 Jingquan Jia1†
Jingquan Jia1† Michael A. Morse
Michael A. Morse John H. Strickler
John H. Strickler
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 28 August 2018
Sec. Gastrointestinal Cancers
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00305
This article is part of the Research Topic Impact of Circulating Tumor DNA (ctDNA) in Patients with Gastrointestinal Malignancies View all 19 articles
MET amplification is rare in treatment-naïve metastatic colorectal cancer (CRC) tumors, but can emerge as a mechanism of resistance to anti-EGFR therapies. Preclinical and clinical data suggest that patients with MET amplified tumors benefit from MET-targeted therapy. Cabozantinib is an inhibitor of multiple tyrosine kinases, included c-MET. Panitumumab is an inhibitor of EGFR. This report describes a patient with KRAS, NRAS, and BRAF wild-type metastatic CRC who experienced disease progression on all standard chemotherapy and anti-EGFR antibody therapy. The patient was enrolled in a clinical trial evaluating the combination of cabozantinib plus panitumumab. After only 6 weeks of treatment, the patient experienced a significant anti-tumor response. Although tumor tissue was negative for MET amplification, molecular profiling of cell-free DNA (cfDNA) revealed MET amplification. This case represents the first report showing the activity of cabozantinib in combination with panitumumab in a patient with metastatic CRC, and suggests that MET amplification in cfDNA may be a biomarker of response. A clinical trial targeting MET amplified metastatic CRC is currently underway.
The receptor tyrosine kinase c-MET (mesenchymal-epithelial transition factor), is implicated in tumorigenesis, proliferation, invasiveness, metastasis, and resistance to cancer treatment (1). Encoded by the MET proto-oncogene, c-MET is a disulfide-linked glycoprotein consisting of an extracellular α-subunit and a membrane spanning β-subunit (1). Hepatocyte growth factor (HGF) is the only known ligand for c-MET, and is predominantly secreted in a paracrine fashion by stromal cells. HGF binding induces c-MET receptor dimerization which in turn activates various downstream signaling pathways (2). HGF/c-MET signaling plays an essential role in diverse physiological processes such as embryonic development, epithelial branching morphogenesis and postnatal organ regeneration (3). Aberrant MET activation can occur via multiple mechanisms, including MET gene amplification (4).
MET gene amplification has been observed in multiple tumor types, including colorectal cancer (CRC) (5, 6), gastric cancer (7, 8), genitourinary cancers (9), head and neck cancer (10), non-small cell lung cancer (NSCLC) (11, 12), neuroblastoma (13), and ovarian cancer (14, 15). MET amplification is one of the key mechanisms mediating both primary (16) and acquired resistance (17) to epidermal growth factor receptor (EGFR) inhibition in patients with NSCLC. It has been shown that MET amplification leads to acquired resistance to EGFR tyrosine kinase inhibitors (TKI)s by persistent activation of ERBB3 signaling (18) and MET amplification can be detected with or without the presence of the EGFR T790M “gatekeeper” mutation (19). The prevalence of MET amplification is low (~3 %) in patients with untreated NSCLC, but increases to 5–22% in patients who develop acquired resistance to EGFR TKI therapy (17, 19, 20). The emergence of MET amplification under the selective pressure of anti-EGFR therapy supports the notion that MET amplification is a driver of acquired treatment resistance (21).
In patients with metastatic CRC, MET amplification is associated with resistance to anti-EGFR antibodies, including cetuximab and panitumumab. In mice engrafted with MET amplified CRC tumors, treatment with cetuximab is ineffective, suggesting that MET amplification may be responsible for intrinsic resistance to anti-EGFR antibodies (22). Functional crosstalk between c-MET and EGFR provides compensatory signal transduction leading to constitutive activation of downstream MAPK and PI3K pathways, thereby circumventing upstream EGFR blockade (23). MET amplification is found in less than 3% of patients with metastatic CRC who have not been exposed to anti-EGFR antibodies. Given the fitness advantage of MET amplification under the selective pressure of anti-EGFR therapies, MET amplification is much more common after exposure to anti-EGFR antibodies. Bardelli et al. (22) found that MET amplification emerged in post-treatment tumor biopsies of 3 out of 7 patients with metastatic CRC who developed acquired resistance to cetuximab or panitumumab (22). In a separate cohort of 22 patients with RAS and BRAF wild-type, HER2/MET negative metastatic CRC who developed resistance to anti-EGFR therapy, in situ hybridization (ISH) of the tumor tissue biopsies identified MET amplification as one of the most common genomic alterations (24).
