 Keisuke Yaku1
Keisuke Yaku1 Takashi Nakagawa
Takashi Nakagawa
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 12 December 2018
Sec. Cancer Molecular Targets and Therapeutics
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00622
Cancer cells have a unique energy metabolism for sustaining rapid proliferation. The preference for anaerobic glycolysis under normal oxygen conditions is a unique trait of cancer metabolism and is designated as the Warburg effect. Enhanced glycolysis also supports the generation of nucleotides, amino acids, lipids, and folic acid as the building blocks for cancer cell division. Nicotinamide adenine dinucleotide (NAD) is a co-enzyme that mediates redox reactions in a number of metabolic pathways, including glycolysis. Increased NAD levels enhance glycolysis and fuel cancer cells. In fact, nicotinamide phosphoribosyltransferase (Nampt), a rate-limiting enzyme for NAD synthesis in mammalian cells, is frequently amplified in several cancer cells. In addition, Nampt-specific inhibitors significantly deplete NAD levels and subsequently suppress cancer cell proliferation through inhibition of energy production pathways, such as glycolysis, tricarboxylic acid (TCA) cycle, and oxidative phosphorylation. NAD also serves as a substrate for poly(ADP-ribose) polymerase (PARP), sirtuin, and NAD gylycohydrolase (CD38 and CD157); thus, NAD regulates DNA repair, gene expression, and stress response through these enzymes. Thus, NAD metabolism is implicated in cancer pathogenesis beyond energy metabolism and considered a promising therapeutic target for cancer treatment. In this review, we present recent findings with respect to NAD metabolism and cancer pathogenesis. We also discuss the current and future perspectives regarding the therapeutics that target NAD metabolic pathways.
Cancer cells involve a unique energy metabolism that promotes their rapid cell proliferation (1). One of the characteristic features of cancer metabolism is the preference for anaerobic glycolysis under normal aerobic conditions, known as the Warburg effect (2, 3). Although aerobic glycolysis efficiently yields 32 molecules of ATP from one molecule of glucose, only 2 molecules of ATP are generated from one molecule of glucose during anaerobic glycolysis (4). This shift to anaerobic glycolysis under normal oxygen conditions in cancer cells appears inefficient; however, the advantages of anaerobic glycolysis include a faster rate of ATP production and reduced generation of reactive oxygen species (ROS) that are mainly produced by ETC during respiration (4). Therefore, cancer cells appear to utilize these advantages instead of focusing on the efficient ATP generation.
Another advantage of shifting energy metabolism to anaerobic glycolysis is the production of the building blocks required for cancer proliferation (5). Glucose 6-phosphate (G6P), the first product of glycolysis that is generated by hexokinase, bypasses glycolysis through the pentose phosphate pathway (PPP). 5-phosphoribose-1-pyrophosphate (PRPP) is an intermediate product of the PPP and is used in biosynthesis of purine and pyrimidine nucleotides. Thus, PPP is activated in cancer cells to maintain sufficient nucleotide pools to support DNA replication and RNA production (6). In some cancer cells, a large proportion of glucose is used in the serine de novo synthesis pathway, wherein 3-phosphoglycerate is used by D-3-phosphoglycerate dehydrogenase (PHGDH) (7). Serine metabolism is associated with the synthesis of ceramide, a component of the cellular membrane (8). Serine is also converted to glycine and connected to the folic acid and methionine metabolism (9, 10). Thus, the serine biosynthesis pathway is also considered critical for sustaining the growth of cancer cells.
Nicotinamide adenine dinucleotide (NAD) is a co-enzyme that mediates redox reactions in various metabolic pathways, including glycolysis, tricarboxylic acid (TCA) cycle, oxidative phosphorylation, and serine biosynthesis (11). Continuous replenishment of NAD promotes the proliferation and survival of fast-dividing cancer cells because elevated NAD levels enhance glycolysis via glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and lactate dehydrogenase (LDH) that require NAD as a co-enzyme (12, 13). PHGDH, a rate-limiting enzyme of the serine biosynthesis pathway, also uses NAD as a co-enzyme, and the intracellular level of NAD is considered to be an important regulator for serine biosynthesis in cancer cells (9, 14). Furthermore, NAD serves as a substrate for poly(ADP-ribose) polymerase (PARP) and sirtuins (NAD-dependent deacetylases) and mediates poly-ADP-ribosylation and deacetylation, respectively. Thus, NAD metabolism is involved in energy metabolism, DNA repair, gene expression, and stress response via the action of these enzymes (15). Recently, several studies have indicated that NAD metabolism is involved in cancer development and progression and is considered a promising therapeutic target in cancer treatment. In this review, we summarize the roles of NAD metabolism in cancer pathogenesis. We also focus on the inhibitors of NAD-synthesis enzymes, and describe their implications in cancer treatment.
NAD is synthesized through the de novo, Preiss-Handler, and salvage pathways from tryptophan, nicotinic acid (NA), and nicotinamide (NAM), respectively (Figure 1). In mammalian cells, two key enzymes, nicotinamide phosphoribosyltransferase (Nampt) and nicotinamide mononucleotide adenylyltransferase (Nmnat) regulate the salvage pathway that is considered critical in controlling intracellular NAD levels (11). Nampt converts NAM and PRPP to nicotinamide mononucleotide (NMN), and Nmnat generates NAD by transferring the adenylyl moiety from ATP to NMN (16, 17). In the Preiss-Handler pathway, nicotinate phosphoribosyltransferase (Naprt) generates nicotinic acid mononucleotide (NAMN) from NA and PRPP, and then Nmnat conjugates ATP to NAMN to generate NAD (18). In the de novo pathway, tryptophan is used as the source for NAD synthesis; further, tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO) mediates the first step and acts as a rate-limiting enzyme in this pathway. In the salvage pathway, NAD degradation is coupled with NAM recycle (19). PARP and sirtuin use NAD as a substrate for ADP-ribosylation and deacetylation, respectively (20, 21). NAD glycohydrolases, CD38 and CD157, also consume NAD and generate ADP-ribose or cyclic-ADP-ribose (22, 23). All these enzymes generate NAM when they degrade NAD, and Nampt reuses NAM for NAD synthesis. In mammals, there are three Nmnat isozymes (Nmnat1–3) with different subcellular localizations and tissue distributions. Nmnat1, Nmnat2, and Nmnat3 are considered to be in the nucleus, Golgi apparatus, and mitochondria, respectively (24). Additionally, Nampt is primarily located in the cytoplasm, and its inhibition blocks glycolysis (13). Nmnat1 is reported to supply nuclear NAD and sustain the activity of PARP and sirtuin (25, 26). In mitochondria, NAD is utilized in TCA cycle, fatty acid oxidation, and oxidative phosphorylation (27). In fact, overexpression of Nmnat3 in mice increases mitochondrial NAD levels and enhances energy metabolism in mitochondria (28).
