 Kwai Fung Hui
Kwai Fung Hui Stephanie Pei Tung Yiu
Stephanie Pei Tung Yiu Kam Pui Tam1
Kam Pui Tam1 Alan Kwok Shing Chiang
Alan Kwok Shing Chiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 26 February 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00081
This article is part of the Research Topic The Role of Epstein Barr Virus in Lymphoproliferative Disease View all 8 articles
Epstein-Barr virus (EBV) is strongly associated with a spectrum of EBV-associated lymphoproliferative diseases (EBV-LPDs) ranging from post-transplant lymphoproliferative disorder, B cell lymphomas (e.g., endemic Burkitt lymphoma, Hodgkin lymphoma, and diffuse large B cell lymphoma) to NK or T cell lymphoma (e.g., nasal NK/T-cell lymphoma). The virus expresses a number of latent viral proteins which are able to manipulate cell cycle and cell death processes to promote survival of the tumor cells. Several FDA-approved drugs or novel compounds have been shown to induce killing of some of the EBV-LPDs by inhibiting the function of latent viral proteins or activating the viral lytic cycle from latency. Here, we aim to provide an overview on the mechanisms by which EBV employs to drive the pathogenesis of various EBV-LPDs and to maintain the survival of the tumor cells followed by a discussion on the development of viral-targeted strategies based on the understanding of the patho-mechanisms.
Epstein-Barr virus (EBV) is a ubiquitous gamma herpesvirus which establishes life-long persistence in 90% of the human populations (1). This virus is closely associated with nasopharyngeal carcinoma (NPC), a subset of gastric carcinoma and several types of lymphoproliferative diseases (LPDs), such as endemic Burkitt lymphoma (BL), Hodgkin lymphoma (HL), nasal NK/T-cell lymphoma, diffuse large B cell lymphoma (DLBCL), AIDS-associated B-cell lymphoma and post-transplant lymphoproliferative disorder (PTLD) (2, 3). EBV is shown to transform primary B cells and could contribute to the pathogenesis of EBV-LPDs in vitro. In these cancer cells, EBV usually persists in a tightly latent state to escape from the human immune surveillance. Occasionally, the virus can switch from the latent cycle to the lytic cycle in response to various physiologic stimuli. At various pathogenic stages of EBV-LPDs, the virus expresses a number of viral latent or lytic proteins to manipulate cell cycle and cell death processes and promote the survival of the tumor cells. This review will summarize the pathogenic mechanisms which are affected by the EBV latent and lytic proteins for the survival of-EBV-LPDs and discuss on the development of therapeutic strategies targeting the patho-mechanisms associated with EBV-LPDs.
Following EBV infection, the virus is able to establish life-long infection in memory B cells where no EBV protein is expressed (latency 0). In EBV-LPDs, the virus can express four different latency patterns, namely, type I, type II, type III and Wp-restricted latency as characterized by the expression patterns of EBV latent proteins. In type I latency, which is observed in the majority of endemic BL, the expression of viral genes is greatly restricted with only EBV nuclear antigen (EBNA)-1, EBV-encoded small RNAs (EBERs) and BamHI-A rightward transcripts (BARTs) are expressed. The transcription of EBNA-1 is initiated at the BamHI Q promoter (Qp) (4). In addition to type I latency, Wp-restricted latency could also be detected in ~15% of the endemic BL (5). In this latency, EBNA-LP, EBNA-1, EBNA-3A, -3B, and -3C are transcribed from the BamHI W promoter (Wp) (6). In type II latency, which is observed in HL, nasal NK/T-cell lymphoma and DLBCL, more latent genes including EBNA-1, EBNA-LP, latent membrane protein (LMP)-1, -2A, and -2B, EBERs and BARTs are expressed. Type III latency is detected in AIDS-associated B-cell lymphoma, PTLD and lymphoblastoid cell line (LCL), an in vitro model of EBV-LPDs. This is the most immunogenic form of latency in which a full set of latent genes including EBNA-1, -2, -LP, -3A, -3B, -3C, LMP-1, -2A, -2B, EBERs and BARTs are expressed (6, 7). Either BamHI C promoter (Cp) or Wp is activated to drive the expression of the EBV latent genes in this latency (Figure 1).
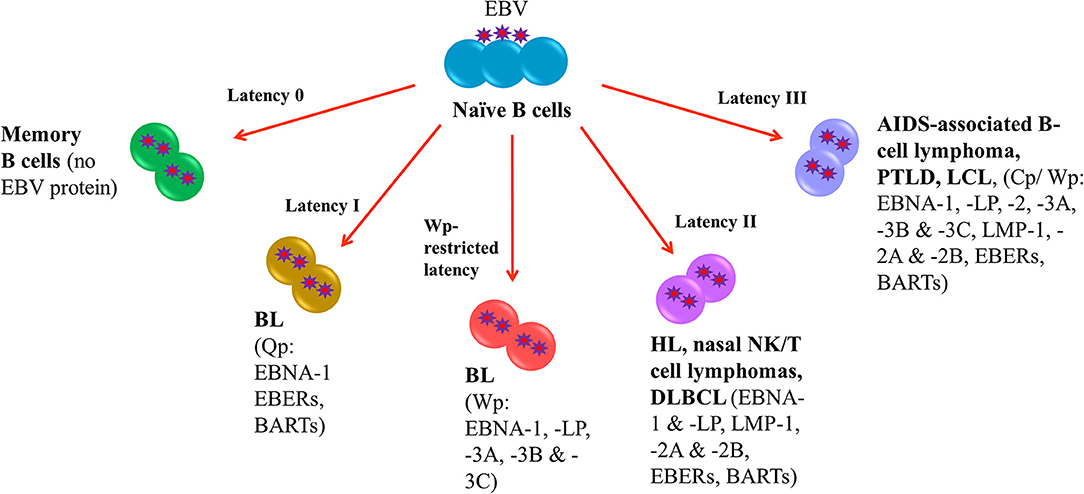
Figure 1. EBV latency in EBV-LPDs. No EBV protein is expressed in Latency 0. Only EBNA-1, EBERs, and BARTs are expressed in Latency I which is associated with endemic BL. The transcription of EBNA-1 is initiated at the BamHI Q promoter. 15% of endemic BL is found to be Wp-restricted latency in which EBNA-LP, EBNA-1, EBNA-3A, -3B, and -3C are transcribed from the BamHI W promoter. HL, nasal NK/T-cell lymphoma and DLBCL are detected in type II latency that EBNA-1, EBNA-LP, latent membrane protein (LMP)-1, -2A, and -2B, EBERs and BARTs are expressed. AIDS-associated B-cell lymphoma, PTLD and lymphoblastoid cell line (LCL), an in vitro model of EBV-LPDs are observed in type III latency. All EBV nuclear antigens (EBNA-1, -2, -LP, -3A, -3B, and -3C), latent membrane proteins (LMP-1, -2A, and -2B), EBERs and BARTs are expressed.
EBV lytic cycle reactivation has been comprehensively studied in the Akata BL cell line, in which the lytic cycle of EBV can be efficiently induced by cross-linking the cell surface receptor with anti-human IgG antibody (8). This model provides an effective way to study the possible physiological mechanisms of viral lytic reactivation in EBV-LPDs. EBV lytic cycle is initiated with the expression of two immediate early proteins, namely Zta and Rta (9–11). Expression of these two immediate early proteins activates the expression of one another and subsequently triggers the expression of a panel of early lytic proteins (e.g., BMRF1, BALF1, BHRF1, etc.,) (3, 12). EBV immediate early and early lytic proteins initiate viral DNA replication and later, the expression of late lytic proteins (e.g., VCA-p18, gp350/220, etc.,) (3). Anti-viral drugs e.g., phosphonoformic acid, which suppress EBV DNA replication can also inhibit expression of EBV late lytic proteins, suggesting that EBV DNA replication is an upstream process that regulates late lytic protein expression (3, 13–15). In case of a complete lytic cycle, the viral DNA is replicated as large head-to-tail molecules which are then cleaved into pieces and packaged into viral progenies for dissemination to the neighboring cells (16). More than 70 EBV lytic genes, which are important for viral replication, dissemination and infection, are expressed during the EBV lytic cycle (Figure 2).
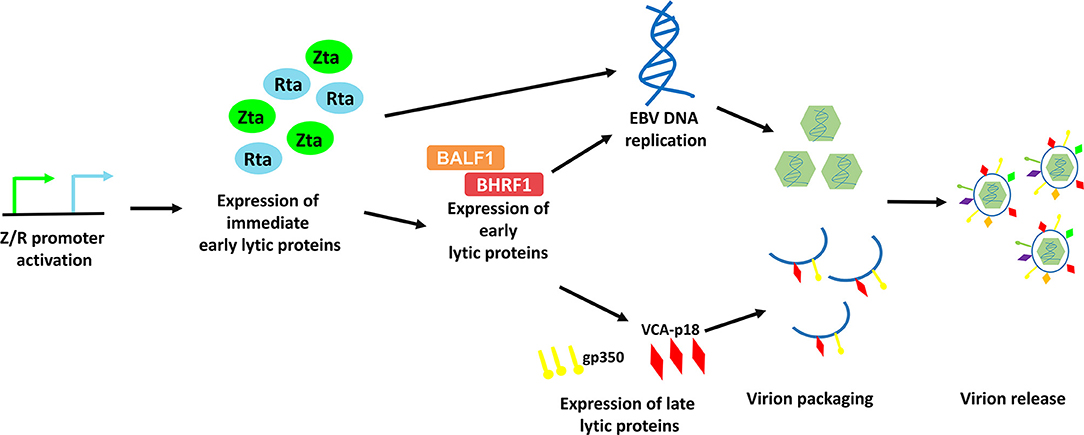
Figure 2. Schematic diagram representing the sequential events occur during EBV lytic reactivation. EBV Z/R promoters are activated upon diverse stimulants e.g., B-cell receptor crosslinking, chemical inductions and cellular stresses, resulting in the expression of immediate early lytic proteins, Zta and Rta. These key drivers of EBV lytic reactivation subsequently induce EBV viral DNA replication and the expression of an array of viral lytic proteins including early lytic proteins e.g., BALF1 and BHRF1 and late lytic proteins e.g., gp350 and VCA-p18. Viral DNA is then being packaged with the help from structural proteins and is assembled into mature virion. Finally, EBV is released via exocytosis.
Both innate and adaptive immunity are responsible for the control of EBV. The phagocytes and natural killer (NK) cells in the innate immunity are responsible for the control of immediate B cell infection and virus replication. The CD4+ and CD8+ T cells in the adaptive immunity are capable of producing interferon (IFN)-γ and other functional cytokines to control the proliferation of EBV-infected B cells during long-term infection. We and others have demonstrated that the presence of EBV-specific polyfunctional T cells (PFCs), which could produce multiple cytokines [e.g., IFN-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-2] simultaneously and readily degranulating, in long-term EBV carriers (17, 18). A clear increase in CD4+ and CD8+ PFC responses against EBV antigens is also demonstrated in infectious mononucleosis (IM) patients, correlating with increased cytotoxicity of T cells against autologous LCLs (19). NK cells play a complementary role with T cells in controlling tumor growths and viral infections. Azzi et al. have demonstrated that a subset of early-differentiated (CD56dimNKG2A+KIR−) NK cells play a more important role than the terminally differentiated (CD56dimNKG2A−KIR+) NK cells in the control of EBV infection in acute IM patients (20). In addition, Hatton et al. have also shown that a NKG2A-expressing subset of NK cells could effectively kill EBV-transformed autologous LCLs (21). We postulate that impairment of EBV-specific PFCs and NKG2A+ NK cells may contribute to the development of EBV-LPDs (Figure 3).
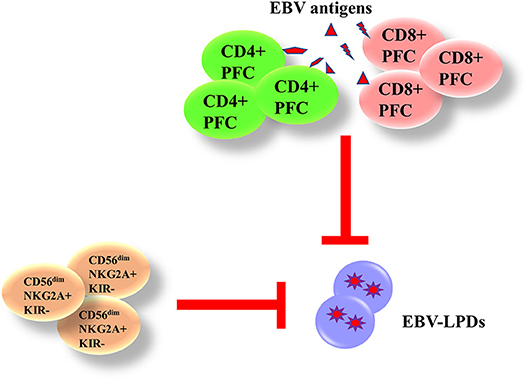
Figure 3. Immunity against EBV-LPDs. (IFN)-γ and other functional cytokines [(TNF)-α and IL-2] are produced from EBV-specific polyfunctional T cells (PFCs) to control the proliferation of EBV-infected B cell during long-term infection. There are increase responses of CD4+ and CD8+ PFCs in infectious mononucleosis (IM) patients. (CD56dimNKG2A+KIR−) NK cells also control EBV infection in acute IM patients and kill LCLs.
EBV effectively infects normal B cells and transforms them into proliferating LCLs in vitro. EBV latent proteins are shown to contribute to the pathogenesis of EBV-LPDs. Besides, there is increasing evidence that shows that EBV lytic proteins can also promote the pathogenesis of EBV-associated diseases. The viral latent and lytic proteins can maintain the proliferation and survival of EBV-positive cancer cells via deregulating the cellular mechanisms that regulate cell cycle, apoptosis and immune recognition of the host cells.
