 Rute M. S. M. Pedrosa1
Rute M. S. M. Pedrosa1 Johan M. Kros
Johan M. Kros
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 May 2018
Sec. Thoracic Oncology
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00159
This article is part of the Research Topic Central Nervous System Metastases in Lung Cancer Patients: From Prevention to Diagnosis and Treatment View all 10 articles
Brain metastases are the most common tumors of the central nervous system (CNS). Incidence rates vary according to primary tumor origin, whereas the majority of the cerebral metastases arise from primary tumors in the lung (40–50%). Brain metastases from lung cancer can occur concurrently or within months after lung cancer diagnosis. Survival rates after lung cancer brain metastasis diagnosis remain poor, to an utmost of 10 months. Therefore, prevention of brain metastasis is a critical concern in order to improve survival among cancer patients. Although several studies have been made in order to disclose the genetic and molecular mechanisms associated with CNS metastasis, the precise mechanisms that govern the CNS metastasis from lung cancer are yet to be clarified. The ability to forecast, which patients have a higher risk of brain metastasis occurrence, would aid cancer management approaches to diminish or prevent the development of brain metastasis and improve the clinical outcome for such patients. In this work, we revise genetic and molecular targets suitable for prediction of lung cancer CNS disease.
Brain metastases are the most common tumors of the central nervous system (CNS). Metastatic brain lesions outnumber primary brain tumors with a 10-fold (1) with incidence rates varying according to the primary tumor origin. The majority of the cerebral metastases arise from primary tumors in the lung (40–50%) and it is estimated that 50% of the patients with small-cell lung cancer (SCLC) or non-small-cell lung cancer (NSCLC) will develop brain metastasis (2, 3). In contrast to cerebral metastases from other primary cancers, where generally a metastatic latency period takes place, brain metastasis from lung cancers often occur months after, or even concurrently, with the diagnosis of the primary tumor (4). Metastatic brain lesions carry a clinical burden of morbidity and mortality, as well as significant neurological deficits, cognitive impairment, and emotional difficulties (5). Despite treatment, lung cancer brain metastases are usually fatal for 90% of patients within two years after the initial diagnosis, with a median survival of 7–10 months five years after diagnosis (2). Previous efforts to characterize patients that are at high risk of developing brain metastasis have been fairly disappointing.
Currently, only clinical and pathologic variables are used to predict the risk of brain metastasis in patients with lung cancer. However, data on predictive parameters are diverse and not clinically usable (Table 1). Identifying patients at highest risk of developing brain metastases on the basis of standard clinical and pathological factors, such as status of primary tumor, tumor histology, nodal involvement, and patient age, may not be reliable due to small hazard ratios and unknown prognostic factors (6). Recently, Hung et al. (7) demonstrated in a study on 182 lung adenocarcinomas with distant metastases that the micropapillary histology subtype was significantly associated with brain metastasis (p = 0.01). However, a more robust method to identify which patients are at risk of developing brain metastasis is urgently needed.
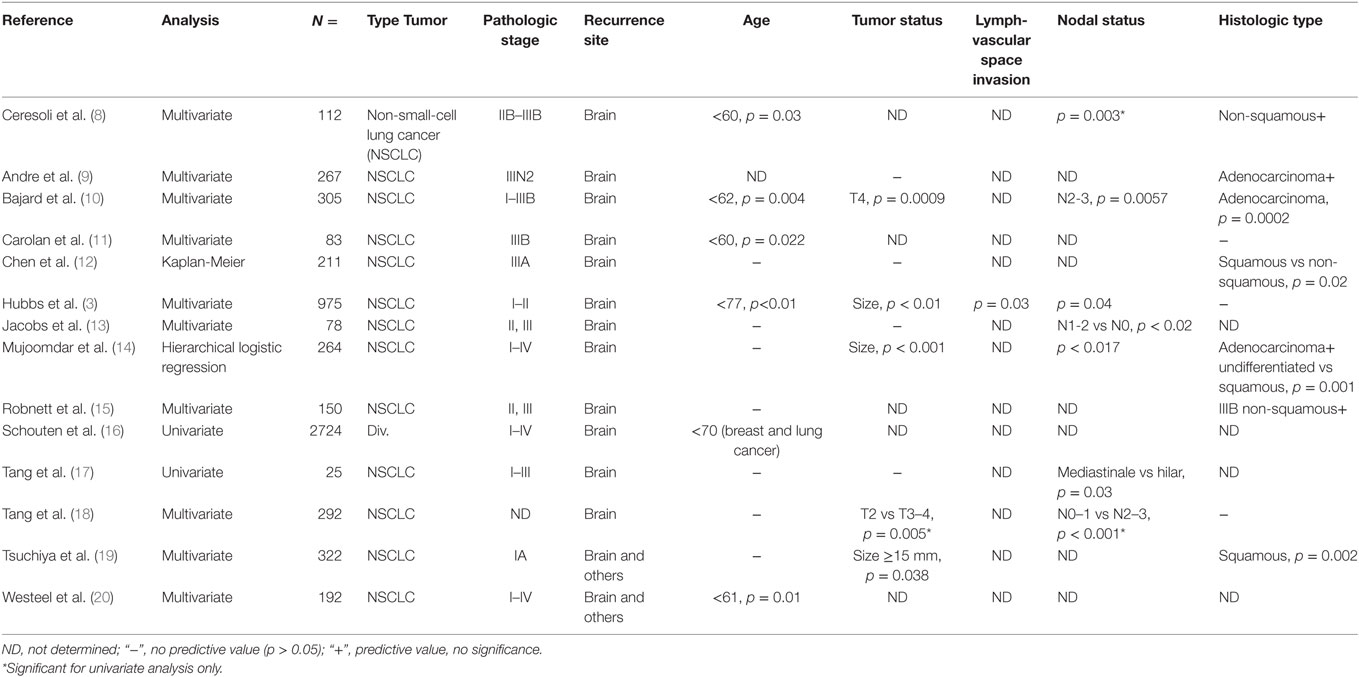
Table 1. Conflicting clinical and pathological risk factors associated with the development of brain metastases.
Molecular classification by correlating distinct molecular markers with oncogenic mechanisms has been practiced to improve risk stratification of the TNM staging system (21). The potential of molecular biologic distinction would direct appropriate therapy, thereby improving patient outcome. Among early-stage (I/II) NSCLC patients, the 5-year overall survival (OS) rate is only 45.1% (22). Many clinical trials have confirmed that postoperative adjuvant therapy can prolong the survival of NSCLC patients. In a recent meta-analysis of 3,923 patients, Chen et al. (23) demonstrated the efficacy of postoperative chemotherapy – both cisplatin based (p < 0.0001) and single tegafur-uracil (UFT, p = 0.002), in stages I–II, IA, and IB NSCLC, and no significant benefit was found in stage IA patients (p = 0.43). In addition, cisplatin was shown to be better than single UFT chemotherapy in OS (p = 0.0005 and p = 0.81, respectively) (23). More trials should be conducted in order to confirm the efficacy of disease-free survival therapies in future clinical practice.
In order to predict the rise of cerebral metastasis of lung cancer, we would need a measurable biomarker that correlates well with brain seeding of the lung cancer cells. Molecular markers may be classified into subgroups based on their mechanism of action in the metastatic cascade to the brain (6). The optimal marker to disclose concealed (brain) metastatic disease would be displayed in primary tumors while not detectable in the serum of control subjects (24). The capacity to identify metastatic disease based on proto-oncogenes such as Kirsten rat sarcoma viral oncogene homolog (KRAS) and tumor suppressor p53 (TP53), present in only half of the lung cancer patients (47 and 50%, respectively) (25), demands a more broaden and deepened spectrum of the investigation of primary lung cancers, the molecular interactions with other cells, and the tumor microenvironment.
The process of metastasis is a selective and refined event called organotropism whereby, apart from an overall tendency to spread and invade, primary tumors show predilection for particular distinct organs (26). Cancers that metastasize to brain need to take a number of anatomic, physiologic, and molecular hurdles. The first requirement is intravasation into the blood stream, dependent upon a reversible epithelial-to-mesenchymal transition (EMT). The epithelial cell traits, such as cell polarity and E-cadherin-mediated cell adhesion, are suppressed and replaced by mesenchymal cell characteristics. The cells become motile, invasive, and resistant to apoptosis (27). Through the EMT process, tumor cells acquire stem cell-like features such as self-renewal, differentiation and ability to seed, justifying the term “tumor-initiating cells” (27). EMT molecular regulation is accomplished through an intricate network arranged by different genes and molecule inducers of EMT (28–30). AXL, a receptor tyrosine kinase belonging to the TAM family, and its ligand GAS6, growth arrest-specific gene 6, have been reported to down-regulate several oncogenic signaling pathways (31), through activation of MAPK/ERK and PI3K/AKT signaling pathways (32, 33). Recently, AXL-GAS6 signal axis has been reported to have a potential key role in NSCLC tumor progression and may be suitable as a prognostic biomarker for identifying high-risk NSCLC brain metastasis patients (34). Tumor cell growth in the brain microenvironment is the result of genetic predisposition and cellular adaptation mechanisms and is largely dependent on cross-talk between tumor and brain-resident cells.
