 Amila Suraweera
Amila Suraweera Kenneth J. O’Byrne
Kenneth J. O’Byrne Derek J. Richard
Derek J. Richard- 1School of Biomedical Research, Institute of Health and Biomedical Innovation at the Translational Research Institute, Queensland University of Technology, Brisbane, QLD, Australia
- 2Princess Alexandra Hospital, Brisbane, QLD, Australia
Genetic and epigenetic changes in DNA are involved in cancer development and tumor progression. Histone deacetylases (HDACs) are key regulators of gene expression that act as transcriptional repressors by removing acetyl groups from histones. HDACs are dysregulated in many cancers, making them a therapeutic target for the treatment of cancer. Histone deacetylase inhibitors (HDACi), a novel class of small-molecular therapeutics, are now approved by the Food and Drug Administration as anticancer agents. While they have shown great promise, resistance to HDACi is often observed and furthermore, HDACi have shown limited success in treating solid tumors. The combination of HDACi with standard chemotherapeutic drugs has demonstrated promising anticancer effects in both preclinical and clinical studies. In this review, we summarize the research thus far on HDACi in combination therapy, with other anticancer agents and their translation into preclinical and clinical studies. We additionally highlight the side effects associated with HDACi in cancer therapy and discuss potential biomarkers to either select or predict a patient’s response to these agents, in order to limit the off-target toxicity associated with HDACi.
Introduction
Genetic and genomic alterations as well as epigenetic modifications of DNA are involved in cancer development and tumor progression. Epigenetic changes modify chromatin structure and the accessibility of DNA, thus regulating patterns of gene expression. Epigenetic mechanisms include covalent histone modifications, DNA methylation, non-covalent mechanisms including the incorporation of histone variants and nucleosome remodeling, and non-coding RNAs. The N-terminal tail of histones can be modified posttranslationally by acetylation, methylation, ubiquitination, phosphorylation, sumoylation, ADP ribosylation, deamination, and proline isomerization (1–3).
Anticancer agents currently used in the clinic, including cytotoxic chemotherapy, targeted therapies, and immunotherapy have played a tremendous role in improving patient survival, symptom control, and quality of life. However, most cytotoxic drugs have a narrow therapeutic index. Cancers eventually develop drug resistance to the majority of systemic therapies. Furthermore, anticancer agents target normal cells in addition to the cancer cells, resulting in toxicities either due to direct cell damage or, in the case of immunotherapy, to inappropriate activation of autoimmune illnesses. Indeed, as cytotoxic drugs target naturally regenerating tissues, mainly the bone marrow and gastrointestinal tract, the formation of secondary hematologic and solid tumors may be seen many years following treatment (4–7). Combining anticancer drugs with other chemotherapeutic agents is often used to maximize efficacy, while reducing toxicity and resistance by administering lower drug doses, and their combination has shown synergistic or additive antitumor effects (8–11). This review focuses on research thus far on histone deacetylase inhibitors (HDACi) in combination therapy with other chemotherapeutic agents and how this understanding can be utilized to optimize their application as anticancer agents in the clinic.
Histone Deacetylases (HDACs) and HDACi
Histone acetylation is one the most extensively studied posttranslational covalent modifications of histones that is regulated by the opposing actions of histone acetyltransferases (HATs) and HDACs. While HATs mediate the acetylation of lysine residues associated with gene transcription, HDACs remove the acetyl group from the positively charged histone lysine residues, enabling the negatively charged DNA to bind to the nucleosome proteins. HDACs are critical regulators of gene expression that act as transcriptional repressors while histone acetylation results in a more relaxed chromatin confirmation, enabling transcriptional activation. This opposing action of HATs and HDACs enable the regulation of gene expression in response to the environment (Figure 1) (12–15). To date, in humans there are 18 HDACs that have been identified which are classified according to their homology to yeast HDACs. The HDAC Classes I, II, and IV are Zn2+-dependent metalloproteins, while Class III HDACs are NAD+-dependent (15, 16). Class I HDACs are related to yeast RPD3 gene and include HDAC1, 2, 3, and 8. On the other hand, HDACs 4, 5, 7, and 9 are classified as Class IIa HDACs, while HDACs 6 and 10 belong to Class IIb and are related to the yeast Hda1 gene (17). Class III (sirtuins) include SIRT1–7 and are related the Sir2 gene while Class IV contains HDAC11 (18, 19).
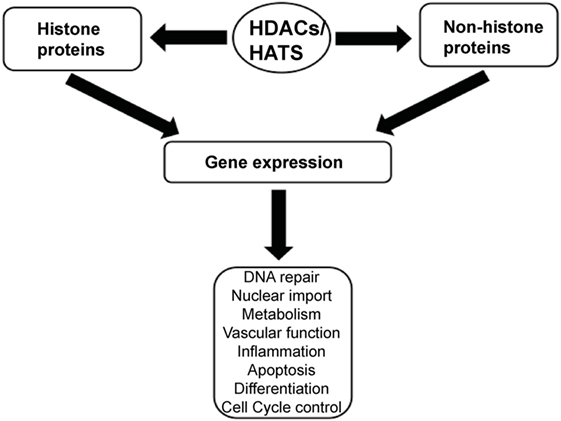
Figure 1. A schematic illustration depicting the central role of histone deacetylases (HDACs) and histone acetyltransferases (HATs) in regulating gene expression.
Histone deacetylases are often dysregulated in numerous disorders, including cancer, thereby influencing gene transcription and affecting normal cellular behavior (13, 20). High expression levels of HDAC1, 2, and 3 have been shown to be associated with poor patient outcomes in gastric and ovarian cancers (21, 22), while high expression of HDAC8 is correlated with poor survival and advanced disease in neuroblastoma (23). Additionally, dysregulation of HDAC1 expression was demonstrated to correlate with poor prognosis in multiple myeloma (24). HDACi are currently used in the clinic as anticancer agents and are a powerful new class of small-molecular therapeutics that alters the regulation of histone and non-histone proteins. HDACi increase the acetylation of core histones, leading to an open chromatin confirmation that is more accessible to DNA-targeting agents. HDACi have pleiotropic cellular effects (Figure 2) and induce the expression of pro-apoptotic genes/proteins, cause cellular differentiation and/or cell cycle arrest (13, 15, 19, 25, 26). The classes of HDACs and the HDACi that are targeted against them are summarized in Table 1.
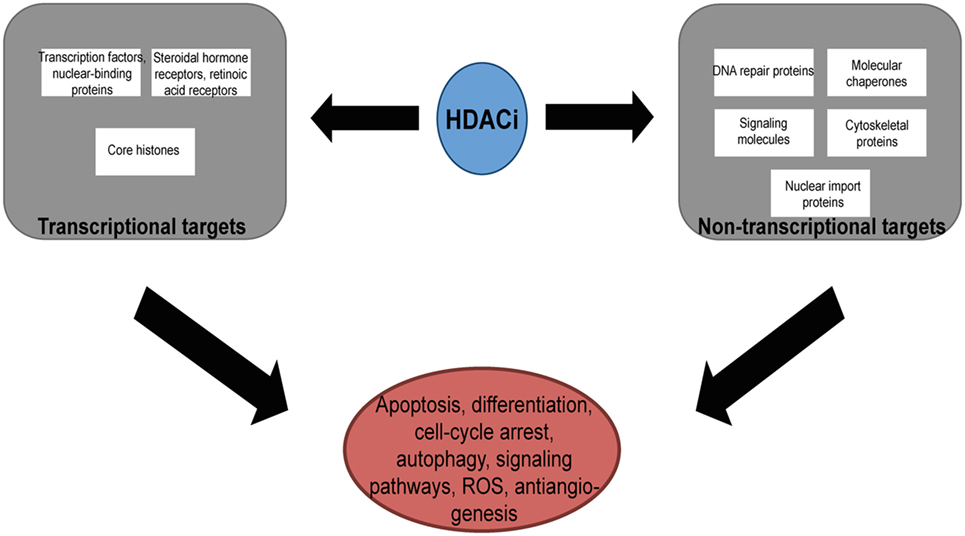
Figure 2. The pleotropic cellular effects of histone deacetylase inhibitors (HDACi). The modification of the acetylation status of cores histones and non-histone proteins result in the multiple cellular effects seen with HDACi. ROS, reactive-oxygen species.
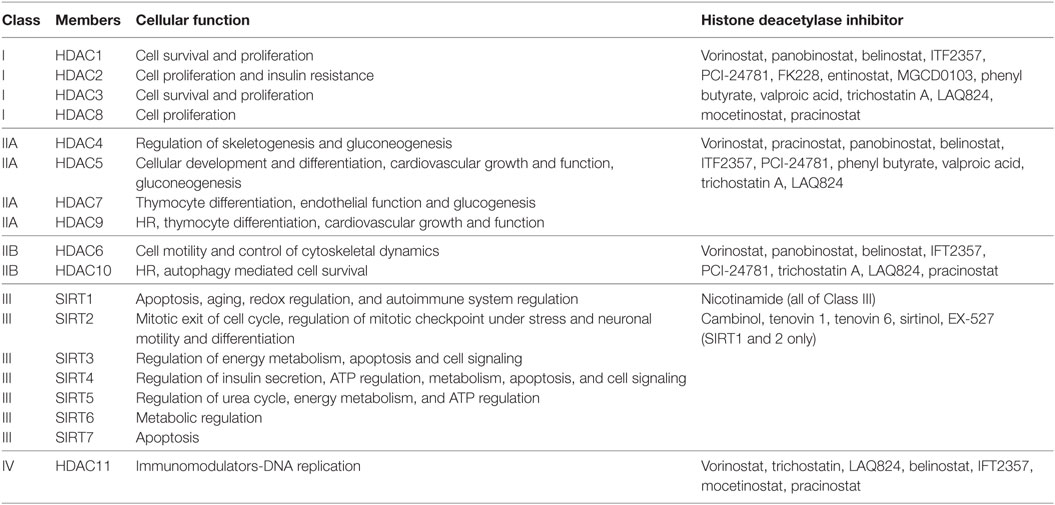
Table 1. Classification of histone deacetylases (HDACs) and the histone deacetylase inhibitors (HDACi) that target them.
