 Praveen Koganti
Praveen Koganti Michael Blank
Michael Blank- Laboratory of Molecular and Cellular Cancer Biology, Azrieli Faculty of Medicine, Bar-Ilan University, Safed, Israel
Protein ubiquitination is an evolutionary conserved highly-orchestrated enzymatic cascade essential for normal cellular functions and homeostasis maintenance. This pathway relies on a defined set of cellular enzymes, among them, substrate-specific E3 ubiquitin ligases (E3s). These ligases are the most critical players, as they define the spatiotemporal nature of ubiquitination and confer specificity to this cascade. Smurf1 and Smurf2 (Smurfs) are the C2-WW-HECT-domain E3 ubiquitin ligases, which recently emerged as important determinants of pivotal cellular processes. These processes include cell proliferation and differentiation, chromatin organization and dynamics, DNA damage response and genomic integrity maintenance, gene expression, cell stemness, migration, and invasion. All these processes are intimately connected and profoundly altered in cancer. Initially, Smurf proteins were identified as negative regulators of the bone morphogenetic protein (BMP) and the transforming growth factor beta (TGF-β) signaling pathways. However, recent studies have extended the scope of Smurfs' biological functions beyond the BMP/TGF-β signaling regulation. Here, we provide a critical literature overview and updates on the regulatory roles of Smurfs in molecular and cell biology, with an emphasis on cancer. We also highlight the studies demonstrating the impact of Smurf proteins on tumor cell sensitivity to anticancer therapies. Further in-depth analyses of Smurfs' biological functions and influences on molecular pathways could provide novel therapeutic targets and paradigms for cancer diagnosis and treatment.
Introduction
Protein ubiquitination is a major posttranslational modification that controls a wide spectrum of biological functions, and is critical in maintaining cellular homeostasis under physiological conditions and in diseases.
Ubiquitination is a multi-step enzymatic process which is mediated by the concerted action of three main types of proteins: (i) ubiquitin-activating enzymes (E1s), which bind, adenylate and activate cognate ubiquitin molecules using the energy of ATP hydrolysis; (ii) ubiquitin-conjugating enzymes (E2s), which accept ubiquitin from E1 in the form of a thioester bond to their active-site cysteine; and (iii) ubiquitin protein ligases (E3s) that recruit ubiquitin-charged E2 enzymes and mediate specific transfers of ubiquitin to protein substrates.
It is estimated that the human genome encodes for more than 630 E3s, ~40 E2s, and only two E1s. E3 ubiquitin ligases are of particular interest since they define the spatio-temporal nature of ubiquitination and, together with other accessory proteins, provide specificity to the cascade.
E3s tightly control protein stability, localization, and function, and thereby regulate a plethora of biological processes. This has instigated intensive investigations of these enzymes as disease biomarkers and drug targets in a variety of human disorders, particularly in cancer (1–3).
Depending on the ubiquitin transfer mechanism and domain characteristics, E3s are classified into three main groups/families: really interesting new gene (RING) family, which is the most abundant in the human genome (~600 family members), homologous to the E6AP carboxyl terminus (HECT) domain E3s (~30 members), and RING-in-between-RING (RBR) E3s (~12 in humans) (4).
Smurf1 and Smurf2 (Smurfs) are two closely related C2-WW-HECT domain E3 ubiquitin ligases, belonging to the NEDD4 subfamily of HECT type E3s. Similar to other NEDD4 family members (nine in total), Smurfs contain: (i) the N-terminal C2 domain, which mediates binding of these E3s to intracellular membranes; (ii) several tryptophan-containing WW domains, which are thought to mediate the protein-protein interactions between the E3s and their interactors and substrates (primarily through association with proline-containing PPxY or LPxY motifs in the binding partners); and (iii) the evolutionary-conserved catalytic HECT domain. Of note, several studies indicate that the HECT domain of NEDD4 E3s are also involved in substrate recognition (5–7).
In mammals, Smurf1 and Smurf2 are encoded by two distinct genes located at chromosomes 7 and 17, respectively (Figure 1). Three isoforms of human Smurf1, resulting from alternative splicing, have been reported, and a single protein product has been confirmed for Smurf2 (9).
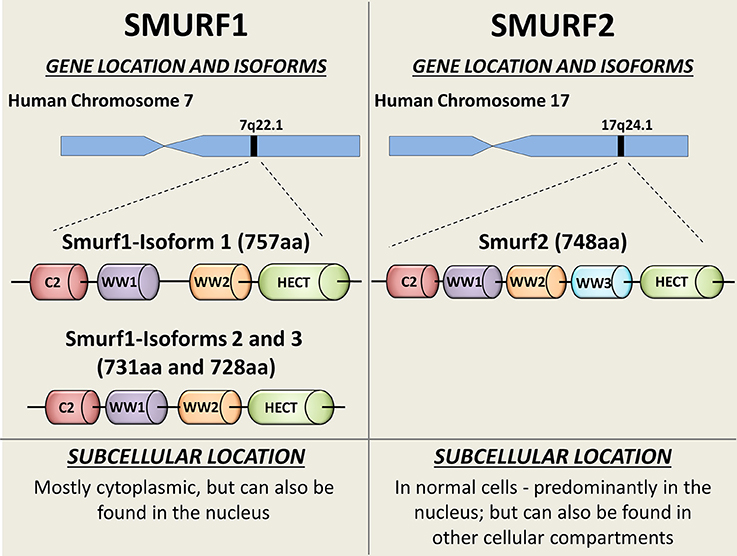
Figure 1. Schematic diagram showing the SMURFs' gene locations, isoforms, and intracellular distribution. Both Smurf1 and Smurf2 possess an N-terminal protein kinase C (PKC)-related C2 domain (Red), 2-3 WW protein interacting domains (purple, orange, and blue) and the catalytical C-terminal HECT domain (green). Smurf1 transcript variant 1 (NP_065162) is the longest Smurf1 isoform (757 aa), and has a 26-residue linker insert between WW domains (8). Catalytical active site—Cys699. Smurf1 transcript variant 2 (NP_851994; 731 aa) lacks an in-frame exon in the coding region, compared to variant 1. Smurf1 isoform 3 (NP_001186776) lacks an in-frame exon in the coding region and uses an alternate in-frame splice site in the 3′ coding region, compared to variant 1. This variant is 728 aa long. Smurf2 (NP_073576) is a 748 aa protein. Catalytical active site—Cys716. Subcellular bio-distributions of Smurf1 and Smurf2 are indicated on the bottom of the figure.
Smurfs share a high sequence homology (>70% amino acid sequence identity) and have similar structural characteristics. Despite these high similarities and some redundancy in their substrate repertoire, these proteins exhibit distinct and in some respects opposite biological functions.
In this review, we will discuss the diverse roles of Smurf proteins in pleotropic cellular functions, including cell proliferation and DNA damage response, chromatin organization, dynamics and genomic integrity maintenance, gene expression, carcinogenesis, and metastases. We also highlight studies implicating Smurfs in cellular responses to anticancer therapies.
Regulatory Roles of Smurfs in the Decisive Cellular Processes
Smurfs in TGF-β/BMP Signaling
Smurf1 and Smurf2 were originally identified as negative regulators of BMP/TGF-β signaling pathways. These pathways play crucial roles in embryogenesis and adult tissue homeostasis, as well as in the pathogenesis of various human diseases (10, 11).
In cancer, these pathways appear to have a dual role: operating in both cancer development and suppression (12, 13). Indeed, the activation and/or inhibition of these pathways are highly related to various aspects of carcinogenesis including epithelial-mesenchymal transition (EMT), angiogenesis, behavior of cancer stem cells, metastases, and tumor cell chemo-refractoriness.
For example, TGF-β signaling exerts in normal cells and at the early-stages of carcinogenesis tumor-suppressor functions, including cell-cycle arrest and triggering of apoptosis. However, at late-stages of carcinogenesis, the role is reversed and TGF-β signaling promotes tumorigenesis, metastases and chemoresistance (13).