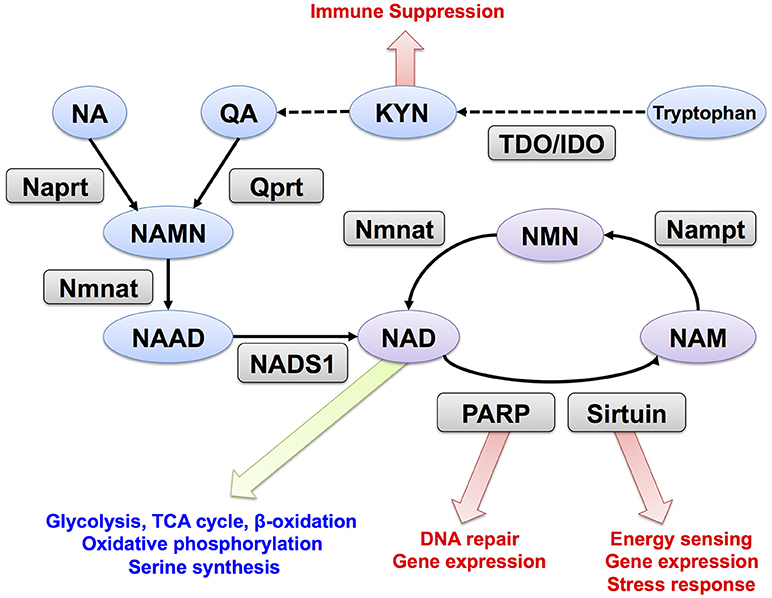
Figure 1. NAD metabolism and its downstream targets. Trp, tryptophan; KYN, kynurenine; NA, nicotinic acid; NAM, nicotinamide; QA, quinolinic acid; NMN, nicotinamide mononucleotide; NAMN, nicotinic acid mononucleotide; NAD, Nicotinamide adenine dinucleotide; NAAD, nicotinic acid adenine dinucleotide; Nampt, nicotinamide phosphoribosyltransferase; Nmnat, nicotinamide mononucleotide adenylyltransferase; Qprt, quinolinic acid phosphoribosyltransferase; Naprt, nicotinic acid phosphoribosyltransferase; NADS1, NAD synthetase; PARP, poly (ADP-ribose) polymerase. TDO, tryptophan 2,3-dioxygenase; IDO, indoleamine 2,3-dioxygenase.
Overexpression of Nampt is frequently observed in several types of malignant tumors, including, colorectal, ovarian, breast, gastric, thyroid, prostate cancers, gliomas, and malignant lymphomas (29–48). Increased NAD levels accompanied by Nampt overexpression sustain rapid cellular proliferation and promote cancer cell survival against anti-cancer cell reagents. In particular, elevated NAD levels boost glycolysis through glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and lactate dehydrogenase (LDH) that require NAD as a co-enzyme and enhance anaerobic glycolysis (12, 13). A well-known oncogene, c-MYC was reported to regulate Nampt expression in cancer cells (49). c-MYC transcriptionally regulates the metabolic reprogramming of cancer cells by enhancing glucose uptake, glycolysis, and lactate production, the increase in Nampt expression by c-MYC may lead to the Warburg effects (50). Several microRNAs regulate Nampt levels and promote cancer cell proliferation. miR26b reportedly suppresses Nampt expression by binding to the 3'-UTR in the Nampt gene. In colorectal cancer cells, miR26b is down regulated, leading to the overexpression of Nampt (51). Similarly, decreased miR206 also induced Nampt expression in breast and pancreatic cancers. In contrast, the exogenous expression of miR-206 reduced the Nampt expression, resulting in a significantly reduced level of NAD and subsequent induction of cell death (44, 52). Other microRNAs, including miR374a, miR451a, and miR568 reportedly regulate Nampt levels in cancer cells (51, 53, 54).
Nampt was originally identified as a cytokine named pre-B-cell colony-enhancing factor (also known as a visfatin), and exists in serum as a secreted form (55, 56). An extracellular form of Nampt (eNampt) is released from multiple types of cells, including mature adipocytes, hepatocytes, myocytes, neurons, and immune cells (57–62). Moreover, eNampt was released from cancer cells, and the eNampt level is reportedly associated with the progression of certain types of cancers (63–68). eNampt induces inflammatory cytokines in the leukocytes, macrophages, gut epithelial cells, and endothelial cells (64, 69, 70) and promotes cancer-related inflammation. Furthermore, eNampt has been reported to polarize the monocytes toward tumor-supporting M2-macrophages. eNampt increases the expression of immunosuppressive molecules, such as IDO, CCL-18, and IL-10, as well as tumor-promoting cytokines, such as IL-1β and IL-6 (64, 69). eNampt also acts as a pro-angiogenic factor by promoting the expressions of VEGF and MMP2/9 through the MAPK and PI3K/Akt signaling pathways (71–73). Overexpression of Nampt also promotes epithelial-to-mesenchymal transition (EMT) by increasing eNampt, and activates the TGFβ signaling pathway (74). In addition, Nampt inhibition can kill the gastric cancer cell lines with EMT-associated gene expression signatures (75). These results suggest that specific neutralization of eNampt can be used as a potential anti-cancer strategy.
A number of studies have demonstrated that the overexpression of Nampt contributes to cancer pathogenesis and development; therefore, Nampt is considered as a potential therapeutic target for cancers. Accordingly, several kinds of Nampt inhibitors have been developed (Table 1). FK866 (also known as APO866) is the first chemical compound discovered to be a highly specific inhibitor of Nampt (76). It inhibits the enzymatic activity of Nampt non-competitively by binding to an allosteric regulatory site (76). The treatment of cultured cancer cells by FK866 significantly reduces intracellular NAD levels followed by the inhibition of the glycolysis pathway catalyzed by GAPDH (13, 93). Thus, FK866 suppresses cancer cell growth by targeting energy metabolism, particularly glycolysis (94). Another research has also shown that FK866 kills blood cancer cells by inducing apoptosis and/or caspase-independent autophagic cell death (95).
