 Leonardo Vinícius Monteiro de Assis1*†
Leonardo Vinícius Monteiro de Assis1*† Gabriela Sarti Kinker2*†
Gabriela Sarti Kinker2*† Maria Nathália Moraes1
Maria Nathália Moraes1 Regina P. Markus2Pedro Augusto Fernandes2Ana Maria de Lauro Castrucci1,3
Regina P. Markus2Pedro Augusto Fernandes2Ana Maria de Lauro Castrucci1,3
- 1Laboratory of Comparative Physiology of Pigmentation, Department of Physiology, Institute of Biosciences, University of São Paulo, São Paulo, Brazil
- 2Laboratory of Neuroimmunemodulation, Department of Physiology, Institute of Biosciences, University of São Paulo, São Paulo, Brazil
- 3Department of Biology, University of Virginia, Charlottesville, VA, United States
Introduction: Melanoma is the most lethal type of skin cancer, with increasing incidence and mortality rates worldwide. Multiple studies have demonstrated a link between cancer development/progression and circadian disruption; however, the complex role of tumor-autonomous molecular clocks remains poorly understood. With that in mind, we investigated the pathophysiological relevance of clock genes expression in metastatic melanoma.
Methods: We analyzed gene expression, somatic mutation, and clinical data from 340 metastatic melanomas from The Cancer Genome Atlas, as well as gene expression data from 234 normal skin samples from genotype-tissue expression. Findings were confirmed in independent datasets.
Results: In melanomas, the expression of most clock genes was remarkably reduced and displayed a disrupted pattern of co-expression compared to the normal skins, indicating a dysfunctional circadian clock. Importantly, we demonstrate that the expression of the clock gene aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1) positively correlates with patient overall survival and with the expression of T-cell activity and exhaustion markers in the tumor bulk. Accordingly, high BMAL1 expression in pretreatment samples was significantly associated with clinical benefit from immune checkpoint inhibitors. The robust intratumoral T-cell infiltration/activation observed in patients with high BMAL1 expression was associated with a decreased expression of key DNA-repair enzymes, and with an increased mutational/neoantigen load.
Conclusion: Overall, our data corroborate previous reports regarding the impact of BMAL1 expression on the cellular DNA-repair capacity and indicate that alterations in the tumor-autonomous molecular clock could influence the cellular composition of the surrounding microenvironment. Moreover, we revealed the potential of BMAL1 as a clinically relevant prognostic factor and biomarker for T-cell-based immunotherapies.
Significance
Here, we provide a first glimpse regarding the impact of a disrupted tumor-autonomous molecular clock on the cellular composition of the tumor microenvironment through the modulation of DNA-repair capacity. Within this line, our data revealed the potential of BMAL1 as a clinically relevant biomarker for immunotherapy response and overall survival of patients with metastatic melanoma.
Introduction
Melanoma is the most lethal type of skin cancer, with increasing incidence and mortality rates worldwide (1, 2). It represents only 4% of skin cancer but accounts for approximately 80% of skin cancer-related death (3). Although complete surgical resection is often curative for melanomas detected at initial stages, patients with metastatic disease have an overall survival of approximately 5 months (4). Therapeutic options for patients with metastatic melanoma have dramatically changed in the past years, with the introduction of more effective agents such as proto-oncogene, serine/theronine kinase (BRAF), mitogen activated protein kinase kinase (MAPK), and immunotherapeutic antibodies directed to cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed cell-death protein 1 (PD-1) and its ligand (PD-L1) (5–8). Melanoma etiology is multifactorial and includes risk factors such as ultraviolet radiation exposure, genetic susceptibility, high nevus density, reduced skin pigmentation, and immunosuppression (9, 10).
Proper temporal control of physiological functions is crucial for maintaining the homeostasis of multi-cellular organisms (11–13). In mammals, the molecular machinery of timekeeping and circadian rhythm generation is based on interconnected positive and negative transcriptional–translational feedback loops. The central hypothalamic clock (suprachiasmatic nuclei, SCN) and clocks located in peripheral tissues share the same molecular architecture, engaging core genes such as aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1 also known as ARNTL), cryptochrome 1 and 2 (CRY1/2), circadian locomotor output cycles kaput (CLOCK), period 1, 2, and 3 (PER1/2/3), receptor subfamily 1, group D, member 1/2 (NRD1/2 also known as REV-ERBα/β), and RAR-related orphan receptor A and B (RORA/B also known as NR1F1/2). In healthy conditions, CLOCK–BMAL1 heterodimers translocate to the nucleus and induce the gene expression of their own inhibitors, PER and CRY proteins. This core oscillatory pathway is augmented and stabilized by a secondary loop involving NRD1/2 and RORA/B, nuclear receptors that modulate BMAL1 expression. Importantly, CLOCK–BMAL1 heterodimers also regulate the expression of several clock-controlled genes, which are tissue- and cell type-specific (11–13).
Many epidemiologic studies have demonstrated that the disturbance of biological rhythms through shift work, increased light exposure at night, and irregular feeding regimens (14–16) is associated with increased risk of developing several types of cancers (17–19). In fact, alterations in the cellular circadian machinery have been shown to affect cancer-related processes such as cell proliferation (20, 21), DNA damage response (22, 23), and metabolism (24–27) in a tumor-specific manner. Accordingly, the aberrant expression of clock core genes such as CRY1, PER1, and PER2 has been shown to impact tumor progression in colorectal, prostate, and breast cancers, respectively (28–30).
In melanoma, mRNA levels and nuclear immunopositivity for CLOCK, CRY1, and PER1 are reduced compared to adjacent non-tumorous skin and present a significant association with clinicopathological features such as Breslow thickness (31). Moreover, the expression of RORA is lower in melanomas than in nevi, and positively correlates with overall survival and disease-free survival (32). Interestingly, enhancing the circadian clock function of melanoma cells impairs cell cycle progression and inhibits tumor growth in vivo (21). In this sense, we have previously demonstrated that the expression of clock core genes in murine melanoma cells can be activated by different stimulus, such as white light exposure (33), UVA radiation (34), estradiol (35), and thermal energy (36). Recently, we have demonstrated that a non-metastatic model of melanoma leads to a systemic chronodisruption in tumor-adjacent skin, lungs, liver, and SCN, as in these tissues the rhythmic expression of Bmal1 was lost in tumor-bearing mice (37). These data reinforce that the modulation of tumor-autonomous clock might represent a novel and promising therapeutic strategy.
To further characterize the pathophysiological relevance of the molecular clock in skin cancer, we investigated the clinical value of clock core genes expression in metastatic melanoma, using public high-throughput molecular data. Overall, we revealed the robust prognostic power of BMAL1 expression and provided evidence into its underlying biological processes.