HDACi That have been Approved by the USA FDA
In 2006, SAHA (Vorinostat, Zolinza™, Merck & Co, Inc., USA) became the first HDACi to be approved by the FDA for the treatment of cutaneous T-cell lymphoma (CTCL) (27). FK228 (Romidepsin, Istodax™, Celgene Corp., USA), a cyclic peptide was later approved in 2009 for treating CTCL in patients who had received at least one prior systemic therapy (28). More recently, belinostat (PXD101, BELEODAQ™, Spectrum Pharmaceuticals, Inc.) was approved by the FDA in 2014 for the use against peripheral T-cell lymphoma (PTCL) (29) and panobinostat (Farydak, Novartis Pharmaceuticals) was licensed in 2015 for the treatment of multiple myeloma (30). A summary of HDACi that are currently approved by the FDA and/or are in clinical trials for the treatment of cancer are shown in Table 2.
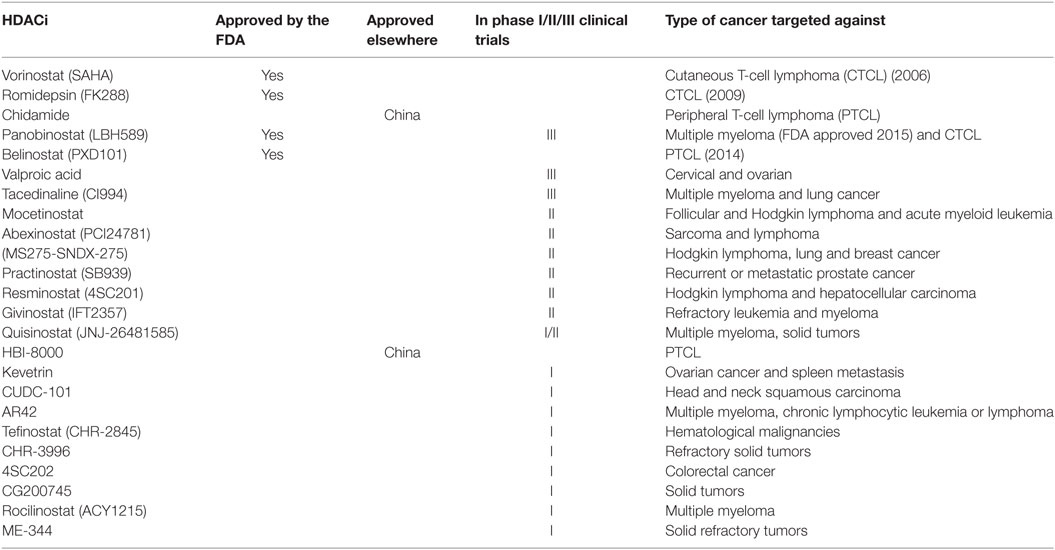
Table 2. Histone deacetylase inhibitors (HDACi) that are currently approved by the FDA or in clinical trials for the treatment of cancer.
HDACi Resistance and Rational for Combination Therapy
Resistance to HDACi is often seen and the basis of this resistance remains largely unknown. High levels of Bcl-2, Trx, and peroxiredoxins have previously been implicated with resistance of transformed cells to chemotherapy and may form the basis of resistance seen with HDACi (31–33). Increased expression of the cell cycle protein, p21, elevated levels of thioredoxin leading to lower reactive-oxygen species (ROS)-mediated DNA damage, constitutive activation of NFκB through p65 acetylation and elevated expression of the signaling proteins, MAPK, PI3K, or STAT3, have additionally been suggested to contribute toward the resistance of HDACi (34). Furthermore, chromatin and epigenetic alterations, changes in the expression levels of HDACs and changes in drug efflux mechanisms have been implicated as factors in the resistance to HDACi (35). It was recently speculated that the resistance to HDACi is a critical evolutionary consequence of environmental exposure to HDACi and that cancers that respond to HDACi have developed mutations that alters how the cancer cell responds to the HDACi. Hence, the identification of these mutations may enable the selective targeting of individual cancers susceptible to HDACi (36). Despite the approval by the FDA for the treatment of certain cancers, HDACi have been shown to have a limited therapeutic efficacy against solid tumors as a single therapeutic agent. HDACi have, however, been shown to function synergistically with a range of structurally and functionally diverse chemical compounds, biologically active polypeptides, and novel immune therapies. Combining HDACi with other cancer therapeutics may thus be an avenue to achieve their full therapeutic potential (19, 37, 38).
HDACi in Combination with Drugs That Target DNA Repair Pathways
DNA double-strand breaks (DSBs) are among the most harmful lesions to cells, as the incorrect repair of just one DSB can lead to chromosomal fragmentation and rearrangements. Upon the induction of DSBs, the DNA damage response plays a crucial role in maintaining genomic integrity. This prevents the development of cancer through cell cycle checkpoint arrest and ensures that the DNA repair occurs prior to the resumption of the cell cycle. Cells have evolved two major pathways to repair DSBs, non-homologous-end joining (NHEJ), and homologous recombination. While NHEJ is a mechanism for rejoining broken DNA ends without the use of extensive homology and thus often regarded as being error-prone, by contrast, HR involves the copying of DNA from a homologous template. The repair mechanisms also act on different stages of the cell cycle. NHEJ is used at every stage of the cell cycle, while cells only use HR during the S and G2 phases of the cell cycle, when a sister chromatid is available. In HR, the protein kinase ataxia-telangiectasia mutated (ATM) is the key regulator of the cellular response to DSBs. Following the induction of DSBs, ATM is activated in a process that involves its auto-phosphorylation by the MRN (MRE11, Rad50, and NBS1) complex. ATM subsequently phosphorylates BRCA1, CHK2, and p53, resulting in the activation of response genes involved with multiple signaling pathways leading to the activation of cell cycle checkpoints, DNA repair, and apoptosis. Upon the induction of DSBs in NHEJ, the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and the Ku70/Ku80 heterodimer initially bind to the two ends of the DSB. These ends are subsequently modified by Artemis, ligated by ligase IV, and finally stabilized by XRCC4 and XLF (39–42).
The upregulation of the DNA damage response has been suggested to contribute toward the resistance of cancer cells to genotoxic therapies and thus, mitigating the DNA damage response represents an avenue to treat cancer cells more effectively. Indeed the selectivity of HDACi in specifically targeting cancer cells can be attributed to HDACi causing DNA damage that normal but not cancer cells can repair (43, 44). For example, the HDACi vorinostat induces DSBs in normal and LNCaP and A549 cancer cells (43). While γH2AX levels, a marker for the appearance of DSBs (45), increased in the cancer cells over time, in continued culture with vorinostat, they were found to decrease in the control, non-cancerous cells. Additionally, the authors showed that vorinostat suppressed the DNA repair proteins, MRE11 and RAD50, in only the cancer cells, collectively leading to cancer cell death.
Histone deacetylases have been recently shown to play a substantial role in attenuating the activation of ATM following DNA damage (46). Ataxia-telangiectasia patients, with defective ATM protein, display increased genomic instability, chromatin decondensation, and radiosensitivity (47). Interestingly, cells treated with HDACi display a similar phenotype (48). There is increasing evidence to suggest that HDACs regulate ATM. For instance, ATM has been shown to interact both in vitro and in vivo with HDAC1 and this interaction increases post-irradiation (46, 49). HDAC2 has been additionally shown to regulate the expression of chromatin remodeling proteins, including SMC1, an ATM substrate (50). More recently, research has shown that HDACs regulate ATM-mediated DNA damage signaling and established that HDAC1 and HDAC2 function as part of the DDR (46). The authors demonstrated that HDACi resulted in a reduced activation of ATM and its expression and decreased p53 activation both in vitro and in vivo. Thus, addition of HDACi followed by a subsequent treatment with DNA damaging agents led to an insufficient induction of the DDR, sustained DNA damage, and an increase in cell death. Olaparib (AZD-2281, Lynparza) is an oral PARP inhibitor, inhibiting the DNA repair protein poly ADP ribose polymerase. Olaparib is approved for the treatment of germline BRCA-mutated advanced ovarian cancer in patients who have received prior chemotherapy (51, 52). A recently reported phase III clinical trial has demonstrated that olaparib monotherapy is significantly superior to physicians choice of chemotherapy in the treatment of metastatic breast cancer (MBC) patients with germline BRAC1/2 mutations in terms of progression-free survival, time to second progression, objective response rate, and global health-related quality of life (ClinicalTrial.gov identifier: NCT02000622) (53). Additionally, there is a phase I/II clinical trial underway with the combination of olaparib and vorinostat. The aim of this study is to determine the safety and effectiveness of the combination of olaparib with high-dose chemotherapy (vorinostat, gemcitabine, busulfan, and melphalan, either with or without rituximab) in patients with refractory lymphomas (ClinicalTrial.gov identifier: NCT03259503).
In addition to their histone substrates, HDACi act on non-histone proteins including p53, Ku70, HSP90, NFκB, and α-tubulin (54). Thus, the acetylation of both histone and non-histone proteins give rise to the pleiotropic antitumor effects seen with HDACi at both epigenetic and cellular levels. By targeting the acetylation of Ku70, HDACi have additionally been shown to sensitize prostate cancer cells to DNA damaging agents. In this study, the authors demonstrated that the prostate cancer cells pretreated with the HDACi, trichostatin A, suberoylanilide hyroxamic acid, MS-275, and OSU-HDAC42, resulted in an increase in Ku70 acetylation which was followed by a lower DNA-binding affinity without the disruption of the Ku70/80 complex. When the prostate cancer cells were treated with the HDACi and the DNA damaging agents, bleomycin, doxorubicin, and etoposide, sequentially, the sensitization of the prostate cancer cells was more pronounced (55). Taken together, the combination of HDACi for sensitizing cancer cells with therapies that induce DNA damage warrants further clinical investigation.