EBNA-3A and -3C proteins are shown to manipulate the cell cycle of the host cells to facilitate successful transformation of B cells and to maintain the proliferation of the transformed cells. The first evidence of the cell cycle regulatory property of EBNA-3 proteins is demonstrated by Allday et al. who found that mutation of EBNA-3C can lead to G1 cell cycle arrest in B cells (22). It is further shown that EBNA-3C can directly interact with cyclin A to stimulate the activity of cyclin-dependent kinase (CDK)-2 and subsequently facilitates LCLs to pass through the retinoblastoma protein (pRb) cell cycle checkpoint (23). EBNA-3C can also stabilize cyclin A and promote the proteasomal degradation of p27KIP1, hence assists the EBV-infected cells to progress to M phase (24, 25). EBNA-3C also mediates the ubiquitin-proteasome degradation of pRb by recruiting SCFSkp2 E3-ubiquitin ligase (26, 27). Consequently, fewer pRb can interact with the transcription factors of E2F family which then suppress the transcription of E2F-dependent cyclin/CDK complexes. More E2F-dependent complexes, including the cyclin-D1/CDK-4/-6 and cyclin-A/-E/CDK-2, will be expressed to allow the cells to enter G1 phase from G0 phase and enter S phase from G1 phase, respectively (27). Furthermore, EBNA-3C also stabilizes Pim-1 protein to promote the phosphorylation and subsequent proteasomal degradation of p21WAF1 for the cells to enter S phase from G1 phase (28). Additionally, EBNA-3C can modulate Skp2 to mediate proteasomal degradation of p27KIP1 which subsequently free the cyclin-A/CDK-2 complex for the cells to enter S phase (29). EBNA-3C also promotes the proteasomal degradation of Bcl-6 which subsequently releases cyclin-D1 for the transition of G1 to S phase (30). Besides, EBNA-3A and -3C are found to co-operate in epigenetic repression of p14ARF and p16INK4a, facilitating the transformation and proliferation of EBV-LPDs (31–33). EBNA-3A and -3C can also facilitate the EBV-LPDs to bypass the G2/M checkpoint regulation upon stimulation by various cytotoxic stresses (34–38) (Figure 4).
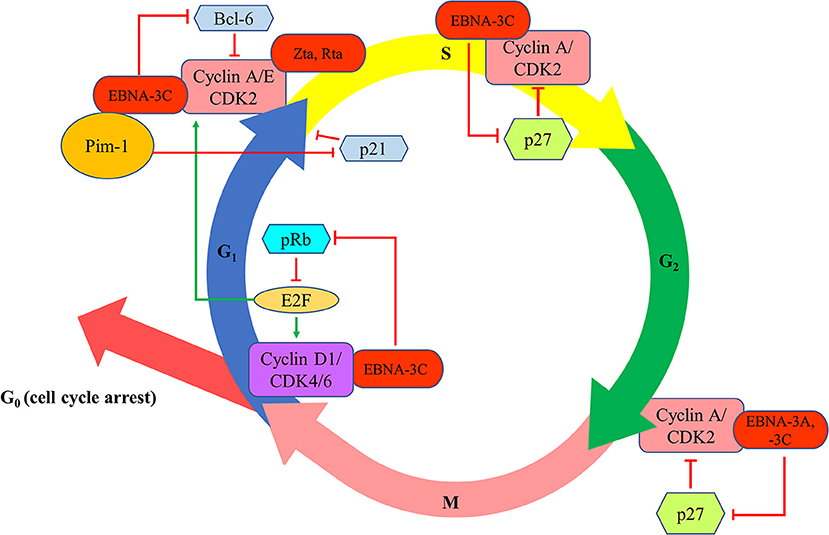
Figure 4. Effects of EBV latent and lytic proteins on the regulation of cell cycle. EBNA-3C interacts with cyclin A/CDK2 and promotes the proteasomal degradation of p27KIP1 to assist the EBV-infected cells to progress to enter S phase and M phase. EBNA-3C mediates the ubiquitin-proteasome degradation of pRb, increasing the transcription of E2F-dependent cyclin/CDK complexes (cyclin-D1/CDK-4/-6 and cyclin-A/-E/CDK-2), to allow the cells to enter G1 phase from G0 phase and enter S phase from G1 phase, respectively. EBNA-3C stabilizes Pim-1 protein to promote the phosphorylation and subsequent proteasomal degradation of p21WAF1 for the cells to enter S phase from G1 phase. EBNA-3C also promotes the proteasomal degradation of Bcl-6 which subsequently releases cyclin-D1 for the transition of G1 to S phase. EBNA-3A and -3C co-operate in epigenetic repression of p14ARF and p16INK4a to facilitate the transformation and proliferation of EBV-LPDs through bypassing the G2/M checkpoint regulation upon stimulation by various cytotoxic stresses.
Reactivation of EBV lytic cycle is also shown to disrupt various cell cycle checkpoints in EBV-infected cells. Zta and Rta can promote the transition from G1 to S phase in BL cells via a mechanism related to the modulation of p53 and p21WAF1 (39). On the other hand, overexpression of Zta protein can induce G1 cell cycle arrest in EBV-positive cells via the interaction with CCAAT/enhancer binding proteins (C/EBP), which subsequently lead to the activation of p53 andthe accumulation of p21WAF1 and p27KIP1 (40). EBV lytic proteins can also activate the cyclin-E/CDK-2 complex for the entry of S phase in hopeto provide an environment suitable for viral DNA replication (41) (Figure 4). Interestingly, treatment of CDK inhibitors, such as purvalanol-A and roscovitine, which inhibit the transition from G1 to S phase of the cell cycle, can block lytic replication of EBV (42). Reactivation of EBV lytic cycle can also lead to G2/M arrest of the host cells. For instance, expression of Zta is shown to induce both G2/M arrest and mitotic block in HeLa cells (43); whilst treatment with 5-azacytidine (5-AZA) can lead to a G2/M phase arrest in Zta-expressing Rael cells (44). We have also reported that a histone deacetylase (HDAC) inhibitor, suberoylanilide hydroxamic acid (SAHA), can reactivate EBV lytic cycle and mediates a pronounced G2/M arrest in EBV-positive epithelial cells (11). We further showed that the induction of G2/M arrest is possibly due to the upregulation of p21WAF1 and downregulation of cycli-D1, p-Rb, cyclin-B1 and p-CDK-1 (45).
EBNA-1 can interact with the herpesvirus associated ubiquitin-specific protease to destabilize and degrade p53 to inhibit apoptosis (46). EBNA-2 antagonizes TGF-β-mediated growth arrest in LCLs (47). LMP-1 also upregulates Bcl-2 and promotes the growth of BL through the activation of NF-kB signaling pathway (48). EBNA-3A upregulates Hsp70 chaperones to suppress apoptosis in exposure to cytotoxic agents (49). EBNA-3C can suppress the p53-mediated cell death by inhibiting the transcription of p53and promoting its degradation (50–52). EBNA-3C also hinders the E2F1-mediated apoptosis induced by DNA damage response throughinhibiting the DNA binding activity of E2F1 and promoting its proteolysis (53). Furthermore, EBNA-3C prevents the proteosomal degradation of MDM2 and to recruit it to initiate the degradation of p53 in order to promote the survival of EBV-LPDs (54–56). EBNA-3C also interacts with Bcl-6 and releases Bcl-2 to suppress apoptosis for lymphomagenesis (30). EBNA-3A and -3C can co-operate to repress the expression of the tumor suppressor gene, p16INK4a, to promote cell proliferation and prevent cell death (31–33). Moreover, they also epigenetically repress the Bim promoter which eventually suppresses the Bim-mediated intrinsic pathway of apoptosis (57, 58) (Figure 5).
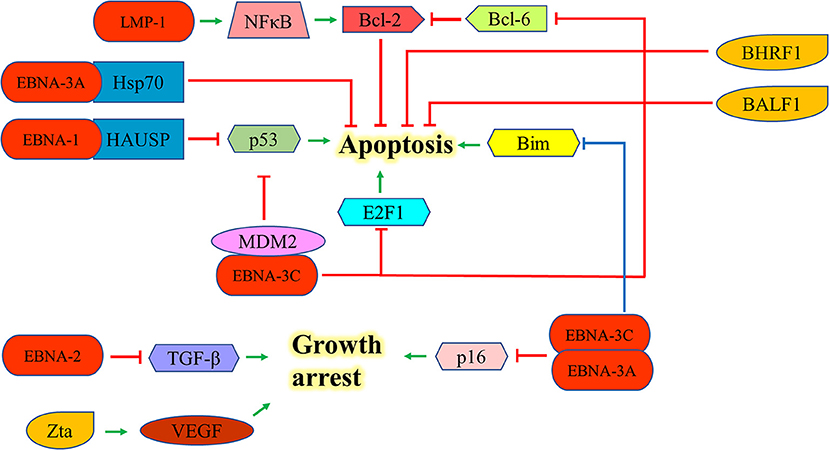
Figure 5. Effects of EBV latent and lytic proteins on inhibition of apoptosis. EBNA-1 interacts with the HAUSP to destabilize and degrade p53. EBNA-2 antagonizes TGF-β-mediated growth arrest in LCLs. LMP-1 upregulates Bcl-2 and promotes the growth of BL through the activation of NF-kB signaling pathway. EBNA-3A upregulates Hsp70 chaperones to suppress the apoptosis in exposure to cytotoxic agents. EBNA-3C can suppress p53-dependent apoptosis through repressing the transcription of p53 and promoting its degradation. EBNA-3C also hinders the E2F1-mediated apoptosis induced by DNA damage response through inhibiting the DNA binding activity of E2F1 and promoting its proteolysis. EBNA-3C also interacts with Bcl-6 and releases the Bcl-2 to suppress apoptosis. EBNA-3A and -3C can co-operate to repress the expression of p16INK4a and Bim to promote cell proliferation. Zta can induce the expression of vascular endothelial growth factor (VEGF) to promote the growth of LCL.
Expression of EBV lytic proteins also plays a role in pathogenesis of EBV-positive cancers by inhibiting apoptotic cell death. Zta can induce the expression of vascular endothelial growth factor (VEGF) to promote the growth of LCL (59). The early lytic genes, BHRF1 and BALF1, which encode Bcl-2 homologs, can inhibit apoptosis of EBV-associated lymphoid cancers and promote the survival of cancer cells during EBV lytic replication (60, 61). Exogenous expression of BHRF1 protein was shown to protect BJAB cells from apoptotic cell death (60); whilst expression of BALF1 protein inhibits Fas ligand-induced apoptosis in HeLa cells (61) (Figure 5). Several EBV lytic genes including BALF3, BARF1, BGLF4, and BGLF5 which induces DNA damage response and genomic instability could also contribute to the carcinogenesis of EBV-associated cancers (62–65).
EBV has developed multiple strategies to escape from the human immune surveillance. In EBV-infected B cells, the presence of glycine-alanine repeats of EBNA-1 rendered it not being able to be processed and presented to CD8+ T cells via class I MHC (66). Zta can induce the secretion of IL-6, -8, -10, and -13 which function to promote the tumorigenesis of different EBV-associated cancers (67). An early lytic protein, BGLF4 protein kinase, can suppress the host innate immune responses and facilitates the production of viral progenies in NPC through inhibiting the interferon regulatory factor 3 and STAT1 (68). Another EBV lytic gene, BCRF1, which encodes for a homolog of cellular IL-10, suppresses INF-γ synthesis from human peripheral blood mononuclear cells, thus allowing the tumor cells to evade from the host immune surveillance (69). BCRF1 can also function as a paracrine growth factor to enhance the transformation of B cells and promote the growth of EBV-LPDs (70). A late EBV lytic gene, BDLF3, can promote the degradation of MHC class I and II molecules, thereby impairing the immune recognition by EBV-specific CD4+ and CD8+ T cells (71) (Figure 6).
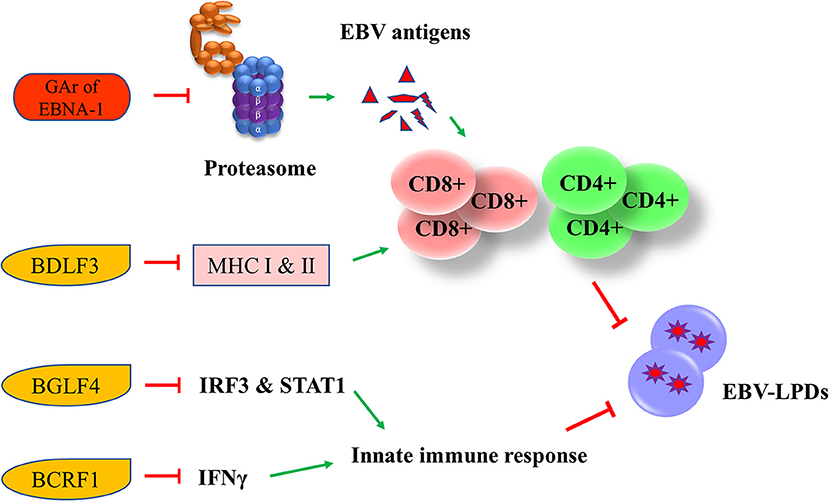
Figure 6. Effects of EBV latent and lytic proteins on immune evasion. Glycine-alanine repeats (GAr) of EBNA-1 render it not be processed and presented to CD8+ T cells via the class I MHC. BDLF3, can promote the degradation of MHC class I and II molecules, impairing the immune recognition by EBV-specific CD4+ and CD8+ T cells. BGLF4 can suppress the host innate immune through the inhibition of interferon regulatory factor 3 (IRF3) and STAT1. BCRF1 suppresses INF-γ synthesis from human peripheral blood mononuclear cells, thus allowing the tumor cells to evade from the host immune surveillance.