Materials and Methods
Datasets of Melanoma and Normal Skin Samples
Gene expression, somatic mutation, and clinical data from 340 metastatic melanomas from The Cancer Genome Atlas (TCGA) and gene expression data from 234 Genotype-Tissue Expression (GTEx) normal skin (not sun exposed) samples were downloaded from the UCSC XENA Browser (http://xena.ucsc.edu) in January of 2017. TCGA and GTEx gene expression data were originally generated by TCGA (38) and GTEx consortia (39), respectively, using the Illumina HiSeq 2000 RNA sequencing platform, quantified using RSEM, upper quartile normalized and log2(x + 1) transformed. TCGA somatic mutation data were generated using the Illumina GAIIx DNA sequencing platform and somatic variants (SNPs and small indels) were identified using MuTect2. Neoantigen load information for TCGA metastatic melanoma samples was obtained from Rooney et al. (40). Briefly, for each metastatic melanoma patient, all novel amino acid 9–10mers resulting from missense mutations in expressed genes (median > 10 TPM) were identified. Mutant peptides with a HLA-binding affinity <500 nM, predicted by NetMHCpan (v2.4), were considered antigenic (41). Clinical information and gene expression data of pretreatment biopsies from 49 patients who received anti-PD1 immunotherapy (nivolumab) were obtained from Riaz et al. (42). Expression data were generated using the Illumina HiSeq 2000 RNA sequencing platform, counted using Rsamtools v3.2, upper quartile normalized and log2(x + 1) transformed. Treatment response for patients was defined by RECIST v1.1.
Co-Expression Network Analysis
Undirected weighted co-expression networks were constructed based on the pairwise Spearman’s correlation coefficients between the expression of clock core genes BMAL1, CRY1, CRY2, NRD1, PER1, PER2, PER3, and RORA. Using the CoGA R package (43), we compared the structural properties of co-expression networks from normal skin and metastatic melanomas by testing the equality in their spectral distributions (44, 45). The spectrum of a graph, defined as the set of eigenvalues of its adjacency matrix, describes several structural features and represents a comprehensive characterization of networks (44, 46). P-values were calculated based on 1,000 phenotype permutations and networks were visualized using the gplots R package.
Gene Set Enrichment Analysis (GSEA)
Genes in the TCGA expression dataset were ranked according to the Spearman’s correlation coefficient between their expression and the expression of BMAL1. GSEA was performed using GSEA v3.0 and Reactome pathways (47, 48). Enrichment scores (ES) were calculated based on a weighted Kolmogorov–Smirnov-like statistic and normalized (NES) to account for the size of each gene set. P-values corresponding to each NES were calculated based on 1,000 phenotype permutations and corrected for multiple comparisons using the false discovery rate (FDR) procedure. Adjusted P-values < 0.05 were considered statistically significant.
Single Sample Gene Set Enrichment Analysis (ssGSEA)
Single sample gene set enrichment analysis, an extension of GSEA, was used to estimate the degree of enrichment of gene sets in individual samples within the TCGA gene expression dataset (49). For each sample, gene expression values were rank-normalized, and ESs were calculated based on the difference between weighted Empirical Cumulative Distribution Functions of genes inside and outside the gene sets. We performed ssGSEA using the GSVA R package (50) and DNA repair-related KEGG pathways (51), namely: base excision repair (hsa03410), nucleotide excision repair (hsa03420), mismatch repair (hsa03430), homologous recombination (hsa03440), and non-homologous end joining (hsa0345).
Statistical Analysis
We used the two-sided Wilcoxon–Mann–Whitney test to perform two-group comparisons, the Spearman’s correlation test to assess ordinal associations, and the Chi-square test to analyze the relationship between two categorical variables. The impact of clock core genes expression on patient overall survival was evaluated using univariate Cox regressions. The prognostic power of BMAL1 expression was further investigated using Kaplan–Meier curves, combined to the log-rank test, and multivariate Cox regressions. Hazard Ratios, including 95% confidence intervals, were calculated. Statistical analyses were performed with GraphPad Prism 6 and R (www.r-project.org). P-values < 0.05 were considered statistically significant. Where indicated, P-values were adjusted for multiple comparisons using the FDR procedure.
Results
Clinical Relevance of Clock Core Genes Expression in Metastatic Melanomas