Genomic instability and mutations are just two of the characteristics of cells associated with the transition from a preneoplastic lesion to an invasive tumor state and consequent progression to metastatic disease. During tumorigenesis, a sequence of genetic modifications such as gene deletions, copy number alterations (CNAs), and chromosomal rearrangements occur. This review focuses on the use of molecular characteristics that are predictive of tumor progression and development of metastatic NSCLC brain metastasis in particular.
Due to the recent discoveries of targetable genetic alterations in the treatment of NSCLC, patients have been stratified according to genetic variations in the primary tumor, including epidermal growth factor receptor (EGFR), KRAS, and anaplastic lymphoma kinase (ALK) (35). A summary of all genetic alterations that will be addressed in this review are presented in Figure 1. There are considerable differences in reported incidence and time to development of brain metastases for these genetic alterations.
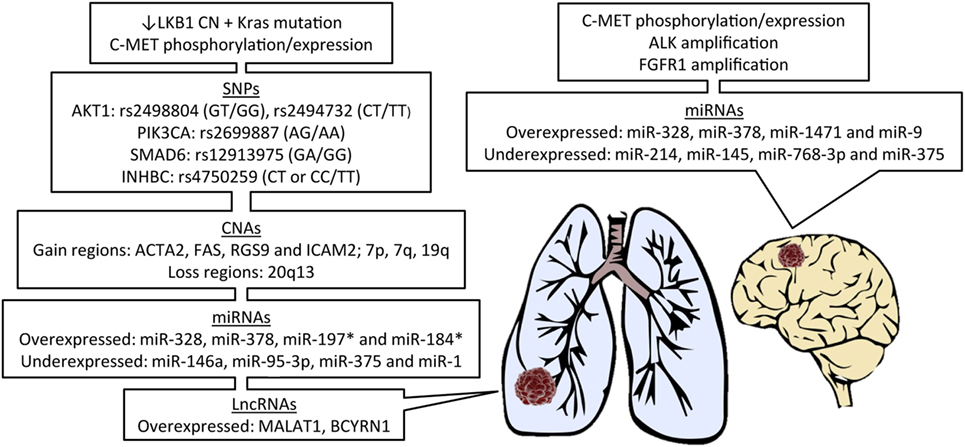
Figure 1. Features of primary lung cancer from patients known to develop brain metastasis and potential biomarker candidates. LKB1, liver kinase B1; CN, copy number; SNPs, single-nucleotide polymorphisms; CNAs, CN alterations; miRNAs, microRNAs; lncRNAs, long non-coding RNAs; *in EGFR-mutant patients.
In the Caucasian population, EGFR-activating mutations are present in 10–15% of adenocarcinomas and in less than 5% of squamous cell carcinomas (36). Roughly 90% of all known EGFR mutations reside in exon 19 (in-frame deletions) and in exon 21 (L858R, point mutation) (37, 38). The prevalence of activating EGFR mutations appears to be dependent on gender, smoking status and ethnicity. In patients from East-Asia, EGFR mutation is reportedly up to five times higher than in Caucasian patients (39–41). The relation between EGFR status, brain metastasis and survival are complex and not fully understood. It has been shown that lung cancer patients suffering from tumors with particular EGFR mutations survive longer, probably due to effective treatment. However, data also suggest that brain metastases arise more frequently in patients with primary lung tumors bearing EGFR mutations (42, 43) and the development of brain metastases is relatively frequent during treatment. There are discordance rates of EGFR mutational status between primary tumors and their CNS metastases that vary from 0 to 32% (44–50). In a series of 55 NSCLC primary tumors with matched cerebral metastases, EGFR was found to be more frequently amplified in the metastatic adenocarcinomas than corresponding primary tumors, with 30 and 10%, respectively (50). Discrepancies regarding the response of the brain metastases may well be due to the timing of administering adjuvant chemotherapy for the primary tumors relative to the occurrence of the brain metastases. The choice of the agents is currently based on the molecular characteristics of the primary, not the metastatic, tumors. Paradoxically, prolonging survival times due to successful response of the primary tumors would create more time for brain metastases to develop as late complication (51, 52). Similar to EGFR, the KRAS status may also be discordant between primary and metastatic tissues (44) and a KRAS mutation in a small subset of tumor cells may confer resistance to EGFR tyrosine kinase inhibitors (TKIs) therapy.
Epidermal growth factor receptor and KRAS mutations are generally mutually exclusive (53, 54), but cases of EGFR and KRAS co-mutations have been identified (55–57). Roughly 15–30% of NSCLCs harbor activating mutations in codons 12 and 13 of the KRAS gene (58). KRAS mutations are associated with advanced tumor progression and clinical aggressiveness (59), forming a persistent risk of lung adenocarcinoma and implying to be an early event in the tumorigenesis process (53). The correlation of the presence of KRAS mutations with a smoking history (60) suggests that KRAS mutations are a sequel of the actions of carcinogens of tobacco products (53). However, in a cohort of 482 lung adenocarcinomas, it was demonstrated that KRAS mutations do occur in patients with lung adenocarcinomas without a smoking history (61), but the mutations are different. Significantly more transition mutations (G>A) are being found in non-smokers than the transversion mutations (G>T or G>C, p < 0.0001) that occur in former- or current smokers (61). This observation supports the idea that the distinct transition profile – replacement of a purine for a purine or a pyrimidine for a pyrimidine (62) – of never smokers is very unlikely to be caused by passive tobacco vulnerability. No specific KRAS targeting treatment has so far shown efficacy. There is little available data on the KRAS mutational status in primary lung cancers as compared to that in their brain metastases (44, 57). In a relatively small series, Munfus-McCray et al. found 23.5% of brain metastatic lung adenocarcinomas with KRAS mutation exclusively in patients with a smoking history (p < 0.01) (59).
Anaplastic lymphoma kinase rearrangements occur in 2–7% of all NSCLC, with predominance in non- or light smokers, younger age, and adenocarcinomas (63, 64). Fusion between EML4 (echinoderm microtubule-associated protein-like 4) and ALK yields at least 15 molecular variants with different biological behaviors and affected signaling pathways and consequences for therapy choice (65). ALK testing is particularly recommended for non-squamous lung cancers in the absence of EGFR mutation, of patients with non- or light smoking history (66). The recommended method for testing the presence of ALK translocation is fluorescent in situ hybridization (FISH) and immunohistochemistry (IHC) as confirmation (67, 68). In a large Western cohort, functional ALK rearrangements appeared to be mutually exclusive with EGFR and KRAS mutations (69). Although ALK translocations seem to be similar in primary tumors and their brain metastases, ALK amplifications are found more frequently in CNS metastasis with discordance rates of only 12.5% (70). Similar to EGFR, ALK rearrangements are predictive of response to TKIs, but the development of brain metastasis in patients with ALK translocations receiving ALK directed TKI is a major clinical problem (71, 72). Recently, second-generation TKI alectinib has shown to delay the development of brain metastases compared to first-generation TKI and also demonstrated promising efficacy in the CNS for crizotinib-resistant ALK-positive NSCLC patients (73, 74). Similar to EGFR and KRAS mutations and ALK rearrangements, several other molecules such as liver kinase B1 (LKB1, also known as STK11), proto-oncogene tyrosine-protein kinase ROS1, and C-MET that encodes the hepatocyte growth factor receptor were found to be implicated in the development of lung cancer (75–79). However, the connection of these molecules with the development of brain metastases is still under investigation and not yet implicated in clinical decision making. KRAS aberrations have a synergistic effect with LKB1 inactivation on lung cancer development and distant metastasis formation (80, 81). In a cohort of 154 NSCLC patients, Zhao et al. demonstrated that a lower LKB1 copy number (CN), along with KRAS mutation, were significantly associated with a higher number of brain metastasis. Moreover, the odds ratio of brain metastasis was ~20 times higher in patients with one decrease in LKB1 CN values (82). LKB1 is observed to be inactivated in ~30% of all NSCLCs (83).
Although several potential targets may not regularly be expressed in a high number of lung cancer brain metastasis, their potential use for personalized treatment of selected lung cancer patients harboring actionable mutations should not be discarded. In a cohort of 874 brain metastases samples, of which 295 NSCLC, Capper et al. showed that, although a total of 51/874 samples harbored a BRAF V600E mutation, only 1/295 NSCLC brain metastases (~0.3%) was BRAF mutant (84). Despite this low frequency of BRAF mutations in lung brain metastasis, regression of both visceral and brain metastases by BRAF inhibitor vemurafenib was reported in a patient with a BRAF V600E-mutated NSCLC (85). While 3% of primary lung cancers harbor ROS1 alterations, only 1/99 adenocarcinomas bore ROS1 translocations and 1/11 squamous cell carcinomas showed ROS1 amplifications (86).