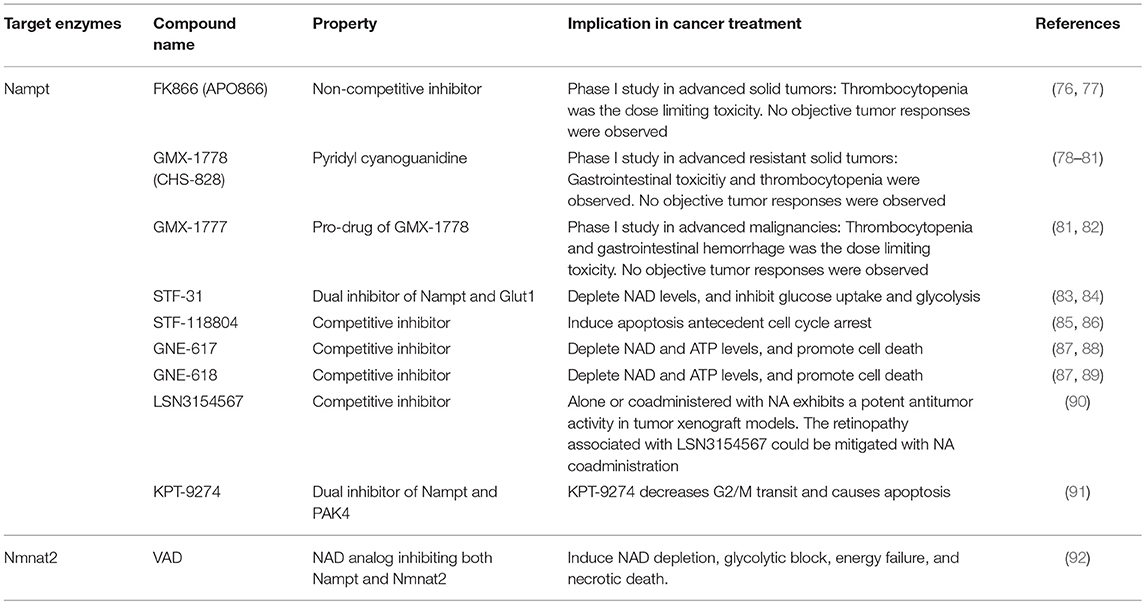
Table 1. Small molecules targeting NAD synthesis enzymes.
Other Nampt-specific inhibitors, such as GMX1777 and GMX1778, have exhibited anti-cancer effects similar to those of FK866 (78). GMX1777 is a cyanoguanidine compound and a pro-drug of GMX1778 (also known as CHS-828) (82). It is rapidly converted into the active form GMX1778, a competitive inhibitor of Nampt (79). The combination of GMX1777 and radiotherapy has demonstrated therapeutic efficacy in a mouse model of head and neck cancer (96). STF-31 was originally identified as a compound that selectively kills renal cell carcinoma with a loss of the von Hippel-Lindau (VHL) tumor suppressor gene (83). STF-31 inhibits glucose uptake and the Warburg effect by targeting Glut1 (83). A more recent study has demonstrated that recurrent NAMPT-H191R mutations confer resistance to STF-31 treatment in cancer cell lines and that STF-31 also exerts an additional inhibitory effect against Nampt (84). Thus, these dual functions are beneficial for countering tumors with a high glucose consumption rate.
Although Nampt inhibitors are promising candidates for preventing tumor cell growth, Nampt is also essential for normal cells. In fact, whole body deletion of Nampt results in embryonic lethality, and muscle-specific Nampt deficient mice exhibit progressive muscle degeneration (57, 97). Moreover, retina-specific Nampt deficient mice exhibit severe vision loss (98). Therefore, it is important to consider these adverse side effects while using Nampt inhibitors in clinical settings.
In contrast to the cases with respect to Nampt, several types of cancer cells reportedly lack in Naprt expression (99, 100). These cells cannot utilize NA for NAD synthesis through Naprt; hence, NAD synthesis solely depends on Nampt. Therefore, the efficacy of FK866 treatment is higher than that in cancer cells that express Naprt. In fact, the DNA hypermethylation of the promoter region of Naprt is frequently observed in certain types of cancers, and this could be used as an effective marker for treatment with FK866 (101). Normal cells usually express both Naprt and Nampt; therefore, co-administration of Nampt inhibitor and NA can preserve NAD levels through NA-Naprt pathway in normal cells. On the other hand, Nampt inhibitor effectively decreases NAD levels in cancer cells lacking Naprt, and subsequent ATP depletion due to impaired glycolysis can selectively kill cancer cells (100). Similarly, selective inhibition of Nampt by GMX1778 blocks NAD production and induces cell death in Naprt-deficient tumors, while NA co-supplementation can rescue NAD depletion in Naprt-positive normal cells, bypassing the NA-Naprt pathway (79). Thus, the Naprt status in tumor cells is a critical indicator in the treatment of cancers using Nampt inhibitors.
Isocitrate dehydrogenase (IDH) mutations in glioma cell lines are reported to decrease the expression of Naprt via increased DNA and histone methylation (102). Mutations in the IDH gene are frequently observed in patients with gliomas and acute myeloid leukemia (AML). Mutated IDH aberrantly generates 2-hydroxyglutarate (2-HG), which inhibits DNA and histone demethylases and promotes the hyper methylation of DNA histones (103). In IDH-mutated cells, NAD levels are significantly decreased due to decreased Naprt, making them sensitive to NAD depletion by Nampt inhibitors, such as 102, FK866 and GMX1778 (104). Decreased NAD levels in FK866-treated IDH-mutant cells suppress the flux in TCA cycle and cause reduction in ATP production, which activates the intracellular energy sensor AMPK, triggers autophagy, and finally leads to both autophagic cell death (102). In fact, 3-methyladenine, an inhibitor of autophagy, partially rescues this cytotoxicity (102, 105). Thus, IDH mutation in tumors, including gliomas and AML, is considered a good indicator of the suitability of Nampt inhibitor therapy.
It is noteworthy that aberrant expression of unconventional prefoldin RPB5 interactor (URI) in hepatocytes causes NAD depletion, ultimately leading to spontaneous liver tumor formation (106). URI downregulates the gene expression of enzymes in the kynurenine pathway through aryl hydrocarbon receptor and estrogen receptors. Depleted NAD levels subsequently promote DNA damage and result in the onset of hepatocellular carcinoma. Notably, nicotinamide riboside (NR) supplementation restores the NAD levels in URI-expressing hepatocytes and prevents DNA damage and tumor formation. In human hepatocellular carcinoma patients, URI expression is negatively correlated with that of the enzymes in the kynurenine pathway and is associated with a poor prognosis (106).
Nmnat inhibitors are expected to exhibit anti-cancer effects via the inhibition of the NAD synthesis pathway (107). Nmnat2 is amplified in colorectal cancer and exhibits a positive correlation with p53 expression. Furthermore, it is reported that the expression of Nmnat2 under DNA damage was induced by p53 (108). Reportedly, SIRT3 activates Nmnat2 through deacetylation and increased NAD levels in non-small cell lung-cancer cells (109). SIRT3-Nmnat2-NAD axis dictates the energy metabolism and cellular proliferation, and disrupting the interaction between SIRT3 and Nmnat2 promotes apoptosis. These results imply that Nmnat2 can be a therapeutic target (109). A recent study has reported that vacor adenine dinucleotide (VAD) inhibits Nmnat2 and Nmapt (92). It is noteworthy that the administration of vacor to Nmnat2-expressing cancer cell leads to VAD formation and depletes NAD pools (92). Furthermore, vacor leads to growth suppression and necrotic cell death via blockage of glycolysis in Nmnat2-positive cancer cells; however, it has no effect on Nmnat2-lacking cells. Overexpression of Nmnat2 in colorectal cancer was reported to enhance cell death induced by tiazofurin, an analog of NAD. Nmnat2 enzymatically converts tiazofurin to thiazole-4-carboxamide adenine dinucleotide, a potent inhibitor of inosine 5'-monophosphate dehydrogenase, required for guanylate synthesis in colorectal cancers (110). Taken together, amplification of Nmnat2 in cancer cells could be a unique target for these compounds.