We first analyzed the expression of clock core genes in normal skin and in metastatic melanomas. Compared to normal skin, metastatic melanomas demonstrated a remarkably decreased expression of BMAL1, CRY1, CRY2, NRD1, PER1, PER2, PER3, and RORA and an increased expression of CLOCK (Figure 1A). In normal skin, we have found a classic pattern of clock gene expression: PERs and CRYs are concomitantly expressed (in phase) and are in antiphase with BMAL1 and CLOCK expression, as expected; on the other hand, in metastatic melanomas such correlations are severely attenuated (Figure 1B), which further corroborates a dysfunctional circadian clock within the tumor. In metastatic melanomas, male presented increased percentage of tumor showing high expression of NRD1, PER2, and PER3 (P = 0.015, P = 0.028, and P < 0.001, respectively; Table 1; Table S1 in Supplementary Material). Patients with high PER3 expression were also significantly older and more frequently diagnosed with stage I–II tumors (P = 0.002 and P = 0.037, respectively; Table 1; Table S1 in Supplementary Material).
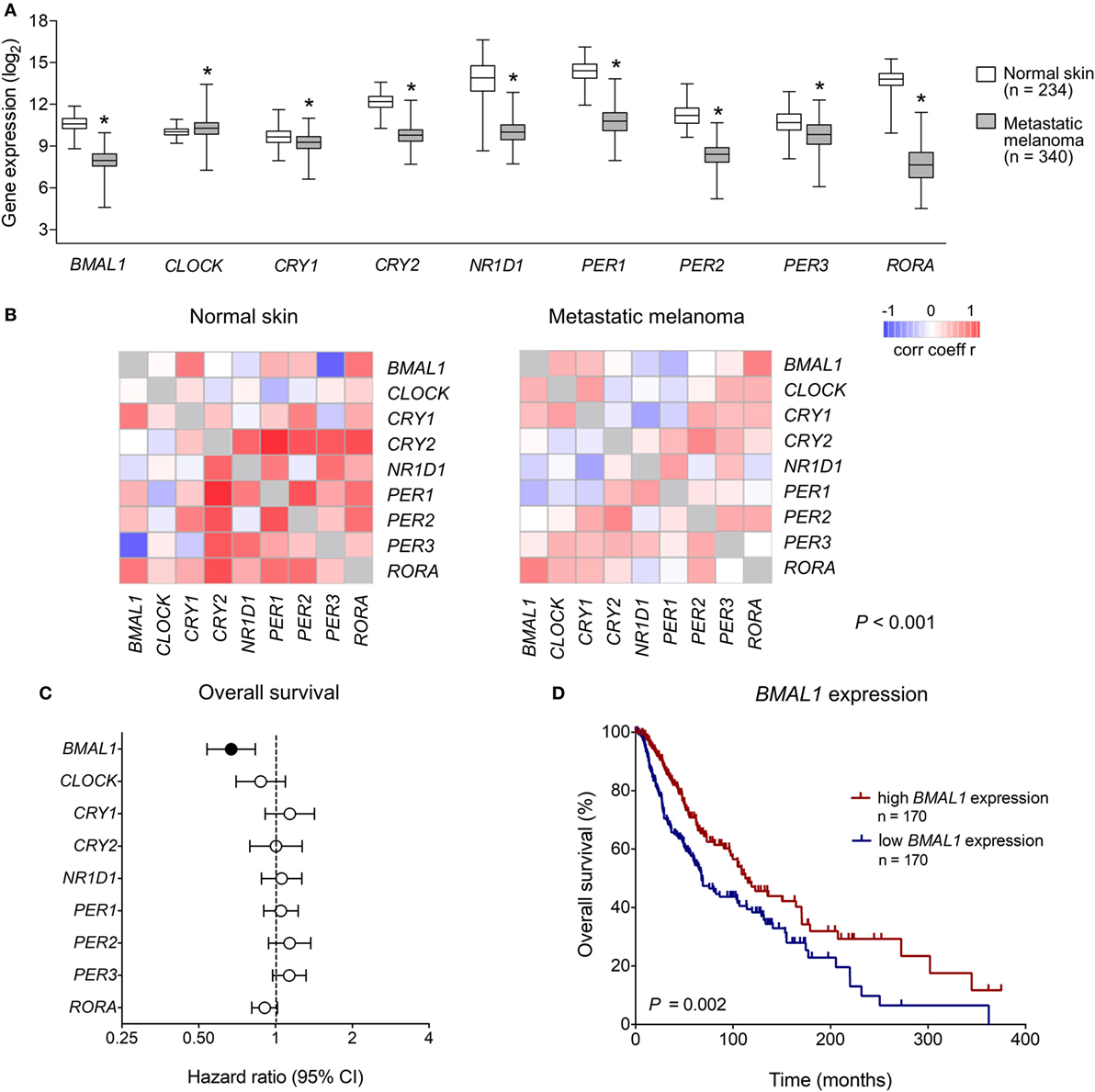
Figure 1. Clinical relevance of clock genes expression in metastatic melanomas. (A) RNAseq analysis of clock genes expression in genotype-tissue expression (GTEx) normal skins (n = 234) and The Cancer Genome Atlas (TCGA) metastatic melanoma (n = 340). Expression values were estimated using RSEM and log2(x + 1) transformed. The boxes extend from the 25th to the 75th percentile, the central bold line shows the median, and whiskers are drawn from minimum to maximum values. Comparisons were performed using the two-sided Wilcoxon–Mann–Whitney test. *Significantly different from normal skin (P < 0.05). (B) Co-expression matrix showing pairwise Spearman’s correlation coefficients of clock core genes in GTEx normal skins (n = 234) and TCGA metastatic melanomas (n = 340). Networks were compared using the CoGA software. (C) Univariate Cox analysis of overall survival according to the expression of clock core genes in TCGA metastatic melanomas. Hazard Ratios including 95% confidence intervals are shown. Genes with a significant prognostic value (P < 0.05) are marked in black. (D) Kaplan–Meier survival curve according to the expression of aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1) in TCGA metastatic melanomas. The median expression of BMAL1 was used as the cutoff to dichotomize the population. Comparisons were performed using the log-rank test.
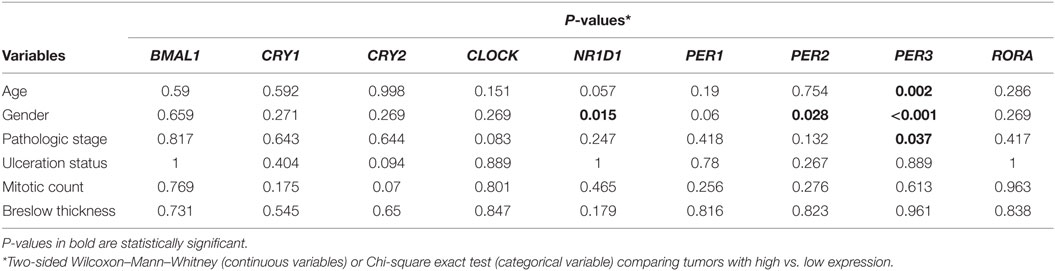
Table 1. Clinicopathological features according to the expression of clock genes in The Cancer Genome Atlas metastatic melanomas.
Next, using univariate Cox regressions we evaluated the clinical relevance of clock core genes in metastatic melanoma. Among all nine genes analyzed, only BMAL1 showed a significant prognostic value: high BMAL1 expression was associated with longer overall survival (HR = 0.678, P = 0.002; Figures 1C,D). Importantly, multivariate Cox regression adjusting for age, gender, tumor pathologic stage, ulceration status, mitotic count, and Breslow thickness revealed BMAL1 expression as an independent prognostic factor (Table 2). Additionally, the prognostic value of BMAL1 expression in metastatic melanomas was confirmed in two other independent datasets (GSE6590 and GSE54467; Figures S1A,B and Table S2 in Supplementary Material).
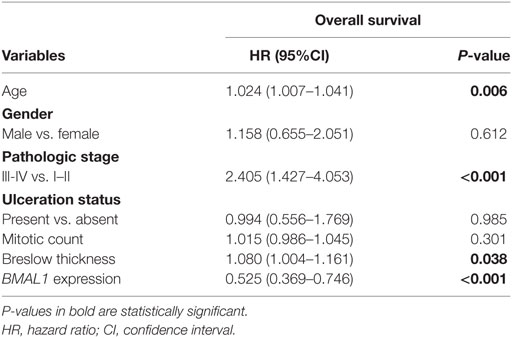
Table 2. Multivariate Cox regression analysis of survival in The Cancer Genome Atlas metastatic melanomas.
BMAL1 Expression and the Overall Biological Profile of Metastatic Melanoma
To investigate the biological mechanisms that likely underlie the impact of BMAL1 expression on patient survival, we performed GSEA using genes ranked according to their Spearman’s correlation with BMAL1 expression. Significantly enriched pathways presented positive NES and were mainly involved in the activation of the immune system (Figure 2A). In fact, in metastatic melanomas, BMAL1 expression exhibited a strong positive correlation with the expression of dendritic cell markers, T-cell markers CD4 and CD8A, and T-cell activation/differentiation markers (Figure 2B). This robust intratumoral activation of leukocytes was accompanied by the expression of T-cells exhaustion markers (Figure 2B), such as CTLA4, PD1, and PDL1, corroborating the fact that T-cell were chronically exposed to antigens (52, 53). Accordingly, patients with high BMAL1 expression in pretreatment biopsies demonstrated improved response to anti-PD1 immunotherapy in comparison to patients expressing low BMAL1 levels (Figure 2C).
