 Benoit Tessoulin1,2,3,4
Benoit Tessoulin1,2,3,4 Geraldine Descamps1,2,3Christelle Dousset1,2,3
Geraldine Descamps1,2,3Christelle Dousset1,2,3 Martine Amiot1,2,3
Martine Amiot1,2,3 Catherine Pellat-Deceunynck1,2,3*
Catherine Pellat-Deceunynck1,2,3*- 1CRCINA, INSERM, CNRS, Université d'Angers, Université de Nantes, Nantes, France
- 2L'Héma-NexT, i-Site NexT, Université de Nantes, Nantes, France
- 3SIRIC ILIAD, Angers, Nantes, France
- 4Service d'Hématologie Clinique, Unité d'Investigation Clinique, CHU de Nantes, Nantes, France
Prima-1Met (APR-246) was previously shown to be dependent on glutathione inhibition and on ROS induction in cancer cells with mutated or deleted TP53. Because this ROS induction was, at least in part, due to a direct interference with the thioredoxin reductase enzyme, we investigated whether activity of Prima-1Met could be mimicked by auranofin, an inhibitor of the thioredoxin reductase. We thus compared the activity of auranofin and Prima-1Met in 18 myeloma cell lines and in 10 samples from patients with multiple myeloma or plasma cell leukemia. We showed that, similar to Prima-1Met, the activity of auranofin was not dependent on either TP53 status or p53 expression; was inhibited by N-acetyl-L-cysteine, a ROS scavenger; displayed a dramatic synergy with L-buthionine sulfoximine, an irreversible inhibitor of glutathione synthesis; and induced cell death that was not dependent on Bax/Bak expression. These data showed that auranofin and Prima-1Met similarly overcome cell death resistance in myeloma cells due to either p53 deficiency or to mitochondrial dysfunction.
Introduction
Mutations and/or deletions of TP53 are associated with resistance to treatments in multiple myeloma as in most B-cell malignancies (1). TP53 mutation and deletion are associated in B-cell malignancies suggesting that bi-allelic alterations of TP53 are involved in resistance, although overall survival of patients with lymphoma or myeloma appears more significantly related to mutations than to deletion (2, 3). Mutations of TP53 induced a more rapid development of spontaneous tumors than the deletion of TP53 (4). Moreover, some mutations are characterized by a gain of function, making mutant forms of the p53 protein interesting therapeutic targets (5). Given the importance of folding for p53 activity and the existence of temperature-dependent mutations, chemical molecules able to change the conformation of p53 and to restore its transcriptional activity were screened (6). During the last 15 years, several molecules were isolated for their efficacy to induce cell death in TP53 mutated cells and some of these molecules were shown to interact with the mutant p53 protein (7, 8). However, the p53 dependency of several p53 reactivating molecules, such as RITA and Prima-1Met, is debated as both drugs killed cancer cells independently from TP53 status and p53 expression (9, 10). Indeed, it was recently demonstrated using CRISPR/Cas9 technology that the cell response to RITA, which is a DNA damaging drug, depended on FANCD2 expression (11). On the other hand, Prima-1Met has been shown to decrease glutathione and to induce ROS independently from p53 expression or mutations (10, 12, 13), at least by directly interfering with thioredoxin reductase, a central enzyme of the detoxifying redox pathway (14). These results prompted us to further investigate whether auranofin, an inhibitor of the thioredoxin reductase, displayed a Prima-1Met-like activity. We therefore investigated activity and death mechanism of auranofin in myeloma cell lines and primary myeloma cells characterized for TP53 status. We showed that activity of auranofin and Prima-1Met correlated in myeloma cells and that both drugs induced a Bax/Bak-independent cell death.