Smurf1 has been shown to ubiquitinate and degrade the BMP receptor-regulated Smad proteins (R-Smads; i.e., Smad1 and Smad5), which form heteromeric complexes with a common-partner Smad (Co-Smad) Smad4. Following formation, this complex translocates into the nucleus to regulate transcription of a variety of target genes, also related to tumorigenesis, cancer progression, and chemoresistance. The ability of Smurf1 to degrade the BMP-specific Smads as well as BMP receptors, provides negative feedback to the BMP signaling pathway. Noteworthy, Smurf1 can cooperate with inhibitory Smad (I-Smad), Smad6, and Smad7, which repress the TGF-β superfamily signaling by several different mechanisms (14–17).
The E3 ubiquitin ligase functions of Smurf2 were primarily associated with its ability to negatively regulate the TGF-β signaling pathway. Following receptor stimulation with the TGF-β ligand, Smurf2 translocates from the nucleus to the cytosol. For this to occur, nuclear Smurf2 needs to bind to I-Smad, in particular of Smad7, which facilitates the nuclear export of Smurf2.
While in the cytosol, Smurf2 interacts with and promotes the proteasomal degradation of the TGF-β receptor (i.e., TGF-βRI) as well as TGF-β-specific R-Smads such as Smad2 and Smad3 (18–21). Of note, although the Smurf1 and Smurf2 activities are primarily attributed to BMP and TGF-β signaling regulation, respectively, experimental data suggest a model where Smurfs can truncate both of these pathways.
Despite these proceedings, studies conducted on Smurf1- and Smurf2-genetically ablated mice question the role of Smurfs in the canonical TGF-β superfamily signaling. Smurf1 knockout mice revealed no significant disruption in the Smad-mediated TGF-β/BMP signaling pathways. Instead, these animals exhibited an age-dependent increase of bone mass due to enhanced osteoblast activity. This activity was related to activation of the MEKK2-JNK signaling cascade (22).
Targeted disruption of Smurf2 in mice has also revealed that the protein levels and stability of the TGF-β receptor and Smad proteins (i.e., Smad2/3) were unaffected by Smurf2 depletion, despite the enhanced cellular response to TGF-β stimulation (23). This phenomenon was explained by the uncovered ability of Smurf2 to monoubiquitinate Smad3 and inhibit the formation of Smad3 complexes (23). The activity of these complexes is required for the TGF-β-mediated transcriptional response.
Collectively, these findings stipulate that at least in mouse experimental models, Smurf proteins do not directly regulate the stability and turnover of the BMP/TGF-β receptors and Smad transducers. Of note, both murine and human Smurf proteins share a very high homology and amino acid identity: 95% for Smurf1 and 99% for Smurf2, suggesting that mouse models can appropriately investigate the biological roles of Smurfs in humans.
In addition, experimental evidence shows that Smurfs are not the only E3 ubiquitin ligases regulating TGF-β signaling. Other NEDD4 E3 ubiquitin ligase family members, including ITCH, NEDD2L, WWP1 and WWP2, can also mitigate this cascade (7). These findings suggest that NEDD4 E3s have overlapping functions in the TGF-β signaling regulation. Moreover, Smurf2 has been reported to ubiquitinate and promote the degradation of Smurf1, introducing further complexities in TGF-β/BMP signaling regulation by Smurfs (24).
Interestingly, a recent study shows that mice deficient for Smurf2 exhibit decreased bone mass due to severe osteoporosis. This phenotype is opposite to the phenotype observed in Smurf1-ablated animals (25). Moreover, the authors demonstrate that elimination of Smurf2, but not Smurf1, significantly increases the expression of RANKL, a key regulator in osteoclastogenesis and bone physiology. Through mechanistic studies, they showed that this phenomenon is associated with the ability of Smurf2 to monoubiquitinate and inactivate transcriptional activity of Smad3, as previously reported (23). In the absence of Smurf2, Smad3 activity remains unrestrained and results in enhanced transactivation activity of the vitamin D receptor (VDR) signaling, ultimately leading to elevated expression of RANKL.
Smurfs in Carcinogenesis
Smurf2 Acts as a Tumor Suppressor
The most surprising and exciting finding on the involvement of Smurfs, in particular of Smurf2, in cancer we, and subsequently another group, obtained using Smurf2-depleted mice (Smurf2−/− mice). We found that while relatively normal in their early lives, these mice developed a wide spectrum of tumors in different organs and tissues as they aged. The majority of tumors appeared in mice older than 90 weeks (7, 26), equivalent to ~70 years of age in humans (27). About 70% of the uncovered tumors were of epithelial origin including hepatocellular carcinoma, lung alveolar carcinoma, mammary gland carcinoma, and others. Hematological malignancies were also detected, in ~30% of the cases.
To best of our knowledge, there are only a few, if any, mouse cancer models which so closely mimic two key characteristics of human cancer: (i) the late cancer onset (~77% of all cancers are diagnosed in persons 55 years of age and older); and (ii) the epithelial origin of tumors. For example, the vast majority of tumors in p53-null mice are hematological malignancies (mostly lymphomas), which develop within a few months after the animal's birth (28, 29).
These findings suggest that the Smurf2-ablated animals are highly relevant to human carcinogenesis model, and could be advantageous when studying cancer-related processes at the whole organism level.
Moreover, Zhang's group further demonstrated that mice heterozygote for Smurf2 (Smurf2+/−) are also susceptible to spontaneous tumorigenesis (30). Further analysis of tumors from these animals revealed the loss of heterozygosity (LOH) at Smurf2. LOH is a common genetic event inactivating residual wild type allele of genes, in particular of tumor suppressors.
Altogether, these findings establish Smurf2 as a potent tumor suppressor, preventing the transformation of normal cells into cancerous ones.
Smurf2 Regulates Chromatin Organization, Dynamics, and Integrity
Our subsequent studies revealed that inactivation of Smurf2 triggers a series of cascading events in cells, and creates the “mutator phenotype,” which under the stress of aging leads to carcinogenesis (26). Mechanistically, we found that Smurf2 regulates chromatin structure landscape and, thereby, affects gene expression, DNA damage response (DDR), and genomic integrity maintenance. We further demonstrated that these Smurf2 activities were associated with and at least in part relied on its ability to ubiquitinate and degrade RNF20 (Figure 2), a RING type E3 ubiquitin ligase responsible for monoubiquitination of histone H2B (ubH2B). The RNF20-ubH2B module regulates chromatin compaction, DNA damage response, and gene expression, and acts both as a tumor suppressor and an oncogene depending on the cellular context (26, 31–39).
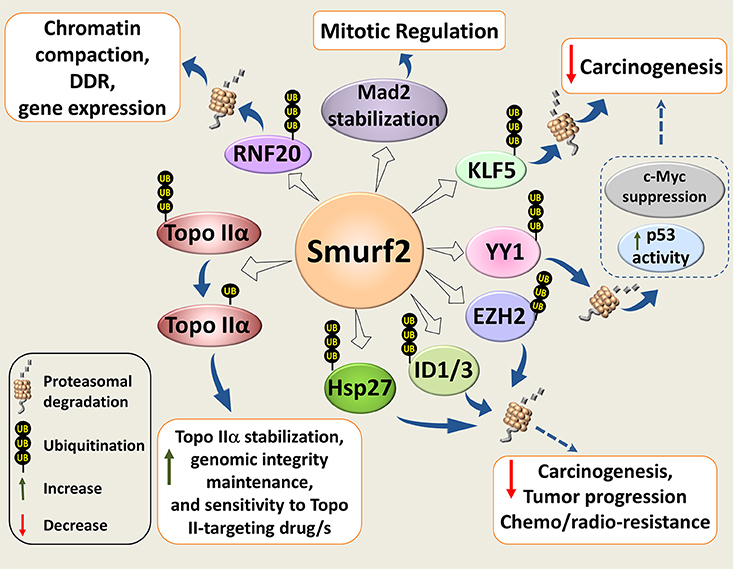
Figure 2. Molecular mechanisms underlying Smurf2 tumor suppressor functions. Smurf2 regulates chromatin compaction, DDR, and gene expression through the ubiquitin-proteasomal degradation of RING-type E3 ubiquitin ligase and histone H2B modifier RNF20. Smurf2 also controls chromatin organization, dynamics and unaltered chromosomal inheritance through stability regulation of Topo IIα. In addition, Smurf2 could affect carcinogenesis, tumor progression and sensitivity to anticancer therapies through the ubiquitin-mediated proteasomal turnover of KLF5, ID1/3, YY1, and others. Degradation of YY1, for example, relieves the suppression of p53 activity by YY1, and decreases the expression of c-Myc. Smurf2-mediated stabilization of the mitotic spindle checkpoint protein Mad2, is also shown in the diagram. Dotted arrows specify potential mechanisms determined from existing evidence.