Combination of HDACi and Radiotherapy
To this day, radiotherapy continues to be among the most widely used cancer treatments, causing cell death by the induction of DSBs. HDACi enhance the radiosensitivity of cancer cells (56, 57). HDACi have been shown in vitro to lower a cell’s ability to repair IR-induced DNA damage, by affecting DNA damage signaling and the NHEJ and HR DSB repair pathways (17). In preclinical studies, HDACi have shown radiosensitizing effects with glioblastoma multiforme, melanoma, and head and neck squamous, colorectal, non-small cell lung, prostate, and MBCs (58). The first group to publish a case report on use of an HDACi with radiotherapy used valproic acid in combination with cisplatin and doxorubicin, 40 Gy radiation, and surgery to successfully treat a patient with anaplastic thyroid carcinoma (59). The novel pan-HDACi, panobinostat (LBH589), is currently being used in phase I and II clinical studies against various hematologic malignancies and solid tumors (60, 61). In combination with radiotherapy, LBH589 was shown to be an effective regimen for the treatment of prostate cancer in vitro. The authors demonstrated that LBH589 at low concentrations (IC20) in combination with radiation induced more apoptosis, resulted in a steady increase of the sub-G1 population of cells and led to the elimination of radiation-induced cell cycle arrest. Additionally, the combination treatment gave rise to more DNA damage and resulted in less activation of NHEJ and HR repair pathways (62). A phase I clinical trial (ClinicalTrial.gov identifier: NCT00455351) studying the treatment of vorinostat with short-term palliative pelvic radiotherapy showed that both treatments were tolerated in gastrointestinal tract carcinoma patients. Additionally, the combined treatment resulted in seven grade 3 adverse events in 16 of the patients receiving the combination. Further research is required to evaluate the efficacy and safety of the combination therapy. There are several completed and ongoing trials combining radiotherapy and various HDACi. These include a phase II trial to determine the efficacy of valproic acid with temozolomide and external beam radiation to treat high-grade gliomas (ClinicalTrial.gov identifier: NCT00302159), a phase I study of vorinostat in combination with palliative radiotherapy for patients with NSCLC (ClinicalTrial.gov identifier: NCT00821951), a phase I study of panobinostat combined with radiation therapy for treating prostate, esophageal, and head and neck cancers (ClinicalTrial.gov identifier: NCT00670553), and capeceitabine, vorinostat, and radiotherapy for the treatment of patients with non-metastatic pancreatic cancer (ClinicalTrial.gov identifier: NCT00983268).
Dual Combination of HDACi and Topoisomerase Inhibitors
Topoisomerases are ubiquitous enzymes that play a vital role in replication, transcription, recombination, DNA repair, and chromatin remodeling, by over-winding or under-winding the DNA helix. There are two groups of topoisomerases, type I and II, which are classified according to their structure and mechanism of action. While type I topoisomerases cleave one strand of the DNA double helix reversibly, type II enzymes mediate strand passage through a double-strand DNA gate (63). Compared to monotherapy, combination therapies with HDACi and topoisomerase II inhibitors have lead to higher nuclear topoisomerase II inhibitor accumulation, an increase in DNA damage, growth inhibition, and cell death (64–67). Preclinical studies have shown greater efficacy when cells were pretreated with HDACi prior to exposure to DNA damaging agents (64, 68), similar to the decondensation of chromatin seen in cells from breast and other cancers (50, 69). The first human phase I dose-escalation clinical study combining valproic acid and the topoisomerase II inhibitor, epirubicin, in solid malignancies has recently been performed (70). This study showed that valproic acid is well tolerated and can be used in a clinical setting. The combination of valproic acid followed by epirubicin, resulted in an objective response rate of 22% and, additionally, 39% of patients had stable disease. Most significantly, this study showed responses in patients with anthracycline-resistant tumors and in heavily pretreated patients (ClinicalTrial.gov identifier: NCT00246103). In a pharmacokinetic and pharmacodynamic phase I/II study of valproic acid in combination with epirubicin/FEC, 64% of breast cancer patients had an objective response in the dose expansion cohort. This study demonstrated that the combination of valproic acid and epirubicin/FEC is safe, tolerable, and feasible (ClinicalTrial.gov identifier: NCT01010854) (71). The authors subsequently carried out a phase I combination trial of vorinostat and doxorubicin in treating patients with metastatic or advanced solid tumors (72). While a modest clinical effect was observed, this study showed that HDAC2 expression is a potential predictive biomarker and a target for developing isotype-specific inhibitors that may lead to greater inhibition with reduced toxicity (ClinicalTrial.gov identifier: NCT00331955). There are several completed and ongoing clinical trials involving HDACi and doxorubicin. A phase I/II dose-escalation trial of belinostat together with doxorubicin in patients with soft tissue sacrcoma (ClinicalTrial.gov identifier: NCT00878800) has just been completed. A dose-finding and dose-escalating phase I/II trial of vorinostat and pegylated liposomal doxorubicin in patients with advanced or refractory lymphoma (ClinicalTrial.gov identifier: NCT00785798) and a phase I trial studying the maximum tolerated dose of vorinostat in combination with bortezomib and doxorubicin hydrochloride liposome for the treatment of relapsed or refractory multiple myeloma (ClinicalTrial.gov identifier: NCT00744354) has just been terminated.
HDACi in Combination with Platinum-Based Chemotherapeutics
Cis-diamminedichloroplatinum(II) (cisplatin) was first approved in 1978 for the treatment of bladder and testicular cancer (73, 74). Cisplatin has since been used as a first-line therapy for many different solid malignancies, including head and neck, ovarian, bladder, testicular, colorectal, bladder, cervical, and lung cancers, as a monotherapy or in combination with chemotherapeutic drugs. Cisplatin exerts its anticancer effects primarily through the generation of DNA lesions, followed by the induction of the DDR and mitochondrial apoptosis. Although cisplatin treatment results in many cases as an initial therapeutic success, chemoresistance eventually develops particularly in non-germ cell solid tumors (25, 75, 76). Thus, combining cisplatin with HDACi is a promising strategy to increase the efficacy of cisplatin (25). Concurrent SAHA (vorinostat) therapy has been shown to enhance tumor cell sensitivity to subtoxic doses of cisplatin in oral squamous cell carcinoma cell lines (77). The authors showed that the combination of both drugs synergistically induced cytotoxicity and apoptosis in the oral squamous cell carcinoma cells, compared to SAHA or cisplatin treatment alone. Trichostatin A was shown to synergistically enhance the antitumor response of cisplatin and additionally resensitize bladder cancer cells that were resistant to cisplatin (78) and valproic acid was shown to induce apoptosis and p16INK4A upregulation, thereby sensitizing melanoma cells to cisplatin and etoposide treatment (79).
Carboplatin (cyclobutane-1,1-dicarboxylate-O,O′) was developed as an analog of cisplatin that results in less nephron and neurotoxicity than cisplatin, but results in the same DNA adducts (80). A phase I clinical study was carried out to determine the safety and pharmacokinetics of the HDACi, belinostat, with carboplatin and/or paclitaxel in solid tumors. Of the 23 patients with solid tumors who were recruited in the study, six patients showed stable disease lasting ≥6 months, two patients had partial responses and one patient had a complete CA-125 response. Overall, belinostat with carboplatin and paclitaxel was well tolerated with no evidence of a pharmacokinetic interaction (81). A phase I pharmacokinetic study was additionally carried out with vorinostat in combination with carboplatin and paclitaxel for advanced solid malignancies (82, 83). This study demonstrated that this combination was tolerated well and of the 19 patients with NSCLC, 10 had partial responses and 4 patients showed stable disease. This study subsequently led to a phase II clinical study involving 94 patients with NSCLC with carboplatin and paclitaxel combined with either a placebo or vorinostat (ClinicalTrial.gov identifier: NCT01413750). The response rate, progression-free survival, and survival overall were significantly higher in those treated with vorinostat-carboplatin-paclitaxel compared to the group who received placebo-carboplatin-paclitaxel. The 1-year survival rate for the vorinostat-carboplatin-paclitaxel group compared to the placebo-carboplatin-paclitaxel group was 51% compared to 33%. Although vorinostat was found to improve the efficacy of carboplatin and paclitaxel, it also led to increased toxicity. Overall, this study demonstrated that vorinostat enhanced the efficacy of carboplatin and paclitaxel in patients with NSCLC patients (84, 85); however, the phase III trial with this combination was terminated due to lack of efficacy (ClinicalTrial.gov identifier: NCT00473889). A phase I trial with high-dose or low-dose vorinostat with carboplatin or paclitaxel for the treatment of advanced solid tumors is currently ongoing (ClinicalTrial.gov identifier: NCT01281176).