Materials and Methods
Human Myeloma Cell Lines (HMCLs) and Primary Samples
Eighteen HMCLs used for this study, i.e., 7 HMCLs with a wild-type TP53 status (MDN, NCI-H929, NAN9, NAN11, XG3, XG6, XG7), 8 HMCLs with a missense TP53 mutation (JIM3, KMS12PE, LP1, NAN10, OPM2, U266, XG2, XG5) and 3 HMCLs with a TP53 indel leading to the lack of mRNA and/or protein expression (JJN3, L363, NAN7). All HMCLs have been extensively characterized (10, 15, 16). TP53 status was performed by direct sequencing of RT-PCR products (16) and by whole exon sequencing (17). After obtaining informed consent, blood or bone marrow samples from patients with MM were collected at the Department of Hematology of the Nantes University Hospital (MYRACLE cohort, ethical approval NCT03807128, Benaniba et al., submitted). Plasma cells were obtained after gradient density centrifugation and FISH was performed as previously described (9, 18).
Reagents and Antibodies
Prima-1Met was purchased from Santa Cruz Biotechnology (CliniSciences, Nanterre, France), L-buthionine sulfoximine (BSO), auranofin and N-acetyl-L-cysteine were purchased from Sigma-Aldrich (Saint-Quentin Fallavier, France). Anti-CD138-PE monoclonal antibody was purchased from Beckman Coulter (Villepinte, France), Annexin V-FITC was purchased from ImmunoTools (Friesoythe, Germany), anti-Bak, anti-Bax and anti-actin were purchased from BD-Biosciences (Le Pont de Claix, France), Cell Signaling (Ozyme, Montigny-le-Bretonneux, France) and Millipore (Guyancourt, France), respectively.
siRNA Experiments
Transient BAX or BAK silencing was performed in LP1 myeloma cells (100 pmol siRNA/3 × 106 cells) using lipofectamine RNAiMax (Thermo Fischer Scientific, Saint-Herblain, France), as previously reported (19).
Cell Death Assay
The cell lines or mononuclear cells from patients' samples (500,000 cells/ml) were incubated with Prima-1Met or auranofin with different concentrations as indicated within the legends of the figures. Cell death was assessed by Annexin V staining in cell lines and by the loss of CD138 staining in primary myeloma cells (10, 19–21). The fluorescence acquisition and analysis were performed using a FACsCalibur with Cell Quest (Becton Dickinson) or FlowJo (Ashland, OR, USA) software, Cytocell core facility (SFR Bonamy, Nantes, France).
Statistical Analyses
The statistical analyses were performed using GraphPad Prism 7.
Results
Sensitivity of Myeloma Cell Lines to Prima-1Met and Auranofin Correlated
We assessed the efficacy of auranofin, an inhibitor of thioredoxin reductase, in comparison with Prima-1Met in 18 HMCLs. We determined the lethal dose 50 (LD50) of auranofin and Prima-1Met to HMCLs using Annexin V staining at day 2, as illustrated in Figure 1A. Figure 1B (left panel) shows a positive correlation between LD50 values for auranofin and Prima-1Met (p = 0.042, r2 = 0.2327, Pearson test); however, auranofin displayed higher activity (about 80-fold) compared to Prima-1Met (median values were 0.4 and 32.5 μM, respectively). Notably, the correlation was essentially supported by HMCLs displaying abnormal TP53 status (Figure 1B, middle and right panels, Pearson test), although activity of auranofin and Prima-1Met was not dependent on p53 mutations or expression (Figure 1C). Using the CellMinerCDB portal (https://discover.nci.nih.gov/cellminercdb/), which provides pharmacologic sensitivity of cancer cell lines, we confirmed that activity of another inhibitor of the thioredoxin reductase (PX-12) also correlated with activity of Prima-1Met in myeloma cell lines (Figure S1).
