
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 19 March 2018
Sec. Molecular and Cellular Oncology
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00072
This article is part of the Research Topic Cancer Models View all 14 articles
 Amit S. Yadav1†
Amit S. Yadav1† Poonam R. Pandey2†
Poonam R. Pandey2† Ramesh Butti1N. N. V. Radharani1
Ramesh Butti1N. N. V. Radharani1 Shamayita Roy1
Shamayita Roy1 Shaileshkumar R. Bhalara1Mahadeo Gorain1Gopal C. Kundu1
Shaileshkumar R. Bhalara1Mahadeo Gorain1Gopal C. Kundu1 Dhiraj Kumar1,3*
Dhiraj Kumar1,3*
Advancements in the early detection of cancer coupled with improved surgery, radiotherapy, and adjuvant therapy led to substantial increase in patient survival. Nevertheless, cancer metastasis is the leading cause of death in several cancer patients. The majority of these deaths are associated with metastatic relapse kinetics after a variable period of clinical remission. Most of the cancer recurrences are thought to be associated with the reactivation of dormant disseminated tumor cells (DTCs). In this review, we have summarized the cellular and molecular mechanisms related to DTCs and the role of microenvironmental niche. These mechanisms regulate the dormant state and help in the reactivation, which leads to metastatic outgrowth. Identification of novel therapeutic targets to eliminate these dormant tumor cells will be highly useful in controlling the metastatic relapse-related death with several cancers.
Metastasis is a continuous biological process consists of an orderly sequence of basic steps including local invasion, intravasation, extravasation, and colonization. These classical events of metastasis help in understanding the complex array of biological properties that are necessary for the progression of primary malignancy to overt metastasis (1, 2). It involves dissemination of malignant cells from the primary tumor to the distant sites and their proliferation at metastatic sites, which leads to failure of vital organs (1, 2). The kinetics of the metastasis have been highly explored in the past decade. Despite significant research efforts and discoveries made in recent years, the precise reasons for tumor relapse remain largely unknown. There has been significant progress in basic cancer research and clinical oncology; however, metastasis remains to be a key challenge in cancer therapy. Systemic studies on understanding the cellular and molecular mechanisms involved in metastasis might be useful in developing novel diagnostic and therapeutic strategies for metastasis prevention. However, biological, clonal and genetic heterogeneity within or between tumors are the biggest challenges in metastasis research (1, 2). The differential progression of certain cancer subtypes under the distinct selective conditions exists in various tissues leads to metastatic speciation. Disseminated cancer cells might exhibit slow growth in order to adapt to the host microenvironment for the metastatic expansion (3–7). These processes are mirrored by several cancer relapse kinetics in a tissue-specific manner and by the manifestation of distinct organ tropism (3–7). Metastasis might be developed without clinical symptoms after a long period of postsurgery (8). During this period, circulating tumor cells (CTCs) or disseminated tumor cells (DTCs) stay in the dormant state through inhibition of cell proliferation and activation of cell survival pathways (9, 10). The dormant tumor cells remain at low numbers after primary tumor resection. These cells are undetectable for long period and may be the reason for continued asymptomatic residual disease progression and treatment resistance (11–14). However, by understanding more about the biology of dormant cancer cells, the potential treatment strategies can be developed to combat the asymptomatic residual disease. The therapies targeting the mechanism of tissue-specific metastasis might open up new clinical avenues for the management of various cancer progression (15). However, to determine whether dormant solitary cells or micrometastases are valid targets for therapy, the cellular and molecular biology of tumor dormancy and reactivation need to be explored. This review emphasizes on the cancer dormancy, metastatic reactivation and the molecular mechanisms underlying these phenomena.
Tumor dormancy is a clinical process that eventually associates with local recurrences or cancer metastases. During this process, the residual disease might be present even after the treatment of primary tumors either in the forms of CTCs, DTCs, and/or micrometastases which have the capability of evading the treatment and survive in a quiescent state. Traditional chemotherapies are most effective on proliferative cells, however, ineffective toward the dormant cells (16). The dormancy nature of the tumor may be reflected by cellular or tumor mass dormancy. In cellular dormancy, cells halt in the G0 phase of the cell cycle and under favorable environmental conditions, they get reactivated by escaping from G0 cell cycle arrest (17). Moreover, during tumor mass dormancy tumor kept constant at a limited size owing to a balance between cell proliferation and cell death. Additionally, angiogenesis and immune response play an important role in maintaining the tumor mass dormancy (18). Dormant cells remain asymptomatic for months, years or even decades and eventually they undergo clinically detectable overt metastatic relapse as shown in Figure 1 (19). Interestingly, dormant cancer cells have also been observed in the primary tumors that undergo epithelial to mesenchymal transition (EMT) to develop migratory and invasive phenotypes (20). In primary tumor dormancy development, somatic mutations play a critical role to withstand apoptosis, senescence, and evade the immune system and trigger neoangiogenesis. In addition, cells undergoing metastatic dormancy are also governed by extracellular matrix (ECM) niches that induce positive signals such as Wnt and Notch and attenuate negative signals like bone morphogenetic protein (BMP) (21). On the contrary, tumor cells at premetastatic sites may undergo dormancy due to delayed adaptation and complex interaction with the local microenvironment (Figure 2).
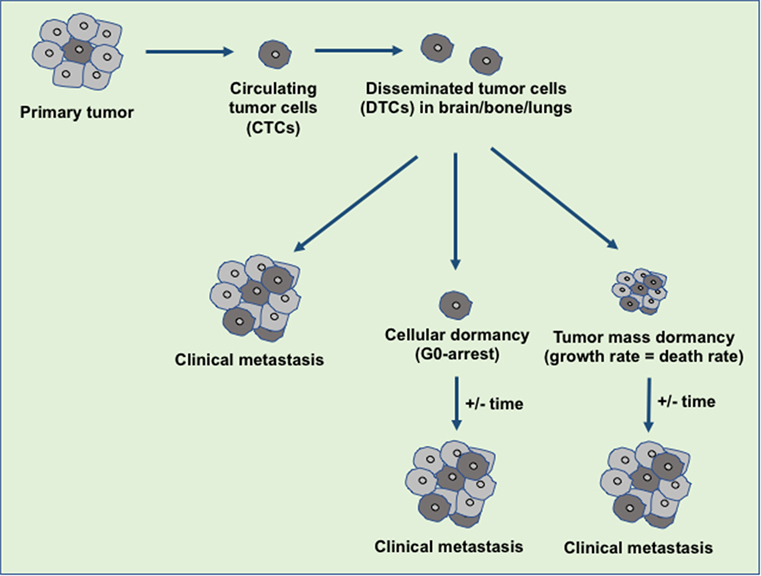
Figure 1. An overview of disseminated tumor cells (DTCs) in dormancy and clinical metastasis relapse. During the metastasis, the disseminated primary tumor cells developed the secondary tumor in the distant organ sites immediately or at a later stage. The tumor microenvironment or the intrinsic factors decide the fate of the DTCs either to develop clinical metastasis or to maintain the dormant state. Over the years, these dormant tumor cells escape from dormancy state and develop the clinical metastasis.