Activating mutations in EGFR are associated with sensitivity to TKI therapy, but nearly 30% of EGFR positive patients show primary resistance to EGFR inhibitor therapy (87). While C-MET amplification is one of the factors commonly associated with disease progression (88), Benedettini et al., in a first cohort of 23 NSCLC samples of patients harboring an EGFR activating mutation, showed that both C-MET phosphorylation and expression were significantly associated with shorter time to progression, correlating with de novo resistance to EGFR TKI. In a second cohort of 40 patients, englobing 18 primary NSCLC from patients who later developed brain metastases and 22 NSCLC from patients that did not develop brain metastases, Benedettini et al. demonstrated that both C-MET expression and phosphorylation, but not C-MET amplification, were significantly higher in the tumors from patients who developed brain metastasis. In 18 matched brain metastasis, amplification was demonstrated (89). In addition, in a cohort of 196 NSCLC brain metastasis samples, Presseur et al. found C-MET gene amplification and overexpression in 21.6 and 44.4%, respectively, confirming that C-MET is commonly activated in brain metastasis manifestation (90). Furthermore, a significant correlation between C-MET and ALK amplification status was observed (p = 0.039). In another study, these authors demonstrated that fibroblast growth factor receptor 1 (FGFR1) amplification in brain metastases of adenocarcinomas – but not squamous cell carcinomas, is fivefold more frequent than reported for primary tumors (~3%). Similar to C-MET, a positive correlation of ALK and FGFR1 amplification status in brain metastasis was reported as significant (p < 0.001) (91). In a recent study, Keap1, Nrf2, and P300, key genes of the Keap1–Nrf2–ARE survival pathway, were found to be mutated in brain metastatic tissue of progressive NSCLC patients (92). Moreover, mutations in Keap1-Nrf2-ARE pathway were found in circulating tumor cells (CTCs), suggesting a role in the ability of CTCs to bear the rough environment in blood-circulation and attain distant organs (92).
An adequate characterization of somatic genetic modifications in human cancers is critical for an optimal diagnosis and subsequent therapy. In brain metastatic tissue, as for all other brain malignancies, repeated biopsies are not a feasible approach to portray the tumor clonal diversity. Several studies have shown that cell-free ctDNA in the plasma could serve to characterize and monitor tumors (93–95). Nevertheless, ctDNA analysis of plasma from patients with brain malignancies has disclosed very low levels of tumor DNA (96). Recently, ctDNA analysis from cerebrospinal fluid (CSF) has been shown promising for brain cancer patients (97–99) and brain metastatic cancer patients (100, 101). CSF is in direct contact with the brain and, therefore, with tumor cells of brain cancer patients. In a comparative study of ctDNA derived from plasma and from CSF of patients with primary or metastatic brain tumors, De Mattos-Arruda et al. showed ctDNA levels of brain malignancies to be more abundantly present in the CSF than in the plasma (100). Moreover, ctDNA from CSF appeared to recapitulate the brain metastasis-specific mutations – private mutations, absent in extracranial tumors of a patient with Her2-positive metastatic breast cancer (100). In addition, the CSF ctDNA proficiency to monitor responses to systemic therapy and brain tumor progression (98, 100), i.e., the capacity of the CSF ctDNA to recapitulate the modulation of mutant allele frequency over time in the brain tumor burden, suggests that genomic CSF analysis may be useful not only in facilitating diagnosis of tumor in the CNS or as guidance to second-line agents choice, but also in pinpointing pathways’ intimate related with cancer spread to the CNS and predictive of brain metastases (98).
Studying SNPs in signaling pathways that regulate cell proliferation and migration and assessing the relationship between multiple SNPs can be used to estimate the risk of brain metastasis. The PI3K–PTEN–AKT–mTOR pathway, important in the control of cell growth, tumorigenesis, and cell invasion, has been shown to be abnormally activated in several cancer types, including NSCLC (102, 103). In a study of genetic variations in the PI3K–AKT–mTOR pathway to predict brain metastasis in NSCLC patients, Quianxia et al. identified three SNPs that appeared to be exclusively associated with higher risk of brain metastasis: the GT/GG (p = 0.006) and CT/TT (p = 0.002) genotypes of AKT1, variant alleles rs2498804 and rs2494732, respectively, and AG/AA (p = 0.010) genotype of PIK3CA, variant allele rs2699887 (103). Furthermore, patients carrying at least one variant allele in PIK3CA had roughly twice the risk of brain metastasis as those without those variants (103). Multiple mechanisms of PI3K activation may be responsible for activation of the PI3K pathway (104), and increased PI3K activity would result in increased metastases. In concordance, Paik et al. reported that patients with aberrant PI3K squamous lung carcinomas (n = 9) had worse survival (median OS: 8.6 vs 19.1 months, p < 0.001), higher metastatic burden (>3 organs, 18 vs 3%, p = 0.025), and higher incidence of brain metastases (27 vs 0%, p < 0.001) (105). Similar to PIK3K–AKT–mTOR pathway, it was hypothesized that common genetic variants in the TGF-β pathway would be associated with the risk of brain metastasis (106). TGF-β pathway has been demonstrated to suppress early-stage tumor development and to stimulate tumor cell growth and invasiveness at later stages of tumorigenesis (107). Quianxia et al. found the GG genotype of SMAD6: rs12913975 (p = 0.014) and the TT genotype of INHBC: rs4750259 (p = 0.024) to be associated with risk of brain metastasis in a cohort of 161 blood samples from NSCLC patients. Furthermore, de combination of both genetic variants was shown to be higher for prediction of brain metastasis (p = 0.001) (106).
Activation or inhibition of a gene occurs through a variety of mechanisms such as, for example, activating mutations and deletions. Gene deletion can be evaluated by CNA. Animal models have given clear evidence that LKB1 haploinsufficiency stimulates KRAS driven lung cancer in mice (81), and a single copy inactivation of LKB1 can considerably ease brain recurrence (82). Although EGFR CN status is still controversial and some of the available data do not support EGFR CN as a prognostic factor (108, 109), Bonanno et al. have shown, despite the less predictive accuracy of FISH analysis compared to EGFR mutation analysis, that patients with EGFR-FISH-positive tumors have better outcomes (median OS: 177 vs 57 weeks, p = 0.048) (110). Considering that primary lung adenocarcinomas with early development of brain metastasis would contain more CNAs predictive of metastatic potential, Lee et al. compared the CN changes of four lung adenocarcinomas with coexistent brain metastasis with 8 lung adenocarcinomas with metachronous brain metastasis (111). Amplification in 5q35.1-2 and 17q23.3-24.1 was detected in 100% and that in 10q23.31 and 17q24.1 was detected in 75% of the cases with synchronous brain metastasis. On the other hand, and in a less frequent ratio, only 5q35.1-2 and 17q24.1 amplification status was found in 12.5% of the metachronous brain metastasis. Moreover, gained regions specific for early (simultaneous) brain metastasis were found to contain ACTA2, FAS, RGS9, and ICAM2 as putative metastasis promoting genes, the latter being most significant (p = 0.002) (111). In the same line, another study compared CNAs of primary NSCLC tumor and matched brain metastasis from one single patient (112). Brain metastatic tissue exhibited a higher degree of genetic heterogeneity when compared with the primary tumor with common regions of gain including 7p, 7q, and 19q and common regions of loss including 20q13 (112). In a stage IV SQCLCs study, four brain metastases and matched archived FFPE primary cancers were shown to have complete loss of PTEN by IHC and whole exome sequencing (105). In an early-stage NSCLC report, 30 (24%) of the total of 125 specimens analyzed for PTEN-IHC showed a lack of staining (113). Although genetic alterations of the PTEN gene are unusual in NSCLC, loss of PTEN protein is not a unique event in early-stage NSCLC and Soria et al. demonstrated that besides being a reversible event, PTEN loss may be partially explained by promotor methylation, in addition to point mutations and homozygous deletions (113).
Recently, molecular studies have stressed the role of miRNAs which are small non-coding endogenous RNAs containing 18–24 nucleotides that regulate gene expression at the post-transcriptional level thereby acting as negative regulators of mRNA translation and/or stability (114). miRNAs appear to regulate several hundred genes and could serve as a better classifier than gene expression profiling (115). miRNAs are known to play a crucial role in normal development, proliferation, differentiation, and apoptosis, and dysregulation of miRNAs has been linked to various pathological conditions, including cancer (116). The role of miRNAs in the development of brain metastases has been recently explored (117, 118).