Intracellular NAD levels also influence the activities of the NAD consuming enzymes, PARP and sirtuin, and regulate cancer initiation and progression. NAD serves as a substrate for PARP that plays crucial roles in DNA repair, chromatin modification, cell transformation, and cell death (20). In particular, DNA repair is targeted by several cancer therapies, including chemotherapy and radiation therapy. Recently, PARP inhibitors have been identified as promising therapeutic agents against breast and ovarian cancers with BRCA mutations. Because BRCA mutations impair the homologous recombination of DNA double-strand breaks (DSBs) in cancer cells, PARP inhibitors increase DNA single-strand breaks (SSBs) and lead to synthetic lethality. Poly ADP-ribosylation during the DNA repair process consumes a large amount of NAD; limiting NAD synthesis may also induce cancer cell death by inhibiting the PARP activity (20). In fact, the combination of a PARP inhibitor and FK866 induced synthetic lethality in triple-negative breast cancer (111). It has also been reported that a combination of PARP inhibitors and β-lapachone, a natural quinone, exhibits synergistic anti-tumor activity against NAD(P)H:quinone oxidoreductase 1 (NQO1)-overexpressing cancers, such as non-small-cell lung cancer, pancreatic cancer, and breast cancer (112). NQO1 is an enzyme that couples NAD(P) oxidation with a reduction of quinone to hydroquinone, and β-lapachone is converted to an unstable hydroquinone that spontaneously generates ROS and induces DNA damages (113). However, β-lapachone also promotes hyper activation of PARP and damages normal cells by depleting NAD and ATP levels (112, 114). Thus, the co-administration of β-lapachone and PARP inhibitors to cancer cells generates considerable amount of hydrogen peroxide without affecting the NAD pools that constantly refuel NQO1 redox cycling and induce massive DNA damage (112). Therefore, the combination of PARP inhibitors and β-lapachone blocks DNA repair and induces tumor-selective apoptosis in NQO1-overexpressing cancers.
The NAD-dependent deacetylases (sirtuins) affect various processes involved in the suppression of tumor initiation and maintenance. Thus, a disturbance in the NAD levels could influence sirtuin-mediated suppression of tumor formation and growth. In fact, dysregulated c-Myc initiates a positive feedback loop between Nampt, the SIRT1 inhibitor DBC1, and SIRT1, that contributes to tumor development and maintenance (49). Recently, it has been reported that Nampt expression is correlated to a large number of cancer-initiating cells in colon cancer patients, and transcription meta-analysis has revealed that Nampt regulates the pathways involved in the maintenance of cancer stemness in colon cancers in a SIRT1- and PARP1-dependent manner (30).
More than 90% of multiple myeloma cells from patients have surface expression of CD38 (115). Daratumumab, an anti-CD38 monoclonal antibody, has been approved as a therapy for multiple myeloma. Daratumumab strongly induces antibody-dependent cellular cytotoxicity and kills myeloma cells. However, the pathological significance of CD38 expression in multiple myeloma cells remains unclear. CD38 reportedly regulates intracellular NAD levels, and CD38 deficiency in mice increases the NAD levels in multiple tissues (116). A recent study has indicated that CD38 expression inversely correlates with the progression of prostate cancer (117, 118). Thus, the inhibition of the enzymatic activity of intracellular CD38 may have adverse effects on tumor proliferation. In myeloma cells, CD38 is overexpressed ectopically, and it is unclear whether extracellular NAD is involved in tumor progression. CD157/BST1 is a paralog of CD38 and shares similar NADase activity. In ovarian cancer, CD157 is often overexpressed and contributes to cell migration and peritoneal invasion (119, 120). Thus, the inhibition of CD157 may also contribute to cancer therapy.
The Warburg effect was discovered more than half a century previously, and the strategy targeting cancer metabolism is regaining popularity. NAD is an important co-factor that mediates a number of metabolic pathways, including glycolysis. In fact, Nampt is overexpressed in several types of malignant tumors and is considered a potential therapeutic target. Various Nampt-specific inhibitors have been developed, and they exhibit effective responses to cancer cells in pre-clinical models (76, 78, 82, 83, 85–91). Although Nampt inhibitor is a promising anti-cancer reagent, several challenges remain with respect to the application in human patients. Most importantly, Nampt is also essential for normal cells, and complete deletion of Nampt in mice has shown to have adverse effects in the retina, bone marrow, liver, and blood platelets. In fact, clinical trials using Nampt inhibitors have encountered hematological toxicities, and their clinical development has been stagnated (77, 80, 81, 121). Therefore, it is important to select patients with therapeutic adaptation to Nampt inhibitor. Thus, the status of Naprt expression is considered a good indicator of the suitability of Nampt inhibitor therapy, and the co-administration of Nampt inhibitor and NA, a substrate for Naprt, is a promising strategy to decrease the toxicity in normal cells. IDH mutation increases Naprt expression in glioma cells, and patients with this mutation may be suitable for Nampt inhibition therapy (102, 105). A recent study has demonstrated that treatment with Nampt inhibitor-containing microparticle successfully extended the survival of mice with IDH-mutated gliomas. Further, the research group has developed a method to determine the presence of mutation in glioma cells using surgical specimens within 30 min. If the test is positive, patients can be treated locally by applying Nampt inhibitor-containing microparticle to the primary lesion during operation (104). Thus, this procedure is considered ideal to avoid adverse side effects and maximize anti-tumor effect to suitable subjects for Nampt inhibitor therapy. Another option is to induce a synthetic lethality using Nampt inhibitors and other anti-cancer drugs. It has been known that the repair process of SSB DNA damage consumes a large amount of NAD through PARP-mediated poly ADP-ribosylation. Thus, the combination with DNA damage inducing reagent and Nampt inhibitor may synergistically induce cell death in cancer cells.
KY, KO, KH, and TN wrote the manuscript. TN and KY revised and edited manuscript.
This work is partially supported by JSPS KAKENHI (Grant Number 17K08653 to TN, 18K17921 to KY, and 18K16193 to KO).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Dr. Michiya Sugimori (University of Toyama) for critical reading of the manuscript.
1. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
4. Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. (2015) 356(2 Pt A):156–64. doi: 10.1016/j.canlet.2014.04.001
5. Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med. (2015) 79:253–63. doi: 10.1016/j.freeradbiomed.2014.08.027
6. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
7. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. (2011) 43:869–74. doi: 10.1038/ng.890
8. Hwang S, Gustafsson HT, O'Sullivan C, Bisceglia G, Huang X, Klose C, et al. Serine-dependent sphingolipid synthesis is a metabolic liability of aneuploid cells. Cell Rep. (2017) 21:3807–18. doi: 10.1016/j.celrep.2017.11.103
9. Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino Acid. J Biol Chem. (2012) 287:19786–91. doi: 10.1074/jbc.R112.357194
10. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. (2014) 39:191–8. doi: 10.1016/j.tibs.2014.02.004
11. Yaku K, Okabe K, Nakagawa T. NAD metabolism: implications in aging and longevity. Ageing Res Rev. (2018) 471:17. doi: 10.1016/j.arr.2018.05.006
12. Yamamoto M, Inohara H, Nakagawa T. Targeting metabolic pathways for head and neck cancers therapeutics. Cancer Metastasis Rev. (2017) 36:503–14. doi: 10.1007/s10555-017-9691-z
13. Tan B, Young DA, Lu ZH, Wang T, Meier TI, Shepard RL, et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. J Biol Chem. (2013) 288:3500–11. doi: 10.1074/jbc.M112.394510
14. Murphy JP, Giacomantonio MA, Paulo JA, Everley RA, Kennedy BE, Pathak GP, et al. The NAD(+) salvage pathway supports PHGDH-driven serine biosynthesis. Cell Rep. (2018) 24:2381–91 e2385. doi: 10.1016/j.celrep.2018.07.086
15. Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, et al. NAD(+) in aging: molecular mechanisms and translational implications. Trends Mol Med. (2017) 23:899–916. doi: 10.1016/j.molmed.2017.08.001
16. Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. (2004) 279:50754–63. doi: 10.1074/jbc.M408388200
17. Jayaram HN, Kusumanchi P, Yalowitz JA. NMNAT expression and its relation to NAD metabolism. Curr Med Chem. (2011) 18:1962–72. doi: 10.2174/092986711795590138
18. Hara N, Yamada K, Shibata T, Osago H, Hashimoto T, Tsuchiya M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J Biol Chem. (2007) 282:24574–82. doi: 10.1074/jbc.M610357200
19. Badawy AA. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res. (2017) 101–10. doi: 10.1177/1178646917691938
20. Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes Dev. (2005) 19:1951–67. doi: 10.1101/gad.1331805
21. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature (2000) 403:795–800. doi: 10.1038/35001622
22. Ceni C, Pochon N, Brun V, Muller-Steffner H, Andrieux A, Grunwald D, et al. CD38-dependent ADP-ribosyl cyclase activity in developing and adult mouse brain. Biochem J. (2003) 370(Pt 1):175–83. doi: 10.1042/BJ20020604
23. Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, et al. CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin Cytom. (2013) 84:207–17. doi: 10.1002/cyto.b.21092
24. Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. (2005) 280:36334–41. doi: 10.1074/jbc.M508660200
25. Zhang T, Berrocal JG, Frizzell KM, Gamble MJ, DuMond ME, Krishnakumar R, et al. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem. (2009) 284:20408–17. doi: 10.1074/jbc.M109.016469
26. Zhang T, Berrocal JG, Yao J, DuMond ME, Krishnakumar R, Ruhl DD, et al. Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. J Biol Chem. (2012) 287:12405–16. doi: 10.1074/jbc.M111.304469
27. Anderson KA, Madsen AS, Olsen CA, Hirschey MD. Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. Biochim Biophys Acta (2017) 1858:991–8. doi: 10.1016/j.bbabio.2017.09.005
28. Gulshan M, Yaku K, Okabe K, Mahmood A, Sasaki T, Yamamoto M, et al. Overexpression of Nmnat3 efficiently increases NAD and NGD levels and ameliorates age-associated insulin resistance. Aging Cell (2018) 2018:e12798. doi: 10.1111/acel.12798
29. Lucena-Cacace A, Otero-Albiol D, Jimenez-Garcia MP, Peinado-Serrano J, Carnero A. NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget (2017) 8:99514–30. doi: 10.18632/oncotarget.20577
30. Lucena-Cacace A, Otero-Albiol D, Jimenez-Garcia MP, Munoz-Galvan S, Carnero A. NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through Sirt1 and PARP. Clin Cancer Res. (2017) 24:1202–15. doi: 10.1158/1078-0432.CCR-17-2575
31. Sawicka-Gutaj N, Waligorska-Stachura J, Andrusiewicz M, Biczysko M, Sowinski J, Skrobisz J, et al. Nicotinamide phosphorybosiltransferase overexpression in thyroid malignancies and its correlation with tumor stage and with survivin/survivin DEx3 expression. Tumour Biol. (2015) 36:7859–63. doi: 10.1007/s13277-015-3506-z
32. Shackelford RE, Bui MM, Coppola D, Hakam A. Over-expression of nicotinamide phosphoribosyltransferase in ovarian cancers. Int J Clin Exp Pathol. (2010) 3:522–7.
33. Zhu Y, Guo M, Zhang L, Xu T, Wang L, Xu G. Biomarker triplet NAMPT/VEGF/HER2 as a de novo detection panel for the diagnosis and prognosis of human breast cancer. Oncol Rep. (2016) 35:454–62. doi: 10.3892/or.2015.4391
34. Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene (2011) 30:907–21. doi: 10.1038/onc.2010.468
35. Bi TQ, Che XM, Liao XH, Zhang DJ, Long HL, Li HJ, et al. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol Rep. (2011) 26:1251–7. doi: 10.3892/or.2011.1378
36. Patel S, Ansari J, Meram A, Abdulsattar J, Cotelingam J, Coppola D, et al. Increased nicotinamide phosphoribosyltransferase and cystathionine-beta-synthase in oral cavity squamous cell carcinomas. Int J Clin Exp Pathol. (2017) 10:702–7.
37. Van Beijnum JR, Moerkerk PT, Gerbers AJ, De Bruine AP, Arends JW, Hoogenboom HR, et al. Target validation for genomics using peptide-specific phage antibodies: a study of five gene products overexpressed in colorectal cancer. Int J Cancer (2002) 101:118–27. doi: 10.1002/ijc.10584
38. Folgueira MA, Carraro DM, Brentani H, Patrao DF, Barbosa EM, Netto MM, et al. Gene expression profile associated with response to doxorubicin-based therapy in breast cancer. Clin Cancer Res. (2005) 11:7434–43. doi: 10.1158/1078-0432.CCR-04-0548
39. Patel ST, Mistry T, Brown JE, Digby JE, Adya R, Desai KM, et al. A novel role for the adipokine visfatin/pre-B cell colony-enhancing factor 1 in prostate carcinogenesis. Peptides (2010) 31:51–7. doi: 10.1016/j.peptides.2009.10.001
40. Neubauer K, Misa IB, Diakowska D, Kapturkiewicz B, Gamian A, Krzystek-Korpacka M. Nampt/PBEF/visfatin upregulation in colorectal tumors, mirrored in normal tissue and whole blood of colorectal cancer patients, is associated with metastasis, hypoxia, IL1beta, and anemia. Biomed Res Int. (2015) 2015:523930. doi: 10.1155/2015/523930
41. Li D, Chen NN, Cao JM, Sun WP, Zhou YM, Li CY, et al. BRCA1 as a nicotinamide adenine dinucleotide (NAD)-dependent metabolic switch in ovarian cancer. Cell Cycle (2014) 13:2564–71. doi: 10.4161/15384101.2015.942208
42. Hufton SE, Moerkerk PT, Brandwijk R, de Bruine AP, Arends JW, Hoogenboom HR. A profile of differentially expressed genes in primary colorectal cancer using suppression subtractive hybridization. FEBS Lett. (1999) 463:77–82.