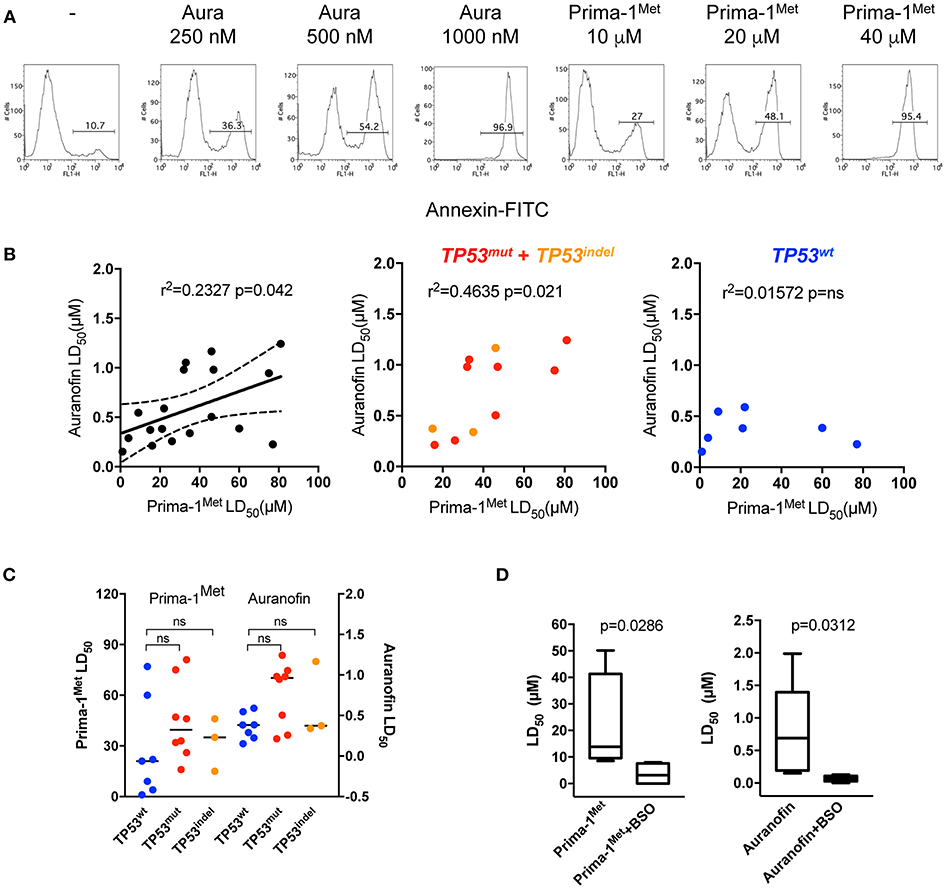
Figure 1. HMCLs were similarly sensitive to Prima-1Met and to auranofin. (A) OMP2 cells (500 000/ml) were incubated with or without (-) increasing concentrations of auranofin or Prima-1Met for 2 days as indicated in the figure, and cell death was assessed by Annexin V staining. (B) The LD50 (lethal dose 50) values of auranofin were plotted against the LD50 values of PRIMA-1Met in 18 HMCLs (left). The graphs of the middle and right panels represent the correlations between the two drugs in TP53Abn and TP53wt HMCLs, respectively. The statistical analyses were performed using the Pearson test. (C) The LD50 values of auranofin and PRIMA-1Met were analyzed according to TP53wt, TP53mut, and TP53indel statuses. The statistical analyses were performed using the Mann-Whitney test. (D) HMCLs were incubated with increasing concentrations of each drug with or without BSO (500 μM) for 2 days. Cell death was assessed by Annexin V staining. The statistical analyses were performed using the Wilcoxon matched-pairs signed-rank test.
Because it was previously shown that Prima-1Met synergized with BSO, an irreversible inhibitor of GSH synthesis, we determined whether auranofin also synergized with BSO (10, 12). Six HMCLs (JJN3, MDN, OPM2, XG5, XG6, and U266) were cultured with increasing concentrations of Prima-1Met or auranofin with or without BSO (500 μM) and LD50 values were determined. Both Prima-1Met and auranofin strongly synergized with BSO, and LD50 values were decreased by ~ 4- and 10-fold, respectively (Figure 1D).