Several studies have addressed the miRNA expression as biomarkers to predict the occurrence of brain metastases in lung cancer. miRNA-328 appeared to be significantly overexpressed in both primary tumor samples and cerebral metastases of patients with NSCLC, when compared with NSCLC patients without brain metastasis. Moreover, miRNA-328 overexpression has been found to promote migration and subsequent brain metastasis formation of NSCLC cells through PRKCA deregulation (119). PRKCA mediates the expression of urokinase plasminogen activator, leading to the migration of the tumor cells (120). Similar to miRNA-328, miRNA-378 has also been demonstrated as a potential biomarker to assist clinicians in stratifying patients for high-risk of brain metastasis, because miRNA-378 was also found to be overexpressed in NSCLC primary tumor samples and matched brain metastasis of NSCLC patients (121). Also, miRNA-378 promotes cell migration, invasion, tumor growth, and angiogenesis, in vitro and in vivo (121). Recently, Remon et al. have identified miRNA-197 and miRNA-184 as two significantly overexpressed miRNAs in EGFR-mutant patients with brain metastases, when compared with EGFR-mutant patients with no brain metastasis (122). However, because of lack of patients with EGFR wild-type (EGFRwt) tumors without BM, no comparison between patients with EGFRwt tumors, with and without BM, could be made. Therefore, the effects of these miRNAs, irrespective of the EGFR status, need further scrutiny.
MicroRNAs’ expression status varies according to their targeted genes. Zhao et al. have reported the significant up-regulation of miRNA-1471 and miRNA-9 and down-regulation of miRNA-214 and miRNA-145 in 11 brain metastatic lung cancer samples, when compared with 40 primary lung adenocarcinomas (p < 0.001 for all four miRNAs) (123). The up-regulation of miRNA-145 in primary lung adenocarcinomas was shown to suppress proliferation of tumor cells (123), consistent with other reports that show inhibition of cell proliferation in human lung adenocarcinomas through miRNA-145 targeting c-Myc, EGFR and NUDT1 (124, 125). Subramani et al. have shown the miRNA-768-3p to be underexpressed in several brain metastases, compared to matched primary tumors (126). miRNA-768-3p was found to be underexpressed in in vitro lung cancer cells after co-culture with astrocytes, driving to increased KRAS protein and downstream effectors ERK1/2 and BRAF, thereby boosting tumor cell viability and promoting metastasis. From various studies, it appears that miRNAs regulate the growth of metastases either by under- or overexpression, within the tumor tissue or in the tumor environment. The brain microenvironment negatively regulates miRNA-768-3p to enhance KRAS expression that promotes the propagation of lung cancer brain metastasis (126). miRNA-146 was shown to be significantly up-regulated in NSCLC tissue when compared to healthy adjacent lung tissue (p < 0.05) (127). In another study, miRNA-146a expression in primary NSCLC was correlated with advanced clinical TNM stages and distant metastasis (p < 0.05). The patients with a high miRNA-146a expression showed longer progression-free-survival times than those with a low expression of miRNA-146a (25.6 and 4.8 weeks, respectively, p < 0.05) (128). In the same line with these findings, in a xenograft model, Hwang et al. showed high expression of miRNA-146a in parental cells, while diminished expression in the brain-seeking cells. Moreover, miRNA-146a overexpression in the brain-seeking cancer cells suppressed their metastatic potential, which was correlated to the up-regulation of β-catenin and down-regulation of heterogeneous nuclear ribonucleoprotein C1/C2 (129). Taken together, these findings suggest that miRNA-146a serve as a valid clinical biomarker for prediction of brain metastasis in lung cancer patients. However, validation of miRNA-146a expression levels in a large cohort of human matched primary and brain metastatic lung tumors is essential to confirm this finding. Similar to miRNA-146a, overexpression of miRNA-95-3p suppresses brain metastasis of lung adenocarcinoma through down-regulation of cyclin D1 (130). miRNA-95-3p is decreased in brain metastases of lung cancers as compared to the primary tumors and higher cyclin D1 expression correlates with poorer prognoses (130). In a recent study, Chen et al. reported miRNA-375 deregulation to be associated with NSCLC brain metastasis (131). miRNA-375 is another miRNA documented to be down-regulated in primary tumors of NSCLC patients with brain metastasis. miRNA-375 expression was significantly decreased in matched brain metastatic NSCLC tissues (p < 0.05) and significantly correlates with total number of brain metastasis (p < 0.001). In addition, VEGF and MMP9 – which roles have been extensively studied in the development of brain metastasis – were over-expressed in down-regulated miRNA-375 tumors (131).
MicroRNAs are linked with several molecular pathways. Several studies have correlated the overexpression of ADAM9 in NSCLC patients with brain metastases (4, 132). ADAM9 has been demonstrated to enhance the ability of tissue plasminogen activator to cleave and stimulate the function of CUB domain containing protein 1 (CDCP1) – promigratory protein, to promote brain metastasis (4). Recently, Chiu et al. reported that ADAM9 down-regulates miRNA-1 via EGFR signaling pathways activation, enhancing CDCP1 expression to promote lung cancer progression (133). miRNA-1 expression was shown to be down-regulated in primary lung tumors but increased in ADAM9-knockdown lung cancer cells. Moreover, miRNA-1 negatively correlates with CDCP1 expression and with migration ability of lung cancer cells (133). Another study has identified miRNA-21 as a target of signal transducers and activators of transcription 3 (STAT3) pathway activity in lung-derived brain metastasis initiating cells (134). STAT3 is admitted as a central regulator in the metastatic process (135), and STAT3-knockdown has been demonstrated to reduce expression of known downstream targets of miRNA-21, while STAT3 and miRNA-21 act as cooperative regulators of stemness, migration and tumor initiation in lung-derived brain metastasis (134). miRNAs appear very promising as diagnostics, prognostics and therapeutics to improve cancer patient outcome; however, the clinical use of miRNA therapeutics to treat brain metastases has yet to be achieved. Advances in pre-clinical and translational studies to identify miRNAs that change after growth in the brain microenvironment have been made, but validation of large cohorts from patient tumor samples is required.
Long non-coding RNAs have been recently identified as effective players in tumorigenesis. lncRNAs represent a class of non-protein coding transcripts longer than 200 nucleotides (136) that covers a broad spectrum of physiological and pathological functions by implementing different modes of action (137). Similar to miRNAs that regulate several hundred genes, lncRNAs are involved in the regulation of multiple miRNAs, impacting the expression of thousands of genes (136). Besides performing a single function, some lncRNAs act at multiple functional levels in different types of cells. Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), localized in nuclear speckles and highly conserved among mammals, regulates alternative splicing (138) and gene expression through additional splicing-independent mechanisms in lung cancer metastasis (139). In a recent study, Shen et al. have shown lncRNA-MALAT1 levels to be significantly higher in primary NSCLC from patients who developed brain metastasis when compared with primary NSCLC from patients without brain metastasis (p < 0.001) (140). Additional in vitro functional studies showed overexpression of vimentin in a highly invasive subline of brain metastasis lung cancer cells overexpressing MALAT1, while overexpression of E-cadherin was observed when MALAT1 was silenced, indicating that MALAT1 overexpression promotes lung cancer brain metastasis by inducing EMT (140). Accordingly, RNAi-mediated suppression of MALAT1-RNA, negatively influenced migration and clonogenic growth in established human NSCLC cell lines. Forced expression of MALAT1 in mouse NIH 3T3 fibroblasts significantly increased migration (141). Concordantly, long non-coding MALAT1 expression was found to enhance cell motility through transcriptional and post-transcriptional regulation of motility related gene expression (142), displaying the strongest association with genes involved in cancer, like cellular growth, movement, proliferation, signaling and immune regulation genes (141). MALAT1 and thymosin β4 expression levels were identified as prognostic parameters for patient survival in stage I NSCLC that are at high risk to develop metastasis (p = 0.04 and p = 0.01, respectively) (143). Tumorigenesis and metastases may be driven by tumor suppressive and oncogenic pathways deregulation through aberrant expression of cancer metastasis-associated lncRNA (144). In a recent in vitro study, the lncRNA brain cytoplasmatic RNA 1 (BCYRN1) was found up-regulated and targeted by c-MYC in human NSCLC cell lines (145). c-MYC is a commonly inhibited oncogene and becomes activated in oncogenic pathways, and correlates with metastasis of NSCLC (146). Besides demonstrating that IncRNA BCYRN1 is essential in the c-MYC-regulated cell migration and invasion, BCYRN1 positively correlates with the expression levels of MMP9 and MMP13 (145). MMP9 and MMP13, two members of the matrixin subfamily of the metzincin superfamily of Zn-dependent metalloproteinases (147), are extracellular matrix degrading proteins proven to induce migration and invasion of tumor cells (147, 148), thereby regulating cancer cell metastasis (149).
Lung adenocarcinoma establishes distant clinical detectable metastasis within months of initial diagnosis (26, 150). This short abeyance indicates that metastatic ability would arise from early oncogenic events that stimulate primary tumor growth rather than late-arising, scarce genomic alterations specific for metastasis (151). Thus, monitoring persistent chromosomal changes in the primary NSCLC alongside with prospective multicenter studies of patient-matched primary and CNS metastatic lesions could help identify targetable approaches for brain metastasis-specific signatures.
RP, DM, JA, and JK wrote and revised the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This paper was carried out in the framework of a project on cerebral metastases of common cancers by the Dutch cancer society (KWF).