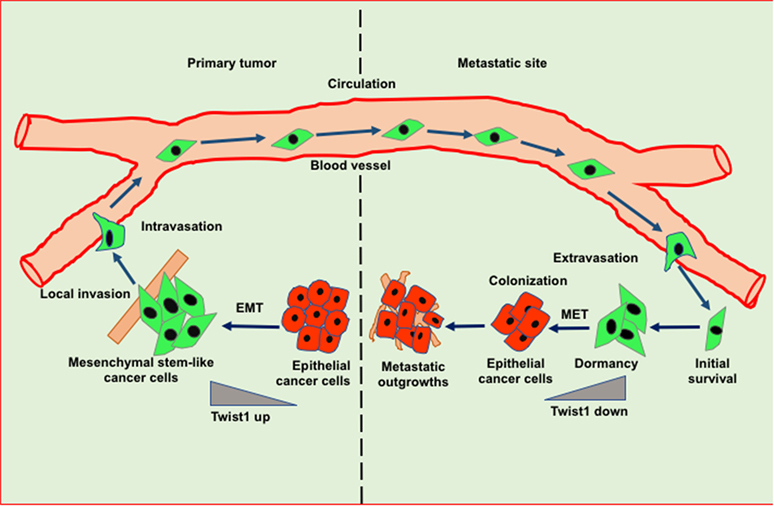
Figure 2. Role of epithelial to mesenchymal transition (EMT) and mesenchymal to epithelial transition (MET) in invasion-metastasis cascade. Cancer cells undergo EMT to acquire stemness and invasion potential leading to cancer cell dissemination. In the target organ, disseminated cancer cells encounter inhibitory signals resulted in the arrest in cell cycle thereby leading to dormancy. Cancer cells undergo MET in order to acquire epithelial features such as proliferation to form metastatic outgrowth in the target organs.
Several signaling pathways such as RAS-MEK-ERK/MAPK and PI3K-Akt play a crucial role during the process of cancer dormancy (17). Additionally, stress signals, including oxidative response and activation of unfolded protein response (UPR) also have a major contribution to metastatic dormancy and reactivation (21). In addition to various signaling pathways, DNA repair mechanism, and genomic instability also contribute to cancer dormancy (22). It is reported that primary tumor microenvironment may generate a dormant subpopulation, which is capable of evading therapy and responsible for metastatic relapse. It has been shown that metabolic pathway plays a crucial role in dormancy. Studies have demonstrated that altered lipid metabolism coupled with accumulation of reactive oxygen species helps in metastatic recurrence (23).
Several reports suggest that cancer cells undergo a protracted period of dormancy by the inhibitory molecular cues derived from primary tumors or restrains caused by target organ microenvironment (24–27). Bidirectional interaction of metastatic tumor cells with microenvironmental niches are imperative for the reactivation of dormant metastatic cells as well as the induction of mesenchymal to epithelial transition (MET) to sculpt the formation of macrometastases. Premetastatic niche provides a favorable microenvironment during metastasis development. Metastatic niche formation includes inflammation, immunosuppression, angiogenesis/vascular permeability, organotropism, lymphangiogenesis, and cellular reprogramming (28). Further, attachment of tumor cells to native basement membrane facilitates their survival, functional differentiation and growth arrest. This suggests that basement membrane is able to contribute to dormancy properties of DTCs. The DTCs often home to the distant organs where the primary basement membrane is mostly vascular in nature such as bone marrow, lung, liver, and brain (29, 30). Studies have shown a close association between DTCs and vascular basement membrane by using the mouse models of breast tumor dissemination (31). Ghajar et al. have shown that endothelial-derived thrombospondin-1 (TSP-1) induces the quiescence in breast cancer cells and this suppressive cue lost during neovasculature. The time-lapse analysis showed that sprouting vessels permit and accelerate breast cancer cell outgrowth (31). Further, they have shown that recreation of the organotypic microvascular niche of lung and bone marrow promotes dormancy and quiescence (31). It has also been shown that attachment with microvasculature in the perivascular niche is necessary for DTCs survival in mouse brain (32). These data support that endothelial cells help in dormancy induction whereas neovascularization in perivascular niche supports reactivation of dormant cells that leads to metastatic outgrowth.
A recent report suggests that a subset of macrophages (TAMs), known as metastasis-associated macrophages (MAMs), are enriched in metastatic breast cancer as compared to primary tumors. Flt1-regulated signaling in these MAMs upregulates inflammatory gene signature which is imperative for cancer cell survival during metastatic seeding (33). In addition to TAMs, circulating VEGFR1+ and bone marrow-derived CD11b+Gr1+ myeloid cells are involved in premetastatic niche formation (34–36). Myeloid cells expressed versican, an ECM proteoglycan, plays a key role in inducing proliferation of cancer cells to form metastatic outgrowth in the lung (35). CYP4A-induced TAMs promote premetastatic niche formation and metastasis in the lung by recruitment of VEGFR1+ myeloid cells (36). Moreover, induction of TGF-β in myeloid cells by natural killer T cell-derived interleukin (IL)-3 suppresses immune responses and controls tumor recurrence (37). Other stromal cells such as fibroblasts and endothelial cells present at premetastatic niche also play an important role in this phenomenon. Cancer-associated fibroblasts show activated phenotype and are integral components of premetastatic niche. Studies show that breast cancer metastasis-associated fibroblasts secrete higher level of IL-6 that promotes malignant growth (38). Furthermore, systemic factors derived from primary tumors induce the fibronectin synthesis by fibroblasts to form premetastatic niche by recruiting a fibronectin-binding integrin α4β1+ hematopoietic progenitor cells. These hematopoietic progenitor cells remodel the local microenvironment by producing MMP-9 and other factors and stimulating angiogenesis (34, 39, 40). Hence, the metastatic niche plays a pivotal role in the survival, maintenance, and reactivation of DTCs.
