared antigens or neoantigens may be more effective. Furthermore, in nine patients whose recurrent tumors expressed GPC3, the frequencies of GPC3-specific CTLs tended to be lower than those in the aforementioned two patients. Although peptide-specific CTLs were induced by vaccination, the reduction of GPC3-positive HCC recurrence due to the peptide vaccine might depend on the strength of CTL induction. We also considered identifying helper T (Th) cell epitopes in the vaccinated patients (40). Our study revealed that GPC3-derived long peptides-specific and HLA class II-restricted CD4± T-cell responses were observed in 14 of 20 patients, and the presence of the specific Th cells was correlated with prolonged overall survival (40). We expect clinical trials to confirm the recurrence prevention effects of peptide vaccines after resection in patients with GPC3-positive HCC.
We also observed the antitumor effects of GPC3 peptide vaccine in advanced OCCC in clinical trials performed at Nagoya University (UMIN Clinical Trials Registry: 000003696) (Table 1) (32, 41). The primary endpoint was disease control rate (DCR) at 6 months. Two cases showed partial response (PR) and one showed stable disease (SD); DCR at 6 months was 9.4% (3/32 cases). While response rates tended to be higher than for HCC, this may be due to differences in tumor quantity. OCCC is extremely difficult to cure with existing anticancer drugs, lending urgency to the development of effective cancer immunotherapies.
As mentioned above, GPC3 is expressed in some pediatric cancers, including hepatoblastoma, nephroblastoma (Wilms' tumor), and yolk sac tumors. We performed a multicenter clinical trial that included GPC3-positive refractory pediatric cancer cases (UMIN Clinical Trials Registry: 000006357) (Table 1) (34); 18 patients received GPC3 peptide vaccination. DLT—the primary endpoint—was not observed, and the vaccine induced a GPC3-specific CTL response in 7/18 patients (39%), nearly all of whom belonged to the remission group and were hepatoblastoma patients. In contrast, GPC3-specific CTL frequency was not increased in the refractory advanced progression group. These results suggest that the vaccine can prevent recurrence of hepatoblastoma after the second remission, a period in which relapse is generally considered as unavoidable. Furthermore, recurrence-free survival of more than 4 years was observed in all five patients with hepatoblastoma.
Tumor cells reduce their antigen presentation to escape host immune surveillance mechanisms (42), which is a major challenge in the development of effective cancer vaccines. HLA class I expression is reduced or absent in 16–50% of various malignancies (43). To circumvent this problem, we developed an intratumoral injection method for peptide vaccines that has been tested in mice. This mode of delivery enhanced anti-tumor activity as compared to conventional subcutaneous injection and induced systemic immune responses that inhibited the growth of metastasized tumors (44). Moreover, combining intratumoral peptide vaccine injection and anti-PD-1 blocking antibody could elicit enhanced antitumor effects by inducing the upregulation of PD-1 on the surface of CTLs (45–47). This may be applicable not only to primary tumors but also to distant metastatic sites, which could be targeted by loading peptide into HLA class I of tumor cells.
In our clinical trials of GPC3 peptide vaccine, we successfully established multiple types of GPC3 peptide-specific CTL clones derived from the peripheral blood and cancer tissue of vaccinated patients (34, 38, 48). Some of these clones have a strong ability to kill cancer cells presenting GPC3 peptide in vitro. By cloning these TCRs, we are currently developing an adoptive immunotherapy approach based on these TCR-transduced T cells. GPC3 peptide-specific TCRs were cloned from CTLs obtained from patients who showed no adverse reactions to the peptide vaccine other than reddening and local swelling at the administration site, thus guaranteeing vaccine safety. Adoptive immunotherapy with TCR-transduced T cells is generally considered as having superior antitumor effects to peptide vaccine therapy, and their application to advanced cancers presenting GPC3 peptide is highly anticipated.