1. Gril B, Evans L, Palmieri D, Steeg PS. Translational research in brain metastasis is identifying molecular pathways that may lead to the development of new therapeutic strategies. Eur J Cancer (2010) 46:1204–10. doi:10.1016/j.ejca.2010.02.033
2. Castrucci WA, Knisely JP. An update on the treatment of CNS metastases in small cell lung cancer. Cancer J (2008) 14:138–46. doi:10.1097/PPO.0b013e318172d6e1
3. Hubbs JL, Boyd JA, Hollis D, Chino JP, Saynak M, Kelsey CR. Factors associated with the development of brain metastases: analysis of 975 patients with early stage nonsmall cell lung cancer. Cancer (2010) 116:5038–46. doi:10.1002/cncr.25254
4. Lin CY, Chen HJ, Huang CC, Lai LC, Lu TP, Tseng GC, et al. ADAM9 promotes lung cancer metastases to brain by a plasminogen activator-based pathway. Cancer Res (2014) 74:5229–43. doi:10.1158/0008-5472.CAN-13-2995
5. Laack NN, Brown PD. Cognitive sequelae of brain radiation in adults. Semin Oncol (2004) 31:702–13. doi:10.1053/j.seminoncol.2004.07.013
6. D’Amico TA, Aloia TA, Moore MB, Conlon DH, Herndon JE II, Kinch MS, et al. Predicting the sites of metastases from lung cancer using molecular biologic markers. Ann Thorac Surg (2001) 72:1144–8. doi:10.1016/S0003-4975(01)02979-4
7. Hung JJ, Jeng WJ, Wu YC, Chou TY, Hsu WH. Factors predicting organ-specific distant metastasis in patients with completely resected lung adenocarcinoma. Oncotarget (2016) 7:58261–73. doi:10.18632/oncotarget.11338
8. Ceresoli GL, Reni M, Chiesa G, Carretta A, Schipani S, Passoni P, et al. Brain metastases in locally advanced nonsmall cell lung carcinoma after multimodality treatment: risk factors analysis. Cancer (2002) 95:605–12. doi:10.1002/cncr.10687
9. Andre F, Grunenwald D, Pujol JL, Girard P, Dujon A, Brouchet L, et al. Patterns of relapse of N2 non-small cell lung carcinoma patients treated with preoperative chemotherapy: should prophylactic cranial irradiation be reconsidered? Cancer (2001) 91:2394–400. doi:10.1002/1097-0142(20010615)91:12<2394::AID-CNCR1273>3.0.CO;2-6
10. Bajard A, Westeel V, Dubiez A, Jacoulet P, Pernet D, Dalphin JC, et al. Multivariate analysis of factors predictive of brain metastases in localised non-small cell lung carcinoma. Lung Cancer (2004) 45:317–23. doi:10.1016/j.lungcan.2004.01.025
11. Carolan H, Sun AY, Bezjak A, Yi Q-L, Payne D, Kane G, et al. Does the incidence and outcome of brain metastases in locally advanced non-small cell lung cancer justify prophylactic cranial irradiation or early detection? Lung Cancer (2005) 49:109–15. doi:10.1016/j.lungcan.2004.12.004
12. Chen AM, Jahan TM, Jablons DM, Garcia J, Larson DA. Risk of cerebral metastases and neurological death after pathological complete response to neoadjuvant therapy for locally advanced nonsmall-cell lung cancer: clinical implications for the subsequent management of the brain. Cancer (2007) 109:1668–75. doi:10.1002/cncr.22565
13. Jacobs RH, Awan A, Bitran JD, Hoffman PC, Little AG, Ferguson MK, et al. Prophylactic cranial irradiation in adenocarcinoma of the lung. A possible role. Cancer (1987) 59:2016–9.
14. Mujoomdar A, Austin JH, Malhotra R, Powell CA, Pearson GD, Shiau MC, et al. Clinical predictors of metastatic disease to the brain from non-small cell lung carcinoma: primary tumor size, cell type, and lymph node metastases. Radiology (2007) 242:882–8. doi:10.1148/radiol.2423051707
15. Robnett TJ, Machtay M, Stevenson JP, Algazy KM, Hahn SM. Factors affecting the risk of brain metastases after definitive chemoradiation for locally advanced non-small-cell lung carcinoma. J Clin Oncol (2001) 19:1344–9. doi:10.1200/JCO.2001.19.5.1344
16. Schouten LJ, Rutten J, Huveneers HA, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer (2002) 94:2698–705. doi:10.1002/cncr.10541
17. Tang SG, Lin FJ, Leung WM. Impact of prophylactic cranial irradiation in adenocarcinoma of the lung. J Formos Med Assoc (1993) 92:413–9.
18. Tang SG, Tseng CK, Tsay PK, Chen CH, Chang JW, Pai PC, et al. Predictors for patterns of brain relapse and overall survival in patients with non-small cell lung cancer. J Neurooncol (2005) 73:153–61. doi:10.1007/s11060-004-3725-4
19. Tsuchiya T, Akamine S, Muraoka M, Kamohara R, Tsuji K, Urabe S, et al. Stage IA non-small cell lung cancer: vessel invasion is a poor prognostic factor and a new target of adjuvant chemotherapy. Lung Cancer (2007) 56:341–8. doi:10.1016/j.lungcan.2007.01.019
20. Westeel V, Choma D, Clement F, Woronoff-Lemsi MC, Pugin JF, Dubiez A, et al. Relevance of an intensive postoperative follow-up after surgery for non-small cell lung cancer. Ann Thorac Surg (2000) 70:1185–90. doi:10.1016/S0003-4975(00)01731-8
21. Goldstraw P, Chansky K, Crowley J, Rami-Porta R, Asamura H, Eberhardt WE, et al. The IASLC Lung Cancer Staging Project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol (2016) 11:39–51. doi:10.1016/j.jtho.2015.09.009
22. Chansky K, Sculier JP, Crowley JJ, Giroux D, Van Meerbeeck J, Goldstraw P, et al. The International Association for the Study of Lung Cancer Staging Project: prognostic factors and pathologic TNM stage in surgically managed non-small cell lung cancer. J Thorac Oncol (2009) 4:792–801. doi:10.1097/JTO.0b013e3181a7716e
23. Chen YY, Wang LW, Wang SY, Wu BB, Wang ZM, Chen FF, et al. Meta-analysis of postoperative adjuvant chemotherapy without radiotherapy in early stage non-small cell lung cancer. Onco Targets Ther (2015) 8:2033–43. doi:10.2147/OTT.S88700
24. D’Amico TA. Molecular biologic substaging of non-small cell lung cancer. J Thorac Cardiovasc Surg (2002) 123:409–10. doi:10.1067/mtc.2002.120339
25. Ahrendt SA, Yang SC, Wu L, Roig CM, Russell P, Westra WH, et al. Molecular assessment of lymph nodes in patients with resected stage I non-small cell lung cancer: preliminary results of a prospective study. J Thorac Cardiovasc Surg (2002) 123:466–473;discussion473–464. doi:10.1067/mtc.2002.120343
26. Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, et al. Metastatic patterns in adenocarcinoma. Cancer (2006) 106:1624–33. doi:10.1002/cncr.21778
27. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi:10.1016/j.cell.2008.03.027
28. Jeevan DS, Cooper JB, Braun A, Murali R, Jhanwar-Uniyal M. Molecular pathways mediating metastases to the brain via epithelial-to-mesenchymal transition: genes, proteins, and functional analysis. Anticancer Res (2016) 36:523–32.
