 Daniele Lettieri-Barbato
Daniele Lettieri-Barbato Katia Aquilano
Katia Aquilano- Department of Biology, University of Rome Tor Vergata, Rome, Italy
The standard cancer treatments include chemotherapy, radiotherapy, or their combination, which are generally associated with a multitude of side effects ranging from discomfort to the development of secondary tumors and severe toxicity to multiple systems including immune system. Mounting evidence has highlighted that the fine-tuning of nutrients may selectively sensitize cancer cells to conventional cancer therapies, while simultaneously protecting normal cells from their side effects. Nutrient modulation through diet also improves cancer immunesurveillance in a way that severe immunosuppression could be avoided or even the immune response or immune-based cancer therapies be potentiated also through patient microbiota remodeling. Here, we review recent advances in cancer therapy focusing on the effects of adjuvant dietary interventions (e.g., ketogenic diets, fasting) on the metabolic pathways within cancer cells and tumor environment (e.g., microbiota, immune system, tumor microenvironment) that are involved in cancer progression and resistance as well as cancer cell death. Finally, based on the overall literature data, we designed a nutritional intervention consisting in a plant-based moderate ketogenic diet that could be exploited for future preclinical research in cancer therapy.
An Overview on the Control of Tumor Progression by Dietary Interventions
A plethora of epidemiological and experimental data demonstrated the efficacy of geroprotective dietary regimens (e.g., fasting, calorie, proteins, or single amino acids restrictions) in cancer prevention (1–3). Furthermore, such dietary patterns are emerging to be effective in selectively killing cancer cells, whereas increasing resistance of normal cells to the toxic effects of the anticancer therapeutics.
Calorie restriction (CR), defined as 30–60% less of daily calorie requirement without malnutrition, is known to extend healthy life span from yeast to mammals (4). The anticancer effects of CR are known since several years (5). CR is particularly effective in reducing the incidence, mass, and metastasis of breast cancer cells (6, 7). Remarkably, applying CR in combination with radiotherapy enhanced the radiotherapy efficacy inducing a more pronounced apoptosis of breast cancer cells than radiotherapy alone (7). In human, however, CR requires high compliance challenges to be maintained for adequate therapeutic period. For these reasons, short period of fasting without malnutrition have been proposed as potentially safe interventions to be associated with cancer treatments (8).
Fasting is commonly defined as a time-controlled deprivation of all kinds of foods and dietary nutrients. Differently to nocturnal fasting, time-controlled fasting leads to a profound metabolic reprogramming building up adaptive stress responses that are involved in life and health span extension (9–13). However, the adaptive stress responses induced by fasting occurring in normal cells differ from those activated by cancer cells because oncogenes might limit the activation of nutrient-sensing pathways while increasing chemotherapy vulnerability (8). Notably, proto-oncogenes such as IGF1R, PI3K, and AKT activate growth signaling and addict cancer cells to nutrient such as glucose and amino acids to meet their high proliferative rate (8). It has been shown that different cycles of fasting are effective in limiting tumor progression in several murine cancer models (14–17). However, the greatest effects were observed when fasting was combined with the conventional chemotherapy or radiotherapy (14–18). Interestingly, in these studies, fasting interventions alone do not cause clear signs of discomfort, but rather improve the animal condition. When fasting was combined with conventional therapies (e.g., temozolomide), most of the mice appeared healthy with the tumor-size below the controls, indicating that the combination of both treatments is well tolerated and improve tumor-bearing survival (14). The protective role of fasting against the side effects of anticancer therapy was confirmed in another study in which fasting was able to improve the overall cardiac response (maintenance of diastolic/systolic volumes and left ventricle wall thickness) to high-dose of doxorubicin (19). Fasting also exerted a significant protection against reduced mobility, ruffled hair, and hunched back posture caused by high dose of etoposide in mice (20). The anticancer effects of fasting might also rely on ketone bodies increase (21, 22). In support of this assumption, meta-analysis on ketogenic diets (KD), low in carbohydrates and high in fats, suggested a salutary impact on survival in animal models, with benefits prospectively linked to the magnitude of ketosis, time of diet initiation, and tumor location (23). Other evidence also demonstrated that KD might be safely used as adjuvant therapies to conventional radiation and chemotherapies (24). In particular, KD together with conventional radiotherapy led to increased radiation sensitivity in pancreatic cancer xenografts in mice (25). Similar results were obtained in mice bearing lung cancer xenografts (26). However, patients have demonstrated difficulty to comply with a KD while receiving concurrent radiation and chemotherapy in advanced lung and pancreatic cancer (25). Therefore, as better tolerated with respect to CR and KD, fasting appears to be more promising as adjuvant treatment in cancer therapy. Finally, it has been demonstrated that fasting could be replaced by the administration of CR mimetics, which showed the capability to improve the efficacy of chemotherapy as well. However, the objective response rates with metformin (27–30) or rapalogs (31) in clinical trials are still unclear and comparative analyses delineating a selective effectiveness of these drugs in cancer treatment and patient tolerability have to be more deeply elucidated.
Nutrient Modulation in Proliferating/Resilient Cancer Cells: A Molecular View
The reduced levels of nutrients and growth factors observed during fasting led to hypothesize their mandatory role in governing the differential stress responses in normal and cancer cells (10, 14, 16, 18). The different responses of normal and cancer cells to fasting shed light on their different sensitivity to nutrients and growth factors (18).
IGF-1/IGF-1R signaling is strongly dependent on nutrient availability and involves intensification of cancer cell proliferation, through the direct effects on PI3K/Akt signaling, and resistance to cell death imposed by chemotherapeutics and radiotherapy (Figure 1) (32). Indeed, fasting reduces circulating IGF-1 levels and this event protects mice deficient in the liver production of IGF-1 against chemotherapy drugs (16). Accordingly, restoration of IGF-1 was sufficient to reverse the protective effect of fasting (16). Reducing IGF-1 protects primary glia, but not glioma cells, against cyclophosphamide and mouse embryonic fibroblasts against doxorubicin (16). In the opposite manner, IGF-1 supplementation in starved breast cancer cells reversed drug sensitization. Overall, these findings strongly indicate that the fasting-mediated sensitization of cancer cells to chemotherapeutic drugs is conferred by the decrease of IGF-1 levels (15).
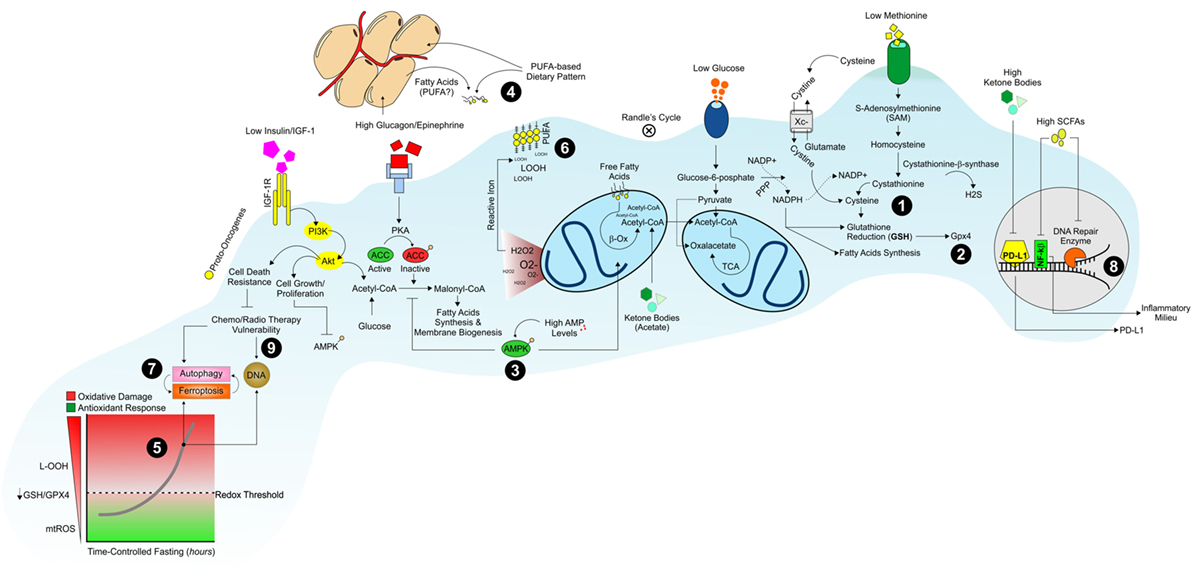
Figure 1. Dietary strategy to promote a hostile metabolism in proliferating/resilient cancer cells. Dietary patterns low in proteins, starch, and sugars promote an environment poor in sulfur amino acids (i.e., methionine, cysteine), glucose, and growth factors (insulin/IGF-1) that could limit NADPH/GSH production (1) and GPX4 activity (2). The diminished levels of glucose and glucagon/insulin ratio switch-off lipid synthesis and switch-on AMPK-driven lipid oxidation pathways in proliferating cancer cells (3). Under such metabolic conditions, cancer cells build-up their membranes by using extracellular dietary-derived and/or white adipose tissues-released fatty acids (4). The concomitant activation of OxPHOS metabolism and reduction in GSH levels are causative of oxidative stress (5) culminating in a massive lipid peroxidation (LOOH) (6) and ferroptosis in cancer cells (7). Diet low in starch, sugars, and proteins but rich in fatty acids also increases ketone bodies and modulates gut microbiota features by producing short-chain fatty acids (SCFAs). Ketone bodies and SCFAs affect PD-L1, nuclear factor-kb, and DNA repair enzymes genes transcription (8) promoting the chemo/radiotherapy vulnerability of cancer cells (9).