43. Zhao W, Chen R, Zhao M, Li L, Fan L, Che XM. High glucose promotes gastric cancer chemoresistance in vivo and in vitro. Mol Med Rep. (2015) 12:843–50. doi: 10.3892/mmr.2015.3522
44. Ju HQ, Zhuang ZN, Li H, Tian T, Lu YX, Fan XQ, et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. (2016) 379:1–11. doi: 10.1016/j.canlet.2016.05.024
45. Olesen UH, Hastrup N, Sehested M. Expression patterns of nicotinamide phosphoribosyltransferase and nicotinic acid phosphoribosyltransferase in human malignant lymphomas. APMIS (2011) 119:296–303. doi: 10.1111/j.1600-0463.2011.02733.x
46. Vora M, Ansari J, Shanti RM, Veillon D, Cotelingam J, Coppola D, et al. Increased nicotinamide phosphoribosyltransferase in rhabdomyosarcomas and leiomyosarcomas compared to skeletal and smooth muscle tissue. Anticancer Res. (2016) 36:503–7.
47. Shackelford R, Hirsh S, Henry K, Abdel-Mageed A, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase and SIRT3 expression are increased in well-differentiated thyroid carcinomas. Anticancer Res. (2013) 33:3047–52.
48. Shackelford RE, Abdulsattar J, Wei EX, Cotelingam J, Coppola D, Herrera GA. Increased nicotinamide phosphoribosyltransferase and cystathionine-beta-synthase in renal oncocytomas, renal urothelial carcinoma, and renal clear cell carcinoma. Anticancer Res. (2017) 37:3423–7. doi: 10.21873/anticanres.11709
49. Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Luscher B, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci USA. (2012) 109:E187–96. doi: 10.1073/pnas.1105304109
50. Tarrado-Castellarnau M, de Atauri P, Cascante M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget (2016) 7:62726–53. doi: 10.18632/oncotarget.10911
51. Zhang C, Tong J, Huang G. Nicotinamide phosphoribosyl transferase (Nampt) is a target of microRNA-26b in colorectal cancer cells. PLoS ONE (2013) 8:e69963. doi: 10.1371/journal.pone.0069963
52. Hesari Z, Nourbakhsh M, Hosseinkhani S, Abdolvahabi Z, Alipour M, Tavakoli-Yaraki M, et al. Down-regulation of NAMPT expression by mir-206 reduces cell survival of breast cancer cells. Gene (2018) 673:149–58. doi: 10.1016/j.gene.2018.06.021
53. Adyshev DM, Elangovan VR, Moldobaeva N, Mapes B, Sun X, Garcia JG. Mechanical stress induces pre-B-cell colony-enhancing factor/NAMPT expression via epigenetic regulation by miR-374a and miR-568 in human lung endothelium. Am J Respir Cell Mol Biol. (2014) 50:409–18. doi: 10.1165/rcmb.2013-0292OC
54. Yamada Y, Arai T, Sugawara S, Okato A, Kato M, Kojima S, et al. Impact of novel oncogenic pathways regulated by antitumor miR-451a in renal cell carcinoma. Cancer Sci. (2018) 109:1239–53. doi: 10.1111/cas.13526
55. Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. (1994) 14:1431–7.
56. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science (2005) 307:426–30. doi: 10.1126/science.1097243
57. Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. (2007) 6:363–75. doi: 10.1016/j.cmet.2007.09.003
58. Garten A, Petzold S, Barnikol-Oettler A, Korner A, Thasler WE, Kratzsch J, et al. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem Biophys Res Commun. (2010) 391:376–81. doi: 10.1016/j.bbrc.2009.11.066
59. Kover K, Tong PY, Watkins D, Clements M, Stehno-Bittel L, Novikova L, et al. Expression and regulation of nampt in human islets. PLoS ONE (2013) 8:e58767. doi: 10.1371/journal.pone.0058767
60. Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, et al. SIRT1-mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD+ and function in mice. Cell Metab. (2015) 21:706–17. doi: 10.1016/j.cmet.2015.04.002
61. Curat CA, Wegner V, Sengenes C, Miranville A, Tonus C, Busse R, et al. Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia (2006) 49:744–7. doi: 10.1007/s00125-006-0173-z
62. Jing Z, Xing J, Chen X, Stetler RA, Weng Z, Gan Y, et al. Neuronal NAMPT is released after cerebral ischemia and protects against white matter injury. J Cereb Blood Flow Metab. (2014) 34:1613–21. doi: 10.1038/jcbfm.2014.119
63. Grolla AA, Torretta S, Gnemmi I, Amoruso A, Orsomando G, Gatti M, et al. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is a tumoural cytokine released from melanoma. Pigment Cell Melanoma Res. (2015) 28:718–29. doi: 10.1111/pcmr.12420
64. Audrito V, Serra S, Brusa D, Mazzola F, Arruga F, Vaisitti T, et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood (2015) 125:111–23. doi: 10.1182/blood-2014-07-589069
65. Lin YC, Wu HC, Liao CC, Chou YC, Pan SF, Chiu CM. Secretion of one adipokine Nampt/Visfatin suppresses the inflammatory stress-induced NF-kappaB activity and affects Nampt-dependent cell viability in Huh-7 cells. Mediators Inflamm. (2015) 2015:392471. doi: 10.1155/2015/392471
66. Sun Y, Zhu S, Wu Z, Huang Y, Liu C, Tang S, et al. Elevated serum visfatin levels are associated with poor prognosis of hepatocellular carcinoma. Oncotarget (2017) 8:23427–35. doi: 10.18632/oncotarget.15080
67. Nakajima TE, Yamada Y, Hamano T, Furuta K, Gotoda T, Katai H, et al. Adipocytokine levels in gastric cancer patients: resistin and visfatin as biomarkers of gastric cancer. J Gastroenterol. (2009) 44:685–90. doi: 10.1007/s00535-009-0063-5
68. Audrito V, Manago A, Zamporlini F, Rulli E, Gaudino F, Madonna G, et al. Extracellular nicotinamide phosphoribosyltransferase (eNAMPT) is a novel marker for patients with BRAF-mutated metastatic melanoma. Oncotarget (2018) 9:18997–9005. doi: 10.18632/oncotarget.24871
69. Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. (2007) 178:1748–58. doi: 10.4049/jimmunol.178.3.1748
70. Gerner RR, Klepsch V, Macheiner S, Arnhard K, Adolph TE, Grander C, et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut (2018) 67:1813–23. doi: 10.1136/gutjnl-2017-314241
71. Adya R, Tan BK, Punn A, Chen J, Randeva HS. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc Res. (2008) 78:356–65. doi: 10.1093/cvr/cvm111
72. Kim JG, Kim EO, Jeong BR, Min YJ, Park JW, Kim ES, et al. Visfatin stimulates proliferation of MCF-7 human breast cancer cells. Mol Cells (2010) 30:341–5. doi: 10.1007/s10059-010-0124-x
73. Bae YH, Bae MK, Kim SR, Lee JH, Wee HJ, Bae SK. Upregulation of fibroblast growth factor-2 by visfatin that promotes endothelial angiogenesis. Biochem Biophys Res Commun. (2009) 379:206–11. doi: 10.1016/j.bbrc.2008.12.042
74. Soncini D, Caffa I, Zoppoli G, Cea M, Cagnetta A, Passalacqua M, et al. Nicotinamide phosphoribosyltransferase promotes epithelial-to-mesenchymal transition as a soluble factor independent of its enzymatic activity. J Biol Chem. (2014) 289:34189–204. doi: 10.1074/jbc.M114.594721
75. Lee J, Kim H, Lee JE, Shin SJ, Oh S, Kwon G, et al. Selective cytotoxicity of the NAMPT inhibitor FK866 toward gastric cancer cells with markers of the epithelial-mesenchymal transition, due to loss of NAPRT. Gastroenterology (2018) 155:799–814 e713. doi: 10.1053/j.gastro.2018.05.024
76. Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. (2003) 63:7436–42.
77. Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs (2008) 26:45–51. doi: 10.1007/s10637-007-9083-2
78. Olesen UH, Christensen MK, Bjorkling F, Jaattela M, Jensen PB, Sehested M, et al. Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem Biophys Res Commun. (2008) 367:799–804. doi: 10.1016/j.bbrc.2008.01.019
79. Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol. (2009) 29:5872–88. doi: 10.1128/MCB.00112-09
80. Ravaud A, Cerny T, Terret C, Wanders J, Bui BN, Hess D, et al. Phase I study and pharmacokinetic of CHS-828, a guanidino-containing compound, administered orally as a single dose every 3 weeks in solid tumours: an ECSG/EORTC study. Eur J Cancer (2005) 41:702–7. doi: 10.1016/j.ejca.2004.12.023
81. von Heideman A, Berglund A, Larsson R, Nygren P. Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharmacol. (2010) 65:1165–72. doi: 10.1007/s00280-009-1125-3
82. Hjarnaa PJ, Jonsson E, Latini S, Dhar S, Larsson R, Bramm E, et al. CHS 828, a novel pyridyl cyanoguanidine with potent antitumor activity in vitro and in vivo. Cancer Res. (1999) 59:5751–7.
83. Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. (2011) 3:94ra70. doi: 10.1126/scitranslmed.3002394
84. Adams DJ, Ito D, Rees MG, Seashore-Ludlow B, Puyang X, Ramos AH, et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem Biol. (2014) 9:2247–54. doi: 10.1021/cb500347p
85. Matheny CJ, Wei MC, Bassik MC, Donnelly AJ, Kampmann M, Iwasaki M, et al. Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens. Chem Biol. (2013) 20:1352–63. doi: 10.1016/j.chembiol.2013.09.014
86. Espindola-Netto JM, Chini CCS, Tarrago M, Wang E, Dutta S, Pal K, et al. Preclinical efficacy of the novel competitive NAMPT inhibitor STF-118804 in pancreatic cancer. Oncotarget (2017) 8:85054–67. doi: 10.18632/oncotarget.18841
87. O'Brien T, Oeh J, Xiao Y, Liang X, Vanderbilt A, Qin A, et al. Supplementation of nicotinic acid with NAMPT inhibitors results in loss of in vivo efficacy in NAPRT1-deficient tumor models. Neoplasia (2013) 15:1314–29. doi: 10.1593/neo.131718
88. Zheng X, Bauer P, Baumeister T, Buckmelter AJ, Caligiuri M, Clodfelter KH, et al. Structure-based discovery of novel amide-containing nicotinamide phosphoribosyltransferase (nampt) inhibitors. J Med Chem. (2013) 56:6413–33. doi: 10.1021/jm4008664
89. Zheng X, Bair KW, Bauer P, Baumeister T, Bowman KK, Buckmelter AJ, et al. Identification of amides derived from 1H-pyrazolo[3,4-b]pyridine-5-carboxylic acid as potent inhibitors of human nicotinamide phosphoribosyltransferase (NAMPT). Bioorg Med Chem Lett. (2013) 23:5488–97. doi: 10.1016/j.bmcl.2013.08.074
90. Zhao G, Green CF, Hui YH, Prieto L, Shepard R, Dong S, et al. Discovery of a highly selective NAMPT inhibitor that demonstrates robust efficacy and improved retinal toxicity with nicotinic acid coadministration. Mol Cancer Ther. (2017) 16:2677–88. doi: 10.1158/1535-7163.MCT-16-0674
91. Abu Aboud O, Chen CH, Senapedis W, Baloglu E, Argueta C, Weiss RH. Dual and specific inhibition of NAMPT and PAK4 By KPT-9274 decreases kidney cancer growth. Mol Cancer Ther. (2016) 15:2119–29. doi: 10.1158/1535-7163.MCT-16-0197
92. Buonvicino D, Mazzola F, Zamporlini F, Resta F, Ranieri G, Camaioni E, et al. Identification of the nicotinamide salvage pathway as a new toxification route for antimetabolites. Cell Chem Biol. (2018) 25:471–82 e477. doi: 10.1016/j.chembiol.2018.01.012
93. Tan B, Dong S, Shepard RL, Kays L, Roth KD, Geeganage S, et al. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, leads to altered carbohydrate metabolism in cancer cells. J Biol Chem. (2015) 290:15812–24. doi: 10.1074/jbc.M114.632141
94. Del Nagro C, Xiao Y, Rangell L, Reichelt M, O'Brien T. Depletion of the central metabolite NAD leads to oncosis-mediated cell death. J Biol Chem. (2014) 289:35182–92. doi: 10.1074/jbc.M114.580159
95. Gehrke I, Bouchard ED, Beiggi S, Poeppl AG, Johnston JB, Gibson SB, et al. On-target effect of FK866, a nicotinamide phosphoribosyl transferase inhibitor, by apoptosis-mediated death in chronic lymphocytic leukemia cells. Clin Cancer Res. (2014) 20:4861–72. doi: 10.1158/1078-0432.CCR-14-0624
96. Kato H, Ito E, Shi W, Alajez NM, Yue S, Lee C, et al. Efficacy of combining GMX1777 with radiation therapy for human head and neck carcinoma. Clin Cancer Res. (2010) 16:898–911. doi: 10.1158/1078-0432.CCR-09-1945
97. Frederick DW, Loro E, Liu L, Davila A Jr, Chellappa K, Silverman IM, et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. (2016) 24:269–82. doi: 10.1016/j.cmet.2016.07.005
98. Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, et al. NAMPT-mediated NAD(+) biosynthesis is essential for vision in mice. Cell Rep. (2016) 17:69–85. doi: 10.1016/j.celrep.2016.08.073
99. Duarte-Pereira S, Pereira-Castro I, Silva SS, Correia MG, Neto C, da Costa LT, et al. Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors. Oncotarget (2016) 7:1973–83. doi: 10.18632/oncotarget.6538