29. Risolino M, Mandia N, Iavarone F, Dardaei L, Longobardi E, Fernandez S, et al. Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-beta-SMAD3 pathway in non-small cell lung adenocarcinoma. Proc Natl Acad Sci U S A (2014) 111:E3775–84. doi:10.1073/pnas.1407074111
30. Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev (2004) 18:1131–43. doi:10.1101/gad.294104
31. Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res (2008) 100:35–83. doi:10.1016/S0065-230X(08)00002-X
32. Hasanbasic I, Cuerquis J, Varnum B, Blostein MD. Intracellular signaling pathways involved in Gas6-Axl-mediated survival of endothelial cells. Am J Physiol Heart Circ Physiol (2004) 287:H1207–13. doi:10.1152/ajpheart.00020.2004
33. Lee WP, Wen Y, Varnum B, Hung MC. Akt is required for Axl-Gas6 signaling to protect cells from E1A-mediated apoptosis. Oncogene (2002) 21:329–36. doi:10.1038/sj.onc.1205066
34. Wu X, Ma W, Zhou Q, Yan H, Lim ZF, Huang M, et al. AXL-GAS6 expression can predict for adverse prognosis in non-small cell lung cancer with brain metastases. J Cancer Res Clin Oncol (2017) 143:1947–57. doi:10.1007/s00432-017-2408-4
35. Wang TJ, Saad S, Qureshi YH, Jani A, Nanda T, Yaeh AM, et al. Does lung cancer mutation status and targeted therapy predict for outcomes and local control in the setting of brain metastases treated with radiation? Neuro Oncol (2015) 17:1022–8. doi:10.1093/neuonc/nov043
36. Perez-Moreno P, Brambilla E, Thomas R, Soria JC. Squamous cell carcinoma of the lung: molecular subtypes and therapeutic opportunities. Clin Cancer Res (2012) 18:2443–51. doi:10.1158/1078-0432.CCR-11-2370
37. Jain D, Iqbal S, Walia R, Malik P, Cyriac S, Mathur SR, et al. Evaluation of epidermal growth factor receptor mutations based on mutation specific immunohistochemistry in non-small cell lung cancer: a preliminary study. Indian J Med Res (2016) 143:308–14. doi:10.4103/0971-5916.182621
38. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer (2007) 7:169–81. doi:10.1038/nrc2088
39. Bauml J, Mick R, Zhang Y, Watt CD, Vachani A, Aggarwal C, et al. Frequency of EGFR and KRAS mutations in patients with non small cell lung cancer by racial background: do disparities exist? Lung Cancer (2013) 81:347–53. doi:10.1016/j.lungcan.2013.05.011
40. Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol (2014) 9:154–62. doi:10.1097/JTO.0000000000000033
41. Tanaka T, Matsuoka M, Sutani A, Gemma A, Maemondo M, Inoue A, et al. Frequency of and variables associated with the EGFR mutation and its subtypes. Int J Cancer (2010) 126:651–5. doi:10.1002/ijc.24746
42. Eichler AF, Kahle KT, Wang DL, Joshi VA, Willers H, Engelman JA, et al. EGFR mutation status and survival after diagnosis of brain metastasis in nonsmall cell lung cancer. Neuro Oncol (2010) 12:1193–9. doi:10.1093/neuonc/noq076
43. Li Z, Lu J, Zhao Y, Guo H. The retrospective analysis of the frequency of EGFR mutations and efficacy of gefitinib in NSCLC patients with brain metastases. J Clin Oncol (2011) 29:e18065. doi:10.1200/jco.2011.29.15_suppl.e18065
44. Cortot AB, Italiano A, Burel-Vandenbos F, Martel-Planche G, Hainaut P. KRAS mutation status in primary nonsmall cell lung cancer and matched metastases. Cancer (2010) 116:2682–7. doi:10.1002/cncr.25014
45. Daniele L, Cassoni P, Bacillo E, Cappia S, Righi L, Volante M, et al. Epidermal growth factor receptor gene in primary tumor and metastatic sites from non-small cell lung cancer. J Thorac Oncol (2009) 4:684–8. doi:10.1097/JTO.0b013e3181a52359
46. Gomez-Roca C, Raynaud CM, Penault-Llorca F, Mercier O, Commo F, Morat L, et al. Differential expression of biomarkers in primary non-small cell lung cancer and metastatic sites. J Thorac Oncol (2009) 4:1212–20. doi:10.1097/JTO.0b013e3181b44321
47. Gow CH, Chang YL, Hsu YC, Tsai MF, Wu CT, Yu CJ, et al. Comparison of epidermal growth factor receptor mutations between primary and corresponding metastatic tumors in tyrosine kinase inhibitor-naive non-small-cell lung cancer. Ann Oncol (2009) 20:696–702. doi:10.1093/annonc/mdn679
48. Italiano A, Vandenbos FB, Otto J, Mouroux J, Fontaine D, Marcy PY, et al. Comparison of the epidermal growth factor receptor gene and protein in primary non-small-cell-lung cancer and metastatic sites: implications for treatment with EGFR-inhibitors. Ann Oncol (2006) 17:981–5. doi:10.1093/annonc/mdl038
49. Matsumoto S, Takahashi K, Iwakawa R, Matsuno Y, Nakanishi Y, Kohno T, et al. Frequent EGFR mutations in brain metastases of lung adenocarcinoma. Int J Cancer (2006) 119:1491–4. doi:10.1002/ijc.21940
50. Sun M, Behrens C, Feng L, Ozburn N, Tang X, Yin G, et al. HER family receptor abnormalities in lung cancer brain metastases and corresponding primary tumors. Clin Cancer Res (2009) 15:4829–37. doi:10.1158/1078-0432.CCR-08-2921
51. Stanic K, Zwitter M, Hitij NT, Kern I, Sadikov A, Cufer T. Brain metastases in lung adenocarcinoma: impact of EGFR mutation status on incidence and survival. Radiol Oncol (2014) 48:173–83. doi:10.2478/raon-2014-0016
52. Zhou Q, Zhang XC, Chen ZH, Yin XL, Yang JJ, Xu CR, et al. Relative abundance of EGFR mutations predicts benefit from gefitinib treatment for advanced non-small-cell lung cancer. J Clin Oncol (2011) 29:3316–21. doi:10.1200/JCO.2010.33.3757
53. Ahrendt SA, Decker PA, Alawi EA, Zhu Yr YR, Sanchez-Cespedes M, Yang SC, et al. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer (2001) 92:1525–30. doi:10.1002/1097-0142(20010915)92:6<1525::AID-CNCR1478>3.0.CO;2-H
54. Marks JL, Broderick S, Zhou Q, Chitale D, Li AR, Zakowski MF, et al. Prognostic and therapeutic implications of EGFR and KRAS mutations in resected lung adenocarcinoma. J Thorac Oncol (2008) 3:111–6. doi:10.1097/JTO.0b013e318160c607
55. Fujimoto N, Wislez M, Zhang J, Iwanaga K, Dackor J, Hanna AE, et al. High expression of ErbB family members and their ligands in lung adenocarcinomas that are sensitive to inhibition of epidermal growth factor receptor. Cancer Res (2005) 65:11478–85. doi:10.1158/0008-5472.CAN-05-1977
56. Han SW, Kim TY, Jeon YK, Hwang PG, Im SA, Lee KH, et al. Optimization of patient selection for gefitinib in non-small cell lung cancer by combined analysis of epidermal growth factor receptor mutation, K-ras mutation, and Akt phosphorylation. Clin Cancer Res (2006) 12:2538–44. doi:10.1158/1078-0432.CCR-05-2845
57. Kalikaki A, Koutsopoulos A, Trypaki M, Souglakos J, Stathopoulos E, Georgoulias V, et al. Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br J Cancer (2008) 99:923–9. doi:10.1038/sj.bjc.6604629
58. Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med (1987) 317:929–35. doi:10.1056/NEJM198710083171504
59. Munfus-McCray D, Harada S, Adams C, Askin F, Clark D, Gabrielson E, et al. EGFR and KRAS mutations in metastatic lung adenocarcinomas. Hum Pathol (2011) 42:1447–53. doi:10.1016/j.humpath.2010.12.011
60. Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol (2005) 23:5900–9. doi:10.1200/JCO.2005.02.857
61. Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res (2008) 14:5731–4. doi:10.1158/1078-0432.CCR-08-0646
62. Hainaut P, Pfeifer GP. Patterns of p53 G – >T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis (2001) 22:367–74. doi:10.1093/carcin/22.3.367
63. Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol (2013) 31:1105–11. doi:10.1200/JCO.2012.44.5353
64. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature (2007) 448:561–6. doi:10.1038/nature05945
65. Sabir SR, Yeoh S, Jackson G, Bayliss R. EML4-ALK variants: biological and molecular properties, and the implications for patients. Cancers (Basel) (2017) 9:2–3. doi:10.3390/cancers9090118
66. Peters S, Adjei AA, Gridelli C, Reck M, Kerr K, Felip E, et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol (2012) 23(Suppl 7):vii56–64. doi:10.1093/annonc/mds226
67. Cooper W, Fox S, O’Toole S, Morey A, Frances G, Pavlakis N, et al. National Working Group Meeting on ALK diagnostics in lung cancer. Asia Pac J Clin Oncol (2014) 10(Suppl 2):11–7. doi:10.1111/ajco.12190
68. Houang M, Toon CW, Clarkson A, Sioson L, Watson N, Farzin M, et al. Reflex ALK immunohistochemistry is feasible and highly specific for ALK gene rearrangements in lung cancer. Pathology (2014) 46:383–8. doi:10.1097/PAT.0000000000000114
69. Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res (2013) 19:4273–81. doi:10.1158/1078-0432.CCR-13-0318
70. Preusser M, Berghoff AS, Ilhan-Mutlu A, Magerle M, Dinhof C, Widhalm G, et al. ALK gene translocations and amplifications in brain metastases of non-small cell lung cancer. Lung Cancer (2013) 80:278–83. doi:10.1016/j.lungcan.2013.01.019
71. Peled N, Zach L, Liran O, Ilouze M, Bunn PA Jr, Hirsch FR. Effective crizotinib schedule for brain metastases in ALK rearrangement metastatic non-small-cell lung cancer. J Thorac Oncol (2013) 8:e112–3. doi:10.1097/JTO.0000000000000038
72. Weickhardt AJ, Scheier B, Burke JM, Gan G, Lu X, Bunn PA Jr, et al. Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogene-addicted non-small-cell lung cancer. J Thorac Oncol (2012) 7:1807–14. doi:10.1097/JTO.0b013e3182745948
73. Gandhi L, Ou SI, Shaw AT, Barlesi F, Dingemans AC, Kim DW, et al. Efficacy of alectinib in central nervous system metastases in crizotinib-resistant ALK-positive non-small-cell lung cancer: comparison of RECIST 1.1 and RANO-HGG criteria. Eur J Cancer (2017) 82:27–33. doi:10.1016/j.ejca.2017.05.019
74. Lockney NA, Wu AJ. Alectinib for the management of ALK-positive non-small cell lung cancer brain metastases. J Thorac Dis (2017) 9:E152–4. doi:10.21037/jtd.2017.02.05
75. Demuth C, Andersen MN, Jakobsen KR, Madsen AT, Sorensen BS. Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment. Oncotarget (2017) 8:68221–9. doi:10.18632/oncotarget.19920
76. Ji M, Liu Y, Li Q, Li XD, Zhao WQ, Zhang H, et al. PD-1/PD-L1 pathway in non-small-cell lung cancer and its relation with EGFR mutation. J Transl Med (2015) 13:5. doi:10.1186/s12967-014-0373-0
77. Killock D. Lung cancer: alternative rearrangements – targeting ROS1 in NSCLC. Nat Rev Clin Oncol (2014) 11:624. doi:10.1038/nrclinonc.2014.180
78. Li A, Gao HF, Wu YL. Targeting the MET pathway for potential treatment of NSCLC. Expert Opin Ther Targets (2015) 19:663–74. doi:10.1517/14728222.2014.995093
79. Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene (2007) 26:5911–8. doi:10.1038/sj.onc.1210418
80. Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell (2010) 17:547–59. doi:10.1016/j.ccr.2010.04.026
81. Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature (2007) 448:807–10. doi:10.1038/nature06030
82. Zhao N, Wilkerson MD, Shah U, Yin X, Wang A, Hayward MC, et al. Alterations of LKB1 and KRAS and risk of brain metastasis: comprehensive characterization by mutation analysis, copy number, and gene expression in non-small-cell lung carcinoma. Lung Cancer (2014) 86:255–61. doi:10.1016/j.lungcan.2014.08.013
83. Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res (2002) 62:3659–62.