EBV persists in a tightly latent state in every tumor cell in EBV-LPDs and therefore, could be served as an excellent target for therapeutic treatments. Various viral-targeted therapies targeting the expression of EBV latent and lytic proteins for the treatment of EBV-LPDs are discussed below.
EBV-based gene therapies have been developed to deliver cytotoxic proteins or chemosensitizers to EBV-infected malignancies. Franken et al. have shown that a targeted expression of thymidine kinase in BL using an EBNA2-responsive Cp could selectively enhance the sensitivity of EBNA2-expressing cells to an anti-viral drug, ganciclovir, in vitro, and in vivo (72). An adenovirus vector with the transgene expression regulated by the origin of replication of EBV can precisely deliver p53 into EBV-positive cancer cells and induced apoptosis to the cells (73). The use of such replication-deficient adenovirus vector for EBV-targeted gene therapy is further demonstrated to be feasible in vivo (74).
Several laboratories have demonstrated that the adoptive immunotherapy which employs an ex vivo expanded virus-specific cytotoxic T lymphocytes (CTLs) is a safe and effective treatment strategy for EBV-associated malignancies including HL, NK/T-cell lymphoma, PTLD and NPC (75, 76). Briefly, CTLs which target EBV latent proteins are isolated from patients followed by activation and expansion in vitro and then infused back into the patients (75). Recently, one of such autologous T cell therapies, CMD-003, has been granted fast track designation by the FDA for treating relapsed/refractory lymphoma and PTLD. Since the upregulation of PD-L1 is observed in various EBV-LPDs, another potential immunotherapy for EBV-LPDs could be by blocking the PD1 and PD-L1 pathways (77–79). Development of novel therapeutic strategy using the combination of autologous T cell therapy and PD-L1 inhibitor might potentially yield synergistic effect on the treatment of EBV-LPDs. Other immunological approaches, such as the development of vaccines and specific monoclonal antibodies against EBV are also under investigations (80). For instance, immunization with polyvalent EBV virus-like particle (VLP) vaccines (gH/gL-EBNA-1 and gB-LMP2) without adjuvant is shown to induce high neutralizing antibody titres against EBV in vitro and in vivo (81).
Reactivation of EBV lytic cycle is another potential strategy that exploits the presence of EBV genome in tumor cells. Several reports show that reactivation of viral lytic cycle can directly induce apoptotic cell death in various EBV-infected cell lines (82–85). Kawanishi et al. have demonstrated that reactivation of EBV lytic cycle by tetradecanoyl phorbol acetate (TPA) can result in fragmentation of chromosomal DNA in Raji BL cells (82). Rta can also induce irreversible G1 arrest, cellular senescence and apoptosis in different EBV-positive cancer cell lines (86). Some of the carcinogenic lytic proteins, such as Zta and BGLF5, were reported to have contradictory roles in mediating cancer cell death. For instance, expression of Zta can phosphorylate p53 to mediate a direct killing of EBV-positive cells (43). The EBV early lytic gene, BGLF5, which encodes for EBV alkaline exonuclease, possesses a shutoff activity during lytic cycle reactivation could potentially induce apoptosis (87). We have shown that lytic cycle reactivation by HDAC inhibitors, including trichostatin A, sodium butyrate, valproic acid and SAHA, can lead to enhanced apoptosis in NPC and gastric carcinoma cells (11, 88). The induction of apoptosis is mediated through the upregulation of p21WAF1 and cell cycle arrest at G2/M phase (45).
Oncolytic therapy which intentionally reactivates lytic cycle of EBV to confer susceptibility of EBV-positive cells to the treatment with antiviral drugs could be a potential therapeutic strategy against EBV-LPD and other EBV-associated diseases. Ganciclovir (GCV), which is a nucleoside-type antiviral drug, is shown to mediate enhanced killing of EBV-positive cancer cells when co-administrated with lytic inducers. This combinatorial strategy relies on the expression of BGLF4, a viral protein kinase expressed during EBV lytic reactivation, to convert GCV into its cytotoxic form (89). The cytotoxic GCV is then incorporated into both viral and cellular DNA of the induced cells and neighboring cells, causing a bystander killing of various EBV-associated malignancies through the induction of premature DNA strand termination and apoptosis in the host cells (83, 90, 91). Alternatively, Fu et al. have demonstrated the possibility of directing [125I]2′-fluoro-2′-deoxy-beta-D-5-iodouracilarabinofuranoside ([125I]FIAU) to lytically-induced EBV-positive BL cells (92). The lytic induction therapy which employs valporic acid and gemcitabine as lytic inducers has been recently shown to achieve clinical responses in some NPC patients (93, 94). However, some cell populations were refractory to lytic cycle reactivation upon treatment with any of the available lytic inducers (45). Investigating on the lytic reactivation mechanisms by these lytic inducers is essential for developing an effective oncolytic therapy (Figure 7).
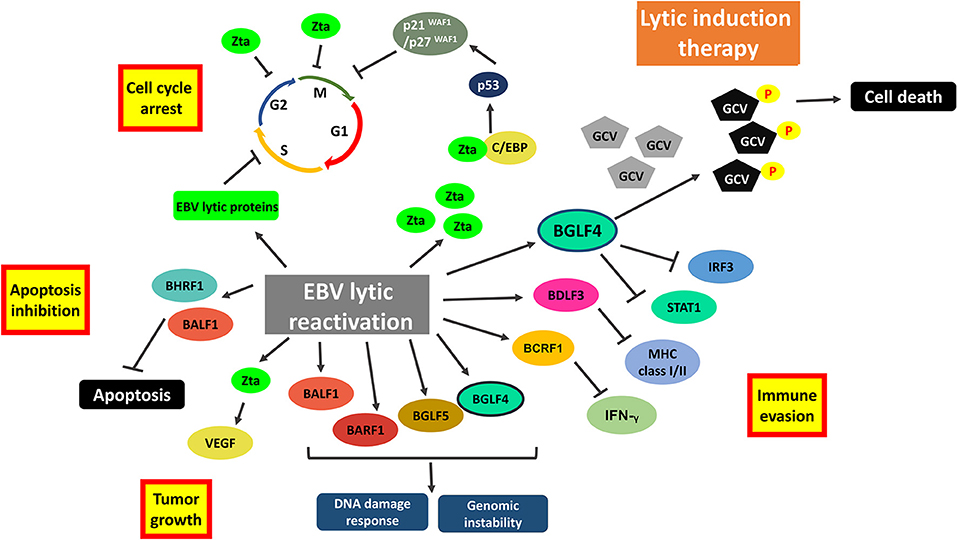
Figure 7. Cellular events associated with EBV lytic reactivation and the rationale of lytic induction therapy. A diverse array of EBV lytic proteins is being expressed during lytic cycle reactivation. Subsequent occurrence of various cellular events include cell cycle arrest, inhibition of apoptosis, tumorigenesis and immune evasion. Expression of viral protein kinase BGLF4 converts antiviral drug e.g., ganciclovir (GCV) from a prodrug to its cytotoxic form, shaping the basis of lytic induction therapy.
EBV latent proteins, particularly EBNA-1, -3A, -3C, LMP-1, and -2A are shown to be important for the pathogenesis of lymphomas. EBNA-1, which is expressed in all types of EBV-positive cancer cells, represents a specific target for the treatment of EBV-LPDs. A number of inhibitors and vaccines have been developed to target EBNA-1 directly in EBV-positive cancer cells (95–97). MDM2 inhibitors e.g., Nutlin-3a, SAR405838, and JNJ-26854165, or c-Abl kinase inhibitor e.g., Nilotinib, can suppress the growth of lymphomas in EμEBNA-1 transgenic mice via the EBNA-1/MDM2/E2F1 pathway (98). LMP-1 is a CD40 homolog that can activate the NF-κB signaling pathway and exert strong oncogenicity in EBV-LPDs and other EBV-associated malignancies (99, 100). It has been reported that bortezomib, which inhibits the NK-κB signaling, can induce apoptosis in EBV-LPDs and EBV-associated epithelial cancer cells (101, 102). LMP-2A is an EBV-encoded membrane protein which acts as a constitutively active B cell receptor through interacting with Lyn kinase to facilitate B cell transformation and proliferation. Dasatinib is found to inhibit splenomegaly and lymphomagenesis in LMP-2A/MYC double transgenic mice via theinhibition of Lyn (103). Moreover, rapamycin significantly reduces tumor growth, splenomegaly and metastasis of B cell lymphoma through theinhibition of the Lyn-activated mTOR pathway (104). LMP-2A also drives lymphomagenesis through enhancing c-Myc expression which subsequently increases the expression of CDK regulatory subunit 1 (Cks1), a cofactor of the SCFSkp2 ubiquitin-ligase complex, leading to the ubiquitination and proteasomal degradation of p27KIP1. Proteasome inhibitors, such as MG-132 can reduce lymphomagenesis by increasing the level of p27KIP1 (105). EBNA-3 proteins, particularly EBNA-3A and -3C, provide important survival advantages to the EBV-infected cells. For instance, BL cells with type III latency are more resistant to the killing by cytotoxic agents, such as taxol and nocodazole when compare to BL cells with type I latency (106). BL cells with Wp-restricted latency which also express EBNA-3 proteins are more resistant to the killing by anti-IgM or ionomycin when compared to BL cells with type I latency (107). Interestingly, we found that HDAC inhibitors and proteasome inhibitors can act synergistically to induce the up-regulation of p21WAF1 and mediate enhanced killing to the BL cells with Wp-restricted or type III latency but not to those with type I latency, suggesting the involvement of EBNA-3 proteins in the cell death mechanism (101). We further tested the mechanism of killing of BL cell lines infected with EBNA3A, -3B, or -3C knockout EBV or with their revertant EBV and found that EBNA3C-expressing cells can bypass the G2/M checkpoint arrest induced by the combination of HDAC and proteasome inhibitors and subsequently become more susceptible to the induction of apoptosis (108). Such enhanced killing is due to the up-regulation of p21WAF1 and down-regulation of p-cdc25c in EBNA3C-expressing cells (108) (Figure 8).
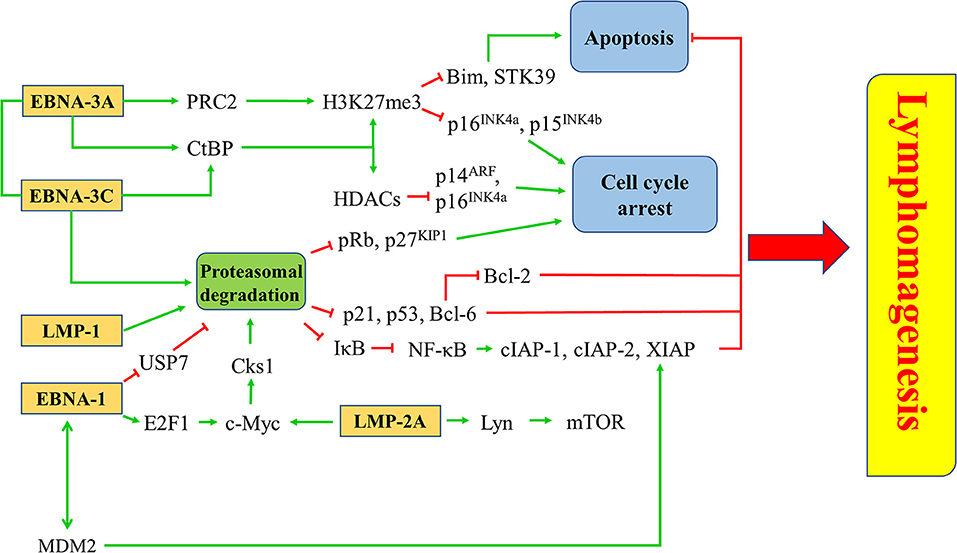
Figure 8. Targeted survival pathways in EBV latency. Several EBV protein-induced survival pathways, such as inhibition of apoptosis and cell cycle arrest through epigenetic repression and/or proteasomal degradation of tumor suppressors for lymphomagenesis can be targeted by novel drugs or drug combinations.
Several FDA-approved drugs and novel compounds are shown to have effects on either inducing EBV lytic cycle or targeting the survival mechanisms delivered by the EBV latent proteins in EBV-positive cancer cells. These compounds work in diverse mechanisms including inhibition of HDAC and proteasome, activation of MAPK pathways, induction of various cellular stress responses (e.g., ER stress, DNA damage response, hypoxia and oxidative stress), autophagy, cell cycle arrest and apoptosis (Table 1, Figures 9, 10).