GC33, a humanized monoclonal antibody against GPC3, has been shown to induce antibody-dependent cell-mediated cytotoxicity against GPC3-positive HCC cell lines and elicit anti-tumor effects in patient-derived xenograft cancer models (49). In a first-in-human phase I trial performed in the United States for patients with advanced HCC, GC33 was well-tolerated and antitumor effects were observed in some patients with high GPC3 expression HCC (Table 1) (36). A phase I trial of GC33 carried out in Japan confirmed its tolerability. While there were no complete response or PR cases, SD was achieved by 7 of 13 patients who received the treatment, three of whom maintained this status for 3 or more months (Table 1) (37). A randomized, placebo-controlled, double-blind, multicenter phase II trial of GC33 is presently underway (Table 2). In addition, the results of a basic research study on ERY974, an anti- GPC3/CD3 bi-specific T cell-redirecting antibody were recently reported (50). A phase I clinical trial to confirm its toxicity is currently underway in which there is no limitation to the cancer type if the primary tumor is GPC3-positive (Table 2).
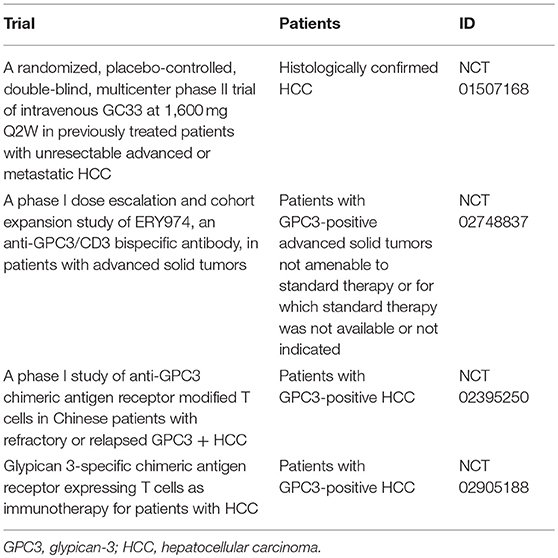
Table 2. Summary of clinical trials for cancer immunotherapy targeting GPC3.
While CAR-T therapy has shown a remarkable response rate exceeding 80% against B cell blood tumors (51, 52), fully promising results have not yet been obtained against solid carcinomas, such as glioblastoma (53), pancreatic cancer (54), and prostate cancer (55, 56). These insufficient responses were thought to be caused by tumor heterogeneity, immunosuppressive mechanism in the tumor microenvironment, and insufficient accumulation of CAR-T cells in the tumor (57). One option for ameliorating these problems is IL-7/CCL19 expressing CAR-T cells (58). These cells promoted the migration and activation of not only CAR-T cells but also T cells and dendritic cells at the tumor locus, thereby demonstrating strong anti-tumor effect on solid tumors in mice (58). Moreover, in order to avoid on-target off-tumor toxicity, it is necessary to select an excellent cancer antigen with high tumor specificity. In this regard, GPC3 is considered an ideal target as described above. The development of CAR-T therapy targeting solid tumors is underway around the world, and therapies based on GPC3 antibody gene (GPC3-CAR) have been developed (59, 60). In China and United States, clinical trials of GPC3-CAR therapy against GPC3-positive HCC have already begun (Table 2). By combining it with new technologies that supplement the weakness of CAR-T therapy, it is desired that the clinical effect against solid tumors would be further improved.
We think that there is significant value to using T cells derived from iPS cells for the following three reasons: (i) they eliminate the effects of effector T cell exhaustion and aging; (ii) they enable reliable gene manipulation at the iPS cell stage; and (iii) they make it possible to continue the treatment over a long period of time. By using an iPS cell bank, we may be able to develop innovative adoptive immunotherapeutics that can be universally adopted at a low cost. In addition, since some patients may have T cells that lack intrinsic anti-tumor potency, an alternative, third party off the shelf T cell product might be a good alternative. Kaneko et al. at Kyoto University in Japan have successfully prevented unwanted reconstruction of TCRs by excluding genes causing TCR remodeling in T cells derived from iPS cells through genome editing, and showed that cytotoxic T cells with high avidity for cancer cells can be induced through this process (61). We are currently developing an iPS cell-derived TCR-T cell (TCR-iPS-T cell) strategy for expressing GPC3-peptide specific TCR in collaboration with their team. In detail, the cell was created by transducing HLA haplotype homologous iPS cell clones with HLA-A*24:02 restricted GPC3298−306 peptide-specific TCR gene using a lentiviral vector and then stimulating differentiation (61). The findings to date indicate that these TCR-iPS-T cells derived from an HLA homozygous iPS cell stock showed both cancer antigen-specific cytotoxic effects caused by CAR and non-specific cytotoxicity due to stimulation of natural killer cell ligands against GPC3-positive cell lines. These results may lead to the development of a novel type of immunotherapy that can prevent the suppression of anti-cancer effects caused by immune editing against a particular antigen.