84. Capper D, Berghoff AS, Magerle M, Ilhan A, Wohrer A, Hackl M, et al. Immunohistochemical testing of BRAF V600E status in 1,120 tumor tissue samples of patients with brain metastases. Acta Neuropathol (2012) 123:223–33. doi:10.1007/s00401-011-0887-y
85. Robinson SD, O’Shaughnessy JA, Cowey CL, Konduri K. BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer (2014) 85:326–30. doi:10.1016/j.lungcan.2014.05.009
86. Preusser M, Streubel B, Birner P. ROS1 translocations and amplifications in lung cancer brain metastases. J Neurooncol (2014) 118:425–6. doi:10.1007/s11060-014-1446-x
87. Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol (2010) 28:357–60. doi:10.1200/JCO.2009.24.7049
88. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316:1039–43. doi:10.1126/science.1141478
89. Benedettini E, Sholl LM, Peyton M, Reilly J, Ware C, Davis L, et al. Met activation in non-small cell lung cancer is associated with de novo resistance to EGFR inhibitors and the development of brain metastasis. Am J Pathol (2010) 177:415–23. doi:10.2353/ajpath.2010.090863
90. Preusser M, Streubel B, Berghoff AS, Hainfellner JA, von Deimling A, Widhalm G, et al. Amplification and overexpression of CMET is a common event in brain metastases of non-small cell lung cancer. Histopathology (2014) 65:684–92. doi:10.1111/his.12475
91. Preusser M, Berghoff AS, Berger W, Ilhan-Mutlu A, Dinhof C, Widhalm G, et al. High rate of FGFR1 amplifications in brain metastases of squamous and non-squamous lung cancer. Lung Cancer (2014) 83:83–9. doi:10.1016/j.lungcan.2013.10.004
92. Aljohani HM, Aittaleb M, Furgason JM, Amaya P, Deeb A, Chalmers JJ, et al. Genetic mutations associated with lung cancer metastasis to the brain. Mutagenesis (2018) 33:137–45. doi:10.1093/mutage/gey003
93. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med (2013) 368:1199–209. doi:10.1056/NEJMoa1213261
94. De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol (2014) 25:1729–35. doi:10.1093/annonc/mdu239
95. Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med (2012) 4:136ra168. doi:10.1126/scitranslmed.3003726
96. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med (2014) 6:224ra224. doi:10.1126/scitranslmed.3007094
97. Pan W, Gu W, Nagpal S, Gephart MH, Quake SR. Brain tumor mutations detected in cerebral spinal fluid. Clin Chem (2015) 61:514–22. doi:10.1373/clinchem.2014.235457
98. Pentsova EI, Shah RH, Tang J, Boire A, You D, Briggs S, et al. Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J Clin Oncol (2016) 34:2404–15. doi:10.1200/JCO.2016.66.6487
99. Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci U S A (2015) 112:9704–9. doi:10.1073/pnas.1511694112
100. De Mattos-Arruda L, Mayor R, Ng CK, Weigelt B, Martinez-Ricarte F, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun (2015) 6:8839. doi:10.1038/ncomms9839
101. Li Y, Pan W, Connolly ID, Reddy S, Nagpal S, Quake S, et al. Tumor DNA in cerebral spinal fluid reflects clinical course in a patient with melanoma leptomeningeal brain metastases. J Neurooncol (2016) 128:93–100. doi:10.1007/s11060-016-2081-5
102. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res (2001) 61:3986–97.
103. Li Q, Yang J, Yu Q, Wu H, Liu B, Xiong H, et al. Associations between single-nucleotide polymorphisms in the PI3K–PTEN–AKT–mTOR pathway and increased risk of brain metastasis in patients with non–small cell lung cancer. Clin Cancer Res (2013) 19:6252–60. doi:10.1158/1078-0432.CCR-13-1093
104. Adamo B, Deal AM, Burrows E, Geradts J, Hamilton E, Blackwell KL, et al. Phosphatidylinositol 3-kinase pathway activation in breast cancer brain metastases. Breast Cancer Res (2011) 13:R125. doi:10.1186/bcr3071
105. Paik PK, Shen R, Won H, Rekhtman N, Wang L, Sima CS, et al. Next-generation sequencing of stage IV squamous cell lung cancers reveals an association of PI3K aberrations and evidence of clonal heterogeneity in patients with brain metastases. Cancer Discov (2015) 5:610–21. doi:10.1158/2159-8290.CD-14-1129
106. Li Q, Wu H, Chen B, Hu G, Huang L, Qin K, et al. SNPs in the TGF-beta signaling pathway are associated with increased risk of brain metastasis in patients with non-small-cell lung cancer. PLoS One (2012) 7:e51713. doi:10.1371/journal.pone.0051713
108. Jeon YK, Sung SW, Chung JH, Park WS, Seo JW, Kim CW, et al. Clinicopathologic features and prognostic implications of epidermal growth factor receptor (EGFR) gene copy number and protein expression in non-small cell lung cancer. Lung Cancer (2006) 54:387–98. doi:10.1016/j.lungcan.2006.08.015
109. Mao C, Qiu LX, Liao RY, Du FB, Ding H, Yang WC, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer (2010) 69:272–8. doi:10.1016/j.lungcan.2009.11.020
110. Bonanno L, Schiavon M, Nardo G, Bertorelle R, Bonaldi L, Galligioni A, et al. Prognostic and predictive implications of EGFR mutations, EGFR copy number and KRAS mutations in advanced stage lung adenocarcinoma. Anticancer Res (2010) 30:5121–8.
111. Lee HW, Seol HJ, Choi YL, Ju HJ, Joo KM, Ko YH, et al. Genomic copy number alterations associated with the early brain metastasis of non-small cell lung cancer. Int J Oncol (2012) 41:2013–20. doi:10.3892/ijo.2012.1663
112. Li F, Sun L, Zhang S. Acquirement of DNA copy number variations in non-small cell lung cancer metastasis to the brain. Oncol Rep (2015) 34:1701–7. doi:10.3892/or.2015.4188
113. Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res (2002) 8:1178–84.