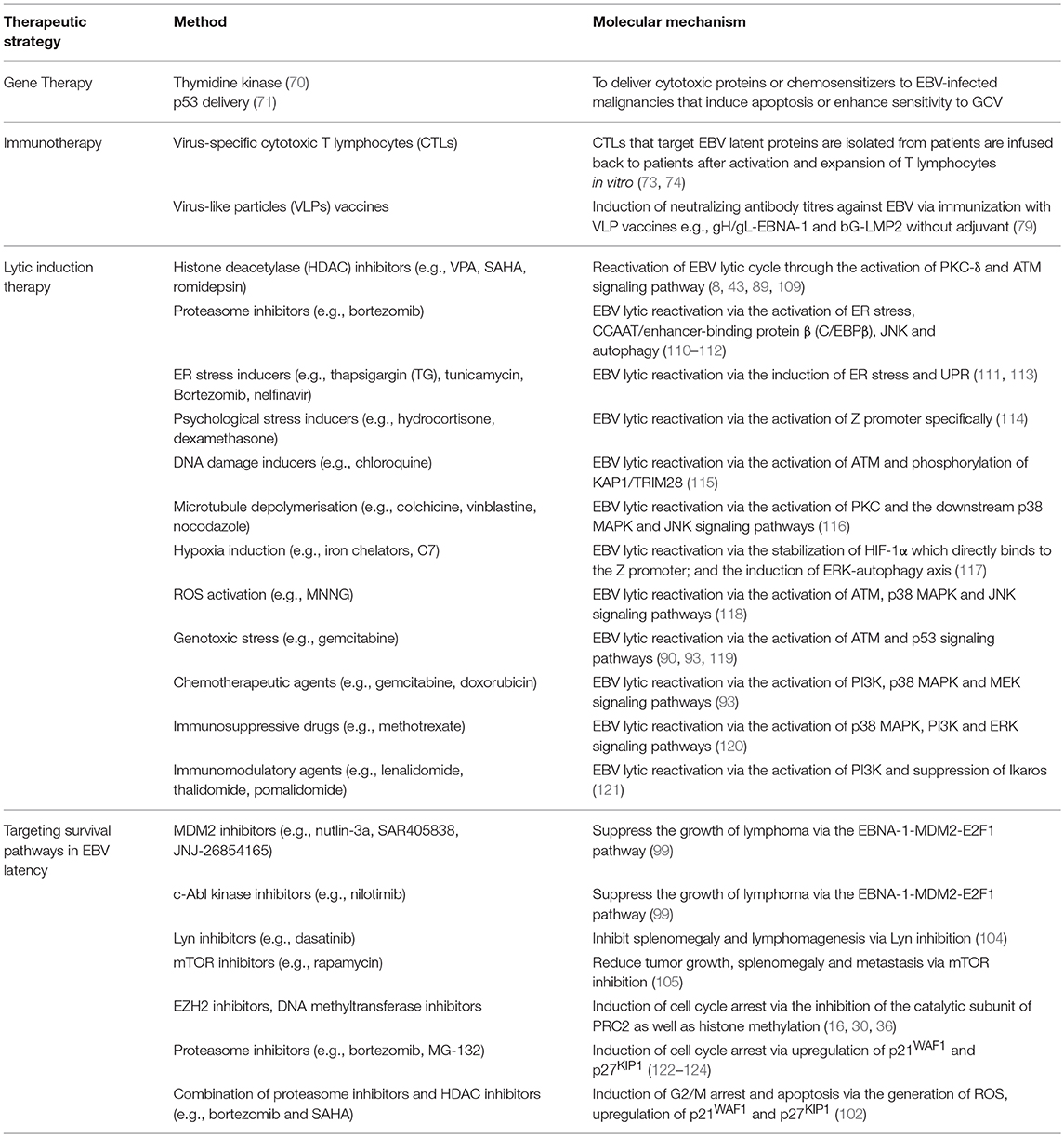
Table 1. Summary on the therapeutic strategies and their corresponding molecular mechanisms against EBV-associated LPDs.
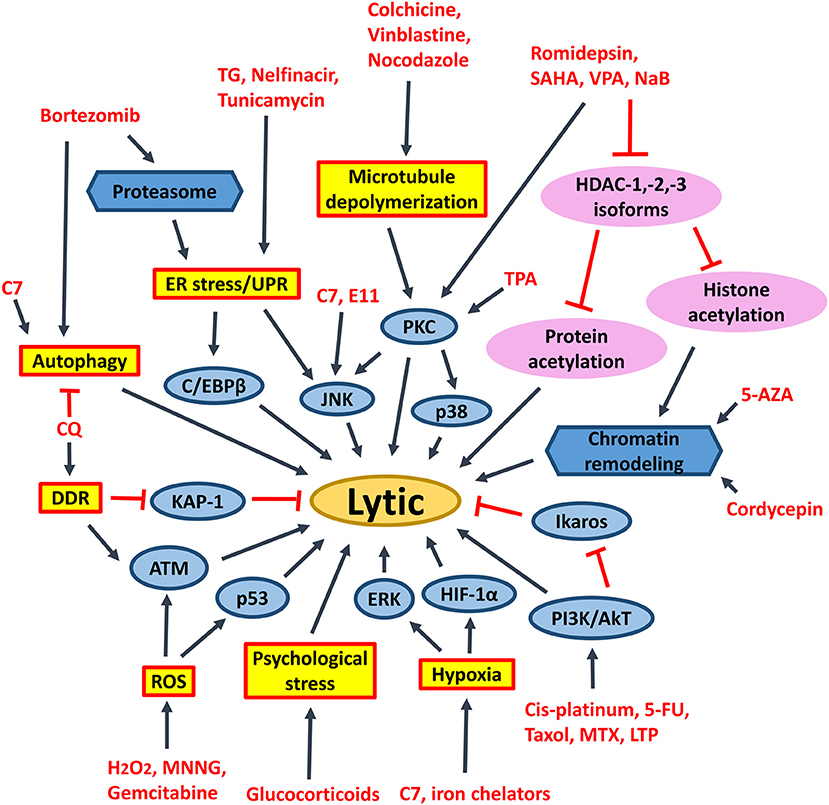
Figure 9. Signaling pathways activated by different chemical lytic inducers for EBV lytic reactivation. EBV lytic reactivation can be achieved through the activation of different cellular signaling pathways e.g., PKC, p38/MAPK, ERK1/2, JNK, PI3K/AKT, DDR, ROS, hypoxia, ATM signaling pathways as well as inhibition of Ikaros and chromatin remodeling. 5-FU, fluorouracil; MTX, methotrexate; 5-AZA, 5-azacytidine; SAHA, suberoylanilide hydroxamic acid; TPA, 12-O-tetradecanoylphorbol-13-acetate; CQ, chloroquine; PKC, protein kinase C; p38/MAPK, P38 mitogen-activated protein kinases; ERK1/2, extracellular signal-regulated protein kinases 1 and 2; JNK, c-Jun N-terminal kinase; C/EBP, CCAAT/enhancer binding proteins; PI3K/AKT, phosphatidylinositol 3-kinase/AKT; TG, thapsigargin; MNNG, methylnitronitrosoguanidine; DDR, DNA damage response; ROS, reactive oxygen species; ER stress, endoplasmic reticulum stress; UPR, unfolded protein response.
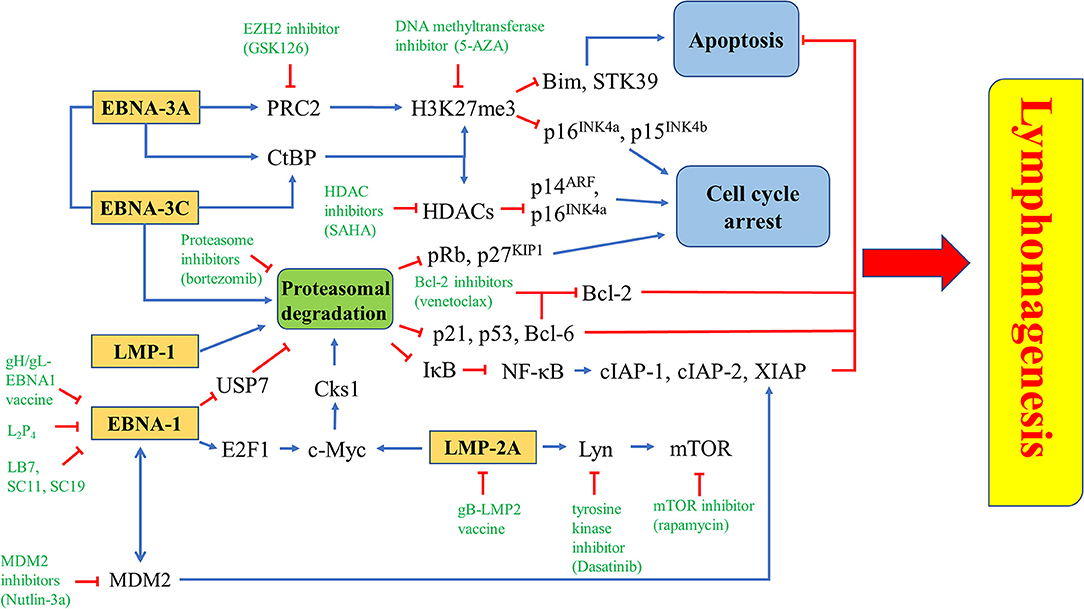
Figure 10. Novel drugs or drug combination targeting EBV latency. EZH2 inhibitior (GSK126), DNA methyltransferase inhibitor (5-AZA) or HDAC inhibitor (SAHA) is used to inhibit the epigenetic repression of Bim, STK39, p14ARF, p15INK4b, and p16INK4a triggered by EBNA-3A and/or EBNA-3C. Proteasome inhibitor (bortezomib) can inhibit proteasomal degradation of tumor suppressors induced by EBNA-1, EBNA-3A, EBNA-3C, LMP-1, or LMP-2A. There are some EBNA-1 inhibitors, including gH/gL-EBNA1 vaccine, L2P4, LB7, SC11 and SC19, while gB-LMP2 vaccine is found to inhibit LMP-2A. Several downstream molecules, such as Lyn, mTOR, and MDM2 can be targeted by tyrosine kinase inhibitor (Dasatinib), mTOR inhibitor (rapamycin), and MDM2 inhibitor (Nutlin-3a), respectively.
HDAC inhibitors of different selectivity are developed to inhibit the actions of various HDAC isoforms in the human cells. Pan-HDAC inhibitors inhibit class I (i.e., HDAC-1/-2/-3/-8), class II (i.e., HDAC-4/-5/-6/-7/-9/-10), and class IV (i.e., HDAC-11) but not class III HDAC isoforms (125). We have shown that several pan-HDAC inhibitors, such as sodium butyrate, valproic acid and SAHA can induce EBV lytic reactivation in EBV-associated epithelial cells (11, 88). We have further demonstrated that selective inhibition of HDAC-1, -2, and -3 by siRNA or specific HDAC inhibitors (e.g., romidepsin, MS-275 and apicidin) is sufficient to reactivate EBV lytic cycle and mediate enhanced killing with ganciclovir in vitro and in vivo (45) (Figure 9). Inhibition of HDAC-2 and -3 can also reactivate the lytic cycle of human immunodeficiency virus (HIV) and Kaposi's sarcoma-associated herpesvirus (KSHV) (126–128). SAHA and romidepsin are FDA-approved drugs for treating several types of malignancies, such as peripheral T-cell lymphoma and cutaneous T-cell lymphoma (129). Both drugs are able to induce EBV lytic cycle in concentrations that are acceptable in the plasma of patients (45, 88). As mentioned in the previous section, the lytic induction therapy that employs valproic acid and gemcitabine as lytic inducers is found to be safe and feasible for the treatment of NPC patients in clinical trials (93, 94). We postulate that substitution of valproic acid with either romidepsin or SAHA would probably lead to a better treatment effect because SAHA or romidepsin has a higher potency in reactivating the EBV lytic cycle in vitro (11, 45).
It was believed that acetylation of histones on Z and R promoters is responsible for the effect of HDAC inhibitors on the reactivation of EBV lytic cycle (130). However, several reports have shown that acetylation of histones alone is not sufficient for EBV lytic cycle reactivation (131–134). Our laboratory has also shown that proteasome inhibitors work synergistically with HDAC inhibitors to induce histone acetylation, but simultaneously suppress the reactivation of EBV lytic cycle in epithelial cells mediated by HDAC inhibitors (102, 135). We postulate that acetylation of non-histone proteins, rather than histone proteins, could directly regulate the reactivation of EBV lytic cycle upon treatment with HDAC inhibitors. The transcription factors which bind to Z promoter, including CREB, C/EBP, Sp1, Sp3, MEF2D, YY1, ZEB1/2, can be modified by lysine acetylation (136–142). It is possible that acetylation of these transcription factors would directly affect their activities on the Z promoter (Figure 9).
Proteasome inhibitors can either covalently or non-covalently bind to 20S proteasome which catalyzes the degradation of ubiquitinated proteins (143). They can induce unfolded protein response (UPR), endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) generation, upregulation of p21WAF1 and p27KIP1, which subsequently lead to cell death in a variety of cancer types (144–146).