Nutrient shortage per se is able to increase mitochondrial reactive oxygen species (ROS) production in cancer cells arguing that limiting nutrient availability could enhance the effectiveness of redox-based cancer therapeutics (Figure 1) (33, 34). Actually, in breast cancer and melanoma cells, nutrient starvation was found to increase superoxide levels and aggravate oxidative stress caused by cyclophosphamide and cisplatin (15, 35). When applied in combination, fasting and chemotherapy act in synergy in elevating ROS levels and triggering DNA damage also in in vivo models of cancer (36). Micro-PET analyses in murine models of colon cancer cells revealed that fasting is effective as oxaliplatin (OXP) in reducing the average tumor glucose consumption and the lowest values were achieved by coupling fasting with OXP. In colon cancer cells, nutrient starvation upregulates oxidative phosphorylation with a significant production in mitochondrial superoxide caused by electron leakage. Consequently, starvation or OXP alone markedly increased ROS generation and their combination (starvation plus OXP) exacerbated ROS production in colon cancer cells (36). The hypothesis that cytotoxicity induced by glucose deprivation in cancer cells is mediated by mitochondrial superoxide and H2O2 was confirmed by exposing glucose-deprived transformed human fibroblasts to electron transport chain blockers (ETCBs), known to increase mitochondrial superoxide and H2O2 production (37). Glucose deprivation in the presence of ETCBs enhanced oxidative stress as well as cell death in several different human cancer cell lines (PC-3, DU145, MDA-MB231, and HT-29). In addition, human osteosarcoma cells lacking functional mitochondrial electron transport chain [rho(0)] were resistant to glucose deprivation-induced cytotoxicity and oxidative stress in the presence of antimycin A (complex III inhibitor), thus highlighting the role of mitochondrial ROS as mediators of cancer cell death (37).
The mechanisms by which KDs act as adjuvants in cancer therapy also seem to be associated with increased oxidative stress within cancer cells (24). Indeed, upon KD, the high level of circulating fatty acids limits the availability of glucose for glycolysis (Randle’s Cycle) (38). This reduces the formation of pyruvate and glucose-6-phosphate and in turn the synthesis of NADPH through the pentose phosphate pathway (PPP) (39). NADPH is necessary for buffering hydroperoxides (LOOH) production via the NADPH-dependent glutathione/glutathione peroxidase (GSH/GPX) system (40, 41). As consequence, an increase of LOOH is likely elicited (24) (Figure 1). Accordingly, hyperketotic diabetic patients have a higher level of lipid peroxidation in erythrocytes membrane and a significant decrease in cellular GSH levels than normal ketonic diabetic patients (42). Treatments with the ketone body acetoacetate elevated the levels of lipid peroxidation in human endothelial cells inhibiting their proliferation (42). This evidence suggests a direct role of ketone bodies in directly affecting GSH levels.
The main non-enzymatic cellular antioxidant GSH acts as an electron donor to reduce oxidized macromolecules, becoming itself oxidized in the process. Oxidized glutathione (GSSG) may then be restored in GSH through the action of the NADPH-dependent glutathione reductase (43). This enzymatic process generates NADP+, which may be reconverted to NADPH using electrons obtained from different biochemical pathways (44). Thus, proliferating cancer cells develop a peculiar metabolic flexibility to maintain a functional redox threshold by regulating NADPH levels through glycolytic flux modulation (33). Indeed, glucose-addicted human cancer cells cultured in a low-glucose medium without serum and amino acids are able to reprogram their metabolism by shifting toward PPP, which sustains the production of NADPH to dampen oxidative stress (33). However, during the initial stages of solid tumor development, when cells migrate to the lumen of lymphatic or blood vessels by loss of attachment (LOA) to the extracellular matrix, the glucose availability could not be sufficient to produce an adequate amount of NADPH and proliferation is inhibited (45). Upon such environmental stress, cancer cells induce adaptive responses consisting in the activation of AMPK signaling that inhibits fatty acid synthesis and triggers fatty acids oxidation to maintain energy production and NADPH levels (46, 47). Although cancer cells build up such adaptive responses, it has been observed that during LOA, cancer cells undergo ATP and NADPH drop and increase ROS production (48). Several papers demonstrated that cancer cells experiencing glucose shortage might maintain their proliferative capacity and membrane biogenesis by the uptake of extracellular lipids (49). Accordingly, extracellular saturated fatty acids supplementation supports the proliferative demand for biomass synthesis of proliferating cells (50, 51). Otherwise, supplementation with polyunsaturated fatty acids (PUFA) induced a significant cytotoxic effect on cancer cells either alone (52–54) or in combination with conventional anticancer therapies (55, 56). Differently to saturated fatty acids, PUFA are strongly susceptible to peroxidation (lipid peroxidation) in in vivo systems (57, 58). This appears to be a key mechanism triggering cancer cell death (59). With all this in mind, forcing the changes in the membrane lipids composition by dietary/nutrient enrichment in PUFA might promote an intrinsic sensitivity toward lipid peroxidation (57, 58, 60) and cancer cell death (Figure 1).
Nutrient-Mediated Commitment to Ferroptosis in Cancer Cells
By preserving NADPH levels, cancer cells sustain GPX/GSH activity during nutrient limitation, and this may confer resistance to redox-based chemotherapeutics (61–63). Indeed, many rebel cancer cells use a common trick to evade annihilation; they enter into what is known as a mesenchymal state that is “epithelial-to-mesenchymal” transition, which provides cancer cell resistance to conventional therapeutic regimens (64). It has been demonstrated that high therapy-resistant mesenchymal cancer cells strictly rely on the selenium-dependent GPX4 for survival (65). By using the reducing power of GSH, GPX4 converts potentially toxic L-OOH to non-toxic lipid alcohols (L-OH) (Figure 1) (66–68). Accordingly, inactivation of GPX4 through GSH depletion with erastin, or with a direct GPX4 inhibitor, ultimately results in lipid peroxidation in cancer cells (69). It is thus provocative to hypothesize that the evolutionary pressure to maintain the selenium protein GPX4 might correlate with an organism’s requirement for an increased PUFA content, which, in turn, renders complex biological activities possible (70).