Molecular profiling of blood-based circulating cell-free DNA (cfDNA) also supports MET amplification as a driver of EGFR antibody resistance. In a study by Siravegna et al. MET amplification was detected in 3 out of 16 patients who developed acquired resistance to anti-EGFR therapy (25). In another cohort of 53 patients with metastatic CRC, MET amplification was detected in in 22.6% (12/53) of patients with RAS wild-type tumors after exposure to anti-EGFR antibody therapy, but not found at an elevated frequency in anti-EGFR antibody-naïve patients (26). In addition, MET amplification was uncommon in RAS mutated patients (26). These findings have two major implications. First, it supports the utility of MET amplification as a biomarker of treatment resistance in patients with RAS wild-type EGFR antibody refractory metastatic CRC. Second, it demonstrates that MET amplification can be detected in cfDNA, thus supporting the clinical validity of cfDNA profiling to select patients for MET-targeted therapy.
The efficacy of MET inhibition in anti-EGFR antibody refractory metastatic CRC has been demonstrated in many preclinical studies. For example, in MET amplified patient-derived colorectal cancer xenograft models, MET tyrosine kinase inhibitors (TKIs) reversed resistance to EGFR blockade (22). Synergistic inhibitory effects between MET TKI and EGFR blockade was shown in a CRC xenograft mouse model expressing human HGF, where more pronounced tumor regression with concomitant MET TKI and cetuximab was observed in vivo in comparison to MET inhibition or cetuximab alone (27).
Cabozantinib is an orally bioavailable TKI that targets c-MET and VEGFR2, as well as RET, ROS1, AXL, KIT, and TIE-2. Cabozantinib is approved by the United States Food and Drug Administration (FDA) for use as monotherapy for metastatic medullary thyroid cancer1 and advanced renal cell carcinoma2. Panitumumab is an anti-EGFR monoclonal antibody FDA-approved for use in patients with KRAS and NRAS wild-type metastatic CRC3. Here we present a case report of a dramatic response to cabozantinib and panitumumab in a patient with MET amplified, EGFR antibody refractory metastatic CRC.
A 57-year-old male was initially diagnosed with locally advanced rectal cancer (T3N1M0) and treated with neoadjuvant chemoradiation followed by surgical resection (Figure 1). He subsequently received adjuvant modified (m) FOLFOX6 followed by colostomy reversal.
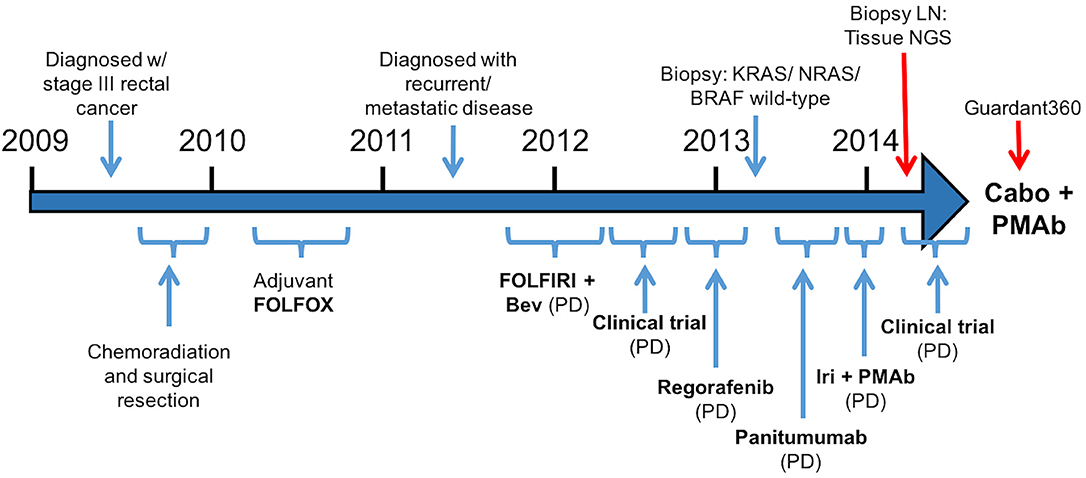
Figure 1.Treatment course (Bev, bevacizumab; Iri, irinotecan; PMAb, panitumumab; LN, lymph node; Tissue NGS, tissue next generation sequencing; Guardant360, cell free DNA profiling; Cabo, cabozantinib).