As described above, GPC3 is released into the serum of HCC patients, and its utility as a tumor marker has been reported (62, 63). We established a novel sandwich enzyme-linked immunosorbent assay system for predicting HCC recurrence after surgery based on post-operative elevation of serum GPC3 level (64). In immunohistochemical analyses of GPC3-positive HCC specimens, a few surrounding normal cells also weakly expressed GPC3, which we believe contribute to post-operative GPC3 secretion and recurrence (64). In partnership with a private company, we have developed an assay to quantify serum full-length GPC3 level, which is the most physiologically relevant parameter. In combination with existing tumor markers such as alpha-fetoprotein and protein induced by vitamin K absence II, we successfully predicted early recurrence of HCC after surgery (Miura, manuscript in preparation). We are presently examining whether this assay can predict hepatocellular carcinogenesis in patients with chronic hepatitis and cirrhosis as well as response to anti-GPC3 therapy.
We are in the process of developing various treatments and diagnostic methods targeting GPC3 (Figure 2) (65). The subcellular localization of GPC3 and its presence of serum are expected to affect the effects of each treatment approach. Soluble full-length GPC3 protein could block anti-GPC3 antibody and CAR-T cells, thereby reducing cytotoxicity and leading to unexpected side effects. Based on these considerations, serum GPC3 could serve as a biomarker for evaluating treatment effect or be used to assess the eligibility of patients for antibody or GPC3-CAR-T therapy. In addition, the clinical trial for GC33 revealed a correlation between the localization of GPC3 and clinical effects (36). We have also described multiple distinct GPC3 expression patterns (Figure 1C). Therefore, it is important to analyze the localization of GPC3 and not only its presence or absence. To this end we are currently developing a method for investigating the co-expression of GPC3 and class I HLA—a cell membrane-associated molecule—by multiple immunolabeling that can be used as a companion diagnostic tool (Figure 3).
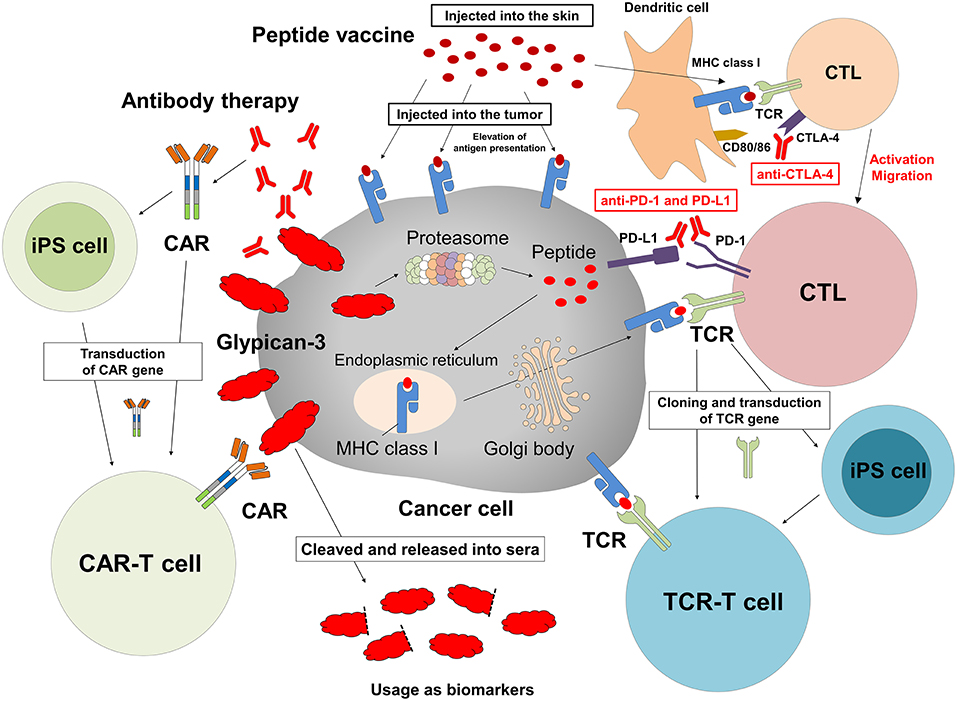
Figure 2. Cancer immunotherapy targeting GPC3. Therapeutic approaches that target intracellular GPC3 include GPC3 peptide vaccine and adoptive immunotherapy based on T cells transduced with a suitable TCR. In contrast, antibody therapy and anti-GPC3-CAR-transduced T cell therapy target membrane-bound GPC3. We are currently developing such T cells and CAR-T cells from iPS cells. In addition, intratumoral vaccination and combination with immune checkpoint inhibitors might enhance effects of these treatments, and serum GPC3 could be a biomarker.