Whilst EBNA-1, LMP-2A and LMP-1 inhibit the proteasomal degradation pathway for maintaining viral latency; EBNA-3C utilizes the proteasome system to promote proteasomal degradation of tumor suppressors (e.g., pRb, p21WAF1, p27KIP1, p53 and Bcl-6) which regulate the cell cycle and apoptosis in the host cells (26, 28–30, 54, 122–124, 143, 147, 148). In our previous study, we have found that bortezomib can induce cell cycle arrest at G2/M phase with a higher percentage of BL cells when compared with LCLs which express a higher level of EBNA-3C protein (101). We have further demonstrated that when EBNA-3C knockout or EBNA-3C revertant BL cells are treated with bortezomib, there is a G2/M arrest in the EBNA-3C knockout cell lines whilst the G2/M arrest is bypassed in the EBNA-3C revertant cell lines (108). In these studies, bortezomib in combination with SAHA can induce a stronger apoptotic effect in the EBNA-3C revertant than EBNA-3C knockout BL cells (101). We postulate that disruption of survival signaling conferred by EBNA-3C, such as the suppression of p53, p21WAF1 and Bcl-6 might be responsible for the induction of apoptosis (54–56). In addition, bortezomib can also induce EBV lytic cycle in BL cells (149, 150). The mechanism of lytic induction by bortezomib is mediated via the activation of ER stress, C/EBP-β, JNK and autophagy (150, 151). However, the effect of bortezomib on EBV lytic cycle reactivation is limited to BL cells but not EBV-positive epithelial cancer cells (102, 152) (Figure 10).
Endoplasmic reticulum (ER) stress inducers, including hapsigargin (TG), tunicamycin, bortezomib and nelfinavir, are shown to induce EBV lytic cycle via the induction of ER-stress or UPR (110, 150). Induction of psychological stress by hydrocortisone and dexamethasone can also activate the Z promoter in BL cells (111). Induction of DNA damage response by chloroquine can reactivate EBV lytic cycle via the activation of ATM and phosphorylation of KAP1/TRIM28 in BL cells (112). Activation of ATM is further shown to be essential for the lytic cycle reactivation by conventional lytic inducers, such as HDAC inhibitors, transforming growth factor β (TGF-β) and anti-IgG in several EBV-positive cell lines (153). Induction of microtubule depolymerization by colchicine, vinblastine and nocodazole can reactivate EBV lytic cycle through the activation of PKC and the downstream p38 MAPK and JNK signaling pathways in NPC cells (113). Induction of hypoxia by iron chelators is reported to reactivate EBV lytic cycle through the direct binding of HIF-1α to the HRE elements within Z promoter (114). Generation of ROS upon treatment with methylnitronitrosoguanidine (MNNG) can induce EBV lytic cycle through the activation of ATM, p38 MAPK and JNK signaling pathways (115). Activation of the ATM/p53 genotoxic stress pathway by gemcitabine is found to induce the lytic cycle of EBV in BL, gastric carcinoma and NPC cells (90, 93, 109). TPA in combination with sodium butyrate activates PCK-θ and the subsequent p38 MAPK pathway to reactivate the EBV lytic cycle (116). Chemotherapeutic agents including gemcitabine and doxorubicin can reactivate the lytic cycle of EBV in BL cells and LCLs via the activation of PI3K, p38 MAPK, and MEK (90). An immunosuppressive drug, methotrexate (MTZ), is also shown to induce EBV lytic cycle via the activation of PI3K, p38 MAPK, and ERK signaling pathways in EBV-positive lymphoma cells (117). Immunomodulatory agents, including lenalidomide, thalidomide and pomalidomide reactivate the lytic cycle of EBV in EBV-positive BL cells via PI3K stimulation and ikaros suppression (118) (Figure 9). Upon induction of these stress signaling pathways and the subsequent reactivation of EBV lytic cycle, increased cell death is concomitantly observed (11, 90, 93, 109, 117, 118).
Activation of ERK and autophagy by Rta is shown to be essential for the EBV lytic progression in BL cells (119). Induction of autophagy via the activation of PKC-θ and p38 MAPK is also demonstrated to be essential for the reactivation of EBV lytic cycle in B cells upon treatment with the combination of TPA and sodium butyrate (116). Granato et al. have shown that reactivation of EBV lytic cycle by bortezomib also requires autophagy initiation (151). Recently, chloroquine is shown to activate ATM to phosphorylate KAP1/TRIM28, which is normally involved in repairing double-strand breaks in heterochromatin, to reactivate EBV lytic cycle in BL cells (112). However, the lytic proteins expressed in early phase of EBV lytic cycle will block autolysosome formation to prevent viral degradation and enhance the viral replication in B cells (120, 121). We have recently demonstrated that a novel compound C7 can reactivate EBV lytic cycle via an autophagy-dependent mechanism in EBV-associated epithelial cells (154) (Figure 9). On the other hand, constitutive activation of autophagy is found to confer resistance to nutlin-3 induced apoptosis in the BL cells with type III latency but not in the BL cells with type I latency, suggesting a possible role of EBV latent proteins in the regulation of autophagy (155).
EBNA-3A, together with EBNA-3C, can recruit polycomb repressor complex 2 (PRC2) or interact with C-terminal Binding Protein (CtBP) to epigenetically down-regulate tumor suppressor genes. Transcription of cell cycle regulatory factors, such as p14ARF, p16INK4a, and p15 INK4b, are repressed by methylation and deacetylation of histones mediated by ,EBNA-3A and EBNA-3C (32, 156). Treatment with enhancer of zeste homolog 2 (EZH2) inhibitors and DNA methyltransferase inhibitors, which inhibit the catalytic subunit of PRC2 and histone methylation, might probably induce cell cycle arrest in EBV-LPDs. EBNA-3A and -3C are known to directly interact with HDAC-1 and -2 to repress the expression of p14ARF and p16INK4a (32, 38, 157). EBNA-3C is also shown to recruit Pim-1 to phosphorylate and suppress pRb, p21WAF1 and p27KIP1 via the proteasomal degradation system in B cells (26, 28, 29). Interestingly, we have shown that combination of proteasome and HDAC inhibitors can upregulate p16INK4a and p21WAF1, and mediate G2/M arrest in EBV-LPDs (101). The G2/M arrest is further demonstrated to be related to the downregulation of p-cdc25c (108) (Figure 10).
LMP-1 can activate both canonical (acts through the p50/RelA dimer) and non-canonical (acts through the p52/RelB dimer) NF-κB pathways to promote the pathogenesis of EBV-positive cancer cells (158, 159). Bortezomib can inhibit the proteasomal degradation of IκBα, thus, inhibit the activation of NF-κB pathways. Consequently, the expression of anti-apoptotic proteins including X-chromosome-linked inhibitor-of-apoptosis protein (XIAP), cellular inhibitor-of-apoptosis protein 1 (cIAP-1) and c-IAP-2, are suppressed (149). EBNA-3A and -3C can work together to epigenetically down-regulate tumor suppressor genes including STK39 and Bim to inhibit apoptosis (58, 160–162). It has been demonstrated that the EZH2 inhibitor GSK126 can inhibit the PRC2 complex and increase the expression of STK39 whereas the DNA methyltransferase inhibitor 5-AZA can significantly increase the mRNA level of STK39 in LCLs (160). The EZH2 inhibitor can also significantly induce the expression of Bim and apoptosis in BL cells (162). EBNA-3C promotes proteasomal degradation of certain tumor suppressors, such as p21WAF1, p53, and Bcl-6 (28, 30, 54). We have reported that combining HDAC and proteasome inhibitors can upregulate p21WAF1 and mediate synergistic killing of BL cells and LCLs via an EBNA-3C-dependent mechanism (101, 108). As inhibition of Bcl-6 by EBNA-3C releases Bcl-2 to suppress apoptosis, it is possible that combination of Bcl-2 inhibitors, such as venetoclax and proteasome inhibitors, such as bortezomib could synergistically induce apoptosis in EBV-LPDs (Figure 10).
The abovementioned treatment strategies, such as HDAC inhibitors, proteasome inhibitors and other stress inducers could all affect multiple signaling pathways and thus, resulting in non-specific effects to the host cells. Identification of novel compounds which can specifically target EBV latent and lytic cycles is critical for further development of viral-targeted therapy against EBV-LPDs. Using computational docking programs, Li et al. have identified 4 structurally related compounds (coded SC7, SC11, SC19 and SC27) which can inhibit the DNA binding of EBNA-1 and reduce the viral genome copy in BL cells (96). In a separate study, the same research team has identified 4 additional EBNA-1 specific inhibitors (coded LB2, LB3, LB7 and LC7) via a cell-based screening of 14,000 small molecule compounds (97). Recently, a peptide-based inhibitor, L2P4, which can bind to EBNA-1 and inhibit EBNA-1 homodimer formation is found to selectively inhibit the proliferation of EBV-positive BL and NPC cells in vitro and in vivo (95) (Figure 10). Tikhmyanova et al. have identified 5 structurally related tetrahydrocarboline derivatives coded C09, C50, C53, C60, and C67, which can significantly reactivate the lytic cycle of EBV and mediate enhanced killing with GCV in both lymphoma and epithelial cancer cells from a high-throughput screening of 66,840 small molecule compounds (163). These compounds do not induce acetylation of histone and phosphorylation of p38 MAPK, S6, p53, and p90RSK, suggesting a distinct lytic reactivation mechanism from those induced by HDAC inhibitors or TPA (163). Our laboratory has also identified 5 hit compounds (coded C7, E11, E7, C8, and A10) through a high-throughput screening of 50,240 small organic compounds (164). These compounds also do not phosphorylate the PKCδ which is utilized by many conventional lytic inducers and do not cause acetylation of histone, suggesting a mechanism of action distinct from HDAC inhibitors or TPA (164). E11 and C7 are further investigated for their mechanisms of EBV lytic cycle reactivation. We have found that the lytic cycle reactivation by E11 requires the JNK signaling pathway whilst the lytic cycle reactivation by C7 requires the activation of both ERK and JNK pathways (164). We have further demonstrated that C7 can reactivate EBV lytic cycle in epithelial cells via chelation of iron and induction of autophagy (154). Recently, another new class of lytic inducer, curcuminoids, which might reactivate EBV lytic cycle through modulation of NK-κB signaling, is identified (165). Since most of these novel compounds can reactivate EBV lytic cycle via mechanisms distinct from conventional inducers, such as HDAC inhibitors and TPA, it would be interesting to test whether combination of conventional inducers with novel inducers can mediate synergistic reactivation of EBV lytic cycle in EBV-LPDs (Figure 9).
In this review, we have summarized the mechanisms by which EBV-LPDs employ to drive the pathogenesis and maintain the survival of the tumor cells. We have shown that the viral latent and lytic proteins can maintain the proliferation and survival of EBV-positive tumor cells via deregulating the mechanisms which control the cell cycle, apoptosis and immune recognition in the host cells. Potential viral-targeted strategies based on the understanding of the patho-mechanisms of EBV-LPDs are also discussed. A number of clinical relevant drugs and novel compounds which can either target the EBV latent proteins or reactivate EBV lytic cycle are reviewed. These compounds work through diverse mechanisms including inhibition of HDAC and proteasome, activation of MAPK pathways, induction of various cellular stress responses (e.g., ER stress, DNA damage response, hypoxia and oxidative stress), autophagy, cell cycle arrest and apoptosis. The effect of combining pharmaceutic compounds which act on multiple signaling pathways in EBV-LPDs should be explored.
AC conceived the project. KH and AC wrote the manuscript with the help of SY and KT.
This work is supported by Health and Medical Research Fund (#17160712) grant of KH, URC-Seed Fund for Basic Research (#104004504), Health and Medical Research Fund (#16150472), NPC Area of Excellence (AoE/M 06/08 Center for Nasopharyngeal Carcinoma Research) and Epstein-Barr virus research (#20004525) grants of AC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Epstein A. Why and how Epstein-Barr virus was discovered 50 years ago. Curr Top Microbiol Immunol. (2015) 390(Pt1):3–15. doi: 10.1007/978-3-319-22822-8_1
2. Hui KF, Chan TF, Yang W, Shen JJ, Lam KP, Kwok H, et al. High risk Epstein-Barr virus variants characterized by distinct polymorphisms in the EBER locus are strongly associated with nasopharyngeal carcinoma. Int J Cancer (2018). doi: 10.1002/ijc.32049. [Epub ahead of print].
3. Rickinson AB, Kieff E. In: Fields B, Knipe DM, Howley PM, editors. Epstein-Barr Virus. 4th ed. Philadelphia, PA: Lippincott Williams and Wilkins (2001). p. 2575–627.
4. Hochberg D, Middeldorp JM, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Demonstration of the Burkitt's lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc Natl Acad Sci USA. (2004) 101:239–44. doi: 10.1073/pnas.2237267100
5. Kelly GL, Stylianou J, Rasaiyaah J, Wei W, Thomas W, Croom-Carter D, et al. Different patterns of Epstein-Barr virus latency in endemic Burkitt lymphoma (BL) lead to distinct variants within the BL-associated gene expression signature. J Virol. (2013) 87:2882–94. doi: 10.1128/JVI.03003-12
6. Rowe M, Kelly GL, Bell AI, Rickinson AB. Burkitt's lymphoma: the Rosetta Stone deciphering Epstein-Barr virus biology. Semin Cancer Biol. (2009) 19:377–88. doi: 10.1016/j.semcancer.2009.07.004
7. Carbone A, Gloghini A, Dotti G. EBV-associated lymphoproliferative disorders: classification and treatment. Oncologist (2008) 13:577–85. doi: 10.1634/theoncologist.2008-0036
8. Sinclair AJ, Brimmell M, Shanahan F, Farrell PJ. Pathways of activation of the Epstein-Barr virus productive cycle. J Virol. (1991) 65:2237–44.