Furthermore, an interesting finding in Smurf2−/− cell genome was the accumulation of multiple chromosomal abnormalities, with translocations being the most notable hallmark (26). Subsequent investigation of this phenomenon showed that Smurf2 expression is essential in preventing the formation of pathological chromatin bridges, also known as anaphase bridges (40). These bridges are a major cause of chromosomal translocations, and are often an output of the compromised decatenation checkpoint.
The decatenation checkpoint is normally mediated by DNA topoisomerase IIα (Topo IIα), a core enzyme in chromatin organization, dynamics and unaltered chromosome inheritance (41).
We found that Smurf2 operates as a molecular editor of Topo IIα, switching its ubiquitination code from the degradation-promoting K48 polyubiquitination to monoubiquitination, and stabilizing the enzyme (40) (Figure 2). Unaltered E3 ubiquitin ligase functions of Smurf2 were indispensable for this regulation. Moreover, we showed that Smurf2 depletion phenocopied Topo IIα depletion and increased the formation of anaphase bridges. Introduction of Topo IIα into Smurf2-depleted cells rescued this phenomenon. Our studies also uncovered that Smurf2 is a determinant of Topo IIα protein levels in cancer cells and tissues, and is a factor affecting tumor cell sensitivity to the Topo II-targeting drug, etoposide (40).
Collectively, these findings establish Smurf2 as a key cellular factor governing chromatin organization, dynamics and genome integrity maintenance. They also indicate Smurf2 as a potent tumor suppressor.
Other Putative Mechanisms for Smurf2-mediated Tumor Suppression
In addition to RNF20 and Topo IIα, Smurf2 has been shown to regulate stability and/or subcellular localization of other decisive cellular proteins implicated in carcinogenesis and drug resistance. These molecules include the molecular chaperone and apoptosis inhibitor HSP27, transcription factors KLF5, YY1, ID1/ID3, histone methyltransferase EZH2, and others (Figure 2).
HSP27 (heat shock protein 27) is one of the central molecules shown to upregulate EMT and affect activities of the matrix metalloproteinases (MMPs), stimulate tumor cell proliferation, migration, invasion, as well as to mediate chemo- and radio-resistance (42). Smurf2 overexpression was reported to alter HSP27 subcellular distribution and induce its ubiquitin-dependent degradation in the human lung adenocarcinoma A549 cell model (43). However, it currently remains unknown whether these Smurf2 activities are pertinent to its tumor suppressor functions in lung cancer, and/or in other types of tumors.
Smurf2 was also shown to promote the degradation of a few principal transcription factors whose activities are associated with carcinogenesis, drug resistance, patient prognosis and survival. Krüppel-like factor 5 (KLF5) is one of these factors with cell growth-promoting and pro-survival activities. KLF5 is also implicated in cell differentiation, migration and stemness and its expression levels are frequently abnormal in different types of cancer (44). Smurf2 was shown to polyubiquitinate and promote the proteasomal degradation of KLF5 in a Smurf2 E3 ligase-dependent manner, thereby inhibiting the transcriptional and pro-proliferative activities of KLF5 (45). Interestingly, KLF5 levels were specifically reduced by Smurf2, but not by Smurf1.
Yin Yang 1 (YY1) is another example of the Krüppel-like zinc finger transcriptional factors negatively regulated by Smurf2. YY1 is overexpressed in multiple cancer types, and its overexpression correlates with poor clinical outcomes, although several studies suggested that in some types of cancer YY1 acts as a tumor suppressor (46). Two research groups reported that Smurf2 ubiquitinates and promotes the degradation of YY1 (47, 48). The outcomes of these Smurf2-mediated effects were a decrease in the YY1-mediated suppression of p53 activity (47), and a reduction of B-cell proliferation and lymphomagenesis (48). The latter was supposedly mediated via the suppression of YY1-c-Myc regulatory axis (Figure 2).
The ability of Smurf2 to ubiquitinate and degrade two dominant inhibitors of helix-loop-helix transcription factors ID1 and ID3 (49) might also be relevant to Smurf2's tumor suppressor activities. Overexpression of these IDs was shown to facilitate tumor growth, angiogenesis, stem cell maintenance, invasiveness, metastasis, as well as correlating with unfavorable clinical prognoses (50, 51). Moreover, ID1 has been shown to confer chemoresistance to different types of cancer (52–55).
In addition, Smurf2 was shown to polyubiquitinate and induce a proteasome-mediated degradation of EZH2, the catalytic subunit of the polycomb repressive complex 2 (PRC2) and histone H3-K27 methyltransferase (56). This was reported in human mesenchymal stem cells during their neuronal differentiation. If this finding is corroborated in tumor cell models it might be highly pertinent to the ability of Smurf2 to interfere with carcinogenesis, as EZH2 was documented as a pro-oncogenic factor involved in neoplastic transformation, cancer cell stemness, metastases and immune evasion. However, it should be mentioned that several studies show that under some circumstances EZH2 also exhibits tumor suppressive activities (57).
Smurf2 has also been implicated in the formation of the functional mitotic spindle checkpoint by regulating the localization and stability of the MAD2 protein (58). Knockdown of Smurf2 or overexpression of its E3 ligase-deficient mutant generated misaligned and lagging chromosomes, premature anaphase onset, and defective cytokinesis in human cervix carcinoma HeLa cells (58). Interestingly, in our study, Smurf2 depletion did not affect the formation of lagging chromosomes, but instead increased the formation of anaphase bridges in osteosarcoma U2OS cells (40). These discrepancies could be explained by different types of cancer cell models used in these studies: HeLa vs. U2OS cells, implying that biological effects of Smurf2 should be very carefully interpreted taking into account cellular context, genetic make-up, and experimental settings.
Altogether, these findings designate Smurf2 as a pleotropic cellular factor that regulates a wide spectrum of molecular pathways and networks to control transcription, DNA damage response and genomic integrity maintenance. When these pathways are compromised, carcinogenic processes can be set in motion, leading to cell transformation and the development of a wide spectrum of tumors, as observed in Smurf2-null mice (7, 26).
Furthermore, the Smurf2-Smad3-RANKL axis described in the previous section could also potentially be involved in tumor formation in Smurf2-deficient animals. This appears to be most relevant to mammary gland carcinomas developed in Smurf2−/− mice, as the upregulated RANKL/RANK signaling pathway could promote mammary stem cell expansion, proliferation and the formation of hormone-induced breast cancer (59).
Interestingly, genomic studies showed that the SMURF2 gene is not frequently mutated in human malignancies (https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=SMURF2). However, changes in Smurf2 expression are common in many cancers (7, 26, 40), similar to some other cancer-related genes such as the two TP53 paralogs, TP63 and TP73 (60), and the members of the FOXO transcription factors family (61).
The Duality of Smurf2 in Cancer
As described above, evidence points to Smurf2 as a potent tumor suppressor operating in normal cells to prevent cell transformation and carcinogenesis. However, results obtained in established cancer cell models argue that Smurf2 has a dual role and under some circumstances acts as an oncogene rather than a tumor suppressor (7, 9, 62). Additionally, the expression levels of Smurf2 were reported to be significantly elevated in several types of cancers including esophageal squamous cell carcinoma tumors (63) and chemo-refractory tumors such as recurrent hepatocellular carcinomas (64). The data available in the COSMIC dataset portal also indicate that Smurf2 is overexpressed in ~49% of ovarian cancer, about 18% of breast cancer, and in ~17% of soft tissue neoplasms.