Two years later, CT imaging demonstrated new retroperitoneal lymphadenopathy suspicious for metastatic disease, and retroperitoneal lymph node (LN) biopsy revealed metastatic adenocarcinoma consistent with CRC primary. He received first-line treatment with FOLFIRI plus bevacizumab, but eventually experienced disease progression. He then progressed on a clinical trial combining capecitabine with an investigational therapy, followed by progression on regorafenib.
As his tumor was KRAS and NRAS wild-type, he was then treated with anti-EGFR antibody therapy (panitumumab). After 7 months of disease control, imaging revealed a new hypermetabolic LN at the right common iliac chain, and irinotecan was added to panitumumab. This treatment was eventually discontinued due to disease progression. A new biopsy of a mediastinal LN was performed and next generation sequencing (NGS) revealed that the tumor was still KRAS, NRAS, and BRAF wild-type, and there was no evidence of MET amplification (see Table 1). After progression on another phase I clinical trial with an investigational therapy, he was then enrolled in a phase Ib clinical trial combining cabozantinib and panitumumab (NCT02008383). At the time that he started treatment, he was increasingly symptomatic due to extensive pulmonary metastases, with worsening cough and shortness of breath. After ~6 weeks of treatment, CT demonstrated dramatic improvement in his pulmonary tumor burden (see Figure 2), as well as resolution of dyspnea and cough. As part of the trial protocol, plasma-EDTA was collected before the start of treatment to explore potential drivers of treatment response and/or resistance. CfDNA profiling utilizing a 54-gene targeted NGS panel (Guardant 360™) was performed on this sample. Blood-based profiling revealed subclonal EGFR, KRAS and BRAF resistance mutations. Additionally, EGFR amplification and MET amplification were observed in cfDNA, but not in tissue obtained 3 months prior (Table 1). Unfortunately his treatment course was complicated by anastomotic dehiscence and leak with abscess evolution. Because the dehiscence was apparently related to marked treatment response and tumor involution, treatment was discontinued. CfDNA profiling performed after 28 days of treatment revealed loss of MET and EGFR amplification (Figure 3A), while the mutant allele frequency (MAF) of KRAS G13D increased from 0.3 to 0.6%. There was also a nearly 10-fold decrease in the MAF of TP53 R213* post treatment, likely correlating with the dramatic reduction of tumor burden (Figure 3B).
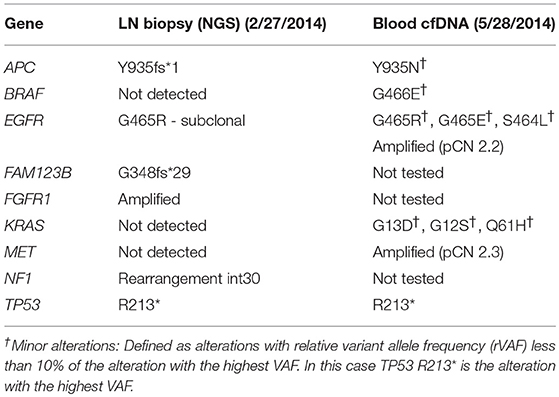
Table 1.Tissue-based next-generation sequencing (NGS) and blood-based cfDNA NGS.
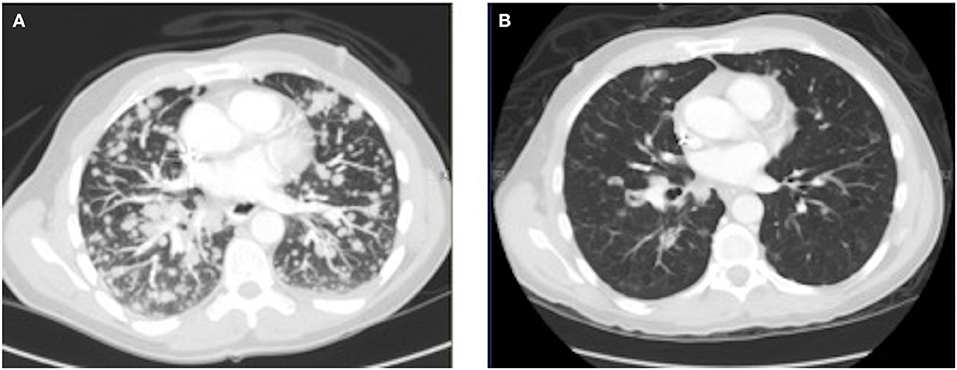
Figure 2.Chest CT image (A) before the start of cabozantinib plus panitumumab and (B) after 42 days of cabozantinib plus panitumumab.