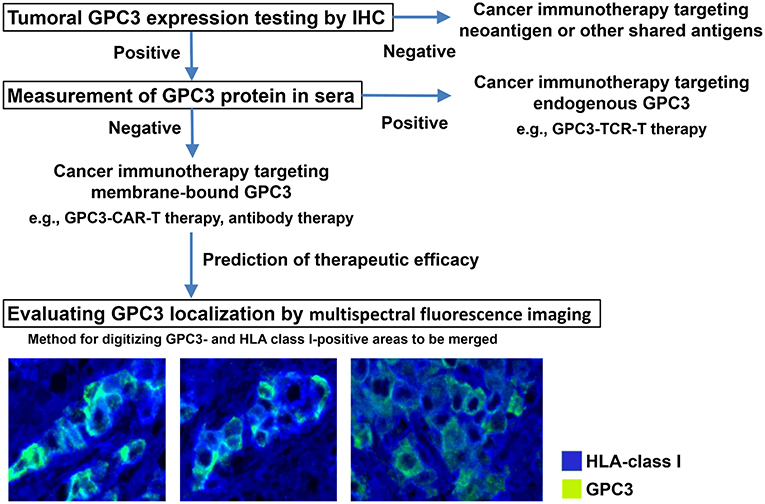
Figure 3. Appropriate treatments targeting GPC3. Analyzing tumor GPC3 expression in combination with serum GPC3 level can reveal the most effective therapeutic strategy. To determine whether GPC3 is predominantly expressed at the cell membrane—which is thought to influence the efficacy of therapies targeting membrane-bound GPC3—we are developing a method of digitizing GPC3- and HLA class I-positive areas to be merged using a multispectral fluorescence imaging system.
GPC3 has unprecedented cancer specificity and is being studied as a target for cancer immunotherapy worldwide. However, there remain many open questions regarding its natural history, function, and dynamics. Clarifying these aspects of GPC3 is necessary for the development of more effective treatments. Since Boon et al. first identified a cancer-specific antigen for melanoma in 1991, many studies have been undertaken to search for new cancer antigens (66). We identified GPC3 as a tumor-associated antigen and have developed GPC3 peptide vaccine as cancer immunotherapy. Based on findings from our clinical trials, we are now developing the next generation of GPC3-targeting therapeutic approaches such as CAR-T and TCR-engineered T cell therapy. On the other hand, neoantigen is gaining global attention, with significant advances in the establishment of immunotherapies targeting these molecules. We are also developing personalized cancer immunotherapies such as cancer vaccines using peptides derived from neoantigens or individualized adoptive T cell therapies using next-generation sequencers. We look forward to advances in research on neoantigens or shared antigens such as GPC3 that can demonstrate which of these can best serve as immunotherapeutic targets.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This study was supported in part by the National Cancer Center Research and Development Fund (25-A-7 and 28-A-8); Health and Labor Science Research Grants for Clinical Research, Japan; and joint research funding from Takeda Pharmaceutical Co, Ltd.; Noile-Immune Biotech Inc.; Ono Pharmaceutical Co., Ltd.; BrightPath Biotherapeutics Co., Ltd.; and Sysmex Co., Ltd. This study was performed as part of a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct), Ministry of Education, Culture, Sports, Science and Technology of Japan.