Epithelial to mesenchymal transition-driven mesenchymal features in cancer cells enable them to invade and metastasize to the distant organs. Several studies suggest that EMT-inducing transcription factors such as Twist and Snail show inhibitory effects on cancer cell proliferation, however, these factors induce migratory potential by downregulating the cadherin junctions (41). A reverse phenomenon of EMT known as MET helps in the tumor relapse or dormancy reactivation through the restoration of epithelial features. Interestingly, during MET tumor cells actively proliferate and regain adhesive junctions to communicate with the surrounding niche of the metastatic sites (Figure 2) (42). Recent reports have shown that blockade of the TGFβ/Smad2 pathway by versican promotes MET phenotype (43). Induction of MET in breast cancer cells is associated with increased metastatic colonization. Tsai et al. have found that attenuation of Twist1 expression promotes the metastatic outgrowth by inducing MET and proliferation of cancer cells (44). Additionally, Prrx1 another EMT transcription factor confers the migratory and invasive properties of cancer cells. Various studies showed that loss of Prrx1 contributes to metastatic colonization by stimulating MET phenotype. Moreover, downregulation of Prrx1 is associated with metastatic disease and poor survival of patients (45). Several studies showed that accumulation of genetic and epigenetic changes in tumor cells facilitated them to revert dormancy and undergo metastatic reactivation. Posttranslational modification of histones is extensively studied epigenetic change which has been observed in transcriptional activation of various EMT/MET-associated genes. The recent report suggests that H3K27me3-demethylase KDM6A expression toggles during EMT and MET processes. KDM6A catalytically removes di- and tri-methyl groups from H3K27me3 suppressive mark of the H3K4me3/H3K27me3 bivalent promoters; to promote the expression of target genes associated with differentiation, proliferation and cellular adhesion (46). Collectively, these studies suggest that the stromal cell signaling and MET contribute to metastatic reactivation.
Metastatic dormancy is a result of growth arrest either in a single DTC termed as cellular dormancy or in micrometastatic lesions called as tumor mass dormancy. Cellular dormancy marked by a quiescent state in DTCs is associated with the decline in Ki67 expression, a proliferation marker or G0/G1 cell cycle arrest. There are various cellular and molecular mechanisms through which DTCs undergo dormancy which is discussed below.
Mitogen, stress signal, and other factors present in the premetastatic niche may be responsible for cell cycle arrest and dormancy. Crosstalk between mitogen and stress-induced signaling pathways are crucial for cellular dormancy. Studies have shown that a set of genes selectively affects the growth at the secondary site including MKK4, MKK6, and Nm23-H1. Interestingly, MKK4 and MKK6 are upstream activators of p38 while, Nm23-H1 indirectly downregulates ERK1/2 by inhibiting EDG2 LPA receptor, a strong activator of ERK1/2 (47). Hence, ERK/p38 signaling ratio seems to have a crucial role in cancer cell dormancy and reactivation (Figure 3). Several studies showed that the enhanced levels of p38 MAPK over ERK1/2 upon downregulation of uPAR induces dormancy in squamous cell carcinoma (48, 49). Researchers have demonstrated that Minibrain-related kinase/dual specificity tyrosine phosphorylation-regulated kinase 1B (Mirk/DYRK1B) blocks cyclin D1 and CDK4 which further regulates the survival signals and cell cycle arrest in pancreatic and ovarian cancer cells (50–52). Likewise, MAPKK4 has been shown to exert dormancy by the upregulation of JNK pathway in prostate and ovarian cancer cells (53, 54).
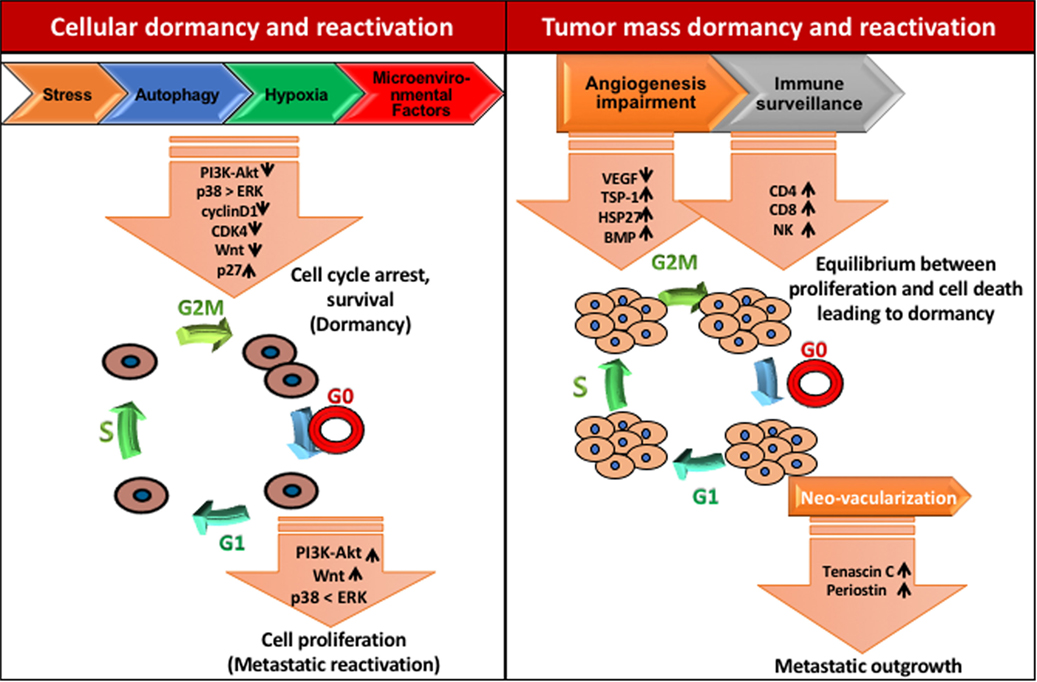
Figure 3. Mechanisms of tumor dormancy. Solitary cell dormancy (cellular dormancy, left) is caused by cell cycle arrest and induction of survival mediated by various signaling cascades including downregulation of PI3K-Akt, ERK, and Wnt signaling and upregulation of p38 MAPK signaling. Tumor mass dormancy (right) is a result of the balance between proliferation and cell death due to less blood supply and immune surveillance.
Several reports have shown that the upregulation of various UPR-associated genes like Grp78, Grp94, PDI, heat shock protein 47 (HSP47), and cyclophilin B in dormant cells play a crucial role in metastatic dormancy (55–58). Ranganathan et al. showed that stress-induced p38 activation leads to upregulation of the endoplasmic reticulum (ER) chaperone BiP. Further, this factor increases the activation of the ER stress-activated PERK signaling that results in higher survival and therapy resistance in dormant cells (56). Additionally, p38 kinase-mediated activation UPR also induces the expression of the ER stress-regulated transcription factor ATF6 and promotes mTOR-mediated survival of the dormant cells (59). Moreover, the mechanistic analysis in DTCs derived from bone marrow of breast cancer patient revealed that the expression of Grp78, a UPR protein, upregulated in low oxygen and glucose conditions and promotes higher proliferation and sustained survival (58).