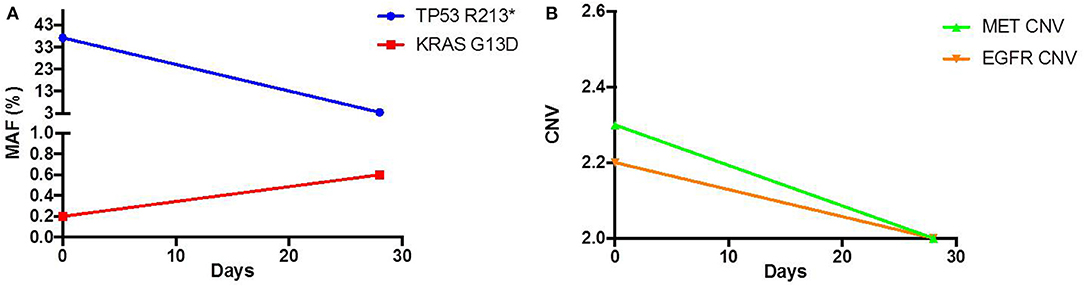
Figure 3.Pre and Post treatment cfDNA profile of (A) mutant allele frequency (MAF) and (B) copy number variation (CNV).
After 2 months off therapy, his CEA increased and his dyspnea and cough returned. Capecitabine was initiated and panitumumab was added 2 months later for additional control. He experienced brief stabilization of disease on capecitabine and panitumumab, with subjective improvement of his pulmonary symptoms. He then experienced disease progression and was transitioned to hospice. He died ~10 months after discontinuing cabozantinib and panitumumab.
Despite advances in the treatment of CRC, it remains the second leading cause of cancer-related death in the United States (28). Patients with RAS wild-type metastatic CRC are eligible for treatment with the anti-EGFR antibodies panitumumab or cetuximab (29, 30). The clinical benefit of anti-EGFR antibodies is modest, with a single agent response rate of ~20% and a median progression free survival of 4 months (31). Even among patients who experience benefit from EGFR antibodies, acquired resistance is nearly universal (32, 33).
Multiple mechanisms of acquired resistance to anti-EGFR therapy have been identified in metastatic CRC, including BRAF mutations, acquired mutations in the EGFR extracellular domain (34, 35), KRAS and NRAS mutations (36, 37), and MET amplification (22) and these mutations often co-occur (38). Of these, MET amplification is potentially treatable with tyrosine kinase inhibitors and antibodies in development. Previous preclinical studies have demonstrated the potential activity of MET inhibitors in treating cetuximab or panitumumab refractory metastatic CRC. For example, treatment with a selective MET TKI successfully restored sensitivity to cetuximab in two cetuximab-resistant human colon cancer cell lines in vitro. The two cell lines displayed MET signaling pathway activation but MET amplification was not examined (39). Using a CRC cell-line harboring MET amplification in a murine xenograft model derived from a patient who developed acquired resistance to anti-EGFR therapy, tumor growth in vivo was effectively inhibited by crizotinib, a MET/ALK inhibitor (22).
To date, several MET TKIs have been developed with variable kinase selectivity against c-MET. Many of these are under different stages of clinical evaluation, either alone or in combination with other targeted therapy in patients with advanced solid tumors (40, 41). The MErCuRIC phase I/II clinical trial aims to assess the safety and efficacy of the combination of crizotinib and a MEK1/2 inhibitor, binemetinib, in patients with MET over-expressing, RAS-mutant or RAS wild-type metastatic CRC (42). Subgroup analysis from this study suggested potential benefit in patients with high c-MET expression (43).
Although the mechanisms of treatment response in this case are not fully known, the response to cabozantinib and panitumumab may be explained by the restoration of sensitivity to panitumumab or potentially synergy from dual MET and EGFR inhibition. Of note, other objective responses to small molecule MET inhibitors have been reported in patients with metastatic NSCLC and gastric cancer who had MET amplification detected by cfDNA profiling (44, 45). Alternatively, the anti-angiogenic properties of cabozantinib may have contributed to the overall response. To better understand whether treatment with cabozantinib alone drives response for patients with MET amplified metastatic CRC, this trial has been expanded to treat patients with MET amplified metastatic CRC with cabozantinib monotherapy.