9. Robertson E, Kieff E. Reducing the complexity of the transforming Epstein-Barr virus genome to 64 kilobase pairs. J Virol. (1995) 69:983–93.
10. Binne UK, Amon W, Farrell PJ. Promoter sequences required for reactivation of Epstein-Barr virus from latency. J Virol. (2002) 76:10282–9 doi: 10.1128/JVI.76.20.10282-10289.2002
11. Hui KF, Chiang AK. Suberoylanilide hydroxamic acid induces viral lytic cycle in Epstein-Barr virus-positive epithelial malignancies and mediates enhanced cell death. Int J Cancer (2010) 126:2479–89. doi: 10.1002/ijc.24945
12. Mainou BA, Everly DN Jr, Raab-Traub N. Unique signaling properties of CTAR1 in LMP1-mediated transformation. J Virol. (2007) 81:9680–92. doi: 10.1128/JVI.01001-07
13. Haddad RS, Hutt-Fletcher LM. Depletion of glycoprotein gp85 from virosomes made with Epstein-Barr virus proteins abolishes their ability to fuse with virus receptor-bearing cells. J Virol. (1989) 63:4998–5005.
14. Lake CM, Molesworth SJ, Hutt-Fletcher LM. The Epstein-Barr virus (EBV) gN homolog BLRF1 encodes a 15-kilodalton glycoprotein that cannot be authentically processed unless it is coexpressed with the EBV gM homolog BBRF3. J Virol. (1998) 72:5559–64.
15. Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. (2006) 13:927–34. doi: 10.1038/sj.cdd.4401912
16. Tsurumi T, Fujita M, Kudoh A. Latent and lytic Epstein-Barr virus replication strategies. Rev Med Virol. (2005) 15:3–15. doi: 10.1002/rmv.441
17. Ning RJ, Xu XQ, Chan KH, Chiang AK. Long-term carriers generate Epstein-Barr virus (EBV)-specific CD4(+) and CD8(+) polyfunctional T-cell responses which show immunodominance hierarchies of EBV proteins. Immunology (2011) 134:161–71. doi: 10.1111/j.1365-2567.2011.03476.x
18. Smith C, Beagley L, Khanna R. Acquisition of polyfunctionality by Epstein-Barr virus-specific CD8+ T cells correlates with increased resistance to galectin-1-mediated suppression. J Virol. (2009) 83:6192–8. doi: 10.1128/jvi.00239-09
19. Lam KP, Hui KF, Ning RJ, Xu XQ, Chan KH, Chiang AK. Emergence of CD4+ and CD8+ polyfunctional T cell responses against immunodominant lytic and latent EBV antigens in children with primary EBV infection. Front Microbiol. (2018) 9:416. doi: 10.3389/fmicb.2018.00416
20. Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood (2014) 124:2533–43. doi: 10.1182/blood-2014-01-553024
21. Hatton O, Strauss-Albee DM, Zhao NQ, Haggadone MD, Pelpola JS, Krams SM, et al. NKG2A-expressing natural killer cells dominate the response to autologous lymphoblastoid cells infected with Epstein-Barr virus. Front Immunol. (2016) 7:607. doi: 10.3389/fimmu.2016.00607
22. Allday MJ, Farrell PJ. Epstein-Barr virus nuclear antigen EBNA3C/6 expression maintains the level of latent membrane protein 1 in G1-arrested cells. J Virol. (1994) 68:3491–8.
23. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer (2009) 9:550–62. doi: 10.1038/nrc2664
24. Vardy L, Pesin JA, Orr-Weaver TL. Regulation of Cyclin A protein in meiosis and early embryogenesis. Proc Natl Acad Sci USA. (2009) 106:1838–43. doi: 10.1073/pnas.0813237106
25. Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. (1992) 11:961–71. doi: 10.1002/j.1460-2075.1992.tb05135.x
26. Knight JS, Sharma N, Robertson ES. Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc Natl Acad Sci USA. (2005) 102:18562–6. doi: 10.1073/pnas.0503886102
27. Kashuba E, Yurchenko M, Yenamandra SP, Snopok B, Isaguliants M, Szekely L, et al. EBV-encoded EBNA-6 binds and targets MRS18-2 to the nucleus, resulting in the disruption of pRb-E2F1 complexes. Proc Natl Acad Sci USA. (2008) 105:5489–94. doi: 10.1073/pnas.0801053105
28. Banerjee S, Lu J, Cai Q, Sun Z, Jha HC, Robertson ES. EBNA3C augments Pim-1 mediated phosphorylation and degradation of p21 to promote B-cell proliferation. PLoS Pathog. (2014) 10:e1004304. doi: 10.1371/journal.ppat.1004304
29. Knight JS, Sharma N, Robertson ES. SCFSkp2 complex targeted by Epstein-Barr virus essential nuclear antigen. Mol Cell Biol. (2005) 25:1749–63. doi: 10.1128/MCB.25.5.1749-1763.2005
30. Pei Y, Banerjee S, Jha HC, Sun Z, Robertson ES. An essential EBV latent antigen 3C binds Bcl6 for targeted degradation and cell proliferation. PLoS Pathog. (2017) 13:e1006500. doi: 10.1371/journal.ppat.1006500
31. Maruo S, Zhao B, Johannsen E, Kieff E, Zou J, Takada K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc Natl Acad Sci USA. (2011) 108:1919–24. doi: 10.1073/pnas.1019599108
32. Skalska L, White RE, Franz M, Ruhmann M, Allday MJ. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. (2010) 6:e1000951. doi: 10.1371/journal.ppat.1000951
33. Skalska L, White RE, Parker GA, Turro E, Sinclair AJ, Paschos K, et al. Induction of p16(INK4a) is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. (2013) 9:e1003187. doi: 10.1371/journal.ppat.1003187
34. Krauer KG, Burgess A, Buck M, Flanagan J, Sculley TB, Gabrielli B. The EBNA-3 gene family proteins disrupt the G2/M checkpoint. Oncogene (2004) 23:1342–53. doi: 10.1038/sj.onc.1207253
35. Choudhuri T, Verma SC, Lan K, Murakami M, Robertson ES. The ATM/ATR signaling effector Chk2 is targeted by Epstein-Barr virus nuclear antigen 3C to release the G2/M cell cycle block. J Virol. (2007) 81:6718–30. doi: 10.1128/JVI.00053-07
36. Wade M, Allday MJ. Epstein-Barr virus suppresses a G(2)/M checkpoint activated by genotoxins. Mol Cell Biol. (2000) 20:1344–60 doi: 10.1128/MCB.20.4.1344-1360.2000
37. Parker GA, Touitou R, Allday MJ. Epstein-Barr virus EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis. Oncogene (2000) 19:700–9. doi: 10.1038/sj.onc.1203327
38. White RE, Groves IJ, Turro E, Yee J, Kremmer E, Allday MJ. Extensive co-operation between the Epstein-Barr virus EBNA3 proteins in the manipulation of host gene expression and epigenetic chromatin modification. PLoS ONE (2010) 5:e13979. doi: 10.1371/journal.pone.0013979
39. Guo Q, Qian L, Guo L, Shi M, Chen C, Lv X, et al. Transactivators Zta and Rta of Epstein-Barr virus promote G0/G1 to S transition in Raji cells: a novel relationship between lytic virus and cell cycle. Mol Immunol. (2010) 47:1783–92. doi: 10.1016/j.molimm.2010.02.017
40. Wu FY, Wang SE, Chen H, Wang L, Hayward SD, Hayward GS. CCAAT/enhancer binding protein alpha binds to the Epstein-Barr virus (EBV) ZTA protein through oligomeric interactions and contributes to cooperative transcriptional activation of the ZTA promoter through direct binding to the ZII and ZIIIB motifs during induction of the EBV lytic cycle. J Virol. (2004) 78:4847–65 doi: 10.1128/JVI.78.9.4847-4865.2004
41. Huang SY, Hsieh MJ, Chen CY, Chen YJ, Chen JY, Chen MR, et al. Epstein-Barr virus Rta-mediated transactivation of p21 and 14-3-3sigma arrests cells at the G1/S transition by reducing cyclin E/CDK2 activity. J Gen Virol. (2012) 93(Pt 1):139–49. doi: 10.1099/vir.0.034405-0
42. Kudoh A, Daikoku T, Sugaya Y, Isomura H, Fujita M, Kiyono T, et al. Inhibition of S-phase cyclin-dependent kinase activity blocks expression of Epstein-Barr virus immediate-early and early genes, preventing viral lytic replication. J Virol. (2004) 78:104–15 doi: 10.1128/JVI.78.1.104-115.2004
43. Mauser A, Holley-Guthrie E, Simpson D, Kaufmann W, Kenney S. The Epstein-Barr virus immediate-early protein BZLF1 induces both a G(2) and a mitotic block. J Virol. (2002) 76:10030–7. doi: 10.1128/JVI.76.19.10030-10037.2002
44. Rodriguez A, Jung EJ, Flemington EK. Cell cycle analysis of Epstein-Barr virus-infected cells following treatment with lytic cycle-inducing agents. J Virol. (2001) 75:4482–9. doi: 10.1128/JVI.75.10.4482-4489.2001
45. Hui KF, Cheung AK, Choi CK, Yeung PL, Middeldorp JM, Lung ML, et al. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int J Cancer (2016) 138:125–36. doi: 10.1002/ijc.29698
46. Holowaty MN, Zeghouf M, Wu H, Tellam J, Athanasopoulos V, Greenblatt J, et al. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J Biol Chem. (2003) 278:29987–94. doi: 10.1074/jbc.M303977200
47. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
48. Horst D, van Leeuwen D, Croft NP, Garstka MA, Hislop AD, Kremmer E, et al. Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of HLA class I-restricted antigen presentation. J Immunol. (2009) 182:2313–24. doi: 10.4049/jimmunol.0803218
49. Young P, Anderton E, Paschos K, White R, Allday MJ. Epstein-Barr virus nuclear antigen (EBNA) 3A induces the expression of and interacts with a subset of chaperones and co-chaperones. J Gen Virol. (2008) 89(Pt 4):866–77. doi: 10.1099/vir.0.83414-0
50. Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget (2012) 3:228–35 doi: 10.18632/oncotarget.443
51. Gu B, Zhu WG. Surf the post-translational modification network of p53 regulation. Int J Biol Sci. (2012) 8:672–84. doi: 10.7150/ijbs.4283
52. Yi F, Saha A, Murakami M, Kumar P, Knight JS, Cai Q, et al. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology (2009) 388:236–47. doi: 10.1016/j.virol.2009.03.027
53. Saha A, Lu J, Morizur L, Upadhyay SK, Aj MP, Robertson ES. E2F1 mediated apoptosis induced by the DNA damage response is blocked by EBV nuclear antigen 3C in lymphoblastoid cells. PLoS Pathog. (2012) 8:e1002573. doi: 10.1371/journal.ppat.1002573
54. Saha A, Murakami M, Kumar P, Bajaj B, Sims K, Robertson ES. Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J Virol. (2009) 83:4652–69. doi: 10.1128/jvi.02408-08
55. Picksley SM, Vojtesek B, Sparks A, Lane DP. Immunochemical analysis of the interaction of p53 with MDM2;–fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene (1994) 9:2523–9.
56. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature (1997) 387:296–9. doi: 10.1038/387296a0.
57. Anderton E, Yee J, Smith P, Crook T, White RE, Allday MJ. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt's lymphoma. Oncogene (2008) 27:421–33. doi: 10.1038/sj.onc.1210668
58. Paschos K, Parker GA, Watanatanasup E, White RE, Allday MJ. BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res. (2012) 40:7233–46. doi: 10.1093/nar/gks391
59. Hong GK, Kumar P, Wang L, Damania B, Gulley ML, Delecluse HJ, et al. Epstein-Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. J Virol. (2005) 79:13984–92. doi: 10.1128/JVI.79.22.13984-13992.2005
60. Celkan T, Alhaj S, Civilibal M, Elicevik M. Control of bleeding associated with hemophagocytic syndrome in children: an audit of the clinical use of recombinant activated factor VII. Pediatr Hematol Oncol. (2007) 24:117–21. doi: 10.1080/08880010601094102
61. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. (2009) 10:29–37. doi: 10.1038/ni.1679
62. Chiu SH, Wu CC, Fang CY, Yu SL, Hsu HY, Chow YH, et al. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget (2014) 5:8583–601. doi: 10.18632/oncotarget.2323
63. Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, et al. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe (2011) 10:390–400. doi: 10.1016/j.chom.2011.08.013
64. Wu CC, Liu MT, Chang YT, Fang CY, Chou SP, Liao HW, et al. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. (2010) 38:1932–49. doi: 10.1093/nar/gkp1169
65. Sakka E, Zur Hausen A, Houali K, Liu H, Fiorini S, Ooka T. Cellular localization of BARF1 oncoprotein and its cell stimulating activity in human epithelial cell. Virus Res. (2013) 174:8–17. doi: 10.1016/j.virusres.2013.01.016
66. Levitskaya J, Sharipo A, Leonchiks A, Ciechanover A, Masucci MG. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc Natl Acad Sci USA. (1997) 94:12616–21. doi: 10.1073/pnas.94.23.12616
67. Li H, Liu S, Hu J, Luo X, Li N, A MB, et al. Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int J Biol Sci. (2016) 12:1309–18. doi: 10.7150/ijbs.16564
68. Yao X, Li X, Toledo F, Zurita-Lopez C, Gutova M, Momand J, et al. Sub-attomole oligonucleotide and p53 cDNA determinations via a high-resolution surface plasmon resonance combined with oligonucleotide-capped gold nanoparticle signal amplification. Anal Biochem. (2006) 354:220–8. doi: 10.1016/j.ab.2006.04.011
69. Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. (2012) 8:e1002704. doi: 10.1371/journal.ppat.1002704
70. Stuart AD, Stewart JP, Arrand JR, Mackett M. The Epstein-Barr virus encoded cytokine viral interleukin-10 enhances transformation of human B lymphocytes. Oncogene (1995) 11:1711–9.