TN, TS, and TY are supported by fundamental research funding from Takeda Pharmaceutical Co., Ltd. and BrightPath Biotherapeutics Co., Ltd. TN and TS are supported by fundamental research funding from Ono Pharmaceutical Co., Ltd. TN is supported by fundamental research funding from Noile-Immune Biotech Inc. and Sysmex Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol. (2015) 4:385. doi: 10.3389/fonc.2014.00385
2. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. (2013) 369:134–44. doi: 10.1056/NEJMoa1305133
3. Gattinoni L, Powell DJ Jr., Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. (2006) 6:383–93. doi: 10.1038/nri1842
4. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
5. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
6. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. (2017) 547:222–6. doi: 10.1038/nature23003
7. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. (2019) 565:234–9. doi: 10.1038/s41586-018-0792-9
8. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Eng J Med. (2015) 372:311–9. doi: 10.1056/NEJMoa1411087
9. Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. (2017) 541:321. doi: 10.1038/nature21349
10. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. (2007) 13:1050–9. doi: 10.1038/nm1622
11. Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol. (2017) 8:1679. doi: 10.3389/fimmu.2017.01679
12. Toh JW, de Souza P, Lim SH, Singh P, Chua W, Ng W, et al. The potential value of immunotherapy in colorectal cancers: review of the evidence for programmed death-1 inhibitor therapy. Clin Colorectal Cancer. (2016) 15:285–91. doi: 10.1016/j.clcc.2016.07.007
13. Ikeda H. T-cell adoptive immunotherapy using tumor-infiltrating T cells and genetically engineered TCR-T cells. Int Immunol. (2016) 28:349–53. doi: 10.1093/intimm/dxw022
14. Nakatsura T, Yoshitake Y, Senju S, Monji M, Komori H, Motomura Y, et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem Biophys Res Commun. (2003) 306:16–25. doi: 10.1016/S0006-291X(03)00908-2
15. Filmus J, Selleck SB. Glypicans: proteoglycans with a surprise. J Clin Invest. (2001) 108:497–501. doi: 10.1172/JCI200113712
16. Pilia G, Hughes-Benzie RM, MacKenzie A, Baybayan P, Chen EY, Huber R, et al. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat Gen. (1996) 12:241. doi: 10.1038/ng0396-241
17. Capurro MI, Xiang YY, Lobe C, Filmus J. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. (2005) 65:6245–54. doi: 10.1158/0008-5472.CAN-04-4244
18. Gutiérrez J, Brandan E. A novel mechanism of sequestering fibroblast growth factor 2 by glypican in lipid rafts, allowing skeletal muscle differentiation. Mol Cell Biol. (2010) 30:1634–49. doi: 10.1128/MCB.01164-09
19. Cano-Gauci DF, Song HH, Yang H, McKerlie C, Choo B, Shi W, et al. Glypican-3-deficient mice exhibit the overgrowth and renal abnormalities typical of the Simpson–Golabi–Behmel syndrome. J Cell Biol. (1999) 146:255–64.
20. Lin H, Huber R, Schlessinger D, Morin PJ. Frequent silencing of the GPC3 gene in ovarian cancer cell lines. Cancer Res. (1999) 59:807–10.