Auranofin and Prima-1Met Induced ROS-Dependent Bax/Bak-Independent Cell Death
We previously demonstrated that Prima-1Met induced ROS-dependent cell death in myeloma cells that was prevented by N-acetyl-L-cysteine (NAC) (10). Because auranofin has been shown to induce ROS production, we thus assessed whether NAC was also able to inhibit cell death induced by auranofin (22). As shown in Figure 2A in L363 and OPM2, the addition of NAC inhibited cell death induced by auranofin and Prima-1Met by 86% (p = 0.03) and 95% (p = 0.03), respectively. Prima-1Met was shown to induce apoptosis that was independent from Bax/Bak: Prima-1Met induced lipid peroxidation that mediated mitochondrial permeabilization, cytochrome C release and activation of caspases (13). To determine the role of Bax/Bak in auranofin-induced cell death, we performed BAX/BAK1 silencing in LP1 cells. A decrease in Bax or Bak expression did not significantly modify cell responses to either drug (Figure 2B). Moreover, the venetoclax-resistant XG5-199R HMCL, in which the expression of both apoptosis executors was lost, remained as sensitive as the parental XG5 cell line to both drugs (Figure 2C) (23). These results showed that auranofin and Prima-1Met induced ROS-dependent, Bax/Bak-independent cell death.
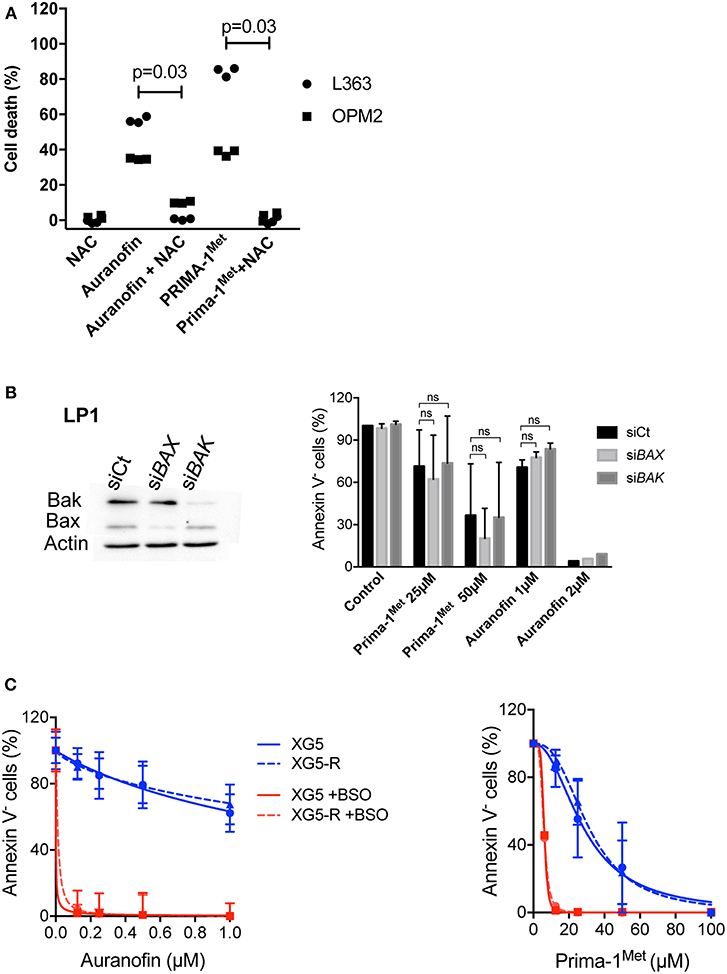
Figure 2. Auranofin and Prima-1Met induced ROS-dependent BAX/BAK-independent cell death. (A) L363 and OPM2 cell lines (500 000 cells/ml) were incubated for 2 days with auranofin (2000 and 1000 nM, respectively) or Prima-1Met (80 and 40 μM, respectively) with or without NAC (0.5 mM). Cell death was assessed by Annexin V staining. The data represent 3 independent experiments. The statistical analyses were performed using the Wilcoxon matched-pairs signed-rank test. (B) LP1 cells were transfected with siRNA against BAX or BAK1 for 2 days and sensitivity to each drug was assessed. The statistical analyses were performed using the two-way ANOVA test with multiple comparisons. (C) Auranofin and Prima-1Met LD50 values were determined in XG5 parental cells and in venetoclax resistant XG5-199R cells. Cells were incubated with increasing concentrations of auranofin or Prima-1Met with or without 500 μM BSO for 2 days, and cell death was assessed by Annexin V staining.