Microenvironmental factors like BMPs and growth arrest-specific 6 (GAS6) derived from mesenchymal cells and osteoclasts, respectively, can curb proliferation and induce dormancy in cancer cells (Figure 3). By using the prostate cancer bone metastasis model, Kobayashi et al. have demonstrated that BMP7 promotes dormancy. BMP7 induces the expression of metastasis suppressor gene N-myc, leading to the activation of p38 MAPK, p21, and cell cycle arrest (60). Moreover, Shiozawa et al. reported that the activation of the GAS6 receptor in prostate cancer cells in the bone marrow environment plays a critical role in establishing metastatic tumor cell dormancy (61). The recent study has demonstrated that latency competent cancer cells from early-stage human lung and breast carcinoma cells can self-imposed in a dormant stage by downregulating Wnt signaling and inducing Sox-dependent stem-like state (62). Altogether, these results suggest that stress signaling helps in the single cell dormancy by arresting proliferation and enhancing survival of DTCs in the premetastatic niche.
In the tumor microenvironment, hypoxia plays a critical role during tumor development and metastasis. Fluegan et al. have explored the influence of hypoxia on the fate of DTCs. They report that hypoxia enhances the expression of key dormancy genes like NR2F1, DEC2, p27 in head and neck squamous cell carcinoma (HNSCC) and primary breast tumor. Posthypoxic solitary DTCs in patient-derived xenografts and transgenic mice show NR2F1hi/DEC2hi/p27hi/TGFβ2hi population with dormant phenotype. NR2F1 and HIF1α involved in the regulation of p27 expression in posthypoxic dormant DTCs. Moreover, hormone receptor-dependent breast cancer cells exhibit higher affinity toward NR2F1-dependent dormancy (63). Harper et al. have delineated the molecular mechanism by which HER2 aberrantly activates a program for early dissemination and generation of DTCs. These early DTCs exhibit p-p38lop-Atf2loTwist1hiE-cadlo expression pattern and an EMT-like dissemination program without complete loss of epithelial feature which was recovered after inhibition of HER2 and Wnt signaling (Figure 3). Interestingly, the dormancy feature in these early DTCs was p38-independent and even after being Twist1hiE-cadlo and dormant, they were able to initiate metastasis (64). These data indicate that the development of dormancy feature is governed by several intrinsic and extrinsic programs and by contextual cues.
miRNAs play an important role in the various biological process. It has been shown that miRNAs may affect the hallmarks of cancer, including sustained proliferation, blocking growth inhibition signals, resisting cell death, inducing invasion, metastasis, and angiogenesis (65). Ono et al. have described the role of miRNA derived from bone marrow mesenchymal stem cells in the induction of dormancy in metastatic breast cancer cells isolated from bone marrow of the mice. This study showed that higher expression of miR-23b in metastatic breast cancer cells leads to dormant phenotype by downregulation of MARCKS gene, associated with cell cycle progression and motility (66). The data also showed a consensus set of 19 miRs with the potential role in governing the phenotypic switch of human dormant breast carcinoma, glioblastoma, osteosarcoma, and liposarcoma to outgrowth. They have shown that loss of dormancy-associated miRs (DmiRs, 16/19) reactivate the fast growth of the dormant tumors. However, reestablishment of a single DmiR (miR-580, 588, or 190) results in the phenotypic switch of fast-growing angiogenic tumor toward prolonged dormancy (67).
Autophagy is an extremely conserved self-degradation process, which has an important role in cancer stem cells (CSCs) regulation and tumor cell survival. Several reports suggest that DTCs possess CSCs properties, which prompted researchers to explore the potential role of autophagy in cancer cell dormancy and stress response. Various groups have shown that autophagy helps in the survival of DTCs for protracted periods (68, 69). Autophagy supports DTCs survival by sustaining amino acid levels, ATP production and blocking energetic catastrophe (69–71). Further, induction of autophagy has linked to dormancy. Liang et al. have shown that the activation of LKB1-AMPK leads to induction of ULK1, which initiates autophagy. Further, this pathway activates p27kip1-dependent growth arrest (G1 arrest) and downregulation of this signaling induces apoptotic cell death (72). They have proposed a novel mechanism which links autophagy stimulation, growth arrest and apoptosis. In recent finding, Lu et al. have shown the role of tumor suppressor protein, aplasia Ras homolog member I (ARH1) which partly induces autophagy by inhibiting PI3K/Akt pathway. This study shows that reexpression of ARH1 in SKOv3 ovarian cancer xenograft results in tumor regression likely due to autophagy. However, the xenograft exhibited prolonged growth arrest indicating the onset of dormancy which was reversed after subsequent knockdown of ARH1 (73). These studies link the onset of autophagy with growth arrest/quiescence program and survival which proposing a key role of autophagy in dormancy.
In contrast to single cell dormancy, tumor mass dormancy is governed by a balance between the rate of proliferation and apoptosis in micrometastatic lesions. The tumor mass dormancy is induced by slow proliferation, restrained blood supply and active immune response. Recent studies reveal that the frequency of osteolytic bone metastasis depends on metastatic niche environment rather than the number of cancer cells (74, 75). Moreover, stromal factors such as TGFβ and BMPs have potential role in the regulation of tumor initiation, proliferation and maintenance of the quiescent state. Bragado et al. have suggested that TGFβ2 induces slow cycling and quiescence in cells by suppressing CDK4 and inducing p27 in HNSCC (76). Interestingly, it has been shown in multiple myeloma that a small population of Ki67+ cells coexists with dormant cells, proposing that for the reactivation defined niches are essential (77, 78). Unfortunately, the mechanisms behind long-term metastatic dormancy are highly unexplored. However, sustenance of tumor mass dormancy relies on the cellular mechanisms that induce slow cycling.
Micrometastatic lesions require the higher blood supply to grow beyond 1–2 mm, which leads to the induction of vessel formation by secretion of angiogenic factors like VEGF (79). Therefore, the antiangiogenic signaling mechanisms could be an interesting factor, which maintains the tumor mass dormancy (31, 80). These studies show that upregulation of TSP-1, an angiogenic inhibitor induces poor vascularization and dormancy in breast cancer, glioblastoma, osteosarcoma, and liposarcoma under in vivo conditions (81). Chaperons like HSP27 also regulate the angiogenesis and dormancy. Ablation of HSP27 in breast cancer prompts the long-term in vivo dormancy while its upregulation results in dormancy exit and enhanced vascular density (80).
Clearance of tumor cells by immune system contributes to another mechanism of tumor mass dormancy. Cancer cells coevolve in a microenvironment where the immune system is suppressed. However, DTCs do not have such support and eventually, most of these cells die due to the natural immune response. It has also been reported that immune system regulates the number of DTCs as well as the size of micrometastatic lesions (82). Additionally, the presence of DTCs in bone marrow of breast cancer patients showed the correlation with the higher immune cell subpopulations including NK cells, macrophages and T lymphocytes. All these cell types are known to be involved in rejection of primary tumors and metastasis, which leads to tumor dormancy (83).