71. Quinn LL, Williams LR, White C, Forrest C, Zuo J, Rowe M. The missing link in Epstein-Barr virus immune evasion: the BDLF3 gene induces ubiquitination and downregulation of major histocompatibility complex class I (MHC-I) and MHC-II. J Virol. (2015) 90:356–67. doi: 10.1128/jvi.02183-15
72. Franken M, Estabrooks A, Cavacini L, Sherburne B, Wang F, Scadden DT. Epstein-Barr virus-driven gene therapy for EBV-related lymphomas. Nat Med. (1996) 2:1379–82. doi: 10.1038/nm1296-1379
73. Li JH, Huang D, Sun BF, Zhang X, Middeldorp J, Klamut H, et al. Efficacy of ionizing radiation combined with adenoviral p53 therapy in EBV-positive nasopharyngeal carcinoma. Int J Cancer (2000) 87:606–10. doi: 10.1002/1097-0215(20000815)87:4<606::AID-IJC23>3.0.CO;2-O
74. Li JH, Chia M, Shi W, Ngo D, Strathdee CA, Huang D, et al. Tumor-targeted gene therapy for nasopharyngeal carcinoma. Cancer Res. (2002) 62:171–8.
75. Lasaro MO, Tatsis N, Hensley SE, Whitbeck JC, Lin SW, Rux JJ, et al. Targeting of antigen to the herpesvirus entry mediator augments primary adaptive immune responses. Nat Med. (2008) 14:205–12. doi: 10.1038/nm1704
76. Smith C, Lee V, Schuessler A, Beagley L, Rehan S, Tsang J, et al. Pre-emptive and therapeutic adoptive immunotherapy for nasopharyngeal carcinoma: phenotype and effector function of T cells impact on clinical response. Oncoimmunology (2017) 6:e1273311. doi: 10.1080/2162402X.2016.1273311
77. Meti N, Esfahani K, Johnson NA. The role of immune checkpoint inhibitors in classical Hodgkin lymphoma. Cancers (Basel) (2018) 10:E204. doi: 10.3390/cancers10060204
78. Li PF, Mao YZ, Bai B, Gao Y, Zhang YJ, Li ZM, et al. Persistent peripheral blood EBV-DNA positive with high expression of PD-L1 and upregulation of CD4+ CD25+ T cell ratio in early stage NK/T cell lymphoma patients may predict worse outcome. Ann Hematol. (2018) 97:2381–9. doi: 10.1007/s00277-018-3467-6
79. Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. (2013) 19:3462–73. doi: 10.1158/1078-0432.CCR-13-0855
80. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. (2017) 27:59–73. doi: 10.1038/cr.2016.153
81. Perez EM, Foley J, Tison T, Silva R, Ogembo JG. Novel Epstein-Barr virus-like particles incorporating gH/gL-EBNA1 or gB-LMP2 induce high neutralizing antibody titers and EBV-specific T-cell responses in immunized mice. Oncotarget (2017) 8:19255–73. doi: 10.18632/oncotarget.13770
82. Kawanishi M. Epstein-Barr virus induces fragmentation of chromosomal DNA during lytic infection. J Virol. (1993) 67:7654–8.
83. Feng W-H, Kenney SC. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expression. Cancer Res. (2006) 66:8762–9. doi: 10.1158/0008-5472.CAN-06-1006
84. Rodriguez A, Armstrong M, Dwyer D, Flemington E. Genetic dissection of cell growth arrest functions mediated by the Epstein-Barr virus lytic gene product, Zta. J Virol. (1999) 73:9029–38.
85. Burrows PD, Kronenberg M, Taniguchi M. NKT cells turn ten. Nat Immunol. (2009) 10:669–71. doi: 10.1038/ni0709-669
86. Chen YL, Chen YJ, Tsai WH, Ko YC, Chen JY, Lin SF. The Epstein-Barr virus replication and transcription activator, Rta/BRLF1, induces cellular senescence in epithelial cells. Cell Cycle (2009) 8:58–65. doi: 10.4161/cc.8.1.7411
87. Horst D, Burmeister WP, Boer IG, van Leeuwen D, Buisson M, Gorbalenya AE, et al. The “Bridge” in the Epstein-Barr virus alkaline exonuclease protein BGLF5 contributes to shutoff activity during productive infection. J Virol. (2012) 86:9175–87. doi: 10.1128/JVI.00309-12
88. Hui KF, Ho DN, Tsang CM, Middeldorp JM, Tsao GS, Chiang AK. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int J Cancer (2012) 131:1930–40. doi: 10.1002/ijc.27439
89. Feng WH, Israel B, Raab-Traub N, Busson P, Kenney SC. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Res. (2002) 62:1920–6.
90. Feng WH, Hong G, Delecluse HJ, Kenney SC. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. J Virol. (2004) 78:1893–902 doi: 10.1128/JVI.78.4.1893-1902.2004
91. Wang JT, Chuang YC, Chen KL, Lu CC, Doong SL, Cheng HH, et al. Characterization of Epstein-Barr virus BGLF4 kinase expression control at the transcriptional and translational levels. J Gen Virol. (2010) 91(Pt 9):2186–96. doi: 10.1099/vir.0.019729-0
92. Fu DX, Tanhehco Y, Chen J, Foss CA, Fox JJ, Chong JM, et al. Bortezomib-induced enzyme-targeted radiation therapy in herpesvirus-associated tumors. Nat Med. (2008) 14:1118–22. doi: 10.1038/nm.1864
93. Wildeman MA, Novalic Z, Verkuijlen SA, Juwana H, Huitema AD, Tan IB, et al. Cytolytic virus activation therapy for Epstein-Barr virus-driven tumors. Clin Cancer Res. (2012) 18:5061–70. doi: 10.1158/1078-0432.CCR-12-0574
94. Stoker SD, Novalic Z, Wildeman MA, Huitema AD, Verkuijlen SA, Juwana H, et al. Epstein-Barr virus-targeted therapy in nasopharyngeal carcinoma. J Cancer Res Clin Oncol. (2015) 141:1845–57. doi: 10.1007/s00432-015-1969-3
95. Jiang L, Lan R, Huang T, Chan C-F, Li H, Lear S, et al. EBNA1-targeted probe for the imaging and growth inhibition of tumours associated with the Epstein–Barr virus. Nat Biomed Eng. (2017) 1:0042. doi: 10.1038/s41551-017-0042
96. Li N, Thompson S, Schultz DC, Zhu W, Jiang H, Luo C, et al. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PLoS ONE (2010) 5:e10126. doi: 10.1371/journal.pone.0010126
97. Thompson S, Messick T, Schultz DC, Reichman M, Lieberman PM. Development of a high-throughput screen for inhibitors of Epstein-Barr virus EBNA1. J Biomol Screen. (2010) 15:1107–15. doi: 10.1177/1087057110379154
98. AlQarni S, Al-Sheikh Y, Campbell D, Drotar M, Hannigan A, Boyle S, et al. Lymphomas driven by Epstein–Barr virus nuclear antigen-1 (EBNA1) are dependant upon Mdm2. Oncogene (2018) 37:3998–4012. doi: 10.1038/s41388-018-0147-x
99. Vaysberg M, Hatton O, Lambert SL, Snow AL, Wong B, Krams SM, et al. Tumor-derived variants of Epstein-Barr virus latent membrane protein 1 induce sustained Erk activation and c-Fos. J Biol Chem. (2008) 283:36573–85. doi: 10.1074/jbc.M802968200
100. Morris MA, Dawson CW, Young LS. Role of the Epstein-Barr virus-encoded latent membrane protein-1, LMP1, in the pathogenesis of nasopharyngeal carcinoma. Future Oncol. (2009) 5:811–25. doi: 10.2217/fon.09.53
101. Hui KF, Leung YY, Yeung PL, Middeldorp JM, Chiang AK. Combination of SAHA and bortezomib up-regulates CDKN2A and CDKN1A and induces apoptosis of Epstein-Barr virus-positive Wp-restricted Burkitt lymphoma and lymphoblastoid cell lines. Br J Haematol. (2014) 167:639–50. doi: 10.1111/bjh.13089
102. Hui KF, Lam BH, Ho DN, Tsao SW, Chiang AK. Bortezomib and SAHA synergistically induce ROS-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of Epstein-Barr virus. Mol Cancer Ther. (2013) 12:747–58. doi: 10.1158/1535-7163.MCT-12-0811
103. Dargart JL, Fish K, Gordon LI, Longnecker R, Cen O. Dasatinib therapy results in decreased B cell proliferation, splenomegaly, and tumor growth in a murine model of lymphoma expressing Myc and Epstein-Barr virus LMP2A. Antiviral Res. (2012) 95:49–56. doi: 10.1016/j.antiviral.2012.05.003
104. Cen O, Longnecker R. Rapamycin reverses splenomegaly and inhibits tumor development in a transgenic model of Epstein-Barr virus-related Burkitt's lymphoma. Mol Cancer Ther. (2011) 10:679–86. doi: 10.1158/1535-7163.Mct-10-0833
105. Fish K, Sora RP, Schaller SJ, Longnecker R, Ikeda M. EBV latent membrane protein 2A orchestrates p27(kip1) degradation via Cks1 to accelerate MYC-driven lymphoma in mice. Blood (2017) 130:2516–26. doi: 10.1182/blood-2017-07-796821
106. Leao M, Anderton E, Wade M, Meekings K, Allday MJ. Epstein-barr virus-induced resistance to drugs that activate the mitotic spindle assembly checkpoint in Burkitt's lymphoma cells. J Virol. (2007) 81:248–60. doi: 10.1128/JVI.01096-06
107. Kelly GL, Milner AE, Tierney RJ, Croom-Carter DS, Altmann M, Hammerschmidt W, et al. Epstein-Barr virus nuclear antigen 2 (EBNA2) gene deletion is consistently linked with EBNA3A,−3B, and−3C expression in Burkitt's lymphoma cells and with increased resistance to apoptosis. J Virol. (2005) 79:10709–17. doi: 10.1128/JVI.79.16.10709-10717.2005
108. Hui KF, Yeung PL, Tam KP, Chiang AKS. Counteracting survival functions of EBNA3C in Epstein-Barr virus (EBV)-driven lymphoproliferative diseases by combination of SAHA and bortezomib. Oncotarget (2018) 9:25101–14. doi: 10.18632/oncotarget.25341
109. Lee HG, Kim H, Kim EJ, Park PG, Dong SM, Choi TH, et al. Targeted therapy for Epstein-Barr virus-associated gastric carcinoma using low-dose gemcitabine-induced lytic activation. Oncotarget (2015) 6:31018–29. doi: 10.18632/oncotarget.5041
110. Taylor GM, Raghuwanshi SK, Rowe DT, Wadowsky RM, Rosendorff A. Endoplasmic reticulum stress causes EBV lytic replication. Blood (2011) 118:5528–39. doi: 10.1182/blood-2011-04-347112
111. Yang EV, Webster Marketon JI, Chen M, Lo KW, Kim SJ, Glaser R. Glucocorticoids activate Epstein Barr virus lytic replication through the upregulation of immediate early BZLF1 gene expression. Brain Behav Immun. (2010) 24:1089–96. doi: 10.1016/j.bbi.2010.04.013
112. Li X, Burton EM, Bhaduri-McIntosh S. Chloroquine triggers Epstein-Barr virus replication through phosphorylation of KAP1/TRIM28 in Burkitt lymphoma cells. PLoS Pathog. (2017) 13:e1006249. doi: 10.1371/journal.ppat.1006249
113. Liu YR, Huang SY, Chen JY, Wang LH. Microtubule depolymerization activates the Epstein-Barr virus lytic cycle through protein kinase C pathways in nasopharyngeal carcinoma cells. J Gen Virol. (2013) 94(Pt 12):2750–8. doi: 10.1099/vir.0.058040-0
114. Kraus RJ, Yu X, Cordes BA, Sathiamoorthi S, Iempridee T, Nawandar DM, et al. Hypoxia-inducible factor-1alpha plays roles in Epstein-Barr virus's natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. (2017) 13:e1006404. doi: 10.1371/journal.ppat.1006404
115. Huang SY, Fang CY, Wu CC, Tsai CH, Lin SF, Chen JY. Reactive oxygen species mediate Epstein-Barr virus reactivation by N-methyl-N′-nitro-N-nitrosoguanidine. PLoS ONE (2013) 8:e84919. doi: 10.1371/journal.pone.0084919
116. Gonnella R, Granato M, Farina A, Santarelli R, Faggioni A, Cirone M. PKC theta and p38 MAPK activate the EBV lytic cycle through autophagy induction. Biochim Biophys Acta (2015) 1853:1586–95. doi: 10.1016/j.bbamcr.2015.03.011
117. Feng WH, Cohen JI, Fischer S, Li L, Sneller M, Goldbach-Mansky R, et al. Reactivation of latent Epstein-Barr virus by methotrexate: a potential contributor to methotrexate-associated lymphomas. J Natl Cancer Inst. (2004) 96:1691–702. doi: 10.1093/jnci/djh313
118. Jones RJ, Iempridee T, Wang X, Lee HC, Mertz JE, Kenney SC, et al. Lenalidomide, thalidomide, and pomalidomide reactivate the Epstein-Barr virus lytic cycle through phosphoinositide 3-kinase signaling and Ikaros expression. Clin Cancer Res. (2016) 22:4901–12. doi: 10.1158/1078-0432.CCR-15-2242
119. Hung CH, Chen LW, Wang WH, Chang PJ, Chiu YF, Hung CC, et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J Virol. (2014) 88:12133–45. doi: 10.1128/JVI.02033-14
120. De Leo A, Colavita F, Ciccosanti F, Fimia GM, Lieberman PM, Mattia E. Inhibition of autophagy in EBV-positive Burkitt's lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. (2015) 6:e1876. doi: 10.1038/cddis.2015.156
121. Granato M, Santarelli R, Farina A, Gonnella R, Lotti LV, Faggioni A, et al. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J Virol. (2014) 88:12715–26. doi: 10.1128/JVI.02199-14
122. Hertle ML, Popp C, Petermann S, Maier S, Kremmer E, Lang R, et al. Differential gene expression patterns of EBV infected EBNA-3A positive and negative human B lymphocytes. PLoS Pathog. (2009) 5:e1000506. doi: 10.1371/journal.ppat.1000506
123. Saha A, Robertson ES. Impact of EBV essential nuclear protein EBNA-3C on B-cell proliferation and apoptosis. Fut Microbiol. (2013) 8:323–52. doi: 10.2217/fmb.12.147
124. Maruo S, Wu Y, Ishikawa S, Kanda T, Iwakiri D, Takada K. Epstein-Barr virus nuclear protein EBNA3C is required for cell cycle progression and growth maintenance of lymphoblastoid cells. Proc Natl Acad Sci USA. (2006) 103:19500–5. doi: 10.1073/pnas.0604919104
125. Butler LM, Webb Y, Agus DB, Higgins B, Tolentino TR, Kutko MC, et al. Inhibition of transformed cell growth and induction of cellular differentiation by pyroxamide, an inhibitor of histone deacetylase. Clin Cancer Res. (2001) 7:962–70.