Furthermore, ours and other studies show that subcellular biodistribution of Smurf2 is prominently altered in cancer vs. normal cells, with a notable accumulation/sequestration of Smurf2 in the cytoplasm of tumor cells (7, 26, 65). It is possible that overexpressed and mislocalized Smurf2 is employed by the carcinogenic machinery to promote oncogenesis, at least in some types of cancer.
The possible pro-oncogenic functions of Smurf2 in genetically-compromised tumor cells could be related to the reported ability of Smurf2 to interfere with the RAS, Wnt/β-catenin, and EGFR-mediated signaling pathways, three central modules in cancer progression and chemoresistance (Figure 3).
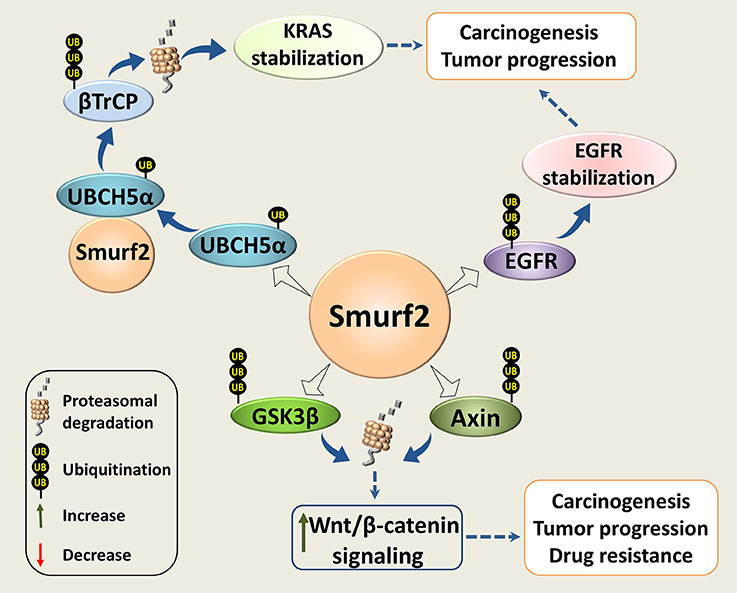
Figure 3. Smurf2 as an oncogene. Smurf2 monoubiquitinates UBCH5α and forms an active complex for the degradation of the KRAS negative regulator, βTrCP. This stabilizes KRAS and could potentiate its pro-oncogenic functions. Smurf2 also ubiquitinates EGFR and protects it from degradation. In addition, Smurf2 through the degradation of GSK3β and Axin could upregulate the Wnt/β-catenin signaling. Through this mechanism, Smurf2 might also increase the pro-oncogenic activities of this pathway. Dotted arrows specify potential mechanisms determined from existing evidence.
It has been reported that Smurf2 together with the E2 ubiquitin-conjugating enzyme UBCH5, stabilize the KRAS oncoprotein (66), the most frequently mutated transforming oncogene in human cancers (67). The authors showed that Smurf2 monoubiquitinates UBCH5 to form an active complex for degradation of β-TrCP, the F-box protein and a component of SCF E3 ligase that negatively regulates KRAS (68). Loss of Smurf2 led to the accumulation of β-TrCP, and KRAS degradation. Interestingly, silencing of Smurf2 mostly affected the mutant form/s of KRAS. In addition, the authors demonstrated that knockdown of Smurf2 reduces the clonogenic survival and prolongs tumor latency in the mutant KRAS-driven tumors generated in nude mice with either human colon or lung carcinoma cells (66).
Under the described experimental setting, Smurf2 appears to act as an oncogene, promoting tumor development. However, it should be mentioned that the SCFβ−TrCP complex mediates the degradation of functionally diverse proteins, and it is capable to downregulate both oncogenes (i.e., RAS, β-catenin, CDC25A, and others) as well as tumor suppressors (e.g., Smad4, IkB, FOXO3 and REST) (69, 70). Thus, the role of the Smurf2/SCFβ−TrCP module in cancer may vary considerably depending on the cell type and molecular composition, and should be determined in a particular context.
Smurf2 was also reported to promote the Wnt/β-catenin signaling through the degradation of its two negative regulators: GSK3β (71) and Axin (72). Through this route, Smurf2 could potentially facilitate the activities of the proto-oncogene β-catenin.
Interestingly, GSK3β phosphorylates and primes RAS proteins for SCFβ−TrCP-mediated degradation (73). Inhibition of this degradation pathway by aberrant Wnt/β-catenin signaling may contribute to Ras-induced transformation in colorectal tumorigenesis (68). In this regard, it will be even more important to investigate the role of the Smurf2/GSK3β/SCFβ−TrCP/Ras module in animal cancer models and in clinical samples.
Smurf2 has also been shown to ubiquitinate and protect from c-Cbl-mediated degradation the epidermal growth factor receptor (EGFR), which is implicated in a wide range of cell responses ranging from cell division to adhesion, motility, and death (74). The authors also reported that the loss of Smurf2 destabilizes EGFR, and reduces the clonogenic survival of EGFR-expressing cancer cell strains. The effects of Smurf2 depletion on EGFR-negative cancer cells, normal fibroblasts, and on normal epithelial cells were minor. In addition, the authors demonstrated that knockdown of Smurf2 reduces the ability of human head and neck squamous cell carcinoma UMSCC74B cells to form tumors in vivo.
Cooperatively, these studies suggest that in immortalized and established cancer cell models Smurf2 operates as an oncogene rather than a tumor suppressor. Of note, Wellbrock's group reported that Smurf2 depletion can significantly increase melanoma cell sensitivity to the cytotoxic effects of the MEK inhibitor selumetinib (AZD6244), both in vitro and in vivo (75).
This finding implies that at least in this type of tumor, inactivating Smurf2 might overcome tumor cell resistance to MAPK pathway inhibitors experienced in clinics.
Smurfs in Tumor Cell Proliferation, Migration, and Invasion
A few studies have shown that Smurf2 is intrinsically involved in these critical for cancer progression processes. However, studies from different groups revealed different and in some respect contradictory results, even when using the same cancer cell model. For example, Zhang's group demonstrated that elevated levels of Smurf2 were required for and promoted migration, invasion and in vivo metastatic dissemination of human breast carcinoma MDA-MB-231 cells. Moreover, the authors demonstrated that Smurf2 E3 ligase-defective mutant (Cys716Gly) decreases the metastatic behavior of these cells (76).
In contrast, Imamura's group showed that Smurf2 knockdown in MDA-MB-231 cells enhances cell migration in vitro and bone metastasis in vivo, implying that under these circumstances Smurf2 is a tumor suppressor (24). The same group also demonstrated that Smurf2 reduces MDA-MB-231 cell migration via Smurf1 degradation. The authors also provided evidence that the motility of Smurf2-knocked down cells is independent of TGF-β-signaling.
In addition, a recent study showed that knock-down of Smurf2 increases the proportion of invasive MDA-MB-231 cell-derived organoids. This group also demonstrated that PIAS3-dependent sumoylation of Smurf2 is important in suppressing the invasive behavior of these cells (77).
The discrepancies in these studies could be explained, at least in part, by different approaches used to manipulate the Smurf2 expression levels (overexpression vs. knock-down). However, a more comprehensive investigation is needed to support this notion. One possible approach is to examine these effects in SMURF2 genetically-ablated MDA-MB-231 cells, as well as in other human cancer cell models. These cells were recently generated by our group using the CRISP/Cas9 gene-editing tool (78), and are currently under investigation.