Sensitivity of Primary Myeloma Cells to Prima-1Met and Auranofin Correlated
We assessed the activity of each drug with or without BSO in 10 samples from patients with either multiple myeloma (MM) or plasma cell leukemia (PCL) with or without the deletion of the short arm of chromosome 17, del(17p). Mononuclear cells were incubated with different concentrations of Prima-1Met or auranofin with or without BSO (250 μM). Cell death was determined by the loss of CD138 expression, Figure 3A. Indeed, cell death could not be monitored by Annexin V staining as the loss of myeloma viability was associated with the loss of expression of the plasma cell specific CD138 expression, as illustrated in Figure S2 (10, 19, 20). Both drugs significantly induced myeloma cell death (Figure 3B; Table 1). The median values of cell death induced by Prima-1Met (5 or 10 μM) were 68% (p = 0.0020) and 97% (p = 0.0039), respectively, and the median values of cell death induced by auranofin (50 or 500 nM) were 20% (p = 0.0391) and 97% (p = 0.0039), respectively. Although BSO (250 μM) induced a weak decrease in cell viability (median cell death 7.5%, p = 0.0312, Figure 3B), it significantly synergized with both Prima-1Met (5 μM) and auranofin (50 nM). The cell death median values of the combination of BSO with Prima-1Met or with auranofin vs. the sum of cell death induced by Prima-1Met or auranofin plus BSO were 85.2% vs. 62.8% (n = 7, p = 0.0156, 1.24-fold increase) and 95.6% vs. 12.7% (n = 7, p = 0.0156, 6.41-fold increase), respectively (Figure 3C). The sensitivity of samples to both drugs was not different in samples with or without the deletion of the short arm of chromosome 17, Figure 3D. Auranofin (50 nM) and Prima-1Met (5 μM) displayed correlated activity in myeloma samples (n = 8, r2 = 0.5963, p = 0.0306), Figure 3E.