MET amplification is not routinely tested in clinical practice due to its low prevalence and unproven actionability. Additionally, access to treatment-refractory tumor tissue and molecular heterogeneity complicates testing efforts (24, 46). Given these limitations, cfDNA profiling may be the optimal approach for detection of MET amplification in the treatment refractory setting (47). In our patient, MET amplification was not detected in a tissue biopsy sample but was detected in plasma cfDNA ~3 months later (Table 1). One explanation for the discrepancy between tissue and blood profiling results is that MET amplification represented a subclonal alteration that was not consistently present throughout the same lesion (intratumoral heterogeneity) or between different lesions throughout the body (intertumoral heterogeneity), as described previously (48, 49). This possibility is supported by the notion that mutations known to mediate acquired anti-EGFR resistance, e.g., KRAS and BRAF mutations, were seen in blood, but not the LN biopsy, suggesting temporal evolution from a common clonal origin. Alternatively, tumor cells harboring MET amplification may not have been present at a sufficiently high allele frequency to be detected by the tissue-based NGS assay.
This is the first case, to our knowledge, showing the activity of cabozantinib in combination with panitumumab in a patient with metastatic CRC. MET amplification, which is an established driver of EGFR antibody resistance, may have played a critical role in sensitizing this refractory tumor to the combination of an anti-MET TKI and anti-EGFR therapy. To further understand the drivers of sensitivity and resistance, studies are ongoing to evaluate the activity of cabozantinib treatment, either alone or in combination with panitumumab, in MET amplified metastatic CRC.
MET amplification is an important driver of EGFR antibody resistance. Anti-MET therapy is active in patients with MET amplified tumors, and may be a clinically actionable target in patients with MET amplified metastatic CRC. Clinical investigations are underway to determine how best to target MET amplified metastatic CRC, and to determine whether targeting MET amplification has meaningful anti-tumor activity. Furthermore, cfDNA profiling is a promising diagnostic technology to detect genomic alterations in the treatment refractory setting. Prospective clinical trials utilizing cfDNA to identify and treat MET amplified metastatic CRC are ongoing.
Written informed consent has been obtained from the next of kin for the publication of this case report.
This study was carried out in accordance with the recommendations and approval of the Duke University Cancer Protocol Committee and the Duke University Institutional Review Board. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
JJ contributed to conception and design, analysis and interpretation of data, writing, review, and revision of the manuscript, and technical, material support. MM, RN, RL, and JS contributed to conception and design, analysis and interpretation of data, writing, review, and revision of the manuscript, and technical/material support.
This work is supported by Exelixis, Inc.
JS is a consultant/advisory board member for Amgen, received commercial research grant support from Exelixis and has a patent pending for the treatment of metastatic colorectal cell carcinoma using cabozantinib plus panitumumab. RL has ownership interest (including patents) in Guardant Health. RN has ownership interest (including patents) in Guardant Health.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^COMETRIQ (cabozantinib) full prescribing information, revised 1/2018.
2. ^CABOMETYX (cabozantinib) full prescribing information, revised: 12/2017.
3. ^VECTIBIX (panitumumab) full prescribing information, revised 6/2017.
1. Maroun CR, Rowlands T. The Met receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacol Ther. (2014) 142:316–38. doi: 10.1016/j.pharmthera.2013.12.014
2. Gherardi E, Sandin S, Petoukhov MV, Finch J, Youles ME, Ofverstedt LG, et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc Natl Acad Sci USA. (2006) 103:4046–51. doi: 10.1073/pnas.0509040103
3. Maina F, Pante G, Helmbacher F, Andres R, Porthin A, Davies AM, et al. Coupling Met to specific pathways results in distinct developmental outcomes. Mol Cell (2001) 7:1293–306. doi: 10.1016/S1097-2765(01)00261-1
4. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. (2010) 11:834–48. doi: 10.1038/nrm3012
5. Jardim DL, Tang C, Gagliato Dde M, Falchook GS, Hess K, Janku F, et al. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson Phase I Clinic. Clin Cancer Res. (2014) 20:6336–45. doi: 10.1158/1078-0432.CCR-14-1293
6. Palma NA, Palmer GA, Ali SM, Stephens PJ, Ross JS, Miller VA, et al. Frequency of MET amplification determined by comprehensive next-generation sequencing (NGS) in multiple solid tumors and implications for use of MET inhibitors. J Clin Oncol. (2013) 31:11068.