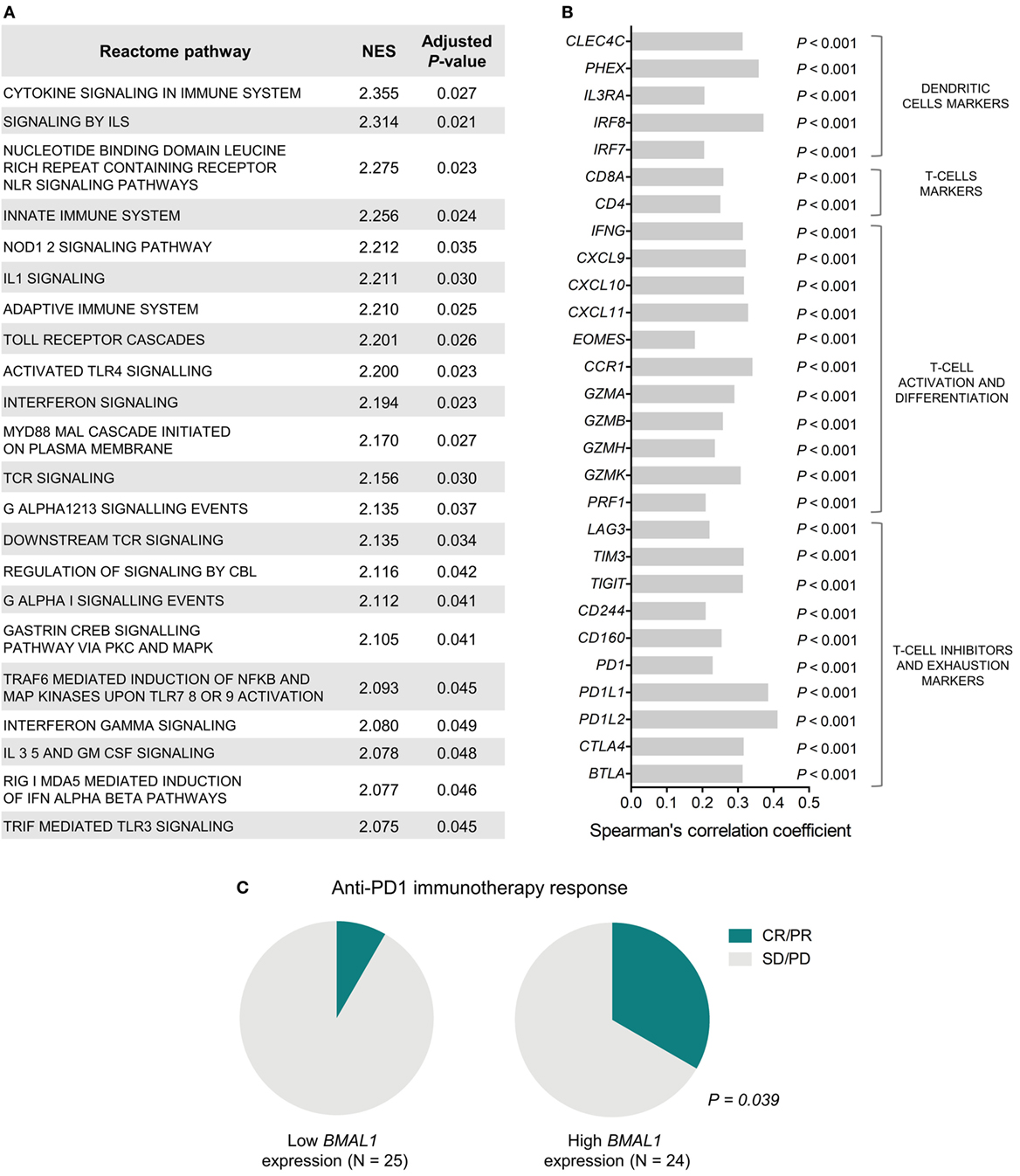
Figure 2. Aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1) expression positively correlates with antitumor immunity in metastatic melanomas. (A) Gene Set Enrichment Analysis (GSEA) using genes ranked according to the Spearman’s correlation coefficient between their expression and the expression of BMAL1 in The Cancer Genome Atlas (TCGA) metastatic melanomas (n = 340). Normalized enrichment scores (NES) and P-values corrected by false discovery rate (FDR) were calculated using GSEA v3.0 and Reactome pathways. Only significantly enriched pathways (adjusted P < 0.05) are shown. (B) Spearman’s correlation coefficient between the expression of BMAL1 and immune cells markers in TCGA metastatic melanomas. P-values were corrected by FDR. (C) Association between BMAL1 expression (pretreatment biopsies) and clinical benefit of melanoma patients from anti-PD1 immunotherapy (nivolumab). RNAseq data and treatment response information were obtained from Ref. (42). Comparisons were performed using the Chi-square test. CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease.
The correlation between BMAL1 expression and antitumor immune response was also confirmed in two additional independent datasets (GSE6590 and GSE54467; Figures S1C,D in Supplementary Material). Importantly, the expression of BMAL1 was a prognostic factor independent of the percentage of leukocyte, monocyte, and neutrophil infiltration in TCGA melanomas (Table 3).
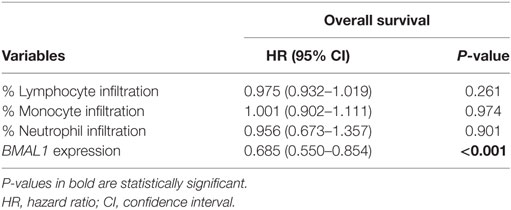
Table 3. Multivariate Cox regression analysis of overall survival in metastatic melanomas adjusted for the percentage of immune cell infiltration in The Cancer Genome Atlas metastatic melanomas.
BMAL1 Expression and the Mutational Load in Metastatic Melanomas
Tumor somatic mutations can generate major histocompatibility complex Class I-associated neoantigens expression that plays a central role in inducing T-cell meditated antitumor cytolytic activity (54, 55). Interestingly, in metastatic melanomas, BMAL1 expression positively correlated with the number of total somatic mutations and predicted neoantigens (Figure 3A). With that in mind, we investigated whether the expression of BMAL1 was associated with the activation of different DNA-repair pathways. Using ssGSEA, we demonstrated that base excision repair is likely impaired in tumors expressing high BMAL1 (Figure 3B). No significant differences were observed regarding the nucleotide excision repair, mismatch repair, homologous recombination, and non-homologous end joining DNA-repairing mechanisms. Importantly, the expression of base excision repair-related genes, such as NTHL1, XRCC1, and SMUG1, and the expression of general DNA repair-related genes, such as POLD1, POLD2, and LIG1, were downregulated in tumors expressing high BMAL1 in all three datasets analyzed (Figure 3C; Figure S2 in Supplementary Material). High BMAL1 expression was also associated with impaired DNA-repair capacity in human melanoma cell lines from the Cancer Cell Line Encyclopedia (Figure S3 in Supplementary Material).