Studies conducted using pancreatic cancer cells also suggest that Smurf2 acts as a tumor suppressor. The authors showed that Smurf2 is downregulated in pancreatic cancer tissues, and its overexpression suppresses migration and invasion of pancreatic cancer cells, while having no effect on cell viability, cell cycle, and senescence (79). Interestingly, the authors also showed that Smurf2 promotes mesenchymal-epithelial transition (MET), and that its expression levels are negatively associated with cancer cell resistance to gemcitabine treatment.
Smurf1 has also been implicated in cancer cell proliferation, migration and invasion (Figure 4). Wrana's group found that Smurf1 plays an important role in regulating protrusive activity and the transformed phenotype of HEK293T cells (80). Mechanistically, the authors demonstrated that Smurf1 is recruited by PKCζ to cellular protrusions, where it controls the protein levels of RhoA, a small GTPase implicated in cell shape, polarity, adhesion, and motility regulation. Smurf2 however, was not involved in the RhoA stability regulation.
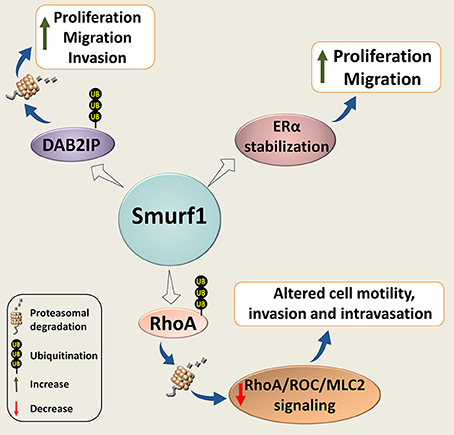
Figure 4. Smurf1 in cancer cell proliferation, motility, and invasion. Smurf1 promotes ubiquitin-dependent degradation of DAB2IP; and stabilizes ERα, resulting in accelerating tumor cell replication, migration and invasion. Smurf1 can also affect cancer cell motility and invasion by proteasomal degradation of RhoA.
Subsequently, Vial's group showed that Smurf1 expression is required for lamellipodia formation, tumor cell plasticity, and motility through the regulation of peripheral RhoA/ROCK/MLC2 signaling. Silencing of Smurf1 or expression of its interfering mutants inhibited cell migration (81). Interestingly, their in vivo studies showed that Smurf1 reduction induces the mesenchymal-amoeboid transition, facilitates cell motility and increases invasion and intravasation. However, this reduction was insufficient to promote metastasis after cells have entered the vessels.
In another study, induction of Smurf1 expression either by EGF or by overexpression of MEK1, as well as Smurf1 overexpression, significantly increased migration and invasion of breast carcinoma MDA-MB-231 cells, whereas knockdown of Smurf1 suppressed the phenotype (82). These findings are in agreement with results reported by Imamura's group (24).
The pro-oncogenic role of Smurf1 was also noted in other types of cancer. For example, suppression of Smurf1 expression in human ovary carcinoma SK-OV-3 and OVCAR-3 cells significantly decreases cell migration and invasion (83). Similar results were also observed in prostate cancer cell models (84). In addition, the authors demonstrated that expression of Smurf1 in prostate cancer cells is regulated through the androgen receptor (AR) signaling, which is critical for prostate cancer growth and survival.
Another recently published study shows that Smurf1 expression is also triggered through estrogen signaling (85). The authors also demonstrated that Smurf1 stabilizes estrogen receptor alpha (ERα) in breast cancer cells, leading to increased estrogen signaling and enhanced cell proliferation. These findings suggest a forward feedback loop in Smurf1-ERα regulation.
Smurf1 is also overexpressed in human gastric cancer (GC) tissues (86). Moreover, Smurf1 expression levels were shown to be positively associated with more advanced tumor-node-metastasis (TNM) stage of GC, and inversely correlated with patient survival. Knockdown of Smurf1 inhibited proliferation, migration and invasion of GC cells, at least in some GC cell models, while Smurf1 overexpression exacerbated these phenotypes. Furthermore, the authors reported that Smurf1-knockdown in GC cells markedly inhibits tumor growth and liver metastasis in vivo. Mechanistically, they linked the Smurf1 pro-oncogenic activities with the ability of Smurf1 to negatively regulate the expression of DAB2IP, a GTPase-activating protein (GAP) and a suggested tumor suppressor (86).
The existence of similar Smurf1-regulated tumor promoting mechanism was also observed in clear cell renal cell carcinoma (ccRCC) cells (87). This mechanism was associated with the ability of Smurf1 to promote proliferation, migration, and invasion of ccRCC cells. In addition, the expression levels of Smurf1 were found to be elevated both in ccRCC cell lines and cancer tissues, and associated with worse patient survival.
Concluding Remarks and Future Directions
In this review, we highlighted and discussed the cancer related biological functions of two C2-WW-HECT E3 ligases, Smurf1 and Smurf2. These proteins surfaced as influential, and under some circumstances, as decisive cellular factors regulating a plethora of cellular processes pertinent to cancer onset, progression and therapy.
The currently available data stipulate that Smurf1 acts as an oncogene, whereas Smurf2 operates both as a tumor suppressor and a tumor promoting molecule, depending on the tumor stage, type, molecular binding partners, and other still unidentified factors.
It is now evident that apart from TGF-β/BMP signaling, Smurfs regulate different signaling pathways and networks. Understanding these networks and the Smurfs' impact on their components is important and should be further investigated.
Another line of investigation is elucidating the mechanisms regulating expression, localization and functions of Smurfs. Currently, these mechanisms remain elusive. Understanding of these mechanisms is imperative in explaining the dual role of Smurf2 in cancer and its impact on cancer progression and treatment. In addition, the full spectrum of mechanisms and networks operating under Smurf1/2 auspices is also currently unknown.
Recently, we have shown that Smurf2 in addition to its ability to control protein homeostasis through the proteasomal breakdown, targets some cellular proteins for autophagic-lysosomal turnover (88). Specifically, we found that Smurf2 regulates selective autophagy of nuclear lamins, in particular lamin A, and its mutant form progerin (Figure 5). The expression of progerin underlies the pathogenesis of the devastating premature aging syndrome, HGPS (Hutchinson-Gilford progeria syndrome). Remarkable, in addition to HGPS, progerin also accumulates in cells during physiological aging and supposedly in cancer, where it could promote genomic instability and increase tumorigenesis (89–91). This association suggests that targeting progerin through the Smurf2-mediated autophagy might be a promising direction to eradicate tumor cells, though more research is needed in this regard.
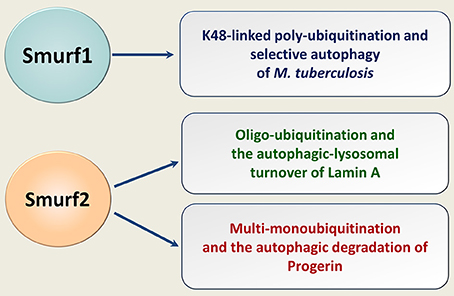
Figure 5. Smurfs in selective autophagy of human pathogens and endogenous cellular proteins. Smurf1 mediates the K48-linked polyubiquitination of human pathogen M. tuberculosis, targeting it for the ubiquitin-dependent selective autophagy. Smurf2 oligo-ubiquitinates lamin A and multi-monoubiquitinates progerin. Smurf2 targets both lamin A and progerin for the autophagic-lysosomal turnover.
Smurf1 has also been implicated in selective autophagy, in particular in eliminating in human cells of Mycobacterium tuberculosis (92) (Figure 5). Whether Smurf1 can also mediate the autophagic degradation of endogenous cellular proteins is currently unknown.
Both the ubiquitin-proteasome and autophagic-lysosomal degradation pathways are intrinsically involved in cancer initiation, progression and cure. Thus, understanding mechanisms operating in the intersection of these protein turnover machineries could provide novel therapeutic targets and paradigms in cancer diagnosis and treatment. Further efforts should be directed to characterize the involvement of Smurf proteins in these processes. Another important direction that we believe should be investigated is elucidating whether and how targeting of Smurfs by pharmacological intervention (e.g., by Smurf catalytic inhibitors) affect the ability of tumor cells to escape the destructive impact of anticancer drugs and therapies used in clinics. This is a more long-term goal as such compounds are currently unavailable.