Combining HDACi and Proteasome Inhibitors
The tightly regulated degradation of ubiquitinated proteins by the proteasome is essential in cells and impairment of the ubiquitin-proteasome system (UPS) leads to several pathological diseases (86). The UPS plays a key role in the cellular protein-degradation machinery and in preventing the accumulation of misfolded proteins (86–88). Cancer cells are dependent on the UPS as they are highly proliferative and have an increased requirement for protein synthesis, hence making them more vulnerable to proteasome inhibitors (89, 90). In 2003, the FDA approved bortezomib (PS-341, Velcade™, Millennium Pharmaceuticals Inc.) to treat relapsed and refractory multiple myeloma (91). However, peripheral neuropathy is often seen and is the most frequently associated dose limiting toxicity. The HDACi, vorinostat, romidepsin, dacinostat, and panobinostat, inhibit proliferation and lead to apoptosis of multiple myeloma cells and murine xenograft models. However, in phase I and phase II clinical trials, is has been observed that as a single agent, the activity of HDACi have been limited (92, 93). In several preclinical studies, synergistic antitumor effects were seen in multiple myeloma, when HDACi were combined with proteasome inhibitors, thus providing the rational for combining HDACi with these agents in clinical trials (94–97). The synergistic effect seen with combination of HDACi and proteasome inhibitors may be attributed to the multiple pathways targeting multiple myeloma cell biology. One prominent mechanism may be by disrupting protein degradation by inhibiting both the proteasome and aggresome with HDAC6i, resulting in the accumulation of significantly more polyubiquitinated proteins and causing increased cellular stress and apoptosis (94, 98, 99). In addition to multiple myeloma, investigators have demonstrated that combination therapy with bortezomib and the Class I HDACi, MS275, apicidin, and romidepsin, resulted in apoptosis in vitro and in vivo of nasopharyngeal carcinoma cells. The authors showed that the synergism observed was independent of HDAC6 but rather through the induction of ROS-dependent estrogen receptor (ER) stress (100). The Class I HDACi, MGCD0103, has also been shown to synergize with proteasome inhibitors and induce apoptosis in Hodgkin lymphoma cell lines by an HDAC6-independent mechanism. The authors demonstrated that MGCD0103 resulted in the upregulation of several inflammatory cytokines, leading to the activation of nuclear factor (NF)-κB (NFκB) and attenuating tumor cell death. The subsequent inhibition of NFκB with proteasome inhibitors enhanced the MGCD0103-induced death of Hodgkin lymphoma cells (101). The combination of HDACi with proteasome inhibitors may additionally be used to treat glioblastomas in order to maximize the therapeutic efficacy and limit the toxicity associated with using proteasome inhibitors as a single agent in this disease (102). Clinical trials with bortezomib in combination with either vorinostat as a third-line treatment in advanced NSCLC (ClinicalTrial.gov identifier: NCT00798720) or with panobinostat for the treatment of relapsed/refractory T-cell lymphoma or NK/T-cell lymphoma following conventional chemotherapy failure (ClinicalTrial.gov identifier: NCT00901147) have been completed.
Carfilzomib is a second-generation proteasome inhibitor that was recently approved by the FDA for the treatment of relapsed and refractory multiple myeloma, in patients who were given at least two prior therapies (103, 104). There is an ongoing phase I/II clinical trial with panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma (ClinicalTrial.gov identifier: NCT01496118) and a completed phase I clinical trial of carfilzomib with vorinostat for the treatment of relapsed/refractory B-cell lymphomas (ClinicalTrial.gov identifier: NCT01276717).
Combining HDACi with Hormonal Therapy
Dysregulation of hormone signaling is seen in many cancers and is an essential component of carcinogenesis. Strategies inhibiting estrogen and androgen signaling in breast and prostate cancers, respectively, have shown clinical success (105, 106). Aberrant acetylation and HDAC expression has been observed in both breast and prostate cancer cell lines and patient tumors (107, 108). Thus, the combination of HDACi and hormonal therapy is under investigation in both clinical and preclinical settings for the treatment of breast and prostate cancers. While vorinostat as a monotherapy was not found to be effective against the treatment of MBC (109), in combination therapy with the anti-estrogen, tamoxifen, it has shown great promise (57). Hormone therapy resistance is a challenging issue for treating ER positive breast cancers and thus, combination therapy with HDACi has been studied. Researchers carried out the first clinical trial combining vorinostat and tamoxifen for hormone therapy-resistant breast cancer (ClinicalTrial.gov identifier: NCT00365599) (67). This study was for advanced breast cancer patients who progressed on prior hormone therapy, as an approach to restore hormone sensitivity to tamoxifen. Vorinostat-induced H4 acetylation and HDAC2 expression was performed in peripheral blood mononuclear cells on the 43 patients recruited in this study. Overall, this study demonstrated that the addition of the HDACi, vorinostat, to tamoxifen hormone receptor-positive breast cancers lead to tumor regression or prolonged disease stabilization in 40% of patients who had progressed on prior hormonal therapy and chemotherapy. Additionally, this study confirms that HDAC2 expression is a predictive marker for response and that histone hyperacetylation may be an effective pharmocodynamic marker for determining the efficacy of vorinostat and tamoxifen combination therapy.
While HDACi as a monotherapy in prostate cancer has not shown promise in clinical trials (110–112), in combination with the antiandrogen, bicalutamide, a synergistic cytotoxicity was observed in preclinical models (113–115). There have been two completed trials evaluating the combination of HDACi and hormonal therapy in prostate cancer (116). The initial trial was a phase II trial with vorinostat in combination with bicalutamide and radical prostatectomy, in patients with localized prostate cancer (ClinicalTrial.gov identifier: NCT00589472) (117). The second clinical trial was a phase I/II clinical trial studying the safety and efficacy of panobinostat and bicalutamide in patients with recurrent prostate cancer after castration (ClinicalTrial.gov identifier: NCT00878436). While the combination of HDACi with androgen-deprivation therapy holds much promise, the advancement to phase III clinical trials will define the fate of HDACi in the treatment of prostate cancer.
Combination of HDACi with Tyrosine Kinase Pathway Inhibitors
Receptor tyrosine kinase (RTK) activation regulates a vast range of biological functions, such as cell growth, survival, organ morphogenesis, neovascularization, and tissue regeneration and repair. While the activity of RTK is regulated in normal cells, RTK signaling is dysregulated in many cancers. Therefore, RTKs are promising therapeutic targets (118, 119). Activation of RTK has been demonstrated to activate RAS-RAF-MEK-MAPK and PI3K-AKT pathways among others. This results in an increased expression of c-Myc and cyclin D1 oncogenes and lowered activation of proteins within the cell cycle checkpoint leading to enhanced cell cycle progression and survival. Monoclonal antibodies or RTK inhibitors of these pathways may block cell cycle progression, inhibit production of pro-angiogenic factors and induce apoptosis in various in vitro and xenograft models (120). Interestingly, cyclin D1 interacts with class I/II HDACs and HDACs regulate the expression of c-Myc and cyclin D1. Additionally, cell treatments with HDACi reduced cyclin D1 transcription and increased c-Myc degradation. Therefore, the combination of RTK targeted therapies and HDACi represents a novel approach for targeted cancer therapy (57, 121). Sorafenib (BAY 43-9006) is a novel multikinase inhibitor which has been shown to block the RAS-RAF-MEK-MAPK pathway. Recent work has demonstrated that vorinostat and sorafenib synergistically kill tumor cells (122). Numerous phase I clinical trials have been conducted with the combination of sorafenib and HDACi for treating patients with advanced/metastatic solid malignancies and refractory/relapsed acute myeloid leukemia (AML), soft tissue sarcomas, and lung, advanced liver, and renal cancers (ClinicalTrial.gov identifier: NCT01159301, NCT00823290, NCT01005797, NCT01075113, NCT00635791). Gefitinib (Iressa) is a selective, first generation, reversible EGFR tyrosine kinase inhibitor (123). Studies have demonstrated that gefitinib combined with HDACi synergistically induces growth inhibition and apoptosis in gefitinib-resistant NSCLC cells (124). Currently, there is a phase I/II clinical trial combining Gefitinib and vorinostat in relapsed/refractory patients with advanced NSCLC (ClinicalTrial.gov identifier: NCT01027676) and phase I study of vorinostat combined with gefitinib in patients with BIM polymorphysim associated resistant EGFR mutant lung cancer (ClinicalTrial.gov identifier: NCT02151721). Additionally, the TRK inhibitor, trastuzumab, a monoclonal antibody, shown to target HER2 and downregulate the PI3K-AKT pathway, was evaluated in combination with panobinostat in a phase I/II clinical trial in adult female patients with HER2 positive MBC whose disease has progressed on or after trastuzumab. Unfortunately, this trial was prematurely terminated as a result of poor efficacy (ClinicalTrial.gov identifier: NCT00567879). The TRK inhibitor, everolimus, which is an mTOR serine/threonine protein kinase inhibitor, is currently being evaluated in the clinic with panobinostat for the treatment of recurrent multiple myeloma, non-Hodgkin and Hodgkin lymphoma (ClinicalTrial.gov identifier: NCT00918333) and in solid tumors/lymphomas with enrichment for EBV-driven tumors (ClinicalTrial.gov identifier: NCT01341834).
HDACi in Combination with Other Epigenetic Modifiers
Aberrant DNA methylation has emerged as a distinct molecular pathway leading to malignant transformation. The hypomethylating agents, azacitidine and decitabine, are active anticancer drugs currently FDA approved for treating AML, chronic myelomonocytic leukemia, and myelodysplatic syndromes (MDSs) (125, 126). The dual inhibition of HDACi with DNA-hypomethylating agents is a growing area of interest in the clinic. Recent in vitro work has demonstrated that the combination of valproic acid and decitabine synergistically inhibits growth and induces apoptosis in the leukemia cells lines, HL-60, and MOLT4 (127). A subsequent phase I/II clinical trial with valproic acid and decitabine included 54 patients with leukemia. In the study, 12 patients showed objective responses including 10 complete remissions and a large cytogenetic response was seen in 6 of the 8 responding patients. Hypomethylation of p15, a central cell cycle regulating gene, proved to be the best indicator of response. This study demonstrated that the combination was safe and resulted in a transient reversal of aberrant epigenetic markers (128) (ClinicalTrial.gov identifier: NCT00075010). Two phase I clinical trials were carried out to assess the pharmacokinetics of 5-azacitidine administered with the HDACi, phenylbutyrate, for the treatment of refractory solid tumors or hematological malignancies (129). 5-azacitidine was administered once daily as a subcutaneous injection, while varying doses of phenylbutyrate were administered as a continuous intravenous infusion. This study showed that 5-azacitidine was rapidly absorbed and eliminated via subcutaneous injection and sufficient 5-azacitidine exposure was seen leading to the pharmacodynamic effects in tumors. Additionally, a phase I clinical study was carried out in MDS or AML patients receiving 5-azacitidine followed by sodium phenylbutyrate. In the trial, 11 of the 29 patients responded; 6 among the 6 responding patients with pretreatment methylation of p15 or CDH-1 promoters saw a reversal of methylation during the first cycle of therapy, while zero among the six non-responding patients studied showed any demethylation. This study showed that the molecular mechanism responsible for responses to DNA methyltransferase and HDACi combination therapy might include the reversal of aberrant epigenetic gene silencing (130) (ClinicalTrial.gov identifier: NCT00004871).