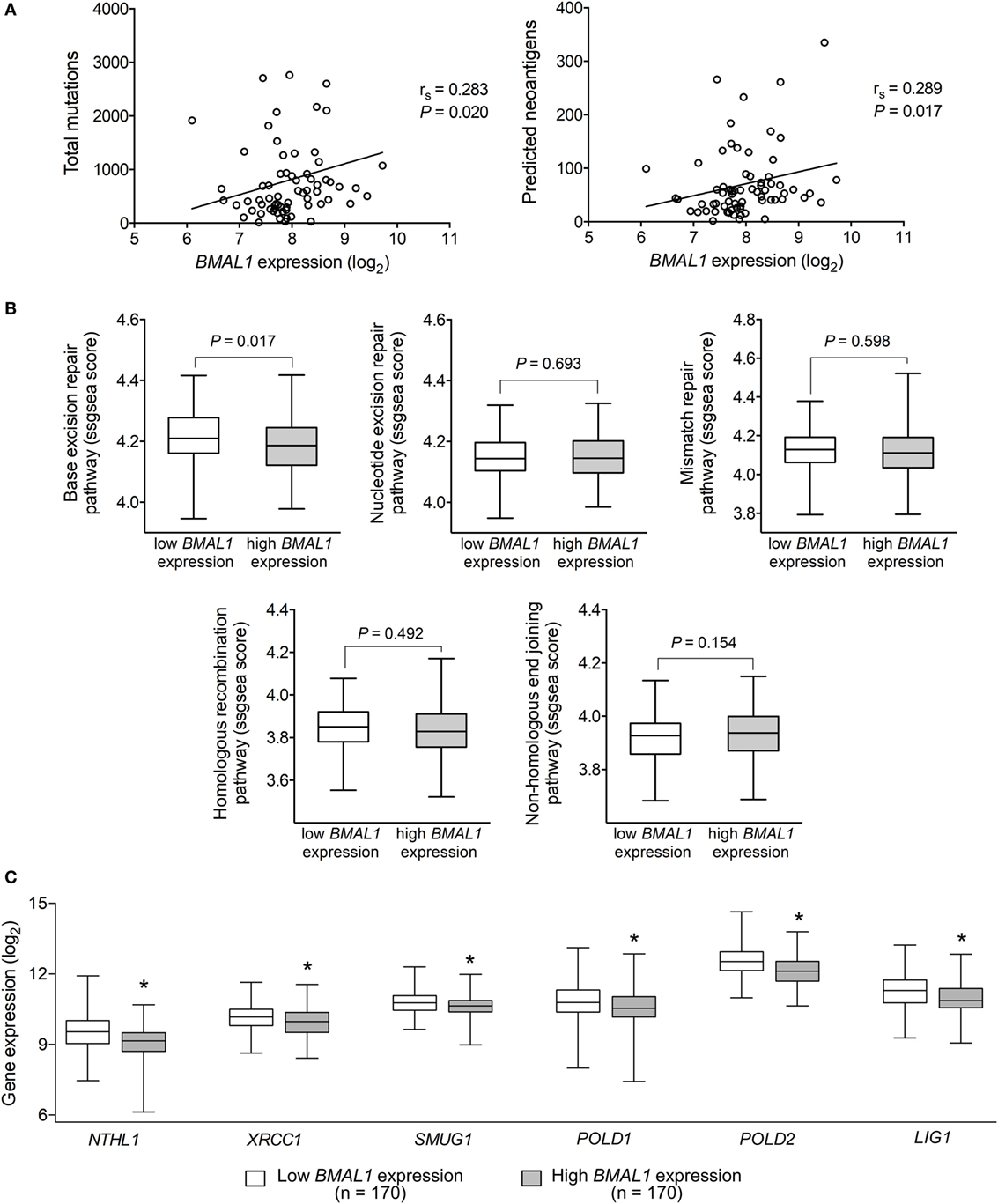
Figure 3. Aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1) expression positively correlates with the mutational and neoantigen load in metastatic melanomas. (A) Spearman’s correlation between the expression of BMAL1 and the number of total mutations and predicted neoantigens in The Cancer Genome Atlas (TCGA) metastatic melanomas (n = 68). NetMHCpan-predicted neoantigens were obtained from Ref. (40). Gene expression of (B) DNA-repair programs and (C) selected base excision repair enzymes according to the expression of BMAL1 in TCGA metastatic melanomas (n = 340). Pathway scores were calculated using single sample Gene Set Enrichment Analysis available in the GSVA R package. The median expression of BMAL1 was used as the cutoff to dichotomize the population. The boxes extend from the 25th to the 75th percentile, the central bold line shows the median, and whiskers are drawn from minimum to maximum values. Comparisons were performed using the two-sided Wilcoxon–Mann–Whitney test. *Significantly different from the low BMAL1 group.
Discussion
Cancer onset, development, and progression have been linked to circadian disruption (17–19); however, the complex role of the tumor-autonomous molecular clock within these processes is yet poorly understood. Here, confirming previous reports in humans and in mice (32, 33, 35, 37), we showed that the expression of core components of the molecular clock machinery is severely repressed in melanomas. Moreover, we demonstrated that, for such tumors, high mRNA levels of BMAL1 are associated with decreased gene expression of base excision repair enzymes and increased mutation load and predicted neoantigen presentation. The high incidence of antigenic peptides observed in metastatic melanomas with high BMAL1 expression was accompanied by increased expression of cytotoxic T-cell activity markers in the tumor bulk and better prognosis. Even though our data do not provide a detailed mechanistic perspective, the present findings strongly support a role for BMAL1 as a clinically relevant biomarker of DNA damage repair deficiency and intratumoral T-cell response. Thus, confirming such findings using common molecular techniques would be of great relevance for prognosis prediction and proposition of personalized therapeutic strategies.
Accumulating evidence implicates cell autonomous-circadian clocks in cancer development, as the disruption of peripheral systems of timekeeping seems to be a common event in malignant tissues (17, 18). As demonstrated here for metastatic melanomas, the expression of most clock core genes is downregulated in several types of cancers when compared to normal tissue (28, 32, 56–61). Moreover, the overexpression of PER1 and PER2 has been shown to impair tumor proliferation and induce apoptosis in lung, prostate, and pancreatic cancer (29, 62, 63), reinforcing the idea that the molecular clock machinery may be considered as a new therapeutic target.
The protein encoded by BMAL1 belongs to the family of the bHLH-PAS structural domain transcription factors and it is estimated to control the expression of more than 150 target genes, including the clock genes CRY1, CRY2, NR1D1, PER1, PER2, and PER3 (64). BMAL1 has also been revealed as a candidate gene for susceptibility to hypertension, diabetes, and obesity, and mutations in BMAL1 have been linked to infertility and metabolic dysfunctions (65–70). Here, we demonstrated that, in metastatic melanomas, the expression of BMAL1 is a robust positive prognostic factor of overall survival and has a negative association with the expression of key DNA-repair enzymes, such as POLD1, POLD2, and LIG1. Accordingly, in colorectal cancer, downregulation of BMAL1 gene expression accelerates cell proliferation in vitro, promotes tumor growth in mice, and decreases DNA damage induced by cisplatin (71). Moreover, high BMAL1 expression is associated with increased sensitivity of colorectal cancer cells to oxaliplatin in vitro and in vivo, and predicts favorable outcome for patients treated with oxaliplatin-based chemotherapy (72). BMAL1 expression also positively correlates with patient survival in pancreatic ductal adenocarcinomas (61), causes growth inhibition in lymphoma/leukemia cells (58), negatively impacts DNA-repair capacity of mice fibroblast (73), but promotes proliferation in malignant pleura mesothelioma (74), suggesting that its role in tumorigenesis is complex and tissue-specific.