Author Contributions
PK and GL-C drafted the initial version of the manuscript, and prepared figures and bibliography. MB conceptualized the study and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dayan Family Foundation for their continued support of our research. We also thank the Israel Cancer Research Fund (grant #00636), Israel Cancer Association (grant #20160073), and FP-7 (Marie-Curie CIG grant #612816).
Abbreviations
aa, amino acids; AR, Androgen receptor; BMP, Bone morphogenetic protein; BMPRI, Bone morphogenetic protein receptor, type 1; β-TrCP, β-transducing repeat containing protein; ccRCC, Clear cell renal cell carcinoma; CDC25A, Cell division Cycle 25A; COSMIC, Catalog of Somatic Mutations in Cancer; Cys, Cystein; DAB2IP, Disabled homolog 2-Interacting Protein; DDR, DNA damage response; E3s, E3 ubiquitin ligases; EGFR, Epidermal growth factor receptor; EMT, Epithelial-mesenchymal transition; ERα, Estrogen receptor alpha; EZH2, Enhancer of zeste homolog 2; GAP, GTPase-activating protein; GC, Gastric cancer; Gly, Glycine; GSK3β, Glycogen synthase kinase 3 beta; GTPase, Guanosine triphosphatases; H2B, Histone H2B; HECT, Homologous to E6-AP carboxyl terminus; HGPS, Hutchinson-Gilford progeria syndrome; HSP27, Heat shock protein 27; ID1, DNA-binding protein inhibitor ID1; ID3, DNA-binding protein inhibitor ID3; I-Smad, Inhibitory Smads; JNK, c-Jun N-terminal protein kinase; KLF5, Krüppel-like factor 5; KRAS, Kirsten rat sarcoma protein; LOH, Loss of heterozygosity; MAD2, Mitotic spindle assembly checkpoint protein; MAPK, Mitogen-activated protein kinase; MAPK, Mitogen activated protein kinase; MLC2, Myosin light chain 2; MEK, Mitogen-activated protein kinase kinase; MEKK2, Mitogen-activated protein kinase kinase kinase 2; MET, Mesenchymal-epithelial transition; NEDD4, Neural precursor cell expressed developmentally down-regulated protein 4; PIAS3, Protein inhibitor of activated STAT3; PKCζ, Protein kinase C zeta; PRC2, Polycomb repressive complex 2; R-Smad, Receptor regulated Smad; RANKL, Receptor activator of nuclear factor kappa-B ligand; RBR, Ring-in-Between-RING fingers; REST, RE1 silencing transcription factor; RhoA, Ras homolog gene family, member A; RING, Really interesting new gene; RNF20, Ring Finger Protein 20; SCF, Skp1–Cullin–F-box proteins; Smurf, Smad ubiquitin regulatory factor; TGF-β, Transforming growth factor beta; TGF-βRI, Transforming growth factor beta receptor I; TNM, Tumor node metastasis; Topo Iiα, DNA Topoisomerase 2 alpha; ubH2B, monoubiquitinated histone H2B; WWP1, WW domain containing E3 ubiquitin protein ligase 1; WWP2, WW domain containing E3 ubiquitin protein ligase 2; YY1, Yin Yang 1.
References
1. Welchman RL, Gordon C, Mayer RJ. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol. (2005) 6:599–609. doi: 10.1038/nrm1700
2. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. (2014) 20:1242–53. doi: 10.1038/nm.3739
3. Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem Sci. (2017) 42:873–86. doi: 10.1016/j.tibs.2017.09.002
4. Morreale FE, Walden H. Types of ubiquitin ligases. Cell (2016) 165:248–248.e1. doi: 10.1016/j.cell.2016.03.003
5. Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene (2004) 23:1972–84. doi: 10.1038/sj.onc.1207436
6. Scheffner M, Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys Acta (2014) 1843:61–74. doi: 10.1016/j.bbamcr.2013.03.024
7. Zou X, Levy-Cohen G, Blank M. Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochim Biophys Acta (2015) 1856:91–106. doi: 10.1016/j.bbcan.2015.06.005
8. Chong PA, Lin H, Wrana JL, Forman-Kay JD. Coupling of tandem Smad ubiquitination regulatory factor (Smurf) WW domains modulates target specificity. Proc Natl Acad Sci USA. (2010) 107:18404–9. doi: 10.1073/pnas.1003023107
9. David D, Nair SA, Pillai MR. Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochim Biophys Acta (2013) 1835:119–28. doi: 10.1016/j.bbcan.2012.11.003
10. Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. (2009) 19:71–88. doi: 10.1038/cr.2008.302
11. Dempke WCM, Fenchel K, Uciechowski P, Chevassut T. Targeting developmental pathways: the achilles heel of cancer? Oncology (2017) 93:213–23. doi: 10.1159/000478703
12. Bach DH, Park HJ, Lee SK. The dual role of bone morphogenetic proteins in cancer. Mol Ther Oncolytics (2017) 8:1–13. doi: 10.1016/j.omto.2017.10.002
13. Colak S, Ten Dijke P. Targeting TGF-β Signaling in Cancer. Trends Cancer (2017) 3:56–71. doi: 10.1016/j.trecan.2016.11.008
14. Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature (1999) 400:687–93.
15. Murakami G, Watabe T, Takaoka K, Miyazono K, Imamura T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol Biol Cell (2003) 14:2809–17. doi: 10.1091/mbc.e02-07-0441
16. Nakao A. Inhibitory Smads: mechanisms of action and roles in human diseases. In: Dijke P, Heldin CH. editors. Smad Signal Transduction. Proteins and Cell Regulation, Vol. 5. Dordrecht: Springer. (2006). p. 379–95.
17. Murakami K, Mathew R, Huang J, Farahani R, Peng H, Olson SC, et al. Smurf1 ubiquitin ligase causes downregulation of BMP receptors and is induced in monocrotaline and hypoxia models of pulmonary arterial hypertension. Exp Biol Med. (2010) 235:805–13. doi: 10.1258/ebm.2010.009383
18. Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature (1997) 389:631–5.
19. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell (2000) 6:1365–75. doi: 10.1016/S1097-2765(00)00134-9
20. Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem. (2000) 275:36818–22. doi: 10.1074/jbc.C000580200
21. Fukuchi M, Imamura T, Chiba T, Ebisawa T, Kawabata M, Tanaka K, et al. Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of ROC1 and associated proteins. Mol Biol Cell (2001) 12:1431–43. doi: 10.1091/mbc.12.5.1431
22. Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell (2005) 121:101–13. doi: 10.1016/j.cell.2005.01.035
23. Tang LY, Yamashita M, Coussens NP, Tang Y, Wang X, Li C, et al. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. (2011) 30:4777–89. doi: 10.1038/emboj.2011.393
24. Fukunaga E, Inoue Y, Komiya S, Horiguchi K, Goto K, Saitoh M, et al. Smurf2 induces ubiquitin-dependent degradation of Smurf1 to prevent migration of breast cancer cells. J Biol Chem. (2008) 283:35660–7. doi: 10.1074/jbc.M710496200
25. Xu Z, Greenblatt MB, Yan G, Feng H, Sun J, Lotinun S, et al. SMURF2 regulates bone homeostasis by disrupting SMAD3 interaction with vitamin D receptor in osteoblasts. Nat Commun. (2017) 8:14570. doi: 10.1038/ncomms14570
26. Blank M, Tang Y, Yamashita M, Burkett SS, Cheng SY, Zhang YE. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat Med. (2012) 18:227–34. doi: 10.1038/nm.2596
27. Dutta S, Sengupta P. Men and mice: relating their ages. Life Sci. (2016) 152:244–8. doi: 10.1016/j.lfs.2015.10.025
28. Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol. (1996) 7:269–78.