Combining HDACi with Immune Checkpoint Inhibitors
Immune checkpoint inhibitors have transformed the way in which solid tumors and hematological malignancies are treated. The immune checkpoint proteins, cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death receptor 1 (PD-1), have been shown to be negative regulators of T-cell immune function. PD-1 regulates T-cell activation through binding to its ligands, PD-L1 and PD-L2. Inhibiting the PD-1 and PD-L1/L2 interaction results in enhanced T cell activation and effector functions, leading to an increased activation of the immune response. At present, there are four immune checkpoint inhibitors approved by the FDA, ipilimumab (an anti-CTLA-4 drug), nivolumab and pembrolizumab (anti-PD-1 drugs), and the anti-PDL1 agents, such as atezolizumab, durvalumab, and avelumab (131, 132). There are over 500 ongoing clinical trials using immune checkpoint inhibitors for the treatment of various cancers including resected, localized, and advanced solid tumors and hematological malignancies. Despite promising results with immune checkpoint blockade therapy, solid and hematological malignancies often evade the host immune system and combination therapy with other chemotherapeutic drugs is an avenue to overcome these limitations. HDACi have been shown to have potent immunomodulatory activity, rationalizing their use in cancer immunotherapies. The combination of immune checkpoint inhibitors with HDACi has shown promising results both in vitro and in vivo (133, 134). HDACi enhance the immunotherapy response (135) and augment immunotherapy with PD-1 blockade in melanoma cells (136), inhibit apoptosis of CD4+ T cells within the tumor, upregulate antitumor immune responses and restrict tumor growth (137). It was recently shown in mice that cancers resistant to the immune checkpoint inhibitors, anti-PD-1 and anti-CTLA-4, could be cured by eliminating myeloid-derived suppressor cells (MDSCs) (138). The authors demonstrated that the HDACi, entinostat, targeted the mouse granulocytic MDSCs, responsible for the resistance to immune checkpoint blockade seen. In a mouse model of lung and renal cell carcinoma, entinostat was additionally shown to improve the antitumor effect of PD-1 targeting by inhibiting MDSC function (139). Furthermore, in breast cancer patients, entinostat reduced MDSCs and the modulation of MDSC CD40 expression and additionally, increased HLA-DR expression on CD14+ monocytes in the cancer patients. These results establish a rationale for combination therapy between entinostat and immune checkpoint inhibitors (140). It was recently shown that the prostate cell line, LNCAP, and the breast cell line, MDA-MB-231, were more sensitive to T-cell mediated lysis following treatment with the HDACi, entinostat, or vorinostat. The authors demonstrated that HDAC1 played a central in the reversal of carcinoma immune escape through the induction of ER stress, resulting in the activation of the unfolded response, subsequently leading to immunogenic modulation and increased tumor sensitivity to T-cell mediated lysis (141).
At present, there are several ongoing clinical trials combing HDACi with immunotherapy strategies. They include the immune checkpoint inhibitor, pembrolizumab, which is currently being evaluated in the clinic in combination with vorinostat for the treatment of advanced renal or urothelial cell carcinoma (ClinicalTrial.gov identifier: NCT02619253). Likewise, pembrolizumab is being evaluated with entinostat for treating metastatic melanoma of the eye (ClinicalTrial.gov identifier: NCT02697630), while the monoclonal anti-PD-1 antibody, nivolumab, is being studied in combination with azacitidine and entinostat in metastatic NSCLC patients (ClinicalTrial.gov identifier: NCT01928576). A phase I/II study assessing entinostat in combination with interleukin 2 for the treatment of metastatic renal carcinoma was recently conducted with promising clinical activity (ClinicalTrial.gov identifier: NCT01038778). Of the 47 patients who took part in the study, an objective response of 37% was observed, a 13.8-month median progression-free survival as seen and 65.3 months was the overall survival (142). Additionally, a phase III trial evaluating endocrine therapy with the HDACi, entinostat, or placebo in hormone receptor-positive breast cancer patients is about to commence (ClinicalTrial.gov identifier: NCT02115282) (140).
A summary of HDACi in combination therapy with anticancer agents in ongoing or completed clinical trials, for the treatment of cancer is discussed in Table 3.
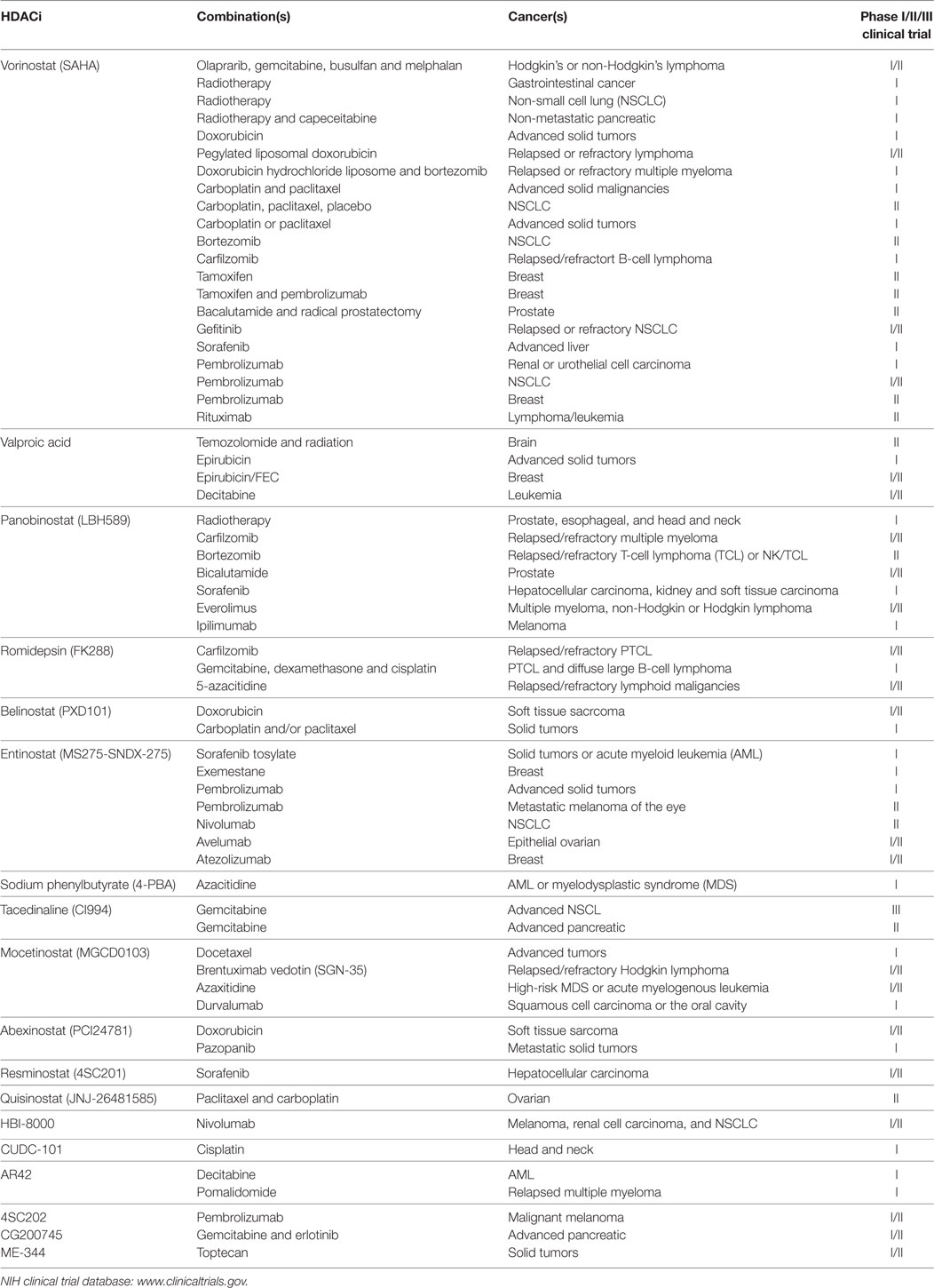
Table 3. Histone deacetylase inhibitors (HDACi) in combination with other anticancer agents: phase I/II/III clinical trials.
Side Effects and the Search for Biomarkers of HDACi
Like other therapeutics, HDACi are associated with side effects. These include thrombocytopenia, neutropenia, nausea, vomiting, diarrhea, and fatigue. The most troubling effect is cardiac toxicity, including ventricular arrhythmia. Indeed, prior to FDA approval, there were six deaths in patients treated with romidepsin (143–145). In order to minimize the off-target toxicity of HDACi, tissue and cell targeted delivery of HDACi and the identification of isoform selective HDACi will become the focus of future research into HDACi.
In an ideal setting, clinical trials would use biomarkers to select patients or predict their responses to HDACi and to limit adverse effects in particular by excluding patients unlikely to benefit from therapy. Recent work has demonstrated that knockdown of HDAC1 and not HDAC2 or HDAC3 resulted in an increased resistance to belinostat-induced cell death in HeLa cells (146). While this data is suggestive for high HDAC1 levels to correlate with sensitivity to HDACi treatment, more experimental data is required to determine whether a tumor-specific HDAC isoenzyme profile may predict the response to individual HDACi. Further research has shown that the molecular profiling of NSCLC cells may be of value in selecting patients for HDACi therapy. The study identified a nine-gene RNA expression signature useful in predicting trichostatin A or vorinostat-induced apoptosis and may lead to individualized treatment for patients with NSCLC (147). Additionally, further studies showed increased nuclear STAT1 and phospho-STAT3 staining in CTCL cells was associated with a lack of clinical response to vorinostat. The authors concluded that blocking this pathway may lead to improvements in the response to vorinostat and may indeed be predictive of response of CTCL patients to the agent (148).