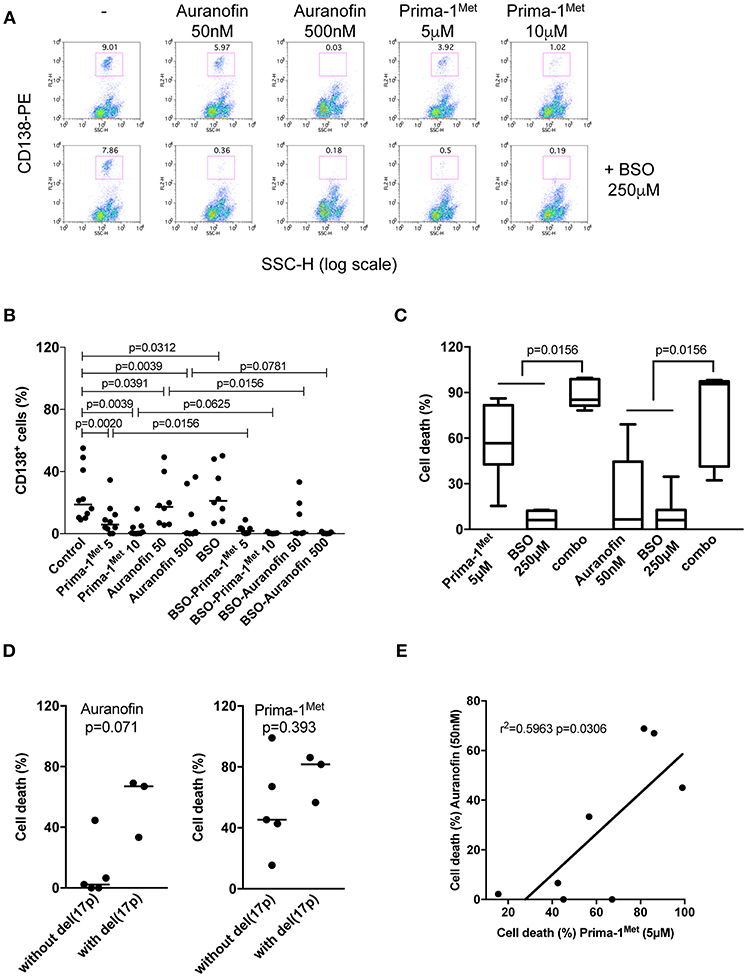
Figure 3. Primary myeloma cells were similarly sensitive to Prima-1Met and to auranofin. (A) Mononuclear cells from patient #4 with myeloma were incubated for 24 hours with or without (-) Prima-1Met (5 or 10 μM) or auranofin (5 or 10 μM) and stained with anti-CD138-PE. Viability was assessed by CD138 expression. (B) Mononuclear cells from 10 samples were incubated for 24 hours with Prima-1Met (5 or 10 μM) or auranofin (5 or 10 μM) with or without BSO (250 μM), and stained with anti-CD138-PE. The statistical analyses were performed using the Wilcoxon matched-pairs signed-rank test. (C) Cell death induced by Prima-1Met (5 μM, samples #3,4,5,6,8,9, and 10), auranofin (50nM, samples #3,4,5,6,7,9, and 10) or BSO (250 μM, samples #3-10) was compared to cell death induced by combinations (combo) of Prima-1Met or auranofin with BSO. The statistical analyses were performed using the Wilcoxon matched-pairs signed-rank test. (D) Cell death induced by auranofin (50 nM) or Prima-1Met (5 μM) was analyzed according to del(17p) status. The statistical analyses were performed using the Mann-Whitney test. (E) Myeloma cell death induced by 5 μM of Prima-1Met was plotted against cell death induced by 50 nM auranofin. Correlation was performed using the Pearson test.
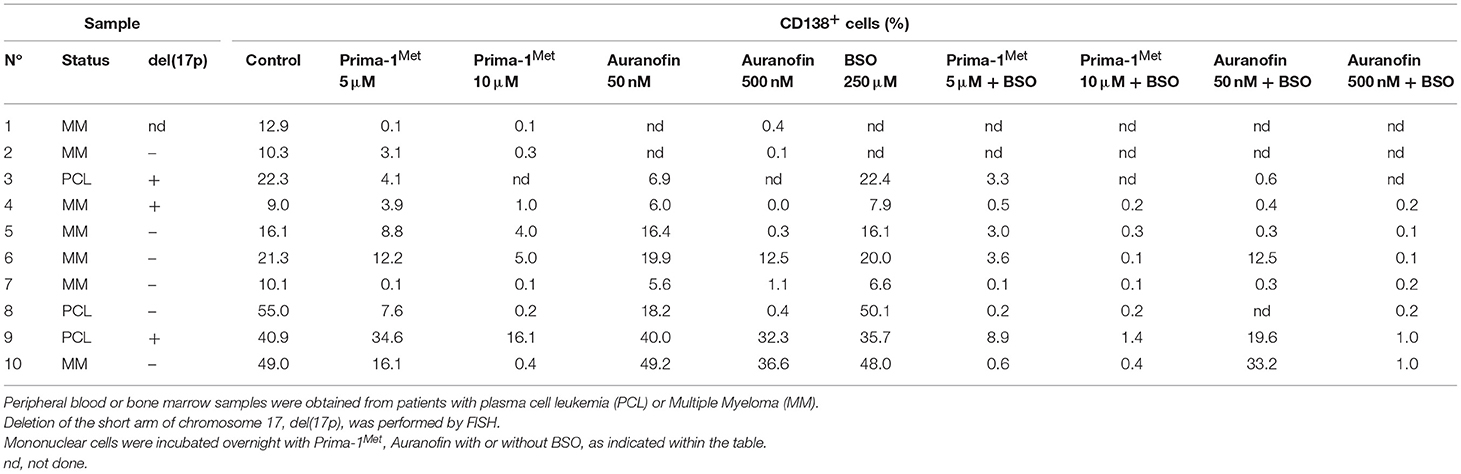
Table 1. Prima-1Met and Auranofin induced cell death in primary myeloma cells.