Although it has been shown that alterations in the tumor molecular clock impact some parameters of tumor progression (28–30, 62, 63), the influence of endogenous oscillatory systems on the cellular composition of the tumor microenvironment is largely unknown. In this sense, our data indicate that the prolonged survival of metastatic melanoma patients with high BMAL1 bulk expression is associated with a robust intratumoral T-cell infiltration/activation, which can be partially explained by the increased neoantigen load that likely reflects the impaired DNA-repair capacity. Previous reports have also linked DNA-repair deficiency to increased mutational load and antitumor immune response in melanomas, lung, colorectal, and endometrial cancers (75–78). It is now clear that DNA repair and genomic instability have a pivotal role in the modulation of antitumor immune responses (79); thus, understanding their interplay with tumor-autonomous clocks may provide clinically relevant insights.
Immunotherapies that boost the ability of T lymphocytes to combat tumor cells have demonstrated therapeutic efficiency in a variety of solid tumors. Monoclonal antibodies against T-cell checkpoint proteins, such as CTLA-4, PD-1, and PD-L1, have now been approved for melanoma treatment and are associated with robust durable responses, but only in a subset of tumors (80–82). Thus, there is a need to identify biomarkers that will allow the selection of treatment-responsive patients, avoid unnecessary toxicity, and help personalize therapy regimens (83). Metastatic melanomas presenting high BMAL1 expression have impaired DNA-repair capacity combined with increased mutation/neoantigen load, T-cell intratumoral infiltration, and T-cell expression of exhaustion markers, all of which have been shown to predict good clinical response to the treatment with immune checkpoint inhibitors (78, 84–87). In fact, we showed that high BMAL1 expression in pretreatment melanoma samples is associated with clinical benefit from anti-PD1 immunotherapy. Considering that whole-genome and -transcriptome sequencing is expensive and time-consuming, profiling a smaller fraction of genes could serve as a useful tool to help translate those findings into routine clinical practices (88). Therefore, the present data indicate that BMAL1 expression in melanoma patients must be considered as a relevant marker for immunotherapy efficacy. Nevertheless, larger clinical studies are necessary to validate the potential of BMAL1 alone, or along with other biomarkers, in discriminating responders from non-responders in immunotherapy regiments.
Conclusion
The molecular characterization of melanomas using high-throughput approaches has the potential to generate insights into their biological heterogeneity, having important implications for prognosis and therapy. In this sense, our data highlight the relevance of further studies focusing on the biological and clinical relevance of the tumor-autonomous molecular clock machinery. Overall, we demonstrated that, in metastatic melanoma, a high bulk BMAL1 expression seems to be associated with a “too tumorigenic” program and could be a marker for immunotherapy response.
Ethics Statement
All data presented in this manuscript are public and freely available. We did not perform any human or animal related experiments. All analyses and conclusions were drawn from the following public datasets: The Cancer Genome Atlas (TCGA), Genotype-Tissue Expression (GTEx), Gene Expression Omnibus, and datasets from Ref. (40, 42). In all mentioned papers, the authors stated that all procedures were carried out according ethical rules.
Author Contributions
LA and GK designed the study, analyzed the data, and drafted the manuscript. All authors provided insightful discussion during data acquisition and aided in the writing process of the manuscript. All authors critically revised the manuscript. All authors have approved the definitive version of the manuscript and agreed to be accountable for all aspects of the study in ensuring that questions related to the accuracy or integrity of any part of the study are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Conflict of Interest Statement
All authors state no conflict of interest that could have impacted the development of this study.
Funding
This work was partially supported by the Sao Paulo Research Foundation (FAPESP, grant 2012/50214-4) and by the National Council of Technological and Scientific Development (CNPq, grants 301293/2011-2 and 303070/2015-3). LA, GK, and MM are fellows of FAPESP (2013/24337-4; 2014/27287-0; and 2014/16412-9, respectively).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fonc.2018.00185/full#supplementary-material.
References
1. Garbe C, Leiter U. Melanoma epidemiology and trends. Clin Dermatol (2009) 27:3–9. doi:10.1016/j.clindermatol.2008.09.001
2. Erdei E, Torres SM. A new understanding in the epidemiology of melanoma. Expert Rev Anticancer Ther (2010) 10:1811–23. doi:10.1586/era.10.170
3. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin (2008) 58:71–96. doi:10.3322/CA.2007.0010
4. Song X, Zhao Z, Barber B, Farr AM, Ivanov B, Novich M. Overall survival in patients with metastatic melanoma. Curr Med Res Opin (2015) 31:987–91. doi:10.1185/03007995.2015.1021904
5. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364:2507–16. doi:10.1056/NEJMoa1103782
6. Grimaldi AM, Simeone E, Ascierto PA. The role of MEK inhibitors in the treatment of metastatic melanoma. Curr Opin Oncol (2014) 26:196–203. doi:10.1097/CCO.0000000000000050
7. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, De Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med (2014) 371:1877–88. doi:10.1056/NEJMoa1406037
8. Redman JM, Gibney GT, Atkins MB. Advances in immunotherapy for melanoma. BMC Med (2016) 14:20. doi:10.1186/s12916-016-0571-0
9. Markovic SN, Erickson LA, Rao RD, Weenig RH, Pockaj BA, Bardia A, et al. Malignant melanoma in the 21st century, part 1: epidemiology, risk factors, screening, prevention, and diagnosis. Mayo Clin Proc (2007) 82:364–80. doi:10.1016/S0025-6196(11)61033-1
10. Vuong K, Armstrong BK, Weiderpass E, Lund E, Adami HO, Veierod MB, et al. Development and external validation of a melanoma risk prediction model based on self-assessed risk factors. JAMA Dermatol (2016) 152:889–96. doi:10.1001/jamadermatol.2016.0939