Uncontrolled lipid peroxidation is causative of the onset of a metabolically regulated cell death called “ferroptosis,” which is characterized by the iron-dependent formation of LOOH leading to cell death (Figure 1) (71). Sulfur amino acids play a key role in ferroptosis. In particular, agents that inhibit cystine uptake via the cystine/glutamate antiporter (XC system), such as sulfasalazine or erastin, arrest tumor growth and induce ferroptosis (72, 73). The uptake of cystine is followed by its NADPH-dependent conversion in cysteine, the rate-limiting amino acid precursor for the GSH biosynthesis (74). Direct depletion of cystine from plasma using an engineered cystine-degrading enzyme conjugate arrests tumor growth and triggers cell death (75). Agents that conjugate to GSH, as well as chemical or genetic inhibition of GSH biosynthesis, disrupt tumor cell growth and induce a ferroptosis-like form of cell death (76). Ferroptosis appears to be an effective cell death mechanism in cancer cells, since lipophilic antioxidant α-tocopherol or iron chelators, such as deferoxamine, efficiently dampen it (77). Hence, the presence of extracellular cysteine and cystine are crucial for growth and proliferation of various types of cancer, as these amino acids maintain GSH levels and prevent oxidative stress (Figure 1) (78–80). Because cysteine is limiting in the biosynthesis of GSH, some cancer cells, under cysteine unavailability, make use of the transsulfuration pathway to biosynthesize cysteine from methionine (Met), a dietary essential sulfur amino acid (81, 82). The essentiality of Met in cancer is supported by the evidence that some cancer cells display a higher sensitivity to Met shortage with respect to normal cells (83–87). The first steps of the transsulfuration pathway are the conversion to S-adenosylmethionine (SAM) and transfer of the methyl group of SAM to a large variety of methyl acceptors with formation of S-adenosylhomocysteine (SAH) (88), which can be then converted to homocysteine (Hcy) by SAH hydrolase (AHCY) (89). Alternatively, Hcy is converted to cystathionine by cystathionine β-synthase (CBS). CBS catalyzes the condensation of Hcy and serine, thereby forming cystathionine, which is subsequently cleaved to cysteine. Furthermore, exogenous cysteine is also essential for several cancer types (glioma, prostate, and pancreatic), as blocking its uptake through the cystine/glutamate antiporter reduces viability due to the cell death caused by uncontrolled oxidative stress (90–92). Similarly, CBS blockage reduces cancer cell proliferation and attenuates growth of patient-derived colon cancer xenografts models (93). Although these findings suggest that fasting or selective nutrient modulation could trigger ferroptotic cell death in cancer cells, a clear evidence linking nutrient availability to ferroptosis is still lacking. Several works demonstrated that starved cancer cells (mainly in amino acids) as well as cells lacking the enzyme producing NADPH from glucose (glucose-6-phosphate dehydrogenase) experience massive ROS production and autophagy-dependent cell death (33, 94, 95). Autophagy is a process described as intracellular removal of damaged organelles by self-degradative process (96). Interestingly, a tight relationship between autophagic cell death and ferroptosis is emerging (97–99). Indeed, it seems that autophagy activation leads to a degradation of ferritin (ferritinophagy) (97), thus increasing the intracellular free iron levels promoting ROS production and ferroptosis (Figure 1) (99).
Dietary Strategies to Boost the Immunometabolic Responses in Cancer Therapy
Short-term fasting has a beneficial impact on cancer immunosurveillance (100). In particular, Pietrocola and co-workers demonstrated that fasting or CR-mimicking drugs, induce the depletion of regulatory T cells (which dampen anticancer immunity), thus igniting autophagic flux in murine models of KRAS-induced lung cancers. Accordingly, the inhibitory effect of fasting on tumor growth is lost in cancers that have been rendered autophagy deficient (100). Recently, also, isocaloric diet with protein restriction has been demonstrated to induce an IRE1α-dependent UPR in cancer cells, enhancing cytotoxic CD8+ T cell (a type of effector T lymphocyte)-mediated response against tumors (101).
Similarly to what observed with prolonged fasting (102), cycles of a fasting-mimicking diet (FMD) are effective in increasing hematopoietic cells proliferation and promoting immune system regeneration and modulation (103). Importantly, FMD has stimulatory effect on common lymphoid progenitor cells and CD8+ T cell-dependent cytotoxicity on breast cancer and melanoma cells (Figure 2) (17, 102). The presence of cytotoxic CD8+ T cells in the tumor environment [tumor infiltrating lymphocytes (TIL)] is considered a positive outcome of the cancer treatment (104, 105).
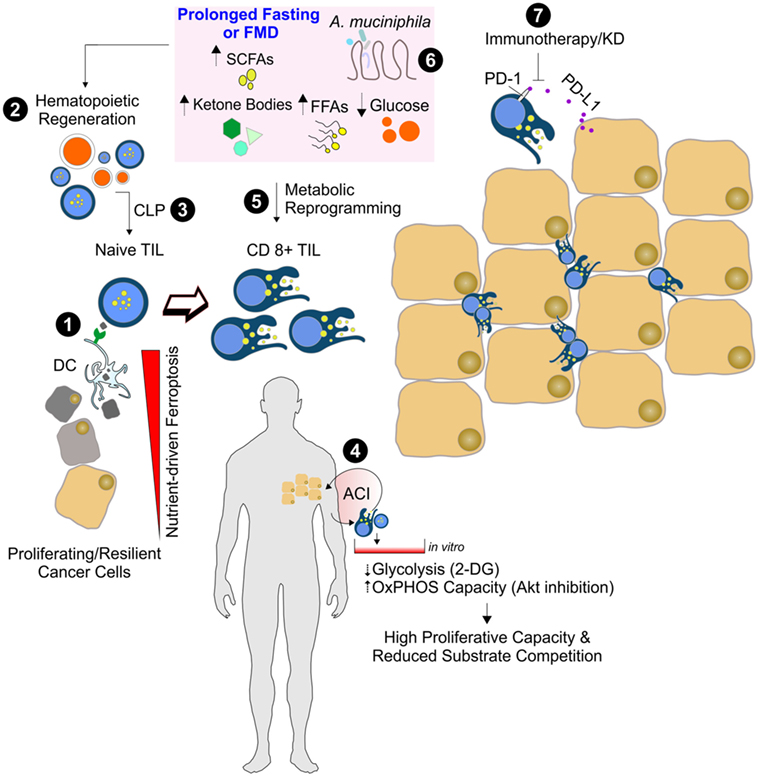
Figure 2. Nutrient manipulation to boost immunometabolic phenotype of CD8+ tumor infiltrating lymphocytes (TILs). Naïve CD8+ T cells recognize the antigen of ferroptotic cancer cells on class I MHC on dendritic cells, thus becoming mature cytotoxic CD8+ T cells (1). After prolonged fasting or fasting-mimicking diet (FMD), an enhanced hematopoietic regeneration rate (2) and enrichment of common lymphoid progenitor cells (CLP) can occur (3). The in vitro adoptive T cells immunotherapy (ACI) (4) and in vivo nutrient changes (5) reset CD8+ TIL metabolism toward mitochondrial oxidative pathways, thus limiting substrate competition with cancer cells and enhancing CD8+ TIL-mediated immunosurveillance. Dietary strategies promoting functional gut microbiota changes (e.g., Akkermansia muciniphila enrichment) (6) might improve the immune-checkpoint inhibitors (anti PD1/PD-L1) efficacy (7).
CD8+ T cells are influenced by nutrients and other supportive signals that are generally available in their environment. Generally, tumor cells inactivate CD8+ T cells. The suppression of oxidative phosphorylation and an upregulated glycolytic flux of proliferating cancer cells create an immunosuppressive microenvironment (106). Indeed, the glucose-dependent CD8+ TIL could undergo a competitive disadvantage for nutrients, and this would negatively affect their immune function. The immunosuppressive metabolic environment could be further enhanced by tumor expression of inhibitory ligands for programmed death 1 receptor (PD-1) which, when bound to their cognate receptors on T cells, limits T cell-intrinsic glucose uptake and glycolysis (107, 108). It has been reported that KD significantly reduces the expression of the inhibitory ligand PD-1 (PD-L1) on CD8+ TIL (109). Additionally, mice fed with KD have reduced expression of PD-L1 on the cancer cells that notoriously inhibits CD8+ T cells activity (109). This suggests that KD may alter tumor-mediated T cell suppression by reducing the number of cells that are susceptible to inhibition through the PD-1 inhibitory pathway (Figure 2).