Conclusion and Future Directions
The HDACi, vorinostat, and romidepsin are FDA approved for the treatment of CTCL, while belinostat and panobinostat are approved for the treatment of PTCL and multiple myeloma, respectively. At present, the most prominent treatment option for cancers is therapy that induces DNA damage. However, lack of response and development of resistance to the treatment is an issue. The approval of vorinostat, romidepsin, belinostat, and panobinostat has promoted HDACi as a routine therapeutic approach for the treatment of an ever-increasing list of cancers. Using HDACi as chemosensitizers that increase the efficiency of other chemotherapeutic compounds has shown great promise in preclinical and clinical trials. The combination of drugs targeting DNA repair pathways and HDACi holds great promise. While there are numerous DNA repair pathway-targeting drugs currently in clinical trials (149), their combination with HDACi have yet to be tested. Radiotherapy, topoisomerase inhibitors, and platinum-based chemotherapeutics, all of which cause cell death through the induction of DNA damage, are currently being evaluated in clinical trials in combination with HDACi. In preclinical and clinical settings, HDACi have shown synergistic or additive antitumor effects with numerous chemotherapeutic agents. The combinations of HDACi with proteasome inhibitors, hormonal therapy, tyrosine kinase inhibitors, DNA-hypomethylating agents, and immune checkpoint inhibitors in clinical trials will define the fate of HDACi in the years to come. In recent years, immune checkpoint inhibitors have played a critical role in the treatment of solid tumors and hematological malignancies. Their combination with HDACi have shown significant synergy in both in preclinical and clinical settings and considered a major breakthrough in the treatment of cancer.
Over the last few decades, we have seen an expansion in our understanding of the epigenetic regulation of normal and cancer cells. As epigenetic changes that occur during the development of cancer are reversible and amenable to pharmacological intervention, HDACi provide a unique avenue of treating cancers (150). Over the last few years we have witnessed the approval of HDACi by the FDA, the approval of the PARP inhibitor, olaparib, the proteasome inhibitor, cafilzomib, and in 2011 the approval of the first immune checkpoint inhibitor, ipilimumab. While HDACi alone and in combination with other anticancer agents have revolutionized the way cancers are being treated, they have been met with limitations. Cancer cell resistance to HDACi and the toxic effects associated with HDACi need to be overcome and larger, multicentric clinical trials are necessary to fully elucidate the benefits of HDACi combination therapy. Future studies will entail improving the selectivity of HDACi to amplify their accumulation in cancer cells at a lower dose and thereby reduce the toxic effect of these drugs on normal healthy cells. Furthermore, the identification of biomarkers for HDACi, alone and in combination with other anticancer agents is imperative in order to predict the response of the individual patient to treatment. In the years to come, genome sequencing may enable the delivery of personalized care to patients by either selecting or predicting an individual patient’s response to the cancer treatment.
Author Contributions
All authors were involved in the conception of the manuscript, the drafting and/or critically reviewing of the manuscript and have approved the final version for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Cancer and Ageing Research Program and grants from the Cancer Council Queensland, National Health and Medical Research Council 1066550, Australian Research Council (DJR, DP 120103099), and the Queensland Health and Medical Research Office.
References
1. Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules (2015) 20(3):3898–941. doi:10.3390/molecules20033898
2. Baxter E, Windloch K, Gannon F, Lee JS. Epigenetic regulation in cancer progression. Cell Biosci (2014) 4:45. doi:10.1186/2045-3701-4-45
3. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis (2010) 31(1):27–36. doi:10.1093/carcin/bgp220
4. Krishnan B, Morgan GJ. Non-Hodgkin lymphoma secondary to cancer chemotherapy. Cancer Epidemiol Biomarkers Prev (2007) 16(3):377–80. doi:10.1158/1055-9965.EPI-06-1069
5. Ismael GF, Rosa DD, Mano MS, Awada A. Novel cytotoxic drugs: old challenges, new solutions. Cancer Treat Rev (2008) 34(1):81–91. doi:10.1016/j.ctrv.2007.08.001
6. Decatris MP, O’Byrne KJ. Immune checkpoint inhibitors as first-line and salvage therapy for advanced non-small-cell lung cancer. Future Oncol (2016) 12(15):1805–22. doi:10.2217/fon-2016-0086
7. Roberts K, Culleton V, Lwin Z, O’Byrne K, Hughes BG. Immune checkpoint inhibitors: navigating a new paradigm of treatment toxicities. Asia Pac J Clin Oncol (2017) 13(4):277–88. doi:10.1111/ajco.12698
8. Mayer LD, Janoff AS. Optimizing combination chemotherapy by controlling drug ratios. Mol Interv (2007) 7(4):216–23. doi:10.1124/mi.7.4.8
9. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res (2010) 70(2):440–6. doi:10.1158/0008-5472.CAN-09-1947
10. Ramsay EC, Dos Santos N, Dragowska WH, Laskin JJ, Bally MB. The formulation of lipid-based nanotechnologies for the delivery of fixed dose anticancer drug combinations. Curr Drug Deliv (2005) 2(4):341–51. doi:10.2174/156720105774370294
11. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol (2016) 17(11):1497–508. doi:10.1016/s1470-2045(16)30498-3
12. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature (1997) 389:349. doi:10.1038/38664
13. Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer (2001) 1(3):194–202. doi:10.1038/35106079
14. Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol (2007) 1(1):19–25. doi:10.1016/j.molonc.2007.01.001
15. Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene (2007) 26(37):5541–52. doi:10.1038/sj.onc.1210620
16. Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science (1996) 272(5260):408–11. doi:10.1126/science.272.5260.408
17. Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer (2013) 108(4):748–54. doi:10.1038/bjc.2013.21
18. Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem (2004) 73:417–35. doi:10.1146/annurev.biochem.73.011303.073651
19. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov (2006) 5(9):769–84. doi:10.1038/nrd2133
20. Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med (2016) 6(10):a026831. doi:10.1101/cshperspect.a026831
21. Weichert W, Denkert C, Noske A, Darb-Esfahani S, Dietel M, Kalloger SE, et al. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia (2008) 10(9):1021–7. doi:10.1593/neo.08474
22. Weichert W, Roske A, Gekeler V, Beckers T, Ebert MP, Pross M, et al. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol (2008) 9(2):139–48. doi:10.1016/s1470-2045(08)70004-4
23. Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res (2009) 15(1):91–9. doi:10.1158/1078-0432.Ccr-08-0684
24. Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A. Dysregulated class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics (2014) 9(11):1511–20. doi:10.4161/15592294.2014.983367
25. Diyabalanage HV, Granda ML, Hooker JM. Combination therapy: histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Lett (2013) 329(1):1–8. doi:10.1016/j.canlet.2012.09.018
26. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci (2017) 18(7):1414. doi:10.3390/ijms18071414
27. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist (2007) 12(10):1247–52. doi:10.1634/theoncologist.12-10-1247
28. Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW, et al. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther (2010) 10(7):997–1008. doi:10.1586/era.10.88
29. Sawas A, Radeski D, O’Connor OA. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: a perspective review. Ther Adv Hematol (2015) 6(4):202–8. doi:10.1177/2040620715592567
30. Moore D. Panobinostat (Farydak): a novel option for the treatment of relapsed or relapsed and refractory multiple myeloma. P T (2016) 41(5):296–300.
31. Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene (2004) 23(16):2934–49. doi:10.1038/sj.onc.1207515
32. Powis G, Mustacich D, Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic Biol Med (2000) 29(3):312–22. doi:10.1016/S0891-5849(00)00313-0
33. Chung YM, Yoo YD, Park JK, Kim YT, Kim HJ. Increased expression of peroxiredoxin II confers resistance to cisplatin. Anticancer Res (2001) 21(2A):1129–33.