11. Brown SA, Azzi A. Peripheral circadian oscillators in mammals. Handb Exp Pharmacol (2013) 217:45–66. doi:10.1007/978-3-642-25950-0_3
12. Buhr ED, Takahashi JS. Molecular components of the mammalian circadian clock. Handb Exp Pharmacol (2013) 217:3–27. doi:10.1007/978-3-642-25950-0_1
13. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet (2017) 18:164–79. doi:10.1038/nrg.2016.150
14. Baron KG, Reid KJ. Circadian misalignment and health. Int Rev Psychiatry (2014) 26:139–54. doi:10.3109/09540261.2014.911149
15. West AC, Bechtold DA. The cost of circadian desynchrony: evidence, insights and open questions. Bioessays (2015) 37:777–88. doi:10.1002/bies.201400173
16. Potter GD, Skene DJ, Arendt J, Cade JE, Grant PJ, Hardie LJ. Circadian rhythm and sleep disruption: causes, metabolic consequences, and countermeasures. Endocr Rev (2016) 37:584–608. doi:10.1210/er.2016-1083
17. Savvidis C, Koutsilieris M. Circadian rhythm disruption in cancer biology. Mol Med (2012) 18:1249–60. doi:10.2119/molmed.2012.00077
18. Kelleher FC, Rao A, Maguire A. Circadian molecular clocks and cancer. Cancer Lett (2014) 342:9–18. doi:10.1016/j.canlet.2013.09.040
19. Masri S, Kinouchi K, Sassone-Corsi P. Circadian clocks, epigenetics, and cancer. Curr Opin Oncol (2015) 27:50–6. doi:10.1097/CCO.0000000000000153
20. Yang X, Wood PA, Ansell CM, Quiton DF, Oh EY, Du-Quiton J, et al. The circadian clock gene Per1 suppresses cancer cell proliferation and tumor growth at specific times of day. Chronobiol Int (2009) 26:1323–39. doi:10.3109/07420520903431301
21. Kiessling S, Beaulieu-Laroche L, Blum ID, Landgraf D, Welsh DK, Storch KF, et al. Enhancing circadian clock function in cancer cells inhibits tumor growth. BMC Biol (2017) 15:13. doi:10.1186/s12915-017-0349-7
22. Gaddameedhi S, Selby CP, Kaufmann WK, Smart RC, Sancar A. Control of skin cancer by the circadian rhythm. Proc Natl Acad Sci U S A (2011) 108:18790–5. doi:10.1073/pnas.1115249108
23. Gaddameedhi S, Selby CP, Kemp MG, Ye R, Sancar A. The circadian clock controls sunburn apoptosis and erythema in mouse skin. J Invest Dermatol (2015) 135:1119–27. doi:10.1038/jid.2014.508
24. Albrecht U. The circadian clock, metabolism and obesity. Obes Rev (2017) 18(Suppl 1):25–33. doi:10.1111/obr.12502
25. Krishnaiah SY, Wu G, Altman BJ, Growe J, Rhoades SD, Coldren F, et al. Clock regulation of metabolites reveals coupling between transcription and metabolism. Cell Metab (2017) 25(5):1206. doi:10.1016/j.cmet.2017.04.023
26. Moraes MN, De Assis LVM, Henriques FDS, Batista ML Jr., Guler AD, Castrucci AML. Cold-sensing TRPM8 channel participates in circadian control of the brown adipose tissue. Biochim Biophys Acta (2017) 1864:2415–27. doi:10.1016/j.bbamcr.2017.09.011
27. Moraes MN, Mezzalira N, De Assis LV, Menaker M, Guler A, Castrucci AM. TRPV1 participates in the activation of clock molecular machinery in the brown adipose tissue in response to light-dark cycle. Biochim Biophys Acta (2017) 1864:324–35. doi:10.1016/j.bbamcr.2016.11.010
28. Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ, Chang JG. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis (2005) 26:1241–6. doi:10.1093/carcin/bgi075
29. Cao Q, Gery S, Dashti A, Yin D, Zhou Y, Gu J, et al. A role for the clock gene per1 in prostate cancer. Cancer Res (2009) 69:7619–25. doi:10.1158/0008-5472.CAN-08-4199
30. Yu H, Meng X, Wu J, Pan C, Ying X, Zhou Y, et al. Cryptochrome 1 overexpression correlates with tumor progression and poor prognosis in patients with colorectal cancer. PLoS One (2013) 8:e61679. doi:10.1371/journal.pone.0061679
31. Lengyel Z, Lovig C, Kommedal S, Keszthelyi R, Szekeres G, Battyani Z, et al. Altered expression patterns of clock gene mRNAs and clock proteins in human skin tumors. Tumour Biol (2013) 34:811–9. doi:10.1007/s13277-012-0611-0
32. Brozyna AA, Jozwicki W, Skobowiat C, Jetten A, Slominski AT. RORalpha and RORgamma expression inversely correlates with human melanoma progression. Oncotarget (2016) 7(39):63261–82. doi:10.18632/oncotarget.11211
33. de Assis LV, Moraes MN, da Silveira Cruz-Machado S, Castrucci AM. The effect of white light on normal and malignant murine melanocytes: a link between opsins, clock genes, and melanogenesis. Biochim Biophys Acta (2016) 1863:1119–33. doi:10.1016/j.bbamcr.2016.03.001
34. de Assis LV, Moraes MN, Castrucci AM. Heat shock antagonizes UVA-induced responses in murine melanocytes and melanoma cells: an unexpected interaction. Photochem Photobiol Sci (2017) 16(5):633–48. doi:10.1039/c6pp00330c
35. Poletini MO, De Assis LV, Moraes MN, Castrucci AM. Estradiol differently affects melanin synthesis of malignant and normal melanocytes: a relationship with clock and clock-controlled genes. Mol Cell Biochem (2016) 421:29–39. doi:10.1007/s11010-016-2781-3
36. Moraes MN, De Assis LVM, Magalhaes-Marques KK, Poletini MO, De Lima L, Castrucci AML. Melanopsin, a canonical light receptor, mediates thermal activation of clock genes. Sci Rep (2017) 7:13977. doi:10.1038/s41598-017-13939-3
37. de Assis LVM, Moraes MN, Magalhães-Marques KK, Kinker GS, da Silveira Cruz-Machado S, Castrucci AM. Non-metastatic cutaneous melanoma induces chronodisruption in central and peripheral circadian clocks. Int J Mol Sci (2018) 19(4):E1065. doi:10.3390/ijms19041065
38. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell (2015) 161:1681–96. doi:10.1016/j.cell.2015.05.044
39. Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science (2015) 348:648–60. doi:10.1126/science.1262110
40. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160:48–61. doi:10.1016/j.cell.2014.12.033
41. Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One (2007) 2:e796. doi:10.1371/journal.pone.0000796
42. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell (2017) 171:934–49. doi:10.1016/j.cell.2017.09.028
43. Santos SDS, Galatro TFDA, Watanabe RA, Oba-Shinjo SM, Nagahashi Marie SK, Fujita A. CoGA: an R package to identify differentially co-expressed gene sets by analyzing the graph spectra. PLoS One (2015) 10:e0135831. doi:10.1371/journal.pone.0135831
44. Takahashi DY, Sato JR, Ferreira CE, Fujita A. Discriminating different classes of biological networks by analyzing the graphs spectra distribution. PLoS One (2012) 7:e49949. doi:10.1371/journal.pone.0049949
45. Kinker GS, Thomas AM, Carvalho VJ, Lima FP, Fujita A. Deletion and low expression of NFKBIA are associated with poor prognosis in lower-grade glioma patients. Sci Rep (2016) 6:24160. doi:10.1038/srep24160