Nowadays, there has been intense interest in developing adoptive T cells immunotherapy (ACI), which consists in reintroducing into a patient T cells that are previously activated and expanded in vitro (110, 111). The success of the ACI depends on the replicative capacity of implanted T cells. A large amount of research has been directed in optimizing T cell activation and using appropriate adjuvants for ACI. However, few experimental studies have been focused on manipulating metabolic pathways that could potentially enhance immunotherapy efficacy. When posed in culture, T cells dispose of a high glucose availability, which is far from the glucose physiological levels especially in the tumor environment (112, 113). Thus, once reintroduced in patients, T cells suffer from low glucose levels and show a moderate survival and replicative capacity. It has been reported that limiting glycolysis in cultured T cells can increase their longevity without inhibiting proliferative capacity (114, 115) (Figure 2). Another potential way to enhance the replicative capacity and longevity of ACI cells is promoting oxidative phosphorylation and mitochondrial biogenesis via the inhibition of glucose-related signaling pathway that ultimately leads to in vivo persistence and improved antitumor immunity (116). The metabolic reprogramming of infiltrating glycolytic lymphocytes toward a catabolic state reliant on fatty acid oxidation appears to assure the success of immunotherapy (113). In line with this assumption, it was recently demonstrated that the enhancement of lipid catabolism in CD8+ T cells increases the efficacy of immunotherapy within a tumor microenvironment low in glucose (117). In a mouse model of malignant glioma, an enhanced cytolysis via tumor-reactive CD8+ T cells was also achieved by ketogenic diet (109). The immunometabolic reprogramming necessary for CD8+ TIL could at least partially explain the mechanism by which KD or fasting enhances cytotoxic effect against cancer cells. Such diets are indeed powerful in inducing a cellular metabolic shift from glycolysis toward FAO.
It is now emerging that CD8+ TIL response to immune checkpoint blockade inhibitor PD1 can be also modulated by gut microbiota (118–120). A very recent paper has revealed that fecal microbiota from patients affected with metastatic melanoma and responsive to anti-PD1 therapy display increased abundance of Akkermansia muciniphila. A. muciniphila introduction into mice receiving human nonresponder fecal microbiota transplant improved antitumor immune CD8+ T cell infiltration and activity and increased anti-PD1 therapy efficacy (120, 121). Another intriguing observation is that Faecalibacterium and Bifidobacterium are associated with anti-inflammatory responses, a regulatory arm of the immune system that aims to prevent overactivation of the immune response and restores host homeostasis (120). Given that changes in host metabolism and microbiota can occur in tandem, it was hypothesized that gut microbial diversity and composition are predictors of the response to cancer therapy (121) (Figure 2). Accordingly, germ-free mice implanted with human tumor cells and transplanted with feces from chemotherapy responders showed an ameliorated response to chemotherapy than mice colonized with microbiota from nonresponder patients (119).
The diet has a strong capacity to rapidly and reproducibly reshape the gut microbiome (122). Indeed, fasting or plant-based diet remodels microbial community structure and overwhelms interindividual differences in microbial gene expression. The animal-based diets are known to increase the abundance of bile-tolerant microorganisms (Alistipes, Bilophila, and Bacteroides) and decrease the levels of the high fermentative Firmicutes that metabolize dietary plant polysaccharides (Roseburia, Eubacterium rectale, and Ruminococcus bromii) (122). More recently, it has been demonstrated that alternate day fasting shifts the gut microbiota composition from Bacteroides to Firmicutes leading to elevation of the fermentation products (123). Plant-based foods are mainly characterized by resistant starches and dietary fibers and promote their gut microbiota-mediated fermentation and decomposition. These processes provide additional amount of short chain fatty acids (SCFAs) to the host (124) (Figure 2). The major SCFAs, i.e., acetate, propionate, and butyrate, have different production ratios and physiological activities. Through 1H NMR-based metabolomics, it was revealed that mice treated with alternate day fasting increased acetate levels both in the cecum and sera (123). Acetate, when ligated to coenzyme A (acetyl-CoA), is among the most central and dynamic metabolites in intermediary metabolism. Under stressful circumstance (e.g., fasting-like conditions), cancer cells may convert extracellular acetate to acetyl-CoA, thus promoting the biogenesis of membrane building blocks that sustain the high proliferative rate. This adaptive response involves the cytosolic form of short-chain acyl-CoA synthetases (ACC2). Accordingly, increased ACC2 protein levels were detected in a subset of human triple negative breast cancer samples, and such an elevation correlates with poor survival (125). Differently to acetate, butyrate shows many regulatory properties including the inhibition of histone deacetylases. Histone deacetylase inhibitors (HDACi’s) are emerging as promising anticancer drugs when administered alone or in combination with chemotherapeutic agents or radiotherapy. Previous research suggests that HDACi’s have a high degree of selectivity for killing cancer cells. For instance, the HDACi sodium butyrate suppresses DNA double strand break repair induced by etoposide more efficiently in MCF-7 cells than in HEK293 cells (126). Sodium butyrate alone also resulted in accumulation of ROS, DNA double-strand breaks, and apoptosis in HCT-116 colon cancer cell lines; when combined with mitomycin C or radiotherapy, sodium butyrate increases sensitivity of cancer cells to the drug (127, 128). In animal models of gastric carcinoma, sodium butyrate was found to inhibit tumor mass formation and increase tumor infiltration by CD8+ TIL (129). Finally, several studies also demonstrated a strong effectiveness of SCFA to inactivate nuclear factor-kb by downregulating the production of the pro-inflammatory cytokine TNFα (130–134), which is commonly activated to promote a pro-carcinogenic environmental milieu (135) (Figure 1).
Conclusion and Perspective
Despite recent advances have been made in cancer therapy, the prognosis for many cancer patients remains poor, and current treatments still show severe adverse events. Thus, finding complementary treatments that have limited patient toxicity and simultaneously enhance therapy responses in cancer versus normal cells is urgent. Diet has a strong capacity to modulate cell responses to environmental stimuli and shows great potential in improving cancer prognosis. The mechanisms by which dietary nutrients enhance anticancer effects of standard anticancer therapies (chemotherapy, radiotherapy, immunotherapy) has not been fully elucidated yet. Preclinical studies have demonstrated the safety and efficacy of specific dietary interventions in counteracting tumor progression during anticancer therapy in murine models. However, most of the data present in the literature take advantage of the use of mice and this may limit the translation to clinical research. Therefore, a huge amount of work is now necessary to confirm these very promising results in humans.
Deprivation of nutrients (e.g., glucose, sulfur amino acids) as well as of nutrient-responsive growth factors (e.g., IGF-1) seems to selectively kill high proliferative/resilient cancer cells by forcing their glycolytic asset toward an oxidative metabolism (i.e., fatty acids and ketone bodies as energy sources) and limiting GPX activity as consequence of reduced GSH levels. Nutrient scarcity also improves immunometabolism enhancing cytotoxic efficiency of CD8+ TIL within the tumor mass through, probably, the concomitant gut microbiota and immunometabolic rearrangements (Figure 3).