114. Markopoulos GS, Roupakia E, Tokamani M, Chavdoula E, Hatziapostolou M, Polytarchou C, et al. A step-by-step microRNA guide to cancer development and metastasis. Cell Oncol (Dordr) (2017) 40:303–39. doi:10.1007/s13402-017-0341-9
115. Liu CG, Calin GA, Volinia S, Croce CM. MicroRNA expression profiling using microarrays. Nat Protoc (2008) 3:563–78. doi:10.1038/nprot.2008.14
116. Chen PS, Su JL, Hung MC. Dysregulation of microRNAs in cancer. J Biomed Sci (2012) 19:90. doi:10.1186/1423-0127-19-90
117. Alsidawi S, Malek E, Driscoll JJ. MicroRNAs in brain metastases: potential role as diagnostics and therapeutics. Int J Mol Sci (2014) 15:10508–26. doi:10.3390/ijms150610508
118. McDermott R, Gabikian P, Sarvaiya P, Ulasov I, Lesniak MS. MicroRNAs in brain metastases: big things come in small packages. J Mol Med (Berl) (2013) 91:5–13. doi:10.1007/s00109-012-0971-3
119. Arora S, Ranade AR, Tran NL, Nasser S, Sridhar S, Korn RL, et al. MicroRNA-328 is associated with (non-small) cell lung cancer (NSCLC) brain metastasis and mediates NSCLC migration. Int J Cancer (2011) 129:2621–31. doi:10.1002/ijc.25939
120. Cheng CY, Hsieh HL, Sun CC, Lin CC, Luo SF, Yang CM. IL-1 beta induces urokinase-plasminogen activator expression and cell migration through PKC alpha, JNK1/2, and NF-kappaB in A549 cells. J Cell Physiol (2009) 219:183–93. doi:10.1002/jcp.21669
121. Chen LT, Xu SD, Xu H, Zhang JF, Ning JF, Wang SF. MicroRNA-378 is associated with non-small cell lung cancer brain metastasis by promoting cell migration, invasion and tumor angiogenesis. Med Oncol (2012) 29:1673–80. doi:10.1007/s12032-011-0083-x
122. Remon J, Alvarez-Berdugo D, Majem M, Moran T, Reguart N, Lianes P. miRNA-197 and miRNA-184 are associated with brain metastasis in EGFR-mutant lung cancers. Clin Transl Oncol (2016) 18:153–9. doi:10.1007/s12094-015-1347-2
123. Zhao C, Xu Y, Zhang Y, Tan W, Xue J, Yang Z, et al. Downregulation of miR-145 contributes to lung adenocarcinoma cell growth to form brain metastases. Oncol Rep (2013) 30:2027–34. doi:10.3892/or.2013.2728
124. Cho WC, Chow AS, Au JS. miR-145 inhibits cell proliferation of human lung adenocarcinoma by targeting EGFR and NUDT1. RNA Biol (2011) 8:125–31. doi:10.4161/rna.8.1.14259
125. Cho WC, Chow AS, Au JS. Restoration of tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung adenocarcinoma patients with epidermal growth factor receptor mutation. Eur J Cancer (2009) 45:2197–206. doi:10.1016/j.ejca.2009.04.039
126. Subramani A, Alsidawi S, Jagannathan S, Sumita K, Sasaki AT, Aronow B, et al. The brain microenvironment negatively regulates miRNA-768-3p to promote K-ras expression and lung cancer metastasis. Sci Rep (2013) 3:2392. doi:10.1038/srep02392
127. Li J, Yang H, Li Y, Liu Y, Chen S, Qi C, et al. MicroRNA-146 up-regulation predicts the prognosis of non-small cell lung cancer by miRNA in situ hybridization. Exp Mol Pathol (2014) 96:195–9. doi:10.1016/j.yexmp.2013.11.004
128. Chen G, Umelo IA, Lv S, Teugels E, Fostier K, Kronenberger P, et al. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PLoS One (2013) 8:e60317. doi:10.1371/journal.pone.0060317
129. Hwang SJ, Seol HJ, Park YM, Kim KH, Gorospe M, Nam DH, et al. MicroRNA-146a suppresses metastatic activity in brain metastasis. Mol Cells (2012) 34:329–34. doi:10.1007/s10059-012-0171-6
130. Hwang SJ, Lee HW, Kim HR, Song HJ, Lee DH, Lee H, et al. Overexpression of microRNA-95-3p suppresses brain metastasis of lung adenocarcinoma through downregulation of cyclin D1. Oncotarget (2015) 6:20434–48. doi:10.18632/oncotarget.3886
131. Chen LJ, Li XY, Zhao YQ, Liu WJ, Wu HJ, Liu J, et al. Down-regulated microRNA-375 expression as a predictive biomarker in non-small cell lung cancer brain metastasis and its prognostic significance. Pathol Res Pract (2017) 213:882–8. doi:10.1016/j.prp.2017.06.012
132. Shintani Y, Higashiyama S, Ohta M, Hirabayashi H, Yamamoto S, Yoshimasu T, et al. Overexpression of ADAM9 in non-small cell lung cancer correlates with brain metastasis. Cancer Res (2004) 64:4190–6. doi:10.1158/0008-5472.CAN-03-3235
133. Chiu KL, Lin YS, Kuo TT, Lo CC, Huang YK, Chang HF, et al. ADAM9 enhances CDCP1 by inhibiting miR-1 through EGFR signaling activation in lung cancer metastasis. Oncotarget (2017) 8:47365–78. doi:10.18632/oncotarget.17648
134. Singh M, Garg N, Venugopal C, Hallett R, Tokar T, McFarlane N, et al. STAT3 pathway regulates lung-derived brain metastasis initiating cell capacity through miR-21 activation. Oncotarget (2015) 6:27461–77. doi:10.18632/oncotarget.4742
135. Devarajan E, Huang S. STAT3 as a central regulator of tumor metastases. Curr Mol Med (2009) 9:626–33. doi:10.2174/156652409788488720
136. Vitiello M, Tuccoli A, Poliseno L. Long non-coding RNAs in cancer: implications for personalized therapy. Cell Oncol (Dordr) (2015) 38:17–28. doi:10.1007/s13402-014-0180-x
137. Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol (2012) 9:703–19. doi:10.4161/rna.20481
138. Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell (2010) 39:925–38. doi:10.1016/j.molcel.2010.08.011
139. Gutschner T, Hammerle M, Eissmann M, Hsu J, Kim Y, Hung G, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res (2013) 73:1180–9. doi:10.1158/0008-5472.CAN-12-2850
140. Shen L, Chen L, Wang Y, Jiang X, Xia H, Zhuang Z. Long noncoding RNA MALAT1 promotes brain metastasis by inducing epithelial-mesenchymal transition in lung cancer. J Neurooncol (2015) 121:101–8. doi:10.1007/s11060-014-1613-0
141. Schmidt LH, Spieker T, Koschmieder S, Schaffers S, Humberg J, Jungen D, et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol (2011) 6:1984–92. doi:10.1097/JTO.0b013e3182307eac
142. Tano K, Mizuno R, Okada T, Rakwal R, Shibato J, Masuo Y, et al. MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett (2010) 584:4575–80. doi:10.1016/j.febslet.2010.10.008
143. Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene (2003) 22:8031–41. doi:10.1038/sj.onc.1206928
144. Singla AK, Thirukkumaran C, Dixit R, Wu W. Genomic landscape of cancer metastasis. In: Wu W, Choudhry H, editors. Next Generation Sequencing in Cancer Research: Volume 1: Decoding the Cancer Genome. New York: Springer (2013). p. 75–90.
145. Hu T, Lu YR. BCYRN1, a c-MYC-activated long non-coding RNA, regulates cell metastasis of non-small-cell lung cancer. Cancer Cell Int (2015) 15:36. doi:10.1186/s12935-015-0183-3
146. Rapp UR, Korn C, Ceteci F, Karreman C, Luetkenhaus K, Serafin V, et al. MYC is a metastasis gene for non-small-cell lung cancer. PLoS One (2009) 4:e6029. doi:10.1371/journal.pone.0006029
147. Nabeshima K, Inoue T, Shimao Y, Sameshima T. Matrix metalloproteinases in tumor invasion: role for cell migration. Pathol Int (2002) 52:255–64. doi:10.1046/j.1440-1827.2002.01343.x
148. Yu T, Wu Y, Helman JI, Wen Y, Wang C, Li L. CXCR4 promotes oral squamous cell carcinoma migration and invasion through inducing expression of MMP-9 and MMP-13 via the ERK signaling pathway. Mol Cancer Res (2011) 9:161–72. doi:10.1158/1541-7786.MCR-10-0386
149. Ghosh S, Basu M, Roy SS. ETS-1 protein regulates vascular endothelial growth factor-induced matrix metalloproteinase-9 and matrix metalloproteinase-13 expression in human ovarian carcinoma cell line SKOV-3. J Biol Chem (2012) 287:15001–15. doi:10.1074/jbc.M111.284034
150. Hoffman PC, Mauer AM, Vokes EE. Lung cancer. Lancet (2000) 355:479–85. doi:10.1016/S0140-6736(99)08003-4
Keywords: brain metastasis, lung cancer, molecular mechanisms, genetic alterations, chemotherapy
Citation: Pedrosa RMSM, Mustafa DAM, Aerts JGJV and Kros JM (2018) Potential Molecular Signatures Predictive of Lung Cancer Brain Metastasis. Front. Oncol. 8:159. doi: 10.3389/fonc.2018.00159
Received: 07 March 2018; Accepted: 25 April 2018;
Published: 11 May 2018
Edited by:
Anne-Marie Dingemans, Academic Hospital Maastricht, NetherlandsReviewed by:
K. Shilo, The Ohio State University, United StatesCopyright: © 2018 Pedrosa, Mustafa, Aerts and Kros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johan M. Kros, ai5tLmtyb3NAZXJhbXVzbWMubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.