21. Nakatsura T, Nishimura Y. Usefulness of the novel oncofetal antigen glypican-3 for diagnosis of hepatocellular carcinoma and melanoma. BioDrugs. (2005) 19:71–7. doi: 10.2165/00063030-200519020-00001
22. Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. (2003) 125:89–97. doi: 10.1016/S0016-5085(03)00689-9
23. Shirakawa H, Suzuki H, Shimomura M, Kojima M, Gotohda N, Takahashi S, et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. (2009) 100:1403–7. doi: 10.1111/j.1349-7006.2009.01206.x
24. Metz CN, Brunner G, Choi-Muira NH, Nguyen H, Gabrilove J, Caras IW, et al. Release of GPI-anchored membrane proteins by a cell-associated GPI-specific phospholipase D. EMBO J. (1994) 13:1741–51. doi: 10.1002/j.1460-2075.1994.tb06438.x
25. Traister A, Shi W, Filums J. Mammalian Notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem J. (2008) 410:503–11. doi: 10.1042/BJ20070511
26. Kakugawa S, Langton PF, Zebisch M, Howell S, Chang TH, Liu Y, et al. Notum deacylates Wnt proteins to suppress signaling activity. Nature. (2015) 519:187–92. doi: 10.1038/nature14259
27. Hippo Y, Watanabe K, Watanabe A, Midorikawa Y, Yamamoto S, Ihara S, et al. Identification of soluble NH2-terminal fragment of glypican-3 as a serological marker for early-stage hepatocellular carcinoma. Cancer Res. (2004) 64:2418–23. doi: 10.1158/0008-5472.CAN-03-2191
28. Nakatsura T, Komori H, Kubo T, Yoshitake Y, Senju S, Katagiri T, et al. Mouse homolog of a novel human oncofetal antigen, glypican-3, evokes T-cell-mediated tumor rejection without autoimmune reactions in mice. Clin Cancer Res. (2004) 10:8630–40. doi: 10.1158/1078-0432.CCR-04-1177
29. Komori H, Nakatsura T, Senju S, Yoshitake Y, Motomura Y, Ikuta Y, et al. Identification of HLA-A2- or HLA-A24-restricted CTL epitopes possibly useful for glypican-3-specific immunotherapy of hepatocellular carcinoma. Clin Cancer Res. (2006) 12:2689–97. doi: 10.1158/1078-0432.CCR-05-2267
30. Sawada Y, Yoshikawa T, Nobuoka D, Shirakawa H, Kuronuma T, Motomura Y, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. (2012) 18:3686–96. doi: 10.1158/1078-0432.CCR-11-3044
31. Sawada Y, Yoshikawa T, Ofuji K, Yoshimura M, Tsuchiya N, Takahashi M, et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology. (2016) 5:e1129483. doi: 10.1080/2162402X.2015.1129483
32. Suzuki S, Sakata J, Utsumi F, Sekiya R, Kajiyama H, Shibata K, et al. Efficacy of glypican-3-derived peptide vaccine therapy on the survival of patients with refractory ovarian clear cell carcinoma. Oncoimmunology. (2016) 5:e1238542. doi: 10.1080/2162402X.2016.1238542
33. Tsuchiya N, Hosono A, Yoshikawa T, Shoda K, Nosaka K, Shimomura M, et al. Phase I study of glypican-3-derived peptide vaccine therapy for patients with refractory pediatric solid tumors. Oncoimmunology. (2017) 7:e1377872. doi: 10.1080/2162402X.2017.1377872
34. Tsuchiya N, Yoshikawa T, Fujinami N, Saito K, Mizuno S, Sawada Y, et al. Immunological efficacy of glypican-3 peptide vaccine in patients with advanced hepatocellular carcinoma. Oncoimmunology. (2017) 6:e1346764. doi: 10.1080/2162402X.2017.1346764
35. Motomura Y, Ikuta Y, Kuronuma T, Komori H, Ito M, Tsuchihara M, et al. HLA-A2 and -A24-restricted glypican-3-derived peptide vaccine induces specific CTLs: preclinical study using mice. Int J Oncol. (2008) 32:985–90. doi: 10.3892/ijo.32.5.985