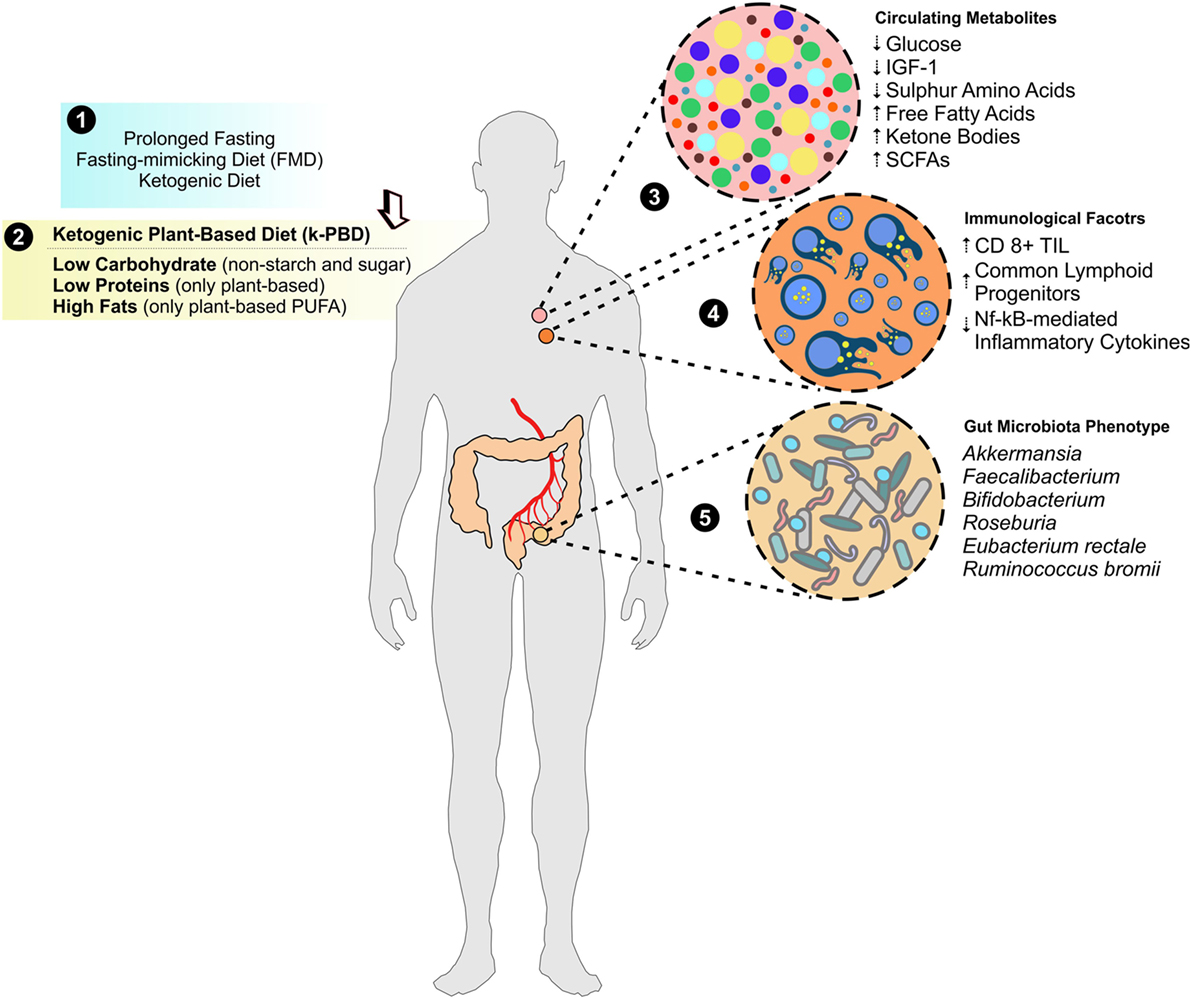
Figure 3. Improving metabolites and immunological anticancer profile by k-PBD. Evidence from prolonged fasting, fasting-mimicking diet (FMD), and ketogenic diet demonstrated a strong usefulness as adjuvants in cancer therapy (1). In this issue, we propose a moderate ketogenic plant-based diet (k-PBD), low in carbohydrates (starch and sugars in particular) and animal proteins (poor in sulfur amino acids and selenium) but rich in fats [mainly in vegetable polyunsaturated fatty acids (PUFA)] (2), which could strongly modulate circulating metabolites (3), immunological factors (4), and gut microbiota asset (5) that overall create a hostile environment to cancer cells.
Herein, we propose weekly cycles of 4 days of a plant-based moderate ketogenic diet (k-PBD) that could reprogram systemic metabolism conferring a hostile environment to cancer cells (Figure 3). In particular, k-PBD should be low in proteins (mainly vegetable proteins low in sulfur amino acids and selenium), carbohydrates (non-starchy vegetables), and high in lipids (mainly unprocessed vegetable oils rich in PUFA). Remarkably, even though not supported by experimental data, it is highly expected that this diet could be able to increase ketonemia as it contains high amounts of fats concomitantly to reduced calories. This diet could increase the efficiency of CD8+ TIL, by reprogramming their metabolism (fat-dependent metabolism) to better counteract the metabolic features of proliferating cancer cells (glucose-dependent metabolism) and sensitize cancer cells to the therapy. The k-PBD could be consumed prior to conventional cancer therapies (e.g., prior each cycle of chemotherapy or prior a single fraction of radiation therapy). With this composition and time of treatment, k-PBD could be effective in: (i) changing the membrane chemistry by PUFA enrichment (high peroxidation index); (ii) reducing the sulfur-dependent antioxidant power (lowering NADPH, GSH, GPX4); (iii) forcing the metabolic shift toward mitochondrial metabolism in cancer cells. Furthermore, the high fermentative fibers of k-PBD could induce a functional microbiota reshaping improving immunotherapy efficacy (e.g., anti-PD1 therapy) (Figure 4).
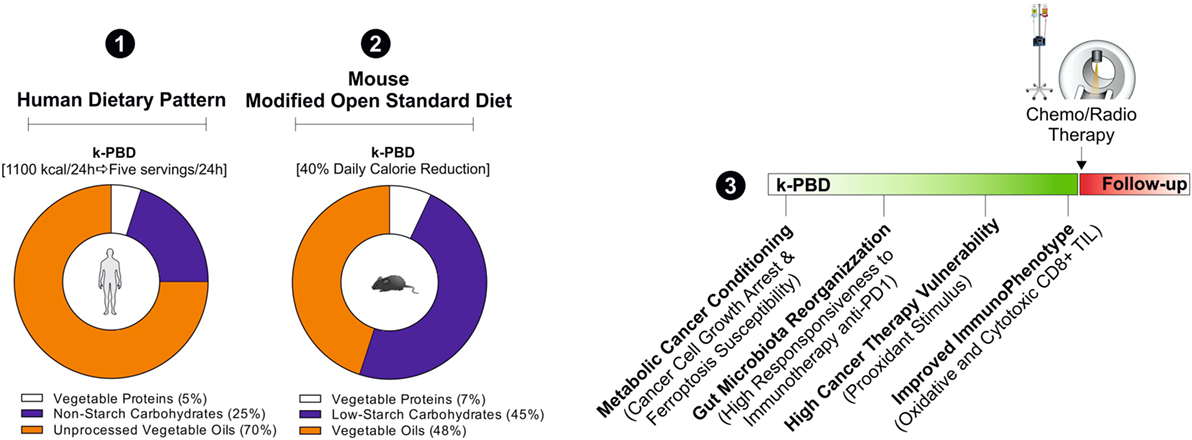
Figure 4. Hypothetical cancer phenotypic responses to k-PBD. For the human dietary intervention studies, k-PBD (for 4 consecutive days; 5 servings each day) should provide about 1,100 kcal/die with 5% of calories from vegetable proteins, 25% from carbohydrates (non-starchy vegetables), and 70% from unprocessed vegetable oils (1). k-PBD proposed for murine cancer models should be reduced in total calories (40% reduction versus ad libitum diet), provide 7% calories from vegetable proteins, 45% from low starch carbohydrates, 48% in vegetable oils (2). The proposed dietary pattern should be started prior to conventional cancer therapies (3).