7. Houldsworth J, Cordon-Cardo C, Ladanyi M, Kelsen DP, Chaganti RS. Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res. (1990) 50:6417–22.
8. Nakajima M, Sawada H, Yamada Y, Watanabe A, Tatsumi M, Yamashita J, et al. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer (1999) 85:1894–902.
9. Jardim DL, de Melo Gagliato DL, Falchook G, Zinner R, Wheler JJ, Janku F, et al. MET abnormalities in patients with genitourinary malignancies and outcomes with c-MET inhibitors. Clin Genitourin Cancer (2015) 13:e19–26. doi: 10.1016/j.clgc.2014.06.017
10. Madoz-Gurpide J, Zazo S, Chamizo C, Casado V, Carames C, Gavin E, et al. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J Transl Med. (2015) 13:282. doi: 10.1186/s12967-015-0633-7
11. Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol. (2008) 3:331–9. doi: 10.1097/JTO.0b013e318168d9d4
12. Schildhaus HU, Schultheis AM, Ruschoff J, Binot E, Merkelbach-Bruse S, Fassunke J, et al. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung. Clin Cancer Res. (2015) 21:907–15. doi: 10.1158/1078-0432.CCR-14-0450
13. Yan B, Lim M, Zhou L, Kuick CH, Leong MY, Yong KJ, et al. Identification of MET genomic amplification, protein expression and alternative splice isoforms in neuroblastomas. J Clin Pathol. (2013) 66:985–91. doi: 10.1136/jclinpath-2012-201375
14. Yamamoto S, Tsuda H, Miyai K, Takano M, Tamai S, Matsubara O. Accumulative copy number increase of MET drives tumor development and histological progression in a subset of ovarian clear-cell adenocarcinomas. Mod Pathol. (2012) 25:122–30. doi: 10.1038/modpathol.2011.143
15. Tang C, Jardim DL, Falchook GS, Hess K, Fu S, Wheler JJ, et al. MET nucleotide variations and amplification in advanced ovarian cancer: characteristics and outcomes with c-Met inhibitors. Oncoscience (2014) 1:5–13. doi: 10.18632/oncoscience.3
16. Cappuzzo F, Varella-Garcia M, Finocchiaro G, Skokan M, Gajapathy S, Carnaghi C, et al. Primary resistance to cetuximab therapy in EGFR FISH-positive colorectal cancer patients. Br J Cancer (2008) 99:83–9. doi: 10.1038/sj.bjc.6604439
17. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. (2011) 3:75ra26. doi: 10.1126/scitranslmed.3002003
18. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316:1039–43. doi: 10.1126/science.1141478
19. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. (2007) 104:20932–7. doi: 10.1073/pnas.0710370104
20. Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. (2011) 17:1169–80. doi: 10.1158/1078-0432.CCR-10-2277
21. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell (2010) 17:77–88. doi: 10.1016/j.ccr.2009.11.022
22. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. (2013) 3:658–73. doi: 10.1158/2159-8290.CD-12-0558
23. Boccaccio C, Luraghi P, Comoglio PM. MET-mediated resistance to EGFR inhibitors: an old liaison rooted in colorectal cancer stem cells. Cancer Res. (2014) 74:3647–51. doi: 10.1158/0008-5472.CAN-14-1088
24. Pietrantonio F, Vernieri C, Siravegna G, Mennitto A, Berenato R, Perrone F, et al. Heterogeneity of Acquired Resistance to Anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res. (2017) 23:2414–2422. doi: 10.1158/1078-0432.CCR-16-1863
25. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. (2015) 21:827. doi: 10.1038/nm0715-827b
26. Raghav K, Morris V, Tang C, Morelli P, Amin HM, Chen K, et al. MET amplification in metastatic colorectal cancer: an acquired response to EGFR inhibition, not a de novo phenomenon. Oncotarget (2016) 7:54627–31. doi: 10.18632/oncotarget.10559
27. Luraghi P, Reato G, Cipriano E, Sassi F, Orzan F, Bigatto V, et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. (2014) 74:1857–69. doi: 10.1158/0008-5472.CAN-13-2340-T
28. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. (2010) 60:277–300. doi: 10.3322/caac.20073
29. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. (2008) 26:1626–34. doi: 10.1200/JCO.2007.14.7116
30. Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. (2008) 359:1757–65. doi: 10.1056/NEJMoa0804385
31. Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. (2014) 15:569–79. doi: 10.1016/S1470-2045(14)70118-4
32. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. (2004) 351:337–45. doi: 10.1056/NEJMoa033025
33. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. (2007) 25:1658–64. doi: 10.1200/JCO.2006.08.1620
34. Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med. (2012) 18:221–3. doi: 10.1038/nm.2609
35. Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. (2011) 3:99ra86. doi: 10.1126/scitranslmed.3002442
36. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature (2012) 486:537–40. doi: 10.1038/nature11219
37. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature (2012) 486:532–6. doi: 10.1038/nature11156
38. Strickler JH, Loree JM, Ahronian LG, Parikh AR, Niedzwiecki DA, Pereira AL, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. (2018) 8:164–173. doi: 10.1158/2159-8290.CD-17-1009
39. Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. (2013) 19:6751–65. doi: 10.1158/1078-0432.CCR-13-0423
40. Ko B, He T, Gadgeel S, Halmos B. MET/HGF pathway activation as a paradigm of resistance to targeted therapies. Ann Transl Med. (2017) 5:4. doi: 10.21037/atm.2016.12.09
41. Bradley CA, Salto-Tellez M, Laurent-Puig P, Bardelli A, Rolfo C, Tabernero J, et al. M.E. consortium, Targeting c-MET in gastrointestinal tumours: rationale, opportunities and challenges. Nat Rev Clin Oncol. (2017) 14:562–76. doi: 10.1038/nrclinonc.2017.40
42. Schaeybroeck SV, Rolfo CD, Elez E, Kelly S, Middleton MR. MErCuRIC1: A Phase I study of MEK1/2 inhibitor PD-0325901 with cMET inhibitor crizotinib in RASMT and RASWT (with aberrant c-MET) metastatic colorectal cancer (mCRC) patients. J Clin Oncol. (2015) 33:3632.
43. Eng C, Bessudo A, Hart LL, Severtsev A, Gladkov O, Muller L, et al. A randomized, placebo-controlled, phase 1/2 study of tivantinib (ARQ 197) in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with wild-type KRAS who have received first-line systemic therapy. Int J Cancer (2016) 139:177–86. doi: 10.1002/ijc.30049
44. Rozenblum AB, Ilouze M, Dudnik E, Dvir A, Soussan-Gutman L, Geva S, et al. Clinical impact of hybrid capture-based next-generation sequencing on changes in treatment decisions in lung cancer. J Thorac Oncol. (2017) 12:258–68. doi: 10.1016/j.jtho.2016.10.021
45. Kim ST, Banks KC, Lee SH, Kim K, Park JO, Park SH, et al. Prospective feasibility study for using cell-free circulating tumor DNA–guided therapy in refractory metastatic solid cancers: an interim analysis. JCO Precis Oncol. (2017) 1:1–15. doi: 10.1200/PO.16.00059
46. Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. (2016) 6:147–153. doi: 10.1158/2159-8290.CD-15-1283
47. Janku F, Angenendt P, Tsimberidou AM, Fu S, Naing A, Falchook GS, et al. Actionable mutations in plasma cell-free DNA in patients with advanced cancers referred for experimental targeted therapies. Oncotarget (2015) 6:12809–21. doi: 10.18632/oncotarget.3373
48. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
Keywords: MET amplification, metastatic colorectal cancer, cabozantinib, cell-free DNA, ctDNA
Citation: Jia J, Morse MA, Nagy RJ, Lanman RB and Strickler JH (2018) Cell-Free DNA Profiling to Discover Mechanisms of Exceptional Response to Cabozantinib Plus Panitumumab in a Patient With Treatment Refractory Metastatic Colorectal Cancer. Front. Oncol. 8:305. doi: 10.3389/fonc.2018.00305
Received: 06 April 2018; Accepted: 19 July 2018;
Published: 28 August 2018.
Edited by:
John James Tentler, University of Colorado, United StatesReviewed by:
Francesco Caiazza, University of California, San Francisco, United StatesCopyright © 2018 Jia, Morse, Nagy, Lanman and Strickler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John H. Strickler, am9obi5zdHJpY2tsZXJAZG0uZHVrZS5lZHU=
†Present Address: Jingquan Jia, Fellow in the Divisions of Hematology, Cellular Therapy, and Medical Oncology, Department of Medicine, Duke University Medical Center, Durham, NC, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.