29. Lozano G. Mouse models of p53 functions. Cold Spring Harb Perspect Biol. (2010) 2:a001115. doi: 10.1101/cshperspect.a001115
30. Ramkumar C, Kong Y, Cui H, Hao S, Jones SN, Gerstein RM, et al. Smurf2 regulates the senescence response and suppresses tumorigenesis in mice. Cancer Res. (2012) 72:2714–9. doi: 10.1158/0008-5472.CAN-11-3773
31. Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol. (2011) 7:113–9. doi: 10.1038/nchembio.501
32. Minsky N, Shema E, Field Y, Schuster M, Segal E, Oren M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat Cell Biol. (2008) 10:483–8. doi: 10.1038/ncb1712
33. Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell (2011) 41:529–42. doi: 10.1016/j.molcel.2011.02.015
34. Liu Z, Oh SM, Okada M, Liu X, Cheng D, Peng J, et al. Human BRE1 is an E3 ubiquitin ligase for Ebp1 tumor suppressor. Mol Biol Cell (2009) 20:757–68. doi: 10.1091/mbc.E08-09-0983
35. Gao Z, Xu MS, Barnett TL, Xu CW. Resveratrol induces cellular senescence with attenuated mono-ubiquitination of histone H2B in glioma cells. Biochem Biophys Res Commun. (2011) 407:271–6. doi: 10.1016/j.bbrc.2011.02.008
36. Jääskeläinen T, Makkonen H, Visakorpi T, Kim J, Roeder RG, Palvimo JJ. Histone H2B ubiquitin ligases RNF20 and RNF40 in androgen signaling and prostate cancer cell growth. Mol Cell Endocrinol. (2012) 350:87–98. doi: 10.1016/j.mce.2011.11.025
37. Wang E, Kawaoka S, Yu M, Shi J, Ni T, Yang W, et al. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc Natl Acad Sci USA. (2013) 110:3901–6. doi: 10.1073/pnas.1301045110
38. Cole AJ, Clifton-Bligh R, Marsh DJ. Histone H2B monoubiquitination: roles to play in human malignancy. Endocr Relat Cancer (2015) 22:T19–33. doi: 10.1530/ERC-14-0185
39. Tarcic O, Granit RZ, Pateras IS, Masury H, Maly B, Zwang Y, et al. RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death Differ. (2017) 24:694–704. doi: 10.1038/cdd.2016.126
40. Emanuelli A, Borroni AP, Apel-Sarid L, Shah PA, Ayyathan DM, Koganti P, et al. Smurf2-mediated stabilization of DNA topoisomerase IIα controls genomic integrity. Cancer Res. (2017) 77:4217–27. doi: 10.1158/0008-5472.CAN-16-2828
41. Chen T, Sun Y, Ji P, Kopetz S, Zhang W. Topoisomerase IIα in chromosome instability and personalized cancer therapy. Oncogene (2015) 34:4019–31. doi: 10.1038/onc.2014.332
42. Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S. Heat shock proteins and cancer. Trends Pharmacol Sci. (2017) 38:226–56. doi: 10.1016/j.tips.2016.11.009
43. Sun Y, Zhou M, Fu D, Xu B, Fang T, Ma Y, et al. Ubiquitination of heat shock protein 27 is mediated by its interaction with Smad ubiquitination regulatory factor 2 in A549 cells. Exp Lung Res. (2011) 37:568–73. doi: 10.3109/01902148.2011.619627
44. Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci. (2009) 66:2691–706. doi: 10.1007/s00018-009-0045-z
45. Du JX, Hagos EG, Nandan MO, Bialkowska AB, Yu B, Yang VW. The E3 ubiquitin ligase SMAD ubiquitination regulatory factor 2 negatively regulates Krüppel-like factor 5 protein. J Biol Chem. (2011) 286:40354–64. doi: 10.1074/jbc.M111.258707
46. Khachigian LM. The Yin and Yang of YY1 in tumor growth and suppression. Int J Cancer (2018) 143:460–5. doi: 10.1002/ijc.31255 [Epub ahead of print].
47. Jeong HM, Lee SH, Yum J, Yeo CY, Lee KY. Smurf2 regulates the degradation of YY1. Biochim Biophys Acta Mol Cell Res. (2014) 1843:2005–11. doi: 10.1016/j.bbamcr.2014.04.023
48. Ramkumar C, Cui H, Kong Y, Jones SN, Gerstein RM, Zhang H. Smurf2 suppresses B-cell proliferation and lymphomagenesis by mediating ubiquitination and degradation of YY1. Nat Commun. (2013) 4:2598. doi: 10.1038/ncomms3598
49. Kong Y, Cui H, Zhang H. Smurf2-mediated ubiquitination and degradation of Id1 regulates p16 expression during senescence. Aging Cell (2011) 10:1038–46. doi: 10.1111/j.1474-9726.2011.00746.x
50. Mern DS, Hoppe-Seyler K, Hoppe-Seyler F, Hasskarl J, Burwinkel B. Targeting Id1 and Id3 by a specific peptide aptamer induces E-box promoter activity, cell cycle arrest, and apoptosis in breast cancer cells. Breast Cancer Res Treat (2010) 124:623–33. doi: 10.1007/s10549-010-0810-6
51. Ling F, Kang B, Sun XH. Id proteins: small molecules, mighty regulators. Curr Top Dev Biol. (2014) 110:189–216. doi: 10.1016/B978-0-12-405943-6.00005-1
52. Li B, Tsao SW, Chan KW, Ludwig DL, Novosyadlyy R, Li YY, et al. Id1-induced IGF-II and its autocrine/endocrine promotion of esophageal cancer progression and chemoresistance–implications for IGF-II and IGF-IR-targeted therapy. Clin Cancer Res. (2014) 20:2651–62. doi: 10.1158/1078-0432.CCR-13-2735
53. Li B, Xu WW, Guan XY, Qin YR, Law S, Lee NP, et al. Competitive binding between Id1 and E2F1 to Cdc20 regulates E2F1 degradation and thymidylate synthase expression to promote esophageal cancer chemoresistance. Clin Cancer Res. (2016) 22:1243–55. doi: 10.1158/1078-0432.CCR-15-1196
54. Zhao Y, Luo A, Li S, Zhang W, Chen H, Li Y, et al. Inhibitor of differentiation/DNA binding 1 (ID1) inhibits etoposide-induced apoptosis in a c-Jun/c-Fos-dependent Manner. J Biol Chem. (2016) 291:6831–42. doi: 10.1074/jbc.M115.704361
55. Yin X, Tang B, Li JH, Wang Y, Zhang L, Xie XY, et al. ID1 promotes hepatocellular carcinoma proliferation and confers chemoresistance to oxaliplatin by activating pentose phosphate pathway. J Exp Clin Cancer Res. (2017) 36:166. doi: 10.1186/s13046-017-0637-7
56. Yu YL, Chou RH, Shyu WC, Hsieh SC, Wu CS, Chiang SY, et al. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol Med. (2013) 5:531–47. doi: 10.1002/emmm.201201783
58. Osmundson EC, Ray D, Moore FE, Gao Q, Thomsen GH, Kiyokawa H. The HECT E3 ligase Smurf2 is required for Mad2-dependent spindle assembly checkpoint. J Cell Biol. (2008) 183:267–77. doi: 10.1083/jcb.200801049
59. Rao S, Cronin SJF, Sigl V, Penninger JM. RANKL and RANK: from mammalian physiology to cancer treatment. Trends Cell Biol. (2018) 28:213–23. doi: 10.1016/j.tcb.2017.11.001
60. Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene (2007) 26:5169–83. doi: 10.1038/sj.onc.1210337
61. Zhang F, Virshup DM, Cheong JK. Oncogenic RAS-induced CK1α drives nuclear FOXO proteolysis. Oncogene (2018) 37:363–76. doi: 10.1038/onc.2017.334
62. Hausmann M, Ilić N, Pilarczyk G, Lee JH, Logeswaran A, Borroni AP, et al. Challenges for super-resolution localization microscopy and biomolecular fluorescent nano-probing in cancer research. Int J Mol Sci. (2017) 18:E2066. doi: 10.3390/ijms18102066
63. Fukuchi M, Fukai Y, Masuda N, Miyazaki T, Nakajima M, Sohda M, et al. High-level expression of the smad ubiquitin ligase Smurf2 correlates with poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Res. (2002) 62:7162–5.