Conclusion
In this paper, we showed that the sensitivity of myeloma cells to auranofin correlated with sensitivity to Prima-1Met in both HMCLs and in primary myeloma cells from patients with MM or PCL, and that auranofin was more efficient than Prima-1Met. Although p53 competent HMCLs were very sensitive to both drugs, the activity of Prima-1Met or auranofin was not dependent on TP53 status, highlighting that targeting the ROS/GSH pathway was efficient in cells expressing mutant p53 protein or lacking p53 expression. Prima-1Met and auranofin induced Bak/Bax-independent cell death and were efficient in myeloma cells resistant to the Bcl2-specific BH3-mimetic venetoclax. These data showed that auranofin, as Prima-1Met, overcomes resistance to cell death mediated by either p53 deficiency or by mitochondrial loss of priming in myeloma cells. Both drugs appear thus of particular interest for resistance in vivo. Prima-1Met (APR-246) is under clinical evaluation in ovarian cancers with mutated TP53 or in refractory (TP53-mutated) myeloid neoplasms, alone or in combination. Auranofin, which was used to treat patients with arthritis, was recently shown to be able to eliminate side populations and to enhance ibrutinib efficacy in solid cancer cells (22, 24). These recent findings are in favor of assessing auranofin in myeloma patients resistant to current therapies as well as resistant to the Bcl2-specific BH3 mimetic venetoclax.
Data Availability
All datasets generated for this study are included in the manuscript and/or the supplementary files.
Author Contributions
BT designed the study, performed experiments, and participated in the writing of the article. GD performed experiments. CD and MA provided XG5 venetoclax-resistant cells. CP-D designed the study and wrote the article. All authors approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Ligue Régionale Contre le Cancer, DHU Oncogreffe, AF3M, Actions Cancer 44, SIRIC ILIAD (INCa-DGOS-Inserm_12558), and i-Site NexT (ANR-16-IDEX-0007). BT was supported by Inserm and ARC (poste d'accueil).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00128/full#supplementary-material
References
1. Tessoulin B, Eveillard M, Lok A, Chiron D, Moreau P, Amiot M, et al. p53 dysregulation in B-cell malignancies: more than a single gene in the pathway to hell. Blood Rev. (2017) 31:251–9. doi: 10.1016/j.blre.2017.03.001
2. Eskelund CW, Dahl C, Hansen JW, Westman M, Kolstad A, Pedersen LB, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. (2017) 130:1903–10. doi: 10.1182/blood-2017-04-779736
3. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. (2019) 33, 159–70. doi: 10.1038/s41375-018-0196-8
4. Xu J, Qian J, Hu Y, Wang J, Zhou X, Chen H, et al. Heterogeneity of Li-Fraumeni syndrome links to unequal gain-of-function effects of p53 mutations. Sci Rep. (2014) 4:4223. doi: 10.1038/srep04223
5. Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. (2013) 20:898–909. doi: 10.1038/cdd.2013.17
6. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. (2018) 18:89–102. doi: 10.1038/nrc.2017.109
7. Zhang Q, Bergman J, Wiman KG, Bykov VJN. Role of Thiol Reactivity for Targeting Mutant p53. Cell Chem Biol. (2018) 25:1219–30 e3. doi: 10.1016/j.chembiol.2018.06.013
8. Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. (2018) 9:439. doi: 10.1038/s41419-018-0463-7
9. Surget S, Descamps G, Brosseau C, Normant V, Maiga S, Gomez-Bougie P, et al. RITA (Reactivating p53 and Inducing Tumor Apoptosis) is efficient against TP53abnormal myeloma cells independently of the p53 pathway. BMC Cancer. (2014) 14:437. doi: 10.1186/1471-2407-14-437