Author Contributions
DL-B conceptualized and wrote the manuscript. KA performed critical revision of the manuscript for intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Berrigan D, Perkins SN, Haines DC, Hursting SD. Adult-onset calorie restriction and fasting delay spontaneous tumorigenesis in p53-deficient mice. Carcinogenesis (2002) 23:817–22. doi:10.1093/carcin/23.5.817
2. Levine ME, Suarez JA, Brandhorst S, Balasubramanian P, Cheng CW, Madia F, et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab (2014) 19:407–17. doi:10.1016/j.cmet.2014.02.006
3. Cavuoto P, Fenech MF. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat Rev (2012) 38:726–36. doi:10.1016/j.ctrv.2012.01.004
4. Fontana L, Partridge L, Longo VD. Extending healthy life span – from yeast to humans. Science (2010) 328:321–6. doi:10.1126/science.1172539
5. Kritchevsky D. Caloric restriction and cancer. J Nutr Sci Vitaminol (Tokyo) (2001) 47:13–9. doi:10.3177/jnsv.47.13
6. Nogueira LM, Dunlap SM, Ford NA, Hursting SD. Calorie restriction and rapamycin inhibit MMTV-Wnt-1 mammary tumor growth in a mouse model of postmenopausal obesity. Endocr Relat Cancer (2012) 19:57–68. doi:10.1530/ERC-11-0213
7. Saleh AD, Simone BA, Palazzo J, Savage JE, Sano Y, Dan T, et al. Caloric restriction augments radiation efficacy in breast cancer. Cell Cycle (2013) 12:1955–63. doi:10.4161/cc.25016
8. Brandhorst S, Longo VD. Fasting and caloric restriction in cancer prevention and treatment. Recent Results Cancer Res (2016) 207:241–66. doi:10.1007/978-3-319-42118-6_12
9. Lettieri-Barbato D, Giovannetti E, Aquilano K. Effects of dietary restriction on adipose mass and biomarkers of healthy aging in human. Aging (Albany NY) (2016) 8:3341–55. doi:10.18632/aging.101122
10. Lettieri Barbato D, Aquilano K. Feast and famine: adipose tissue adaptations for healthy aging. Ageing Res Rev (2016) 28:85–93. doi:10.1016/j.arr.2016.05.007
11. Lettieri Barbato D, Tatulli G, Aquilano K, Ciriolo MR, et al. Mitochondrial hormesis links nutrient restriction to improved metabolism in fat cell. Aging (Albany NY) (2015) 7:869–81. doi:10.18632/aging.100832
12. Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab (2012) 15:451–65. doi:10.1016/j.cmet.2012.02.013
13. Longo VD. Linking sirtuins, IGF-I signaling, and starvation. Exp Gerontol (2009) 44:70–4. doi:10.1016/j.exger.2008.06.005
14. Safdie F, Brandhorst S, Wei M, Wang W, Lee C, Hwang S, et al. Fasting enhances the response of glioma to chemo- and radiotherapy. PLoS One (2012) 7:e44603. doi:10.1371/journal.pone.0044603
15. Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med (2012) 4:124ra127. doi:10.1126/scitranslmed.3003293
16. Lee C, Safdie FM, Raffaghello L, Wei M, Madia F, Parrella E, et al. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res (2010) 70:1564–72. doi:10.1158/0008-5472.CAN-09-3228
17. Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell (2016) 30:136–46. doi:10.1016/j.ccell.2016.06.005
18. Di Biase S, Longo VD. Fasting-induced differential stress sensitization in cancer treatment. Mol Cell Oncol (2016) 3:e1117701. doi:10.1080/23723556.2015.1117701
19. Di Biase S, Shim HS, Kim KH, Vinciguerra M, Rappa F, Wei M, et al. Fasting regulates EGR1 and protects from glucose- and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol (2017) 15:e2001951. doi:10.1371/journal.pbio.2001951
20. Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A (2008) 105:8215–20. doi:10.1073/pnas.0708100105
21. Longo VD, Panda S. Fasting, circadian rhythms, and time-restricted feeding in healthy lifespan. Cell Metab (2016) 23:1048–59. doi:10.1016/j.cmet.2016.06.001
22. Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab (2014) 19:181–92. doi:10.1016/j.cmet.2013.12.008
23. Klement RJ, Champ CE, Otto C, Kämmerer U. Anti-tumor effects of ketogenic diets in mice: a meta-analysis. PLoS One (2016) 11:e0155050. doi:10.1371/journal.pone.0155050
24. Allen BG, Bhatia SK, Anderson CM, Eichenberger-Gilmore JM, Sibenaller ZA, Mapuskar KA, et al. Ketogenic diets as an adjuvant cancer therapy: history and potential mechanism. Redox Biol (2014) 2:963–70. doi:10.1016/j.redox.2014.08.002
25. Zahra A, Fath MA, Opat E, Mapuskar KA, Bhatia SK, Ma DC, et al. Consuming a ketogenic diet while receiving radiation and chemotherapy for locally advanced lung cancer and pancreatic cancer: the university of iowa experience of two phase 1 clinical trials. Radiat Res (2017) 187:743–54. doi:10.1667/RR14668.1
26. Allen BG, Bhatia SK, Buatti JM, Brandt KE, Lindholm KE, Button AM, et al. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin Cancer Res (2013) 19:3905–13. doi:10.1158/1078-0432.CCR-12-0287
27. Zhou PT, Li B, Liu FR, Zhang MC, Wang Q, Li YY, et al. Metformin is associated with survival benefit in pancreatic cancer patients with diabetes: a systematic review and meta-analysis. Oncotarget (2017) 8:25242–50. doi:10.18632/oncotarget.15692
28. Jung YS, Park CH, Eun CS, Park DI, Han DS. Metformin use and the risk of colorectal adenoma: a systematic review and meta-analysis. J Gastroenterol Hepatol (2017) 32:957–65. doi:10.1111/jgh.13639
29. Pernicova I, Korbonits M. Metformin – mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol (2014) 10:143–56. doi:10.1038/nrendo.2013.256
30. Samsuri NAB, Leech M, Marignol L. Metformin and improved treatment outcomes in radiation therapy – a review. Cancer Treat Rev (2017) 55:150–62. doi:10.1016/j.ctrv.2017.03.005
31. Pavel ME, Singh S, Strosberg JR, Bubuteishvili-Pacaud L, Degtyarev E, Neary MP, et al. Health-related quality of life for everolimus versus placebo in patients with advanced, non-functional, well-differentiated gastrointestinal or lung neuroendocrine tumours (RADIANT-4): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2017) 18:1411–22. doi:10.1016/S1470-2045(17)30471-0
32. Kasprzak A, Kwasniewski W, Adamek A, Gozdzicka-Jozefiak A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat Res Rev Mutat Res (2017) 772:78–104. doi:10.1016/j.mrrev.2016.08.007
33. Desideri E, Vegliante R, Cardaci S, Nepravishta R, Paci M, Ciriolo MR, et al. MAPK14/p38alpha-dependent modulation of glucose metabolism affects ROS levels and autophagy during starvation. Autophagy (2014) 10:1652–65. doi:10.4161/auto.29456
34. Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol (2015) 4:184–92. doi:10.1016/j.redox.2014.12.003
35. Antunes F, Pereira GJ, Jasiulionis MG, Bincoletto C, Smaili SS. Nutritional shortage augments cisplatin-effects on murine melanoma cells. Chem Biol Interact (2018) 281:89–97. doi:10.1016/j.cbi.2017.12.027
36. Bianchi G, Martella R, Ravera S, Marini C, Capitanio S, Orengo A, et al. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget (2015) 6:11806–19. doi:10.18632/oncotarget.3688
37. Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, Buettner GR, et al. Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem (2005) 280:4254–63. doi:10.1074/jbc.M411662200
38. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab (2009) 297:E578–91. doi:10.1152/ajpendo.00093.2009
39. Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life (2012) 64:362–9. doi:10.1002/iub.1017
40. Moreno-Sánchez R, Gallardo-Pérez JC, Rodríguez-Enríquez S, Saavedra E, Marín-Hernández Á. Control of the NADPH supply for oxidative stress handling in cancer cells. Free Radic Biol Med (2017) 112:149–61. doi:10.1016/j.freeradbiomed.2017.07.018
41. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med (2003) 34:145–69. doi:10.1016/S0891-5849(02)01197-8
42. Jain SK, Kannan K, Lim G. Ketosis (acetoacetate) can generate oxygen radicals and cause increased lipid peroxidation and growth inhibition in human endothelial cells. Free Radic Biol Med (1998) 25:1083–8. doi:10.1016/S0891-5849(98)00140-3
43. Aquilano K, Baldelli S, Ciriolo MR. Glutathione: new roles in redox signaling for an old antioxidant. Front Pharmacol (2014) 5:196. doi:10.3389/fphar.2014.00196
44. Ciccarese F, Ciminale V. Escaping death: mitochondrial redox homeostasis in cancer cells. Front Oncol (2017) 7:117. doi:10.3389/fonc.2017.00117
45. Qu Q, Zeng F, Liu X, et al. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis (2016) 7:e2226. doi:10.1038/cddis.2016.132
46. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39:347–54. doi:10.1016/j.tibs.2014.06.005
47. Lettieri Barbato D, Vegliante R, Desideri E, Ciriolo MR. Managing lipid metabolism in proliferating cells: new perspective for metformin usage in cancer therapy. Biochim Biophys Acta (2014) 1845:317–24. doi:10.1016/j.bbcan.2014.02.003
48. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer (2013) 13:227–32. doi:10.1038/nrc3483
49. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer (2016) 16:732–49. doi:10.1038/nrc.2016.89
50. Yao CH, Fowle-Grider R, Mahieu NG, Liu GY, Chen YJ, Wang R, et al. Exogenous fatty acids are the preferred source of membrane lipids in proliferating fibroblasts. Cell Chem Biol (2016) 23:483–93. doi:10.1016/j.chembiol.2016.03.007
51. Egnatchik RA, DeBerardinis RJ. Liposuction: extracellular fat removal promotes proliferation. Cell Chem Biol (2016) 23:431–2. doi:10.1016/j.chembiol.2016.04.003
52. Berquin IM, Edwards IJ, Chen YQ. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett (2008) 269:363–77. doi:10.1016/j.canlet.2008.03.044
53. Serini S, Piccioni E, Merendino N, Calviello G. Dietary polyunsaturated fatty acids as inducers of apoptosis: implications for cancer. Apoptosis (2009) 14:135–52. doi:10.1007/s10495-008-0298-2
54. Vaughan VC, Hassing MR, Lewandowski PA. Marine polyunsaturated fatty acids and cancer therapy. Br J Cancer (2013) 108:486–92. doi:10.1038/bjc.2012.586
55. Biondo PD, Brindley DN, Sawyer MB, Field CJ. The potential for treatment with dietary long-chain polyunsaturated n-3 fatty acids during chemotherapy. J Nutr Biochem (2008) 19:787–96. doi:10.1016/j.jnutbio.2008.02.003
56. D’Eliseo D, Velotti F. Omega-3 fatty acids and cancer cell cytotoxicity: implications for multi-targeted cancer therapy. J Clin Med (2016) 5:ii:E15. doi:10.3390/jcm5020015
57. Jenkinson A, Franklin MF, Wahle K, Duthie GG. Dietary intakes of polyunsaturated fatty acids and indices of oxidative stress in human volunteers. Eur J Clin Nutr (1999) 53:523–8. doi:10.1038/sj.ejcn.1600783
58. Diniz YS, Cicogna AC, Padovani CR, Santana LS, Faine LA, Novelli EL. Diets rich in saturated and polyunsaturated fatty acids: metabolic shifting and cardiac health. Nutrition (2004) 20:230–4. doi:10.1016/j.nut.2003.10.012
59. Agmon E, Stockwell BR. Lipid homeostasis and regulated cell death. Curr Opin Chem Biol (2017) 39:83–9. doi:10.1016/j.cbpa.2017.06.002
60. Barelli H, Antonny B. Lipid unsaturation and organelle dynamics. Curr Opin Cell Biol (2016) 41:25–32. doi:10.1016/j.ceb.2016.03.012
61. Ferretti A, Chen LL, Di Vito M, Barca S, Tombesi M, Cianfriglia M, et al. Pentose phosphate pathway alterations in multi-drug resistant leukemic T-cells: 31P NMR and enzymatic studies. Anticancer Res (1993) 13:867–72.
62. Friesen C, Kiess Y, Debatin KM. A critical role of glutathione in determining apoptosis sensitivity and resistance in leukemia cells. Cell Death Differ (2004) 11(Suppl 1):S73–85. doi:10.1038/sj.cdd.4401431
63. Gessner T, Vaughan LA, Beehler BC, Bartels CJ, Baker RM. Elevated pentose cycle and glucuronyltransferase in daunorubicin-resistant P388 cells. Cancer Res (1990) 50:3921–7.
64. Chen JJ, Galluzzi L. Fighting resilient cancers with iron. Trends Cell Biol (2018) 28:77–8. doi:10.1016/j.tcb.2017.11.007
65. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547:453–7. doi:10.1038/nature23007
66. Burk RF. Protection against free radical injury by selenoenzymes. Pharmacol Ther (1990) 45:383–5. doi:10.1016/0163-7258(90)90073-B
67. Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res (1998) 39:1529–42.
68. Thomas JP, Maiorino M, Ursini F, Girotti AW. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J Biol Chem (1990) 265:454–61.
69. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi:10.1016/j.cell.2012.03.042
70. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell (2018) 172:409–422.e21. doi:10.1016/j.cell.2017.11.048
71. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171:273–85. doi:10.1016/j.cell.2017.09.021
72. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife (2014) 3:e02523. doi:10.7554/eLife.02523
73. Toyokuni S, Ito F, Yamashita K, Okazaki Y, Akatsuka S. Iron and thiol redox signaling in cancer: an exquisite balance to escape ferroptosis. Free Radic Biol Med (2017) 108:610–26. doi:10.1016/j.freeradbiomed.2017.04.024
74. Hellmich MR, Coletta C, Chao C, Szabo C, et al. The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer. Antioxid Redox Signal (2015) 22:424–48. doi:10.1089/ars.2014.5933
75. Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med (2017) 23:120–7. doi:10.1038/nm.4232
76. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
77. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Mazière JC, Chauffert B, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer (2013) 133:1732–42. doi:10.1002/ijc.28159
78. Desideri E, Filomeni G, Ciriolo MR. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy (2012) 8:1769–81. doi:10.4161/auto.22037
79. Giustarini D, Dalle-Donne I, Milzani A, Rossi R. Oxidative stress induces a reversible flux of cysteine from tissues to blood in vivo in the rat. FEBS J (2009) 276:4946–58. doi:10.1111/j.1742-4658.2009.07197.x
80. Yu X, Long YC. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci Rep (2016) 6:30033. doi:10.1038/srep30033
81. Stipanuk MH. Metabolism of sulfur-containing amino acids. Annu Rev Nutr (1986) 6:179–209. doi:10.1146/annurev.nu.06.070186.001143
82. Finkelstein JD, Martin JJ, Harris BJ. Methionine metabolism in mammals. The methionine-sparing effect of cystine. J Biol Chem (1988) 263:11750–4.