Dormant cancer cells may be subjected to reactivation to initiate metastasis in response to specific signals from their specialized niche, which maintains the balance between the self-renewal and production of differentiated progeny (84–88). Cancer cells start preconditioning the host microenvironment even before seeding by secreting various soluble factors (39, 89). Heparanase, osteopontin, and lysyl oxidase facilitate the invasion, survival, and proliferation of metastatic breast cancer cells (90–92). After extravasation, DTCs may encounter different niches including perivascular niche. It has been shown that attachment of DTCs on the abluminal surface of mature blood vessels promotes dormancy through endothelium-derived TSP-1, while neovascularization creates a local microenvironment favoring metastatic reactivation. After neovascular sprouting, vessel homeostasis gets disrupted and endothelial cells start secreting tumor-promoting signals and growth factors like ECM proteins, periostin and active TGFβ that leads to micrometastatic outgrowth (31). It has been reported that ECM protein tenascin C activates Notch and Wnt signaling leading to enhanced metastatic outgrowth (93, 94). TGFβ helps in the production of periostin from stromal fibroblasts and endothelial cells in the neovascular area that supports metastatic outgrowth (31, 95). Further, Gao et al. have reported that Coco a secreted antagonist of TGFβ ligand reactivates solitary breast cancer cells at organ-specific metastatic sites by shielding metastasis-initiating cells from inhibitory signals provided by lung-derived BMP proteins. A large group of patients expressing Coco showed predicted relapse to lung but not to brain and bone due to the absence of bioactive BMP (96). Hence, the metastasis-initiating cells may promote the permissive niche comprising of matrix proteins which are involved in activation of specific signaling pathways such as Wnt and Notch that in turn activate their self-renewal. Recent report suggests that the TAM family of receptor tyrosine kinases TYRO3, AXL, and MERTK have a potential role in dormancy regulation in prostate cancer. MERTK stimulates the reactivation of dormant prostate cancer cells through MAP kinase-dependent mechanism, which involves p27, pluripotency transcription factors, and histone methylation (97).
Recent achievements in cancer therapy and increased overall survival motivate the researchers to look for new diagnostics for the patients at high risk of late metastasis and therapeutic system targeting DTCs. The limitation of current conventional and adjuvant therapies to prevent relapse, is they basically target growing tumor cells rather than DTCs. The systemic nature of the metastatic disease along with the heterogeneity of metastatic tumors, complex inter-connected pathways and the resistance against therapy makes its pharmacological management very difficult. Hence, there is a need to focus on preventing metastasis (98, 99). Bone-modifying drugs have been used clinically for management of bone metastasis-related morbidity. However, when they used in the preventive adjuvant setting against cancer, inconclusive results were observed (98–100). A detailed understanding of the mechanism of metastatic dormancy and colonization along with innovative therapeutics must be developed to solve this medical dilemma. For this, therapeutic agents that can inhibit metastasis by targeting metastatic cell-autonomous functions and mechanisms responsible for dormancy and their survival would serve as a new opportunity to prevent minimal residual disease (Figure 4). Since DTCs are highly dependent on signaling, hence targeting these pathways may be helpful in enhancing the efficacy of adjuvant therapy and managing the metastatic relapse. Based on existing reports, targeting Src, Akt, or Tor by using their inhibitors alone or in combination with chemotherapy can be a potential approach for the treatment of minimal residual disease. Studies under in vivo preclinical and 3D in vitro model of dormancy demonstrated that targeting the Src family kinase and MEK1/2 using their specific inhibitors resulted in apoptosis in a large fraction of the dormant cells and delayed metastatic outgrowth in breast cancer (101). Inhibition of Src kinase family signaling or Src knockdown leads to the nuclear localization of cyclin-dependent kinase inhibitor p27 resulting in prevention of metastatic outgrowth; however, it did not affect the survival of the dormant cells. MEK1/2 inhibitors that block the downstream ERK1/2 signaling suppresses DTCs survival. Several studies have shown that the various phenotypic and functional similarities are shared between metastasis-initiating cells and CSCs. Hence, CSCs targeted therapies may be effective in the treatment of metastatic disease. Moreover, stem cell signaling pathways also induce resistance to chemotherapy. Thus, combination therapy targeting stem cell pathways like Notch and Wnt along with canonical oncogenic pathway or reactivating BMP signaling may be effective in metastatic disease therapy. It has been shown that autophagy promotes the survival of the dormant cancer cells. Interestingly, inhibition of autophagy reduces clonogenic survival of lung, cervical, and breast cancer cell (102). Therefore, autophagy can also be considered as a therapeutic target in cancer metastasis.
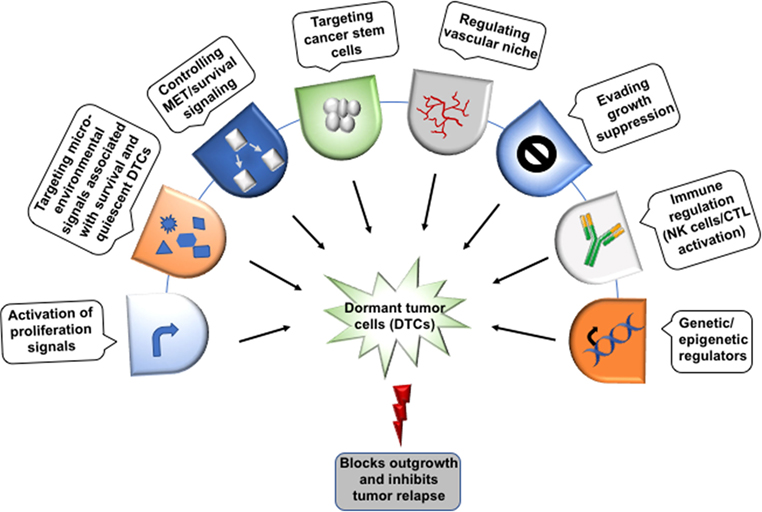
Figure 4. Therapeutic implications of dormant tumor cells. The possible target sites to eliminate the dormant tumor cells (DTCs) in order to regulate metastatic relapse. Though the direct evidence to target the dormant cells yet needs to identify extensively. Moreover, the dormant cells can be targeted at several checkpoints including epigenetic regulators (DNMT1, EZH2), immune cells (NK cells/CTL) activation, evading growth suppression, vascular niche, quiescent cancer stem cells, survival signaling, and the microenvironment signals (bone morphogenetic protein 4/7, CXCL12, TRAIL, growth arrest-specific 6, TGFβ-2, BME, and thrombospondin 1) that help in the maintenance of the dormant state.