10. Tessoulin B, Descamps G, Moreau P, Maiga S, Lode L, Godon C, et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. (2014) 124:1626–36. doi: 10.1182/blood-2014-01-548800
11. Wanzel M, Vischedyk JB, Gittler MP, Gremke N, Seiz JR, Hefter M, et al. CRISPR-Cas9-based target validation for p53-reactivating model compounds. Nat Chem Biol. (2016) 12:22–8. doi: 10.1038/nchembio.1965
12. Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. (2009) 15:376–88. doi: 10.1016/j.ccr.2009.03.003
13. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun. (2017) 8:14844. doi: 10.1038/ncomms14844
14. Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, et al. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. (2013) 4:e881. doi: 10.1038/cddis.2013.417
15. Maiga S, Brosseau C, Descamps G, Dousset C, Gomez-Bougie P, Chiron D, et al. A simple flow cytometry-based barcode for routine authentication of multiple myeloma and mantle cell lymphoma cell lines. Cytometry A. (2015) 87:285–8. doi: 10.1002/cyto.a.22643
16. Moreaux J, Klein B, Bataille R, Descamps G, Maiga S, Hose D, et al. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica. (2011) 96:574–82. doi: 10.3324/haematol.2010.033456
17. Tessoulin B, Moreau-Aubry A, Descamps G, Gomez-Bougie P, Maiga S, Gaignard A, et al. Whole-exon sequencing of human myeloma cell lines shows mutations related to myeloma patients at relapse with major hits in the DNA regulation and repair pathways. J Hematol Oncol. (2018) 11:137. doi: 10.1186/s13045-018-0679-0
18. Lode L, Eveillard M, Trichet V, Soussi T, Wuilleme S, Richebourg S, et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica. (2010) 95: 1973–6. doi: 10.3324/haematol.2010.023697
19. Lok A, Descamps G, Tessoulin B, Chiron D, Eveillard M, Godon C, et al. p53 regulates CD46 expression and measles virus infection in myeloma cells. Blood Adv. (2018) 2:3492–505. doi: 10.1182/bloodadvances.2018025106
20. Surget S, Chiron D, Gomez-Bougie P, Descamps G, Menoret E, Bataille R, et al. Cell death via DR5, but not DR4, is regulated by p53 in myeloma cells. Cancer Res. (2012) 72:4562–73. doi: 10.1158/0008-5472.CAN-12-0487
21. Gomez-Bougie P, Maiga S, Tessoulin B, Bourcier J, Bonnet A, Rodriguez MS, et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood. (2018) 132:2656–69. doi: 10.1182/blood-2018-03-836718
22. Hou GX, Liu PP, Zhang S, Yang M, Liao J, Yang J, et al. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis. (2018) 9:89. doi: 10.1038/s41419-017-0159-4
23. Dousset C, Maiga S, Gomez-Bougie P, Le Coq J, Touzeau C, Moreau P, et al. BH3 profiling as a tool to identify acquired resistance to venetoclax in multiple myeloma. Br J Haematol. (2017) 179:684–8. doi: 10.1111/bjh.14251
Keywords: Prima-1Met, APR-246, auranofin, ROS, venetoclax
Citation: Tessoulin B, Descamps G, Dousset C, Amiot M and Pellat-Deceunynck C (2019) Targeting Oxidative Stress With Auranofin or Prima-1Met to Circumvent p53 or Bax/Bak Deficiency in Myeloma Cells. Front. Oncol. 9:128. doi: 10.3389/fonc.2019.00128
Received: 09 November 2018; Accepted: 13 February 2019;
Published: 06 March 2019.
Edited by:
Massimo Libra, Università degli Studi di Catania, ItalyReviewed by:
Himanshi Bhatia, Washington University in St. Louis, United StatesNadim Mahmud, University of Illinois at Chicago, United States
Copyright © 2019 Tessoulin, Descamps, Dousset, Amiot and Pellat-Deceunynck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine Pellat-Deceunynck, Y2F0aGVyaW5lLnBlbGxhdC1kZWNldW55bmNrQHVuaXYtbmFudGVzLmZy