64. Wang SM, Ooi LL, Hui KM. Identification and validation of a novel gene signature associated with the recurrence of human hepatocellular carcinoma. Clin Cancer Res. (2007) 13:6275–83. doi: 10.1158/1078-0432.CCR-06-2236
65. Liu X, Gu X, Sun L, Flowers AB, Rademaker AW, Zhou Y, et al. Downregulation of Smurf2, a tumor-suppressive ubiquitin ligase, in triple-negative breast cancers: involvement of the RB-microRNA axis. BMC Cancer (2014) 14:57. doi: 10.1186/1471-2407-14-57
66. Shukla S, Allam US, Ahsan A, Chen G, Krishnamurthy PM, Marsh K, et al. KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated β-TrCP1 degradation. Neoplasia (2014) 16:115–28. doi: 10.1593/neo.14184
67. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer (2011) 11:761–74. doi: 10.1038/nrc3106
68. Jeong WJ, Yoon J, Park JC, Lee SH, Lee SH, Kaduwal S, et al. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci Signal. (2012) 5:ra30. doi: 10.1126/scisignal.2002242
69. Wang H, Maitra A, Wang H. The emerging roles of F-box proteins in pancreatic tumorigenesis. Semin Cancer Biol. (2016) 36:88–94. doi: 10.1016/j.semcancer.2015.09.004
70. Lau AW, Fukushima H, Wei W. The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front Biosci. (2012) 17:2197–212. doi: 10.2741/4045
71. Wu Q, Huang J, Sampson E, Kim KO, Zuscik MJ, O'Keefe RJ, et al. Smurf2 induces degradation of GSK-3β and upregulates β-catenin in chondrocytes: a potential mechanism for Smurf2-induced degeneration of articular cartilage. Exp Cell Res. (2009) 315:2386–98. doi: 10.1016/j.yexcr.2009.05.019
72. Kim S, Jho E. The protein stability of axin, a negative regulator of Wnt signaling, is regulated by Smad ubiquitination regulatory factor 2 (Smurf2). J Biol Chem. (2010) 285:36420–6. doi: 10.1074/jbc.M110.137471
73. Kim SE, Yoon JY, Jeong WJ, Jeon SH, Park Y, Yoon JB, et al. H-Ras is degraded by Wnt/beta-catenin signaling via beta-TrCP-mediated polyubiquitylation. J Cell Sci. (2009) 122(Pt 6):842–8. doi: 10.1242/jcs.040493.
74. Ray D, Ahsan A, Helman A, Chen G, Hegde A, Gurjar SR, et al. Regulation of EGFR protein stability by the HECT-type ubiquitin ligase SMURF2. Neoplasia (2011) 13:570–8. doi: 10.1593/neo.11632
75. Smith MP, Ferguson J, Arozarena I, Hayward R, Marais R, Chapman A, et al. Effect of SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J Natl Cancer Inst. (2013) 105:33–46. doi: 10.1093/jnci/djs471
76. Jin C, Yang Y, Anver MR, Morris N, Wang X, Zhang YE. Smad ubiquitination regulatory factor 2 promotes metastasis of breast cancer cells by enhancing migration and invasiveness. Cancer Res. (2009) 69:735–40. doi: 10.1158/0008-5472.can-08-1463
77. Chandhoke AS, Chanda A, Karve K, Deng L, Bonni S. The PIAS3-Smurf2 sumoylation pathway suppresses breast cancer organoid invasiveness. Oncotarget (2017) 8:21001–14. doi: 10.18632/oncotarget.15471
78. Manikoth Ayyathan D, Ilić N, Gil-Henn H, Blank M. Generation of SMURF2 knockout human cells using the CRISPR/Cas9 system. Anal Biochem. (2017) 531:56–9. doi: 10.1016/j.ab.2017.05.024
79. Zhang WL, Zhang JH, Wu XZ, Yan T, Lv W. miR-15b promotes epithelial-mesenchymal transition by inhibiting SMURF2 in pancreatic cancer. Int J Oncol. (2015) 47:1043–53. doi: 10.3892/ijo.2015.3076
80. Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, et al. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science (2003) 302:1775–9. doi: 10.1126/science.1090772
81. Sahai E, Garcia-Medina R, Pouysségur J, Vial E. Smurf1 regulates tumor cell plasticity and motility through degradation of RhoA leading to localized inhibition of contractility. J Cell Biol. (2007) 176:35–42. doi: 10.1083/jcb.200605135
82. Kwon A, Lee HL, Woo KM, Ryoo HM, Baek JH. SMURF1 plays a role in EGF-induced breast cancer cell migration and invasion. Mol Cells (2013) 36:548–55. doi: 10.1007/s10059-013-0233-4
83. Wang W, Ren F, Wu Q, Jiang D, Li H, Peng Z, et al. MicroRNA-497 inhibition of ovarian cancer cell migration and invasion through targeting of SMAD specific E3 ubiquitin protein ligase 1. Biochem Biophys Res Commun. (2014) 449:432–7. doi: 10.1016/j.bbrc.2014.05.053
84. Gang X, Wang G, Huang H. Androgens regulate SMAD ubiquitination regulatory factor-1 expression and prostate cancer cell invasion. Prostate (2015) 75:561–72. doi: 10.1002/pros.22935
85. Yang H, Yu N, Xu J, Ding X, Deng W, Wu G, et al. SMURF1 facilitates estrogen receptor signaling in breast cancer cells. J Exp Clin Cancer Res. (2018) 37:24. doi: 10.1186/s13046-018-0672-z
86. Tao Y, Sun C, Zhang T, Song Y. SMURF1 promotes the proliferation, migration and invasion of gastric cancer cells. Oncol Rep. (2017) 38:1806–14. doi: 10.3892/or.2017.5825
87. Ke M, Mo L, Li W, Zhang X, Li F, Yu H. Ubiquitin ligase SMURF1 functions as a prognostic marker and promotes growth and metastasis of clear cell renal cell carcinoma. FEBS Open Bio. (2017) 7:577–86. doi: 10.1002/2211-5463.12204
88. Borroni AP, Emanuelli A, Shah PA, Ilić N, Apel-Sarid L, Paolini B, et al. Smurf2 regulates stability and the autophagic-lysosomal turnover of lamin A and its disease-associated form progerin. Aging Cell (2018) 17:e12732. doi: 10.1111/acel.12732
89. Butin-Israeli V, Adam SA, Goldman AE, Goldman RD. Nuclear lamin functions and disease. Trends Genet. (2012) 28:464–71. doi: 10.1016/j.tig.2012.06.001
90. Tang Y, Chen Y, Jiang H, Nie D. Promotion of tumor development in prostate cancer by progerin. Cancer Cell Int. (2010) 10:47. doi: 10.1186/1475-2867-10-47
91. Kreienkamp R, Graziano S, Coll-Bonfill N, Bedia-Diaz G, Cybulla E, Vindigni A, et al. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin. Cell Rep. (2018) 22:2006–15. doi: 10.1016/j.celrep.2018.01.090
Keywords: Smurf1, Smurf2, ubiquitination, protein degradation, cell signaling, cancer
Citation: Koganti P, Levy-Cohen G and Blank M (2018) Smurfs in Protein Homeostasis, Signaling, and Cancer. Front. Oncol. 8:295. doi: 10.3389/fonc.2018.00295
Received: 24 May 2018; Accepted: 16 July 2018;
Published: 02 August 2018.
Edited by:
Rami I. Aqeilan, Hadassah Medical Center, IsraelReviewed by:
Zaidoun Salah, Al-Quds University, PalestineDaniele Vergara, University of Salento, Italy
Copyright © 2018 Koganti, Levy-Cohen and Blank. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Blank, bWljaGFlbC5ibGFua0BiaXUuYWMuaWw=