34. Robey RW, Chakraborty AR, Basseville A, Luchenko V, Bahr J, Zhan Z, et al. Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol Pharm (2011) 8(6):2021–31. doi:10.1021/mp200329f
35. Fantin VR, Richon VM. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res (2007) 13(24):7237–42. doi:10.1158/1078-0432.Ccr-07-2114
36. Halsall JA, Turner BM. Histone deacetylase inhibitors for cancer therapy: an evolutionarily ancient resistance response may explain their limited success. Bioessays (2016) 38(11):1102–10. doi:10.1002/bies.201600070
37. Nolan L, Johnson PW, Ganesan A, Packham G, Crabb SJ. Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential? Br J Cancer (2008) 99(5):689–94. doi:10.1038/sj.bjc.6604557
38. Lindemann RK, Gabrielli B, Johnstone RW. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle (2004) 3(6):777–86. doi:10.4161/cc.3.6.927
39. Rodgers K, McVey M. Error-prone repair of DNA double-strand breaks. J Cell Physiol (2016) 231(1):15–24. doi:10.1002/jcp.25053
40. Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet (2001) 27(3):247–54. doi:10.1038/85798
41. Saleh-Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res (2004) 32(12):3683–8. doi:10.1093/nar/gkh703
42. Khanna KK, Lavin MF, Jackson SP, Mulhern TD. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ (2001) 8(11):1052–65. doi:10.1038/sj.cdd.4400874
43. Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A (2010) 107(33):14639–44. doi:10.1073/pnas.1008522107
44. Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene (2012) 31(5):537–51. doi:10.1038/onc.2011.267
45. Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A (2003) 100(9):5057–62. doi:10.1073/pnas.0830918100
46. Thurn KT, Thomas S, Raha P, Qureshi I, Munster PN. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther (2013) 12(10):2078–87. doi:10.1158/1535-7163.MCT-12-1242
47. Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol (1997) 15(1):177–202. doi:10.1146/annurev.immunol.15.1.177
48. Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer Lett (2009) 274(2):169–76. doi:10.1016/j.canlet.2008.06.005
49. Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem (1999) 274(44):31127–30. doi:10.1074/jbc.274.44.31127
50. Marchion DC, Bicaku E, Turner JG, Schmitt ML, Morelli DR, Munster PN. HDAC2 regulates chromatin plasticity and enhances DNA vulnerability. Mol Cancer Ther (2009) 8(4):794–801. doi:10.1158/1535-7163.MCT-08-0985
51. Sistigu A, Manic G, Obrist F, Vitale I. Trial watch – inhibiting PARP enzymes for anticancer therapy. Mol Cell Oncol (2016) 3(2):e1053594. doi:10.1080/23723556.2015.1053594
52. Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, et al. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res (2015) 21(19):4257–61. doi:10.1158/1078-0432.CCR-15-0887
53. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med (2017) 377(6):523–33. doi:10.1056/NEJMoa1706450
54. Lin HY, Chen CS, Lin SP, Weng JR, Chen CS. Targeting histone deacetylase in cancer therapy. Med Res Rev (2006) 26(4):397–413. doi:10.1002/med.20056
55. Chen CS, Wang YC, Yang HC, Huang PH, Kulp SK, Yang CC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res (2007) 67(11):5318–27. doi:10.1158/0008-5472.CAN-06-3996
56. Karagiannis TC, El-Osta A. Modulation of cellular radiation responses by histone deacetylase inhibitors. Oncogene (2006) 25(28):3885–93. doi:10.1038/sj.onc.1209417
57. Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol (2011) 7(2):263–83. doi:10.2217/fon.11.2
58. Shabason JE, Tofilon PJ, Camphausen K. Grand rounds at the National Institutes of Health: HDAC inhibitors as radiation modifiers, from bench to clinic. J Cell Mol Med (2011) 15(12):2735–44. doi:10.1111/j.1582-4934.2011.01296.x
59. Noguchi H, Yamashita H, Murakami T, Hirai K, Noguchi Y, Maruta J, et al. Successful treatment of anaplastic thyroid carcinoma with a combination of oral valproic acid, chemotherapy, radiation and surgery. Endocr J (2009) 56(2):245–9. doi:10.1507/endocrj.K08E-016
60. Prince HM, Bishton MJ, Johnstone RW. Panobinostat (LBH589): a potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol (2009) 5(5):601–12. doi:10.2217/fon.09.36
61. Venturelli S, Armeanu S, Pathil A, Hsieh CJ, Weiss TS, Vonthein R, et al. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer (2007) 109(10):2132–41. doi:10.1002/cncr.22652
62. Xiao W, Graham PH, Hao J, Chang L, Ni J, Power CA, et al. Combination therapy with the histone deacetylase inhibitor LBH589 and radiation is an effective regimen for prostate cancer cells. PLoS One (2013) 8(8):e74253. doi:10.1371/journal.pone.0074253
63. Chen SH, Chan NL, Hsieh TS. New mechanistic and functional insights into DNA topoisomerases. Annu Rev Biochem (2013) 82:139–70. doi:10.1146/annurev-biochem-061809-100002
64. Marchion DC, Bicaku E, Turner JG, Daud AI, Sullivan DM, Munster PN. Synergistic interaction between histone deacetylase and topoisomerase II inhibitors is mediated through topoisomerase IIbeta. Clin Cancer Res (2005) 11(23):8467–75. doi:10.1158/1078-0432.CCR-05-1073
65. Lopez G, Liu J, Ren W, Wei W, Wang S, Lahat G, et al. Combining PCI-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin Cancer Res (2009) 15(10):3472–83. doi:10.1158/1078-0432.CCR-08-2714
66. Das CM, Aguilera D, Vasquez H, Prasad P, Zhang M, Wolff JE, et al. Valproic acid induces p21 and topoisomerase-II (alpha/beta) expression and synergistically enhances etoposide cytotoxicity in human glioblastoma cell lines. J Neurooncol (2007) 85(2):159–70. doi:10.1007/s11060-007-9402-7
67. Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer (2011) 104(12):1828–35. doi:10.1038/bjc.2011.156
68. Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem (2004) 92(2):223–37. doi:10.1002/jcb.20045
69. Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res (2005) 65(9):3815–22. doi:10.1158/0008-5472.can-04-2478
70. Munster P, Marchion D, Bicaku E, Schmitt M, Lee JH, DeConti R, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol (2007) 25(15):1979–85. doi:10.1200/JCO.2006.08.6165
71. Munster P, Marchion D, Bicaku E, Lacevic M, Kim J, Centeno B, et al. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC. Clin Cancer Res (2009) 15(7):2488–96. doi:10.1158/1078-0432.CCR-08-1930
72. Munster PN, Marchion D, Thomas S, Egorin M, Minton S, Springett G, et al. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer (2009) 101(7):1044–50. doi:10.1038/sj.bjc.6605293
73. Prestayko AW, D’Aoust JC, Issell BF, Crooke ST. Cisplatin (cis-diamminedichloroplatinum II). Cancer Treat Rev (1979) 6(1):17–39. doi:10.1016/S0305-7372(79)80057-2
74. Terenzi A, Pirker C, Keppler BK, Berger W. Anticancer metal drugs and immunogenic cell death. J Inorg Biochem (2016) 165:71–9. doi:10.1016/j.jinorgbio.2016.06.021
75. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene (2012) 31(15):1869–83. doi:10.1038/onc.2011.384
76. Basu A, Krishnamurthy S. Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids (2010) 2010:201367. doi:10.4061/2010/201367
77. Shen J, Huang C, Jiang L, Gao F, Wang Z, Zhang Y, et al. Enhancement of cisplatin induced apoptosis by suberoylanilide hydroxamic acid in human oral squamous cell carcinoma cell lines. Biochem Pharmacol (2007) 73(12):1901–9. doi:10.1016/j.bcp.2007.03.009
78. Yoon CY, Park MJ, Lee JS, Lee SC, Oh JJ, Park H, et al. The histone deacetylase inhibitor trichostatin A synergistically resensitizes a cisplatin resistant human bladder cancer cell line. J Urol (2011) 185(3):1102–11. doi:10.1016/j.juro.2010.10.034
79. Valentini A, Gravina P, Federici G, Bernardini S. Valproic acid induces apoptosis, p16INK4Aupregulation and sensitization to chemotherapy in human melanoma cells. Cancer Biol Ther (2014) 6(2):185–91. doi:10.4161/cbt.6.2.3578
80. Harrap KR. Preclinical studies identifying carboplatin as a viable cisplatin alternative. Cancer Treat Rev (1985) 12(Suppl A):21–33. doi:10.1016/0305-7372(85)90015-5
81. Lassen U, Molife LR, Sorensen M, Engelholm SA, Vidal L, Sinha R, et al. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br J Cancer (2010) 103(1):12–7. doi:10.1038/sj.bjc.6605726
82. Ramalingam SS, Parise RA, Ramanathan RK, Lagattuta TF, Musguire LA, Stoller RG, et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res (2007) 13(12):3605–10. doi:10.1158/1078-0432.CCR-07-0162
83. Lee MJ, Kim YS, Kummar S, Giaccone G, Trepel JB. Histone deacetylase inhibitors in cancer therapy. Curr Opin Oncol (2008) 20(6):639–49. doi:10.1097/CCO.0b013e3283127095
84. Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(1):56–62. doi:10.1200/JCO.2009.24.9094
85. Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Mitsui M, et al. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer (2011) 74(2):300–4. doi:10.1016/j.lungcan.2011.02.019
86. Suraweera A, Munch C, Hanssum A, Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol Cell (2012) 48(2):242–53. doi:10.1016/j.molcel.2012.08.003
87. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem (1998) 67(1):425–79. doi:10.1146/annurev.biochem.67.1.425
88. Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem (2009) 78:477–513. doi:10.1146/annurev.biochem.78.081507.101607
89. LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res (2002) 62(17):4996–5000.
90. Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans (2007) 35(Pt 1):12–7. doi:10.1042/bst0350012
91. Field-Smith A, Morgan GJ, Davies FE. Bortezomib (velcadetrade mark) in the treatment of multiple myeloma. Ther Clin Risk Manag (2006) 2(3):271–9. doi:10.2147/tcrm.2006.2.3.271
92. Richardson P, Mitsiades C, Colson K, Reilly E, McBride L, Chiao J, et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma (2008) 49(3):502–7. doi:10.1080/10428190701817258
93. Niesvizky R, Ely S, Mark T, Aggarwal S, Gabrilove JL, Wright JJ, et al. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer (2011) 117(2):336–42. doi:10.1002/cncr.25584
94. Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A (2005) 102(24):8567–72. doi:10.1073/pnas.0503221102
95. Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res (2004) 10(11):3839–52. doi:10.1158/1078-0432.ccr-03-0561
96. Campbell RA, Sanchez E, Steinberg J, Shalitin D, Li ZW, Chen H, et al. Vorinostat enhances the antimyeloma effects of melphalan and bortezomib. Eur J Haematol (2010) 84(3):201–11. doi:10.1111/j.1600-0609.2009.01384.x
97. Ocio EM, Vilanova D, Atadja P, Maiso P, Crusoe E, Fernandez-Lazaro D, et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica (2010) 95(5):794–803. doi:10.3324/haematol.2009.015495
98. Catley L, Weisberg E, Kiziltepe T, Tai YT, Hideshima T, Neri P, et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood (2006) 108(10):3441–9. doi:10.1182/blood-2006-04-016055
99. Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther (2011) 10(11):2034–42. doi:10.1158/1535-7163.MCT-11-0433
100. Hui KF, Chiang AK. Combination of proteasome and class I HDAC inhibitors induces apoptosis of NPC cells through an HDAC6-independent ER stress-induced mechanism. Int J Cancer (2014) 135(12):2950–61. doi:10.1002/ijc.28924
101. Buglio D, Mamidipudi V, Khaskhely NM, Brady H, Heise C, Besterman J, et al. The class-I HDAC inhibitor MGCD0103 induces apoptosis in Hodgkin lymphoma cell lines and synergizes with proteasome inhibitors by an HDAC6-independent mechanism. Br J Haematol (2010) 151(4):387–96. doi:10.1111/j.1365-2141.2010.08342.x
102. Huang WJ, Chen WW, Zhang X. Proteasome inhibitors in glioblastoma. Oncol Lett (2017) 13(3):1058–62. doi:10.3892/ol.2017.5585
103. Kortuem KM, Stewart AK. Carfilzomib. Blood (2013) 121(6):893–7. doi:10.1182/blood-2012-10-459883
104. Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat (2015) 18:18–35. doi:10.1016/j.drup.2014.12.001
105. Bonneterre J, Buzdar A, Nabholtz JM, Robertson JF, Thurlimann B, von Euler M, et al. Anastrozole is superior to tamoxifen as first-line therapy in hormone receptor positive advanced breast carcinoma. Cancer (2001) 92(9):2247–58. doi:10.1002/1097-0142(20011101)92:9<2247::AID-CNCR1570>3.0.CO;2-Y
106. Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res (1998) 58(24):5718–24.
107. Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, et al. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast Cancer Res Treat (2005) 94(1):11–6. doi:10.1007/s10549-005-6001-1
108. Weichert W, Roske A, Gekeler V, Beckers T, Stephan C, Jung K, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer (2008) 98(3):604–10. doi:10.1038/sj.bjc.6604199
109. Luu TH, Morgan RJ, Leong L, Lim D, McNamara M, Portnow J, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res (2008) 14(21):7138–42. doi:10.1158/1078-0432.CCR-08-0122
110. Rathkopf D, Wong BY, Ross RW, Anand A, Tanaka E, Woo MM, et al. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol (2010) 66(1):181–9. doi:10.1007/s00280-010-1289-x
111. Molife LR, Attard G, Fong PC, Karavasilis V, Reid AH, Patterson S, et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann Oncol (2010) 21(1):109–13. doi:10.1093/annonc/mdp270
112. Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith DC, et al. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute trial 6862): trial results and interleukin-6 analysis: a study by the department of defense prostate cancer clinical trial consortium and University of Chicago phase 2 consortium. Cancer (2009) 115(23):5541–9. doi:10.1002/cncr.24597
113. Bjorkman M, Iljin K, Halonen P, Sara H, Kaivanto E, Nees M, et al. Defining the molecular action of HDAC inhibitors and synergism with androgen deprivation in ERG-positive prostate cancer. Int J Cancer (2008) 123(12):2774–81. doi:10.1002/ijc.23885
114. Pfeiffer MJ, Mulders PF, Schalken JA. An in vitro model for preclinical testing of endocrine therapy combinations for prostate cancer. Prostate (2010) 70(14):1524–32. doi:10.1002/pros.21187
115. Marrocco DL, Tilley WD, Bianco-Miotto T, Evdokiou A, Scher HI, Rifkind RA, et al. Suberoylanilide hydroxamic acid (vorinostat) represses androgen receptor expression and acts synergistically with an androgen receptor antagonist to inhibit prostate cancer cell proliferation. Mol Cancer Ther (2007) 6(1):51–60. doi:10.1158/1535-7163.MCT-06-0144
116. Kaushik D, Vashistha V, Isharwal S, Sediqe SA, Lin MF. Histone deacetylase inhibitors in castration-resistant prostate cancer: molecular mechanism of action and recent clinical trials. Ther Adv Urol (2015) 7(6):388–95. doi:10.1177/1756287215597637
117. Marrocco-Tallarigo DL, Centenera MM, Scher HI, Tilley WD, Butler LM. Finding the place of histone deacetylase inhibitors in prostate cancer therapy. Expert Rev Clin Pharmacol (2009) 2(6):619–30. doi:10.1586/ecp.09.44
118. Takeuchi K, Ito F. Receptor tyrosine kinases and targeted cancer therapeutics. Biol Pharm Bull (2011) 34(12):1774–80. doi:10.1248/bpb.34.1774
119. Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther (2005) 315(3):971–9. doi:10.1124/jpet.105.084145
120. Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology (2009) 77(6):400–10. doi:10.1159/000279388
121. Fu M, Rao M, Bouras T, Wang C, Wu K, Zhang X, et al. Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J Biol Chem (2005) 280(17):16934–41. doi:10.1074/jbc.M500403200
122. Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res (2008) 14(17):5385–99. doi:10.1158/1078-0432.CCR-08-0469
123. Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (the IDEAL 1 trial) [corrected]. J Clin Oncol (2003) 21(12):2237–46. doi:10.1200/JCO.2003.10.038
124. Witta SE, Dziadziuszko R, Yoshida K, Hedman K, Varella-Garcia M, Bunn PA Jr, et al. ErbB-3 expression is associated with E-cadherin and their coexpression restores response to gefitinib in non-small-cell lung cancer (NSCLC). Ann Oncol (2009) 20(4):689–95. doi:10.1093/annonc/mdn703
125. Derissen EJ, Beijnen JH, Schellens JH. Concise drug review: azacitidine and decitabine. Oncologist (2013) 18(5):619–24. doi:10.1634/theoncologist.2012-0465
126. Kurkjian C, Kummar S, Murgo AJ. DNA methylation: its role in cancer development and therapy. Curr Probl Cancer (2008) 32(5):187–235. doi:10.1016/j.currproblcancer.2008.08.002
127. Yang H, Hoshino K, Sanchez-Gonzalez B, Kantarjian H, Garcia-Manero G. Antileukemia activity of the combination of 5-aza-2’-deoxycytidine with valproic acid. Leuk Res (2005) 29(7):739–48. doi:10.1016/j.leukres.2004.11.022
128. Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, et al. Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood (2006) 108(10):3271–9. doi:10.1182/blood-2006-03-009142
129. Rudek MA, Zhao M, He P, Hartke C, Gilbert J, Gore SD, et al. Pharmacokinetics of 5-azacitidine administered with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J Clin Oncol (2005) 23(17):3906–11. doi:10.1200/JCO.2005.07.450
130. Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res (2006) 66(12):6361–9. doi:10.1158/0008-5472.CAN-06-0080
131. Romano E, Romero P. The therapeutic promise of disrupting the PD-1/PD-L1 immune checkpoint in cancer: unleashing the CD8 T cell mediated anti-tumor activity results in significant, unprecedented clinical efficacy in various solid tumors. J Immunother Cancer (2015) 3:15. doi:10.1186/s40425-015-0059-z
132. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell (2015) 161(2):205–14. doi:10.1016/j.cell.2015.03.030
133. Mazzone R, Zwergel C, Mai A, Valente S. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics (2017) 9:59. doi:10.1186/s13148-017-0358-y
134. Shen L, Orillion A, Pili R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics (2016) 8(3):415–28. doi:10.2217/epi.15.118
135. Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget (2017) 8(47):83155–70. doi:10.18632/oncotarget.17950
136. Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res (2015) 3(12):1375–85. doi:10.1158/2326-6066.Cir-15-0077-t
137. Cao K, Wang G, Li W, Zhang L, Wang R, Huang Y, et al. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene (2015) 34(49):5960–70. doi:10.1038/onc.2015.46
138. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A (2014) 111(32):11774–9. doi:10.1073/pnas.1410626111
139. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res (2017) 23(17):5187–201. doi:10.1158/1078-0432.Ccr-17-0741
140. Tomita Y, Lee M-J, Lee S, Tomita S, Chumsri S, Cruickshank S, et al. The interplay of epigenetic therapy and immunity in locally recurrent or metastatic estrogen receptor-positive breast cancer: correlative analysis of ENCORE 301, a randomized, placebo-controlled phase II trial of exemestane with or without entinostat. Oncoimmunology (2016) 5(11):e1219008. doi:10.1080/2162402X.2016.1219008
141. Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget (2016) 7(7):7390–402. doi:10.18632/oncotarget.7180
142. Pili R, Quinn DI, Hammers HJ, Monk P, George S, Dorff TB, et al. Immunomodulation by entinostat in renal cell carcinoma patients receiving high-dose interleukin 2: a multicenter, single-arm, phase I/II trial (NCI-CTEP#7870). Clin Cancer Res (2017) 23(23):7199–208. doi:10.1158/1078-0432.Ccr-17-1178
143. Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, et al. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res (2006) 12(13):3997–4003. doi:10.1158/1078-0432.CCR-05-2689
144. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol (2009) 27(32):5459–68. doi:10.1200/JCO.2009.22.1291
145. Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem (2012) 4(4):505–24. doi:10.4155/fmc.12.3
146. Dejligbjerg M, Grauslund M, Litman T, Collins L, Qian X, Jeffers M, et al. Differential effects of class I isoform histone deacetylase depletion and enzymatic inhibition by belinostat or valproic acid in HeLa cells. Mol Cancer (2008) 7:70. doi:10.1186/1476-4598-7-70
147. Miyanaga A, Gemma A, Noro R, Kataoka K, Matsuda K, Nara M, et al. Antitumor activity of histone deacetylase inhibitors in non-small cell lung cancer cells: development of a molecular predictive model. Mol Cancer Ther (2008) 7(7):1923–30. doi:10.1158/1535-7163.MCT-07-2140
148. Fantin VR, Loboda A, Paweletz CP, Hendrickson RC, Pierce JW, Roth JA, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res (2008) 68(10):3785–94. doi:10.1158/0008-5472.CAN-07-6091
149. Jekimovs C, Bolderson E, Suraweera A, Adams M, O’Byrne KJ, Richard DJ. Chemotherapeutic compounds targeting the DNA double-strand break repair pathways: the good, the bad, and the promising. Front Oncol (2014) 4:86. doi:10.3389/fonc.2014.00086
Keywords: cancer, chemotherapeutic drugs, histone deacetylases, histone deacetylase inhibitors, combination therapy
Citation: Suraweera A, O’Byrne KJ and Richard DJ (2018) Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 8:92. doi: 10.3389/fonc.2018.00092
Received: 17 January 2018; Accepted: 16 March 2018;
Published: 29 March 2018
Edited by:
Suzie Chen, Rutgers, The State University of New Jersey, United StatesReviewed by:
K. B. Harikumar, Rajiv Gandhi Centre for Biotechnology, IndiaLucia Altucci, Università degli Studi della Campania “Luigi Vanvitelli” Caserta, Italy
James W. Hodge, National Institutes of Health (NIH), United States
Copyright: © 2018 Suraweera, O’Byrne and Richard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Derek J. Richard, ZGVyZWsucmljaGFyZEBxdXQuZWR1LmF1