Immunotherapy is being explored extensively for cancer management. Saudemont et al. have shown NK cells-based immune therapy targets dormant cells. Their study demonstrated that NK cells activated by CXCL10 can kill dormant tumor cells which are able to resist CTL-mediated lysis (103). As discussed earlier, secretory molecules and cytokines in microenvironment also play a key role in the regulation of dormancy (Figure 4). Osteopontin, an ECM protein has been reported in the progression of various cancers (91, 104, 105). Boyerinas et al. have shown that stromal osteopontin helps in anchoring leukemia cells in bone marrow premetastatic niche and support dormancy by inducing cell cycle arrest. Neutralizing the osteopontin resulted in the proliferation of dormant cells and enable them sensitive to chemotherapy (106). Hence, the better understanding of the mechanism governing dormancy and reactivation and the role of metastatic niche may help in the identification of new potential therapeutic targets for the treatment of minimal residual disease.
Tumor dormancy and reactivation has become an interesting point as a key element of tumor evolution and metastatic relapse. Although metastasis-initiating cells undergo dormancy and ultimately get reactivated under the influence of microenvironment signals, various key questions are still unanswered. It will be interesting to explore the phenotypic and functional similarities between DTCs and CSCs, the role of MET, microenvironmental niches and genetic and epigenetic changes in metastasis-initiating cells in metastatic reactivation. Current approaches and models to investigate the molecular basis of metastasis have been very successful. Nevertheless, new approaches need to be discovered in order to gain an in-depth understanding of tumor dormancy and reactivation. Lineage-tracing studies utilizing newly developed reporter systems can provide critical understanding in this area by using the transgenic mouse models which mimic the natural conditions. Moreover, recently invented genetic screening strategy can be useful in quick identification of mediators involved in dormancy and reactivation. Future studies need to be conducted to assess the efficacy of screening the shRNA libraries for the recognition of regulators of dormancy and their potential use in various tumor types and clinical samples.
Advance strategies for characterization of various aspects of CTCs and better access to samples of metastases will be required to complete this goal. With the current progress in the field of metastasis, these questions will be addressed rapidly by designing and implementing the improved strategies for cancer treatment.
DK, AY, PP, and RB drafted the manuscript, composed the figures, and critically revised the manuscript. DK and GK conceived the manuscript and finalized the draft. NR, SR, SB, and MG have written some part and revised the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Anuradha Bulbule for useful discussion and reading the manuscript. We apologize to many colleagues whose contributions could not site due to lack of space.
1. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell (2006) 127(4):679–95. doi:10.1016/j.cell.2006.11.00
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
3. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature (2009) 459(7249):1005–9. doi:10.1038/nature08021
4. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell (2011) 20(6):701–14. doi:10.1016/j.ccr.2011.11.002
5. Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell (2009) 16(1):67–78. doi:10.1016/j.ccr.2009.05.017
6. Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell (2008) 133(1):66–77. doi:10.1016/j.cell.2008.01.046
7. Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer (2009) 9(4):274–84. doi:10.1038/nrc2622
8. Manjili MH. Tumor dormancy and relapse: from a natural byproduct of evolution to a disease state. Cancer Res (2017) 77(10):2564–9. doi:10.1158/0008-5472.CAN-17-0068
9. Braun S, Naume B. Circulating and disseminated tumor cells. J Clin Oncol (2005) 23(8):1623–6. doi:10.1200/JCO.2005.10.073
10. Ross JS, Slodkowska EA. Circulating and disseminated tumor cells in the management of breast cancer. Am J Clin Pathol (2009) 132(2):237–45. doi:10.1309/AJCPJI7DEOLKCS6F
11. Páez D, Labonte MJ, Bohanes P, Zhang W, Benhanim L, Ning Y, et al. Cancer dormancy: a model of early dissemination and late cancer recurrence. Clin Cancer Res (2012) 18(3):645–53. doi:10.1158/1078-0432.CCR-11-2186
12. Gay LJ, Malanchi I. The sleeping ugly: tumour microenvironment’s act to make or break the spell of dormancy. Biochim Biophys Acta (2017) 1868(1):231–8. doi:10.1016/j.bbcan.2017.05.002
13. Gao XL, Zhang M, Tang YL, Liang XH. Cancer cell dormancy: mechanisms and implications of cancer recurrence and metastasis. Onco Targets Ther (2017) 10:5219–28. doi:10.2147/OTT.S140854
14. Dasgupta A, Lim AR, Ghajar CM. Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol Oncol (2017) 11(1):40–61. doi:10.1002/1878-0261.12022
15. Body JJ, Casimiro S, Costa L. Targeting bone metastases in prostate cancer: improving clinical outcome. Nat Rev Urol (2015) 12(6):340–56. doi:10.1038/nrurol.2015.90
16. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer (2007) 7(11):834–46. doi:10.1038/nrc2256
17. Yeh AC, Ramaswamy S. Mechanisms of cancer cell dormancy-another hallmark of cancer? Cancer Res (2015) 75(23):5014–22. doi:10.1158/0008-5472.CAN-15-1370
18. Todenhöfer T, Hennenlotter J, Faber F, Wallwiener D, Schilling D, Kühs U, et al. Significance of apoptotic and non-apoptotic disseminated tumor cells in the bone marrow of patients with clinically localized prostate cancer. Prostate (2015) 75(6):637–45. doi:10.1002/pros.22947
19. Gomis RR, Gawrzak S. Tumor cell dormancy. Mol Oncol (2016) 11:62–78. doi:10.1016/j.molonc.2016.09.009
20. Patel P, Chen EI. Cancer stem cells, tumor dormancy, and metastasis. Front Endocrinol (2012) 3:125. doi:10.3389/fendo.2012.00125
21. Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell (2013) 155(4):750–64. doi:10.1016/j.cell.2013.10.029
22. Evans EB, Lin SY. New insights into tumor dormancy: targeting DNA repair pathways. World J Clin Oncol (2015) 6(5):80–8. doi:10.5306/wjco.v6.i5.80
23. Havas KM, Milchevskaya V, Radic K, Alladin A, Kafkia E, Garcia M, et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J Clin Invest (2017) 127(6):2091–105. doi:10.1172/JCI89914
24. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson Å, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature (2004) 431(7012):1112–7. doi:10.1038/nature03043
25. Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med (1995) 1(2):149–53. doi:10.1038/nm0295-149
26. Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest (2010) 120(6):2030–9. doi:10.1172/JCI42002
27. Rabinovsky R, Uhr JW, Vitetta ES, Yefenof E. Cancer dormancy: lessons from a B cell lymphoma and adenocarcinoma of the prostate. Adv Cancer Res (2007) 97:189–202. doi:10.1016/S0065-230X(06)97008-0
28. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell (2016) 30(5):668–81. doi:10.1016/j.ccell.2016.09.011
29. Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer (2015) 15(4):238–47. doi:10.1038/nrc3910
30. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer (2002) 2(8):563–72. doi:10.1038/nrc865
31. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol (2013) 15(7):807–17. doi:10.1038/ncb2767
32. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med (2010) 16(1):116–22. doi:10.1038/nm.2072
33. Qian BZ, Zhang H, Li J, He T, Yeo EJ, Soong DY, et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J Exp Med (2015) 212(9):1433–48. doi:10.1084/jem.20141555
34. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438(7069):820–7. doi:10.1038/nature04186
35. Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res (2012) 72(6):1384–94. doi:10.1158/0008-5472.CAN-11-2905