36. Zhu AX, Gold PJ, El-Khoueiry AB, Abrams TA, Morikawa H, Ohishi N, et al. First-in-man phase I study of GC33, a novel recombinant humanized antibody against glypican-3, in patients with advanced hepatocellular carcinoma. Clin Cancer Res. (2013) 19:920–8. doi: 10.1158/1078-0432.CCR-12-2616
37. Ikeda M, Ohkawa S, Okusaka T, Mitsunaga S, Kobayashi S, Morizane C, et al. Japanese phase I study of GC33, a humanized antibody against glypican-3 for advanced hepatocellular carcinoma. Cancer Sci. (2014) 105:455–62. doi: 10.1111/cas.12368
38. Yoshikawa T, Nakatsugawa M, Suzuki S, Shirakawa H, Nobuoka D, Sakemura N, et al. HLA-A2-restricted glypican-3 peptide-specific CTL clones induced by peptide vaccine show high avidity and antigen-specific killing activity against tumor cells. Cancer Sci. (2011) 102:918–25. doi: 10.1111/j.1349-7006.2011.01896.x
39. Sawada Y, Yoshikawa T, Fujii S, Mitsunaga S, Nobuoka D, Mizuno S, et al. Remarkable tumor lysis in a hepatocellular carcinoma patient immediately following glypican-3-derived peptide vaccination: an autopsy case. Hum Vaccin Immunother. (2013) 9:1228–33. doi: 10.4161/hv.24179
40. Sayem MA, Tomita Y, Yuno A, Hirayama M, Irie A, Tsukamoto H, et al. Identification of glypican-3-derived long peptides activating both CD8± and CD4± T cells; prolonged overall survival in cancer patients with Th cell response. Oncoimmunology. (2016) 5:e1062209. doi: 10.1080/2162402X.2015.1062209
41. Suzuki S, Shibata K, Kikkawa F, Nakatsura T. Significant clinical response of progressive recurrent ovarian clear cell carcinoma to glypican-3-derived peptide vaccine therapy: two case reports. Hum Vaccin Immunother. (2014) 10:338–43. doi: 10.4161/hv.27217
42. Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today. (1999) 5:178–86. doi: 10.1016/S1357-4310(99)01451-3
43. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. (2002) 3:999–1005. doi: 10.1038/ni1102-999
44. Nobuoka D, Yoshikawa T, Fujiwara T, Nakatsura T. Peptide intra-tumor injection for cancer immunotherapy: enhancement of tumor cell antigenicity is a novel and attractive strategy. Hum Vaccin Immunother. (2013) 9:1234–6. doi: 10.4161/hv.23990
45. Nobuoka D, Yoshikawa T, Takahashi M, Iwama T, Horie K, Shimomura M, et al. Intratumoral peptide injection enhances tumor cell antigenicity recognized by cytotoxic T lymphocytes: a potential option for improvement in antigen-specific cancer immunotherapy. Cancer Immunol Immunother. (2013) 62:639–52. doi: 10.1007/s00262-012-1366-6
46. Sawada Y, Yoshikawa T, Shimomura M, Iwama T, Endo I, Nakatsura T. Programmed death-1 blockade enhances the antitumor effects of peptide vaccine-induced peptide-specific cytotoxic T lymphocytes. Int J Oncol. (2015) 46:28–36. doi: 10.3892/ijo.2014.2737
47. Fujinami N, Yoshikawa T, Sawada Y, Shimomura M, Iwama T, Sugai S, et al. Enhancement of antitumor effect by peptide vaccine therapy in combination with anti-CD4 antibody: Study in a murine model. Biochem Biophys Rep. (2016) 5:482–91. doi: 10.1016/j.bbrep.2016.02.010
48. Tada Y, Yoshikawa T, Shimomura M, Sawada Y, Sakai M, Shirakawa H, et al. Analysis of cytotoxic T lymphocytes from a patient with hepatocellular carcinoma who showed a clinical response to vaccination with a glypican 3 derived peptide. Int J Oncol. (2013) 43:1019–26. doi: 10.3892/ijo.2013.2044
49. Ishiguro T, Sugimoto M, Kinoshita Y, Miyazaki Y, Nakano K, Tsunoda H, et al. Anti-glypican 3 antibody as a potential antitumor agent for human liver cancer. Cancer Res. (2008) 68:9832–8. doi: 10.1158/0008-5472.CAN-08-1973