100. Cole J, Guiot MC, Gravel M, Bernier C, Shore GC, Roulston A. Novel NAPRT specific antibody identifies small cell lung cancer and neuronal cancers as promising clinical indications for a NAMPT inhibitor/niacin co-administration strategy. Oncotarget (2017) 8:77846–59. doi: 10.18632/oncotarget.20840
101. Shames DS, Elkins K, Walter K, Holcomb T, Du P, Mohl D, et al. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin Cancer Res. (2013) 19:6912–23. doi: 10.1158/1078-0432.ccr-13-1186
102. Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell (2015) 28:773–84. doi: 10.1016/j.ccell.2015.11.006
103. Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med. (2010) 16:387–97. doi: 10.1016/j.molmed.2010.07.002
104. Shankar GM, Kirtane AR, Miller JJ, Mazdiyasni H, Rogner J, Tai T, et al. Genotype-targeted local therapy of glioma. Proc Natl Acad Sci USA. (2018) 115:E8388–94. doi: 10.1073/pnas.1805751115
105. Tateishi K, Higuchi F, Miller JJ, Koerner MVA, Lelic N, Shankar GM, et al. The alkylating chemotherapeutic temozolomide induces metabolic stress in IDH1-mutant cancers and potentiates NAD(+) depletion-mediated cytotoxicity. Cancer Res. (2017) 77:4102–15. doi: 10.1158/0008-5472.CAN-16-2263
106. Tummala KS, Gomes AL, Yilmaz M, Grana O, Bakiri L, Ruppen I, et al. Inhibition of de novo NAD(+) synthesis by oncogenic URI causes liver tumorigenesis through DNA damage. Cancer Cell (2014) 26:826–39. doi: 10.1016/j.ccell.2014.10.002
107. Cui C, Qi J, Deng Q, Chen R, Zhai D, Yu J. Nicotinamide mononucleotide adenylyl transferase 2: a promising diagnostic and therapeutic target for colorectal cancer. Biomed Res Int. (2016) 2016:1804137. doi: 10.1155/2016/1804137
108. Pan LZ, Ahn DG, Sharif T, Clements D, Gujar SA, Lee PW. The NAD+ synthesizing enzyme nicotinamide mononucleotide adenylyltransferase 2 (NMNAT-2) is a p53 downstream target. Cell Cycle (2014) 13:1041–8. doi: 10.4161/cc.28128
109. Li H, Feng Z, Wu W, Li J, Zhang J, Xia T. SIRT3 regulates cell proliferation and apoptosis related to energy metabolism in non-small cell lung cancer cells through deacetylation of NMNAT2. Int J Oncol. (2013) 43:1420–30. doi: 10.3892/ijo.2013.2103
110. Kusumanchi P, Zhang Y, Jani MB, Jayaram NH, Khan RA, Tang Y, et al. Nicotinamide mononucleotide adenylyltransferase2 overexpression enhances colorectal cancer cell-kill by Tiazofurin. Cancer Gene Ther. (2013) 20:403–12. doi: 10.1038/cgt.2013.33
111. Bajrami I, Kigozi A, Van Weverwijk A, Brough R, Frankum J, Lord CJ, et al. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol Med. (2012) 4:1087–96. doi: 10.1002/emmm.201201250
112. Huang X, Motea EA, Moore ZR, Yao J, Dong Y, Chakrabarti G, et al. Leveraging an NQO1 bioactivatable drug for tumor-selective use of Poly(ADP-ribose) polymerase inhibitors. Cancer Cell (2016) 30:940–52. doi: 10.1016/j.ccell.2016.11.006
113. Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of β-lapachone cytotoxicity. J Biol Chem. (2000) 275:5416–24. doi: 10.1074/jbc.275.8.5416
114. Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L, et al. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci USA. (2007) 104:11832–7. doi: 10.1073/pnas.0702176104
115. Atanackovic D, Steinbach M, Radhakrishnan SV, Luetkens T. Immunotherapies targeting CD38 in multiple myeloma. Oncoimmunology (2016) 5:e1217374. doi: 10.1080/2162402X.2016.1217374
116. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. (2016) 23:1127–39. doi: 10.1016/j.cmet.2016.05.006
117. Chmielewski JP, Bowlby SC, Wheeler FB, Shi L, Sui G, Davis AL, et al. CD38 inhibits prostate cancer metabolism and proliferation by reducing cellular NAD(+) pools. Mol Cancer Res. (2018) 16:1687–700. doi: 10.1158/1541-7786.MCR-17-0526
118. Mottahedeh J, Haffner MC, Grogan TR, Hashimoto T, Crowell PD, Beltran H, et al. CD38 is methylated in prostate cancer and regulates extracellular NAD(+). Cancer Metab. (2018) 61:3. doi: 10.1186/s40170-018-0186-3
119. Ortolan E, Arisio R, Morone S, Bovino P, Lo-Buono N, Nacci G, et al. Functional role and prognostic significance of CD157 in ovarian carcinoma. J Natl Cancer Inst. (2010) 102:1160–77. doi: 10.1093/jnci/djq256
120. Morone S, Lo-Buono N, Parrotta R, Giacomino A, Nacci G, Brusco A, et al. Overexpression of CD157 contributes to epithelial ovarian cancer progression by promoting mesenchymal differentiation. PLoS ONE (2012) 7:e43649. doi: 10.1371/journal.pone.0043649
Keywords: NAD, Warburg effect, Nampt, Naprt, CD38, PARP, sirtuin, FK866
Citation: Yaku K, Okabe K, Hikosaka K and Nakagawa T (2018) NAD Metabolism in Cancer Therapeutics. Front. Oncol. 8:622. doi: 10.3389/fonc.2018.00622
Received: 27 October 2018; Accepted: 30 November 2018;
Published: 12 December 2018.
Edited by:
Suzie Chen, Rutgers University, The State University of New Jersey, United StatesReviewed by:
Alessandro Rufini, University of Leicester, United KingdomCopyright © 2018 Yaku, Okabe, Hikosaka and Nakagawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Nakagawa, bmFrYWdhd2FAbWVkLnUtdG95YW1hLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.