36. Chen XW, Yu TJ, Zhang J, Li Y, Chen HL, Yang GF, et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene (2017) 36(35):5045–57. doi:10.1038/onc.2017.118
37. Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med (2003) 198(11):1741–52. doi:10.1084/jem.20022227
38. Studebaker AW, Storci G, Werbeck JL, Sansone P, Sasser AK, Tavolari S, et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6–dependent manner. Cancer Res (2008) 68(21):9087–95. doi:10.1158/0008-5472.CAN-08-0400
39. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer (2009) 9(4):285–93. doi:10.1038/nrc2621
40. van Deventer HW, Wu QP, Bergstralh DT, Davis BK, O’Connor BP, Ting JP, et al. C-C chemokine receptor 5 on pulmonary fibrocytes facilitates migration and promotes metastasis via matrix metalloproteinase 9. Am J Pathol (2008) 173(1):253–64. doi:10.2353/ajpath.2008.070732
41. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139(5):871–90. doi:10.1016/j.cell.2009.11.007
42. Yao D, Dai C, Peng S. Mechanism of the mesenchymal–epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res (2011) 9(12):1608–20. doi:10.1158/1541-7786.MCR-10-0568
43. Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial–mesenchymal transitions in cancer. Cancer Res (2012) 72(19):4883–9. doi:10.1158/0008-5472.CAN-12-1223
44. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell (2012) 22(6):725–36. doi:10.1016/j.ccr.2012.09.022
45. Ocaña OH, Córcoles R, Fabra Á, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell (2012) 22(6):709–24. doi:10.1016/j.ccr.2012.10.012
46. Taube JH, Sphyris N, Johnson KS, Reisenauer KN, Nesbit TA, Joseph R, et al. The H3K27me3-demethylase KDM6A is suppressed in breast cancer stem-like cells, and enables the resolution of bivalency during the mesenchymal-epithelial transition. Oncotarget (2017) 8(39):65548–65. doi:10.18632/oncotarget.19214
47. Sosa MS, Avivar-Valderas A, Bragado P, Wen HC, Aguirre-Ghiso JA. ERK1/2 and p38α/β signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clin Cancer Res (2011) 17(18):5850–7. doi:10.1158/1078-0432.CCR-10-2574
48. Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res (2003) 63(7):1684–95.
49. Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell (2001) 12(4):863–79. doi:10.1091/mbc.12.4.863
50. Deng X, Ewton DZ, Friedman E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res (2009) 69(8):3317–24. doi:10.1158/0008-5472.CAN-08-2903
51. Jin K, Ewton DZ, Park S, Hu J, Friedman E. Mirk regulates the exit of colon cancer cells from quiescence. J Biol Chem (2009) 284(34):22916–25. doi:10.1074/jbc.M109.035519
52. Ewton DZ, Hu J, Vilenchik M, Deng X, Luk KC, Polonskaia A, et al. Inactivation of mirk/dyrk1b kinase targets quiescent pancreatic cancer cells. Mol Cancer Ther (2011) 10(11):2104–14. doi:10.1158/1535-7163.MCT-11-0498
53. Hickson JA, Huo D, Vander Griend DJ, Lin A, Rinker-Schaeffer CW, Yamada SD. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res (2006) 66(4):2264–70. doi:10.1158/0008-5472.CAN-05-3676
54. Vander Griend DJ, Kocherginsky M, Hickson JA, Stadler WM, Lin A, Rinker-Schaeffer CW. Suppression of metastatic colonization by the context-dependent activation of the c-Jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Res (2005) 65(23):10984–91. doi:10.1158/0008-5472.CAN-05-2382
55. Bartkowiak K, Effenberger KE, Harder S, Andreas A, Buck F, Peter-Katalinic J, et al. Discovery of a novel unfolded protein response phenotype of cancer stem/progenitor cells from the bone marrow of breast cancer patients. J Proteome Res (2010) 9(6):3158–68. doi:10.1021/pr100039d
56. Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res (2006) 66(3):1702–11. doi:10.1158/0008-5472.CAN-05-3092
57. Chery L, Lam HM, Coleman I, Lakely B, Coleman R, Larson S, et al. Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget (2014) 5(20):9939–51. doi:10.18632/oncotarget.2480
58. Bartkowiak K, Kwiatkowski M, Buck F, Gorges TM, Nilse L, Assmann V, et al. Disseminated tumor cells persist in the bone marrow of breast cancer patients through sustained activation of the unfolded protein response. Cancer Res (2015) 75(24):5367–77. doi:10.1158/0008-5472.CAN-14-3728
59. Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci U S A (2008) 105(30):10519–24. doi:10.1073/pnas.0800939105
60. Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med (2011) 208(13):2641–55. doi:10.1084/jem.20110840
61. Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia (2010) 12(2):116–27. doi:10.1593/neo.91384
62. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell (2016) 165(1):45–60. doi:10.1016/j.cell.2016.02.025
63. Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol (2017) 19(2):120–32. doi:10.1038/ncb3465
64. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature (2016) 540:588–92. doi:10.1038/nature20609
65. Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther (2016) 1:15004. doi:10.1038/sigtrans.2015.4
66. Ono M, Kosaka N, Tominaga N, Yoshioka Y, Takeshita F, Takahashi RU, et al. Exosomes from bone marrow mesenchymal stem cells contains a microRNA that promotes dormancy in metastatic breast cancer cells. Sci Signal (2014) 7(332):ra63. doi:10.1126/scisignal.2005231
67. Almog N, Ma L, Schwager C, Brinkmann BG, Beheshti A, Vajkoczy P, et al. Consensus micro RNAs governing the switch of dormant tumors to the fast-growing angiogenic phenotype. PLoS One (2012) 7(8):e44001. doi:10.1371/journal.pone.0044001
68. Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cells. J Clin Invest (2008) 118(12):3917–29. doi:10.1172/JCI35512
69. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer (2014) 14(9):611–22. doi:10.1038/nrc3793
70. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell (2014) 159(6):1263–76. doi:10.1016/j.cell.2014.11.006
71. Lum JJ, Bauer DE, Kong M, Harris MH, Li CY, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell (2005) 120(2):237–48. doi:10.1016/j.cell.2004.11.046
72. Liang J, Saho SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1-AMPK pathway regulates p27kip1 phosphorylation mediatiing the decision to enter autophagy or apoptosis. Nat Cell Biol (2007) 9(2):218–24. doi:10.1038/ncb1537
73. Lu Z, Yang H, Sutton MN, Yang M, Clarke CH, Liao WS, et al. ARHI (DIRAS3) induces autophagy in ovarian cancer cells by downregulating the epidermal growth factor receptor, inhibiting PI3K and Ras/MAP signaling and activating the FOXo3a-mediated induction of Rab7. Cell Death Differ (2014) 21(8):1275–89. doi:10.1038/cdd.2014.48
74. Wang N, Reeves KJ, Brown HK, Fowles AC, Docherty FE, Ottewell PD, et al. The frequency of osteolytic bone metastasis is determined by conditions of the soil, not the number of seeds; evidence from in vivo models of breast and prostate cancer. J Exp Clin Cancer Res (2015) 34:124. doi:10.1186/s13046-015-0240-8
75. Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell (2015) 27(2):193–210. doi:10.1016/j.ccell.2014.11.017
76. Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat Cell Biol (2013) 15(11):1351–61. doi:10.1038/ncb2861
77. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature (2015) 526(7571):131–5. doi:10.1038/nature15260
78. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun (2015) 6:8983. doi:10.1038/ncomms9983
79. Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science (2008) 319(5860):195–8. doi:10.1126/science.1150224
80. Straume O, Shimamura T, Lampa MJ, Carretero J, Øyan AM, Jia D, et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc Natl Acad Sci U S A (2012) 109(22):8699–704. doi:10.1073/pnas.1017909109
81. Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med (2002) 6(1):1–12. doi:10.1111/j.1582-4934.2002.tb00307.x
82. Müller M, Gounari F, Prifti S, Hacker HJ, Schirrmacher V, Khazaie K. EblacZ tumor dormancy in bone marrow and lymph nodes: active control of proliferating tumor cells by CD8+ immune T cells. Cancer Res (1998) 58(23):5439–46.