50. Ishiguro T, Sano Y, Komatsu S, Kamata-Sakurai M, Kaneko A, Kinoshita Y, et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci Transl Med. (2017) 9:eaal4291. doi: 10.1126/scitranslmed.aal4291
51. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene xiloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
52. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Eng J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
53. O'rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJ, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Trans Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
54. Beatty GL, O'Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. (2018) 155:29–32. doi: 10.1053/j.gastro.2018.03.029
55. Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo AS, et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate. (2016) 76:1257–70. doi: 10.1002/pros.23214
56. Pettitt D, Arshad Z, Smith J, Stanic T, Holländer G, Brindley D. CAR-T cells: a systematic review and mixed methods analysis of the clinical trial landscape. Mol Ther. (2018) 26:342–53. doi: 10.1016/j.ymthe.2017.10.019
57. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
58. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. (2018) 36:346–51. doi: 10.1038/nbt.4086
59. Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front Immunol. (2017) 7:690. doi: 10.3389/fimmu.2016.00690
60. Li W, Guo L, Rathi P, Marinova E, Gao X, Wu MF, et al. Redirecting T cells to glypican-3 with 4-1BB zeta chimeric antigen receptors results in Th1 polarization and potent antitumor activity. Hum Gene Ther. (2017) 28:437–48. doi: 10.1089/hum.2016.025
61. Minagawa A, Yoshikawa T, Yasukawa M, Hotta A, Kunitomo M, Iriguchi S, et al. Enhancing T cell receptor stability in rejuvenated iPSC-derived T cells improves their use in cancer immunotherapy. Cell Stem Cell. (2018) 6:850–8. doi: 10.1016/j.stem.2018.10.005
62. Yamauchi N, Watanabe A, Hishinuma M, Ohashi K, Midorikawa Y, Morishita Y, et al. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Mod Pathol. (2005) 18:1591–8. doi: 10.1038/modpathol.3800436
63. Chen M, Li G, Yan J, Lu X, Cui J, Ni Z, et al. Reevaluation of glypican-3 as a serological marker for hepatocellular carcinoma. Clin Chim Acta. (2013) 423:105–11. doi: 10.1016/j.cca.2013.04.026
64. Ofuji K, Saito K, Suzuki S, Shimomura M, Shirakawa H, Nobuoka D, et al. Perioperative plasma glypican-3 level may enable prediction of the risk of recurrence after surgery in patients with stage I hepatocellular carcinoma. Oncotarget. (2017) 8:37835–44. doi: 10.18632/oncotarget.14271
65. Shimizu Y, Suzuki T, Yoshikawa T, Tsuchiya N, Sawada Y, Endo I, et al. Cancer immunotherapy-targeted glypican-3 or neoantigens. Cancer Sci. (2018) 109:531–41. doi: 10.1111/cas.13485
Keywords: glypican-3 (GPC3), cancer antigen, cancer immunotherapy, cancer vaccine, cytotoxic T cell, TCR-engineered T cell therapy, CAR-T therapy
Citation: Shimizu Y, Suzuki T, Yoshikawa T, Endo I and Nakatsura T (2019) Next-Generation Cancer Immunotherapy Targeting Glypican-3. Front. Oncol. 9:248. doi: 10.3389/fonc.2019.00248
Received: 04 February 2019; Accepted: 18 March 2019;
Published: 10 April 2019.
Edited by:
Cinzia Lanzi, National Tumor Institute, ItalyReviewed by:
Mariangela Figini, National Tumor Institute, ItalyCopyright © 2019 Shimizu, Suzuki, Yoshikawa, Endo and Nakatsura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tetsuya Nakatsura, dG5ha2F0c3VAZWFzdC5uY2MuZ28uanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.