83. Lien EC, Ghisolfi L, Geck RC, Asara JM, Toker A. Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci Signal (2017) 10:eaao6604. doi:10.1126/scisignal.aao6604
84. Strekalova E, Malin D, Good DM, Cryns VL. Methionine deprivation induces a targetable vulnerability in triple-negative breast cancer cells by enhancing TRAIL receptor-2 expression. Clin Cancer Res (2015) 21:2780–91. doi:10.1158/1078-0432.CCR-14-2792
85. Hoffman RM. Development of recombinant methioninase to target the general cancer-specific metabolic defect of methionine dependence: a 40-year odyssey. Expert Opin Biol Ther (2015) 15:21–31. doi:10.1517/14712598.2015.963050
86. Hoffman RM. Methionine dependence in cancer cells – a review. In Vitro (1982) 18:421–8. doi:10.1007/BF02796353
87. Cao WX, Ou JM, Fei XF, Zhu ZG, Yin HR, Yan M, et al. Methionine-dependence and combination chemotherapy on human gastric cancer cells in vitro. World J Gastroenterol (2002) 8:230–2. doi:10.3748/wjg.v8.i2.230
88. Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem (1990) 1:228–37. doi:10.1016/0955-2863(90)90070-2
89. Finkelstein JD, Kyle WE, Harris BJ. Methionine metabolism in mammals: regulatory effects of S-adenosylhomocysteine. Arch Biochem Biophys (1974) 165:774–9. doi:10.1016/0003-9861(74)90306-3
90. Lo M, Ling V, Wang YZ, Gout PW. The xc- cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer (2008) 99:464–72. doi:10.1038/sj.bjc.6604485
91. Doxsee DW, Gout PW, Kurita T, Lo M, Buckley AR, Wang Y, et al. Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy. Prostate (2007) 67:162–71. doi:10.1002/pros.20508
93. Szabo C, Coletta C, Chao C, Módis K, Szczesny B, Papapetropoulos A, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci U S A (2013) 110:12474–9. doi:10.1073/pnas.1306241110
94. Cardaci S, Rizza S, Filomeni G, Bernardini R, Bertocchi F, Mattei M, et al. Glutamine deprivation enhances antitumor activity of 3-bromopyruvate through the stabilization of monocarboxylate transporter-1. Cancer Res (2012) 72:4526–36. doi:10.1158/0008-5472.CAN-12-1741
95. Vegliante R, Desideri E, Di Leo L, Ciriolo MR. Dehydroepiandrosterone triggers autophagic cell death in human hepatoma cell line HepG2 via JNK-mediated p62/SQSTM1 expression. Carcinogenesis (2016) 37:233–44. doi:10.1093/carcin/bgw003
96. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy (2016) 12:1–222. doi:10.1080/15548627.2015.1100356
97. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res (2016) 26:1021–32. doi:10.1038/cr.2016.95
98. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ III, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy (2016) 12:1425–8. doi:10.1080/15548627.2016.1187366
99. Ma S, Dielschneider RF, Henson ES, Xiao W, Choquette TR, Blankstein AR, et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One (2017) 12:e0182921. doi:10.1371/journal.pone.0182921
100. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell (2016) 30:147–60. doi:10.1016/j.ccell.2016.05.016
101. Rubio-Patino C, Bossowski JP, De Donatis GM, Mondragón L, Villa E, Aira LE, et al. Low-protein diet induces IRE1alpha-dependent anticancer immunosurveillance. Cell Metab (2018) 27(4):828–42.e7. doi:10.1016/j.cmet.2018.02.009
102. Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell (2014) 14:810–23. doi:10.1016/j.stem.2014.04.014
103. Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab (2015) 22:86–99. doi:10.1016/j.cmet.2015.05.012
104. Andre F, Dieci MV, Dubsky P, Sotiriou C, Curigliano G, Denkert C, et al. Molecular pathways: involvement of immune pathways in the therapeutic response and outcome in breast cancer. Clin Cancer Res (2013) 19:28–33. doi:10.1158/1078-0432.CCR-11-2701
105. Issa-Nummer Y, Darb-Esfahani S, Loibl S, Kunz G, Nekljudova V, Schrader I, et al. Prospective validation of immunological infiltrate for prediction of response to neoadjuvant chemotherapy in HER2-negative breast cancer – a substudy of the Neoadjuvant GeparQuinto Trial. PLoS One (2013) 8:e79775. doi:10.1371/journal.pone.0079775
106. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38:633–43. doi:10.1016/j.immuni.2013.04.005
107. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. doi:10.1182/blood-2008-12-195792
108. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
109. Lussier DM, Woolf EC, Johnson JL, Brooks KS, Blattman JN, Scheck AC. Enhanced immunity in a mouse model of malignant glioma is mediated by a therapeutic ketogenic diet. BMC Cancer (2016) 16:310. doi:10.1186/s12885-016-2337-7
110. Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity (2013) 39:49–60. doi:10.1016/j.immuni.2013.07.002
111. Bonini C, Mondino A. Adoptive T-cell therapy for cancer: the era of engineered T cells. Eur J Immunol (2015) 45:2457–69. doi:10.1002/eji.201545552
112. Marelli-Berg FM, Fu H, Mauro C. Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. Immunology (2012) 136:363–9. doi:10.1111/j.1365-2567.2012.03583.x
113. Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol (2016) 17:364–8. doi:10.1038/ni.3415
114. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342:1242454. doi:10.1126/science.1242454
115. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123:4479–88. doi:10.1172/JCI69589
116. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res (2015) 75:296–305. doi:10.1158/0008-5472.CAN-14-2277
117. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32:377–391.e9. doi:10.1016/j.ccell.2017.08.004
118. Vetizou M, Trinchieri G. Anti-PD1 in the wonder-gut-land. Cell Res (2018) 28(3):263–4. doi:10.1038/cr.2018.12
119. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (2018) 359:97–103. doi:10.1126/science.aan4236
120. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science (2018) 359:104–8. doi:10.1126/science.aao3290
121. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (2018) 359:91–7. doi:10.1126/science.aan3706
122. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature (2014) 505:559–63. doi:10.1038/nature12820
123. Li GL, Xie C, Lu SY, Nichols RG, Tian Y, Li L, et al. Intermittent fasting promotes white adipose browning and decreases obesity by shaping the gut microbiota. Cell Metab (2017) 26:672.e–85.e. doi:10.1016/j.cmet.2017.10.007
124. Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes (2016) 7:189–200. doi:10.1080/19490976.2015.1134082
125. Lyssiotis CA, Cantley LC. Acetate fuels the cancer engine. Cell (2014) 159:1492–4. doi:10.1016/j.cell.2014.12.009
126. Li L, Sun Y, Liu J, Wu X, Chen L, Ma L, et al. Histone deacetylase inhibitor sodium butyrate suppresses DNA double strand break repair induced by etoposide more effectively in MCF-7 cells than in HEK293 cells. BMC Biochem (2015) 16:2. doi:10.1186/s12858-014-0030-5
127. Gospodinov A, Popova S, Vassileva I, Anachkova B. The inhibitor of histone deacetylases sodium butyrate enhances the cytotoxicity of mitomycin C. Mol Cancer Ther (2012) 11:2116–26. doi:10.1158/1535-7163.MCT-12-0193
128. Koprinarova M, Botev P, Russev G. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair (Amst) (2011) 10:970–7. doi:10.1016/j.dnarep.2011.07.003
129. Shan YS, Fang JH, Lai MD, Yen MC, Lin PW, Hsu HP, et al. Establishment of an orthotopic transplantable gastric cancer animal model for studying the immunological effects of new cancer therapeutic modules. Mol Carcinog (2011) 50:739–50. doi:10.1002/mc.20668
130. Usami M, Kishimoto K, Ohata A, Miyoshi M, Aoyama M, Fueda Y, et al. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr Res (2008) 28:321–8. doi:10.1016/j.nutres.2008.02.012
131. Kendrick SF, O’Boyle G, Mann J, Zeybel M, Palmer J, Jones DE, et al. Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology (2010) 51:1988–97. doi:10.1002/hep.23572
132. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A (2014) 111:2247–52. doi:10.1073/pnas.1322269111
133. Singh N, Thangaraju M, Prasad PD, Martin PM, Lambert NA, Boettger T, et al. Blockade of dendritic cell development by bacterial fermentation products butyrate and propionate through a transporter (Slc5a8)-dependent inhibition of histone deacetylases. J Biol Chem (2010) 285:27601–8. doi:10.1074/jbc.M110.102947
134. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med (2014) 20:159–66. doi:10.1038/nm.3444
Keywords: mitochondria, diet, fasting, immunemodulation, microbiota and immunity
Citation: Lettieri-Barbato D and Aquilano K (2018) Pushing the Limits of Cancer Therapy: The Nutrient Game. Front. Oncol. 8:148. doi: 10.3389/fonc.2018.00148
Received: 12 February 2018; Accepted: 23 April 2018;
Published: 08 May 2018
Edited by:
Salvatore Rizza, Danish Cancer Society, DenmarkReviewed by:
Colin E. Champ, University of Pittsburgh Cancer Institute, United StatesEnrico Desideri, Universität Wien, Austria
Copyright: © 2018 Lettieri-Barbato and Aquilano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Lettieri-Barbato, ZC5sZXR0aWVyaWJhcmJhdG9AaG90bWFpbC5pdA==