46. Van Mieghem P. Graph Spectra for Complex Networks. Cambridge: Cambridge University Press (2010).
47. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A (2005) 102:15545–50. doi:10.1073/pnas.0506580102
48. Croft D, O’kelly G, Wu G, Haw R, Gillespie M, Matthews L, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res (2011) 39:D691–7. doi:10.1093/nar/gkq1018
49. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature (2009) 462:108–12. doi:10.1038/nature08460
50. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics (2013) 14:7. doi:10.1186/1471-2105-14-7
51. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res (2000) 28:27–30. doi:10.1093/nar/28.1.27
52. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15:486–99. doi:10.1038/nri3862
53. Catakovic K, Klieser E, Neureiter D, Geisberger R. T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun Signal (2017) 15:1. doi:10.1186/s12964-016-0160-z
54. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14:1014–22. doi:10.1038/ni.2703
55. Coulie PG, Van Den Eynde BJ, Van Der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14:135–46. doi:10.1038/nrc3670
56. Yeh KT, Yang MY, Liu TC, Chen JC, Chan WL, Lin SF, et al. Abnormal expression of period 1 (PER1) in endometrial carcinoma. J Pathol (2005) 206:111–20. doi:10.1002/path.1756
57. Kuo SJ, Chen ST, Yeh KT, Hou MF, Chang YS, Hsu NC, et al. Disturbance of circadian gene expression in breast cancer. Virchows Arch (2009) 454:467–74. doi:10.1007/s00428-009-0761-7
58. Taniguchi H, Fernandez AF, Setien F, Ropero S, Ballestar E, Villanueva A, et al. Epigenetic inactivation of the circadian clock gene BMAL1 in hematologic malignancies. Cancer Res (2009) 69:8447–54. doi:10.1158/0008-5472.CAN-09-0551
59. Huisman SA, Ahmadi AR, Jn IJ, Verhoef C, Van Der Horst GT, De Bruin RW. Disruption of clock gene expression in human colorectal liver metastases. Tumour Biol (2016) 37(10):13973–81. doi:10.1007/s13277-016-5231-7
60. Jiang W, Zhao S, Jiang X, Zhang E, Hu G, Hu B, et al. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett (2016) 371:314–25. doi:10.1016/j.canlet.2015.12.002
61. Li W, Liu L, Liu D, Jin S, Yang Y, Tang W, et al. Decreased circadian component Bmal1 predicts tumor progression and poor prognosis in human pancreatic ductal adenocarcinoma. Biochem Biophys Res Commun (2016) 472:156–62. doi:10.1016/j.bbrc.2016.02.087
62. Hua H, Wang Y, Wan C, Liu Y, Zhu B, Wang X, et al. Inhibition of tumorigenesis by intratumoral delivery of the circadian gene mPer2 in C57BL/6 mice. Cancer Gene Ther (2007) 14:815–8. doi:10.1038/sj.cgt.7701061
63. Oda A, Katayose Y, Yabuuchi S, Yamamoto K, Mizuma M, Shirasou S, et al. Clock gene mouse period2 overexpression inhibits growth of human pancreatic cancer cells and has synergistic effect with cisplatin. Anticancer Res (2009) 29:1201–9.
64. Hatanaka F, Matsubara C, Myung J, Yoritaka T, Kamimura N, Tsutsumi S, et al. Genome-wide profiling of the core clock protein BMAL1 targets reveals a strict relationship with metabolism. Mol Cell Biol (2010) 30:5636–48. doi:10.1128/MCB.00781-10
65. Rudic RD, Mcnamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol (2004) 2:e377. doi:10.1371/journal.pbio.0020377
66. Woon PY, Kaisaki PJ, Braganca J, Bihoreau MT, Levy JC, Farrall M, et al. Aryl hydrocarbon receptor nuclear translocator-like (BMAL1) is associated with susceptibility to hypertension and type 2 diabetes. Proc Natl Acad Sci U S A (2007) 104:14412–7. doi:10.1073/pnas.0703247104
67. Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature (2010) 466:627–31. doi:10.1038/nature09253
68. Milagro FI, Gomez-Abellan P, Campion J, Martinez JA, Ordovas JM, Garaulet M. CLOCK, PER2 and BMAL1 DNA methylation: association with obesity and metabolic syndrome characteristics and monounsaturated fat intake. Chronobiol Int (2012) 29:1180–94. doi:10.3109/07420528.2012.719967
69. Hodzic A, Ristanovic M, Zorn B, Tulic C, Maver A, Novakovic I, et al. Genetic variation in circadian rhythm genes CLOCK and ARNTL as risk factor for male infertility. PLoS One (2013) 8:e59220. doi:10.1371/journal.pone.0059220
70. Kettner NM, Katchy CA, Fu L. Circadian gene variants in cancer. Ann Med (2014) 46:208–20. doi:10.3109/07853890.2014.914808
71. Zeng ZL, Wu MW, Sun J, Sun YL, Cai YC, Huang YJ, et al. Effects of the biological clock gene Bmal1 on tumour growth and anti-cancer drug activity. J Biochem (2010) 148:319–26. doi:10.1093/jb/mvq069
72. Zeng ZL, Luo HY, Yang J, Wu WJ, Chen DL, Huang P, et al. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin Cancer Res (2014) 20:1042–52. doi:10.1158/1078-0432.CCR-13-0171
73. Khapre RV, Kondratova AA, Susova O, Kondratov RV. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle (2011) 10:4162–9. doi:10.4161/cc.10.23.18381
74. Elshazley M, Sato M, Hase T, Yamashita R, Yoshida K, Toyokuni S, et al. The circadian clock gene BMAL1 is a novel therapeutic target for malignant pleural mesothelioma. Int J Cancer (2012) 131:2820–31. doi:10.1002/ijc.27598
75. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372:2509–20. doi:10.1056/NEJMoa1500596
76. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi:10.1126/science.aaa1348
77. van Gool IC, Eggink FA, Freeman-Mills L, Stelloo E, Marchi E, De Bruyn M, et al. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin Cancer Res (2015) 21:3347–55. doi:10.1158/1078-0432.CCR-15-0057
78. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165:35–44. doi:10.1016/j.cell.2016.02.065
79. Mouw KW, Goldberg MS, Konstantinopoulos PA, D’andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov (2017) 7:675–93. doi:10.1158/2159-8290.CD-17-0226
80. Hodi FS, O’day SJ, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
81. Topalian SL, Sznol M, Mcdermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol (2014) 32:1020–30. doi:10.1200/JCO.2013.53.0105
82. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol (2015) 33:1889–94. doi:10.1200/JCO.2014.56.2736
83. Spencer KR, Wang J, Silk AW, Ganesan S, Kaufman HL, Mehnert JM. Biomarkers for immunotherapy: current developments and challenges. Am Soc Clin Oncol Educ Book (2016) 35:e493–503. doi:10.14694/EDBK_160766
84. Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother (2012) 61:1019–31. doi:10.1007/s00262-011-1172-6
85. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371:2189–99. doi:10.1056/NEJMoa1406498
86. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res (2014) 20:5064–74. doi:10.1158/1078-0432.CCR-13-3271
87. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (2015) 350:207–11. doi:10.1126/science.aad0095
Keywords: skin cancer, melanoma, circadian rhythms, clock genes, ARNTL/BMAL1 immunotherapy
Citation: de Assis LVM, Kinker GS, Moraes MN, Markus RP, Fernandes PA and Castrucci AML (2018) Expression of the Circadian Clock Gene BMAL1 Positively Correlates With Antitumor Immunity and Patient Survival in Metastatic Melanoma. Front. Oncol. 8:185. doi: 10.3389/fonc.2018.00185
Received: 10 February 2018; Accepted: 10 May 2018;
Published: 12 June 2018
Edited by:
Svetlana Karakhanova, Universität Heidelberg, GermanyReviewed by:
Derre Laurent, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandZsuzsanna Lengyel, University of Pécs, Hungary
Copyright: © 2018 de Assis, Kinker, Moraes, Markus, Fernandes and Castrucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonardo Vinícius Monteiro de Assis, ZGVhc3Npcy5sZW9uYXJkb0B1c3AuYnI=;
Gabriela Sarti Kinker, Z2FicmllbGEua2lua2VyQHVzcC5icg==
†These authors have contributed equally to this work.