83. Feuerer M, Rocha M, Bai L, Umansky V, Solomayer EF, Bastert G, et al. Enrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer patients. Int J Cancer (2001) 92(1):96–105. doi:10.1002/1097-0215(200102)9999:9999<:AID-IJC1152>3.0.CO;2-Q
84. Alvarez-Buylla A, Lim DA. For the long run: maintaining germinal niches in the adult brain. Neuron (2004) 41(5):683–6. doi:10.1016/S0896-6273(04)00111-4
85. Hsu YC, Fuchs E. A family business: stem cell progeny join the niche to regulate homeostasis. Nat Rev Mol Cell Biol (2012) 13(2):103–14. doi:10.1038/nrm3272
86. Kumar D, Gorain M, Kundu G, Kundu GC. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol Cancer (2017) 16(1):7. doi:10.1186/s12943-016-0578-3
87. Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell (2008) 132(4):598–611. doi:10.1016/j.cell.2008.01.038
88. Kumar D, Kumar S, Gorain M, Tomar D, Patil HS, Radharani NN, et al. Notch1-MAPK signaling axis regulates CD133+ cancer stem cell-mediated melanoma growth and angiogenesis. J Invest Dermatol (2016) 136(12):2462–74. doi:10.1016/j.jid.2016.07.024
89. Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer (2011) 11(6):411–25. doi:10.1038/nrc3055
90. Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature (2015) 522(7554):106–10. doi:10.1038/nature14492
91. Bandopadhyay M, Bulbule A, Butti R, Chakraborty G, Ghorpade P, Ghosh P, et al. Osteopontin as a therapeutic target for cancer. Expert Opin Ther Targets (2014) 18(8):883–95. doi:10.1517/14728222.2014.925447
92. Kelly T, Suva LJ, Huang Y, MacLeod V, Miao HQ, Walker RC, et al. Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res (2005) 65(13):5778–84. doi:10.1158/0008-5472.CAN-05-0749
93. O’Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci U S A (2011) 108(38):16002–7. doi:10.1073/pnas.1109493108
94. Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med (2011) 17(7):867–74. doi:10.1038/nm.2379
95. Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature (2011) 481(7379):85–9. doi:10.1038/nature10694
96. Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell (2012) 150(4):764–79. doi:10.1016/j.cell.2012.06.035
97. Cackowski FC, Eber MR, Rhee J, Decker AM, Yumoto K, Berry JE, et al. Mer tyrosine kinase regulates disseminated prostate cancer cellular dormancy. J Cell Biochem (2017) 118(4):891–902. doi:10.1002/jcb.25768
98. Coleman RE. Bone cancer in 2011: prevention and treatment of bone metastases. Nat Rev Clin Oncol (2011) 9(2):76–8. doi:10.1038/nrclinonc.2011.198
99. Coleman RE. Adjuvant bone-targeted therapy to prevent metastasis: lessons from the AZURE study. Curr Opin Support Palliat Care (2012) 6(3):322–9. doi:10.1097/SPC.0b013e32835689cd
100. Smith MR, Coleman RE, Klotz L, Pittman K, Milecki P, Ng S, et al. Denosumab for the prevention of skeletal complications in metastatic castration-resistant prostate cancer: comparison of skeletal-related events and symptomatic skeletal events. Ann Oncol (2015) 26(2):368–74. doi:10.1093/annonc/mdu519
101. El Touny LH, Vieira A, Mendoza A, Khanna C, Hoenerhoff MJ, Green JE. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J Clin Invest (2014) 124(1):156–68. doi:10.1172/JCI70259
102. Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res (2008) 68:1485–94. doi:10.1158/0008-5472.CAN-07-0562
103. Saudemont A, Jouy N, Hetuin D, Quesnel B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood (2005) 105(6):2428–35. doi:10.1182/blood-2004-09-3458
104. Ahmed M, Behera R, Chakraborty G, Jain S, Kumar V, Sharma P, et al. Osteopontin: a potentially important therapeutic target in cancer. Expert Opin Ther Targets (2011) 15(9):1113–26. doi:10.1517/14728222.2011.594438
105. Kumar S, Sharma P, Kumar D, Chakraborty G, Gorain M, Kundu GC. Functional characterization of stromal osteopontin in melanoma progression and metastasis. PLoS One (2013) 8(7):e69116. doi:10.1371/journal.pone.0069116
Keywords: cancer metastasis, dormancy, reactivation, tumor microenvironment, epithelial to mesenchymal transition
Citation: Yadav AS, Pandey PR, Butti R, Radharani NNV, Roy S, Bhalara SR, Gorain M, Kundu GC and Kumar D (2018) The Biology and Therapeutic Implications of Tumor Dormancy and Reactivation. Front. Oncol. 8:72. doi: 10.3389/fonc.2018.00072
Received: 09 November 2017; Accepted: 02 March 2018;
Published: 19 March 2018
Edited by:
Michael Breitenbach, University of Salzburg, AustriaReviewed by:
Markus Schosserer, University of Natural Resources and Life Sciences, Vienna, AustriaCopyright: © 2018 Yadav, Pandey, Butti, Radharani, Roy, Bhalara, Gorain, Kundu and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dhiraj Kumar, ZGt1bWFyQG1kYW5kZXJzb24ub3Jn
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.