126. Barton KM, Archin NM, Keedy KS, Espeseth AS, Zhang YL, Gale J, et al. Selective HDAC inhibition for the disruption of latent HIV-1 infection. PLoS ONE (2014) 9:e102684. doi: 10.1371/journal.pone.0102684
127. Shin HJ, DeCotiis J, Giron M, Palmeri D, Lukac DM. Histone deacetylase classes I and II regulate Kaposi's sarcoma-associated herpesvirus reactivation. J Virol. (2014) 88:1281–92. doi: 10.1128/JVI.02665-13
128. Sogaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. (2015) 11:e1005142. doi: 10.1371/journal.ppat.1005142
129. Harrison SJ, Bishton M, Bates SE, Grant S, Piekarz RL, Johnstone RW, et al. A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin [depsipeptide, Istodax(R)]. Epigenomics (2012) 4:571–89. doi: 10.2217/epi.12.52
130. Samols MA, Skalsky RL, Maldonado AM, Riva A, Lopez MC, Baker HV, et al. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. (2007) 3:e65. doi: 10.1371/journal.ppat.0030065
131. Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. (2007) 81:2545–53. doi: 10.1128/JVI.02021-06
132. Cooper LJ. Persuading natural killer cells to eliminate bad B cells. Clin Cancer Res. (2009) 15:4790–1. doi: 10.1158/1078-0432.CCR-09-0966
133. Jenkins PJ, Binne UK, Farrell PJ. Histone acetylation and reactivation of Epstein-Barr virus from latency. J Virol. (2000) 74:710–20. doi: 10.1128/JVI.74.2.710-720.2000
134. Countryman JK, Gradoville L, Miller G. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein-Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. J Virol. (2008) 82:4706–19. doi: 10.1128/JVI.00116-08
135. Hui KF, Chiang AK. Combination of proteasome and class I HDAC inhibitors induces apoptosis of NPC cells through an HDAC6-independent ER stress-induced mechanism. Int J Cancer (2014) 135:2950–61. doi: 10.1002/ijc.28924
136. Moosmann A, Khan N, Cobbold M, Zentz C, Delecluse HJ, Hollweck G, et al. B cells immortalized by a mini-Epstein-Barr virus encoding a foreign antigen efficiently reactivate specific cytotoxic T cells. Blood (2002) 100:1755–64.
137. Saulquin X, Bodinier M, Peyrat MA, Hislop A, Scotet E, Lang F, et al. Frequent recognition of BCRF1, a late lytic cycle protein of Epstein-Barr virus, in the HLA-B*2705 context: evidence for a TAP-independent processing. Eur J Immunol. (2001) 31:708–15. doi: 10.1002/1521-4141(200103)31:3<708::AID-IMMU708>3.0.CO;2-5
138. Plunkett FJ, Soares MV, Annels N, Hislop A, Ivory K, Lowdell M, et al. The flow cytometric analysis of telomere length in antigen-specific CD8+ T cells during acute Epstein-Barr virus infection. Blood (2001) 97:700–7. doi: 10.1182/blood.V97.3.700
139. Mills KH. Induction, function and regulation of IL-17-producing T cells. Eur J Immunol. (2008) 38:2636–49. doi: 10.1002/eji.200838535
140. Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol. (2009) 9:28–38. doi: 10.1038/nri2451
141. Lunemann A, Lunemann JD, Munz C. Regulatory NK-cell functions in inflammation and autoimmunity. Mol Med. (2009) 15:352–8. doi: 10.2119/molmed.2009.00035
142. Yao YL, Yang WM, Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol. (2001) 21:5979–91 doi: 10.1128/MCB.21.17.5979-5991.2001
143. Hui KF, Tam KP, Chiang AKS. Therapeutic strategies against Epstein-Barr Virus-associated cancers using proteasome inhibitors. Viruses (2017) 9:E352. doi: 10.3390/v9110352
144. Rapino F, Naumann I, Fulda S. Bortezomib antagonizes microtubule-interfering drug-induced apoptosis by inhibiting G2/M transition and MCL-1 degradation. Cell Death Dis. (2013) 4:e925. doi: 10.1038/cddis.2013.440
145. Hong YS, Hong SW, Kim SM, Jin DH, Shin JS, Yoon DH, et al. Bortezomib induces G2-M arrest in human colon cancer cells through ROS-inducible phosphorylation of ATM-CHK1. Int J Oncol. (2012) 41:76–82. doi: 10.3892/ijo.2012.1448.
146. Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia (2007) 21:838–42. doi: 10.1038/sj.leu.2404528
147. Cai Q, Guo Y, Xiao B, Banerjee S, Saha A, Lu J, et al. Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS Pathog. (2011) 7:e1002418. doi: 10.1371/journal.ppat.1002418
148. Tursiella ML, Bowman ER, Wanzeck KC, Throm RE, Liao J, Zhu J, et al. Epstein-Barr virus nuclear antigen 3A promotes cellular proliferation by repression of the cyclin-dependent kinase inhibitor p21WAF1/CIP1. PLoS Pathog. (2014) 10:e1004415. doi: 10.1371/journal.ppat.1004415
149. Zou P, Kawada J, Pesnicak L, Cohen JI. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J Virol. (2007) 81:10029–36. doi: 10.1128/JVI.02241-06
150. Shirley CM, Chen J, Shamay M, Li H, Zahnow CA, Hayward SD, et al. Bortezomib induction of C/EBPbeta mediates Epstein-Barr virus lytic activation in Burkitt lymphoma. Blood (2011) 117:6297–303. doi: 10.1182/blood-2011-01-332379
151. Granato M, Romeo MA, Tiano MS, Santarelli R, Gonnella R, Gilardini Montani MS, et al. Bortezomib promotes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci Rep. (2017) 7:13052. doi: 10.1038/s41598-017-13533-7
152. Hui KF, Yeung PL, Chiang AK. Induction of MAPK- and ROS-dependent autophagy and apoptosis in gastric carcinoma by combination of romidepsin and bortezomib. Oncotarget (2016) 7:4454–67. doi: 10.18632/oncotarget.6601
153. Hagemeier SR, Barlow EA, Meng Q, Kenney SC. The cellular ataxia telangiectasia-mutated kinase promotes epstein-barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J Virol. (2012) 86:13360–70. doi: 10.1128/JVI.01850-12
154. Yiu SPT, Hui KF, Choi CK, Kao RYT, Ma CW, Yang D, et al. Intracellular iron chelation by a novel compound, C7, reactivates Epstein(-)Barr virus (EBV) lytic cycle via the ERK-autophagy axis in EBV-positive epithelial cancers. Cancers (Basel) (2018) 10:E505. doi: 10.3390/cancers10120505
155. Pujals A, Favre L, Pioche-Durieu C, Robert A, Meurice G, Le Gentil M, et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy (2015) 11:2275–87. doi: 10.1080/15548627.2015.1115939
156. Bazot Q, Deschamps T, Tafforeau L, Siouda M, Leblanc P, Harth-Hertle ML, et al. Epstein-Barr virus nuclear antigen 3A protein regulates CDKN2B transcription via interaction with MIZ-1. Nucleic Acids Res. (2014) 42:9700–16. doi: 10.1093/nar/gku697
157. Knight JS, Lan K, Subramanian C, Robertson ES. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J Virol. (2003) 77:4261–72 doi: 10.1128/JVI.77.7.4261-4272.2003
158. Luftig M, Yasui T, Soni V, Kang MS, Jacobson N, Cahir-McFarland E, et al. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-kappaB activation. Proc Natl Acad Sci USA. (2004) 101:141–6. doi: 10.1073/pnas.2237183100
159. Atkinson PG, Coope HJ, Rowe M, Ley SC. Latent membrane protein 1 of Epstein-Barr virus stimulates processing of NF-kappa B2 p100 to p52. J Biol Chem. (2003) 278:51134–42. doi: 10.1074/jbc.M304771200
160. Bazot Q, Paschos K, Allday MJ. Epstein-Barr virus (EBV) latent protein EBNA3A directly targets and silences the STK39 gene in B cells infected by EBV. J Virol. (2018) 92:e01918-17. doi: 10.1128/JVI.01918-17
161. Paschos K, Smith P, Anderton E, Middeldorp JM, White RE, Allday MJ. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. (2009) 5:e1000492. doi: 10.1371/journal.ppat.1000492
162. Wood CD, Veenstra H, Khasnis S, Gunnell A, Webb HM, Shannon-Lowe C, et al. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. Elife (2016) 5:e18270. doi: 10.7554/eLife.18270
163. Tikhmyanova N, Schultz DC, Lee T, Salvino JM, Lieberman PM. Identification of a new class of small molecules that efficiently reactivate latent Epstein-Barr Virus. ACS Chem Biol. (2014) 9:785–95. doi: 10.1021/cb4006326
164. Choi CK, Ho DN, Hui KF, Kao RY, Chiang AK. Identification of novel small organic compounds with diverse structures for the induction of Epstein-Barr virus (EBV) lytic cycle in EBV-positive epithelial malignancies. PLoS ONE (2015) 10:e0145994. doi: 10.1371/journal.pone.0145994
Keywords: Epstein-Barr virus, lymphoproliferative diseases, viral-targeted strategies, EBV latency, lytic cycle reactivation, histone deacetylase inhibitors, proteasome inhibitors
Citation: Hui KF, Yiu SPT, Tam KP and Chiang AKS (2019) Viral-Targeted Strategies Against EBV-Associated Lymphoproliferative Diseases. Front. Oncol. 9:81. doi: 10.3389/fonc.2019.00081
Received: 01 October 2018; Accepted: 29 January 2019;
Published: 26 February 2019.
Edited by:
Vincent Yeung, Thomas Jefferson University, United StatesReviewed by:
Michael Diamantidis, University Hospital of Larissa, GreeceCopyright © 2019 Hui, Yiu, Tam and Chiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alan Kwok Shing Chiang, Y2hpYW5nYWtAaGt1Y2MuaGt1Lmhr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.