 Sanghamitra Mylavarapu
Sanghamitra Mylavarapu Asmita Das
Asmita Das Monideepa Roy
Monideepa Roy- 1Invictus Oncology Pvt. Ltd., Delhi, India
- 2Department of Biotechnology, Delhi Technological University, Delhi, India
Recent years have seen cancer emerge as one of the leading cause of mortality worldwide with breast cancer being the second most common cause of death among women. Individuals harboring BRCA mutations are at a higher risk of developing breast and/or ovarian cancers. This risk is much greater in the presence of germline mutations. BRCA1 and BRCA2 play crucial role in the DNA damage response and repair pathway, a function that is critical in preserving the integrity of the genome. Mutations that interfere with normal cellular function of BRCA not only lead to onset and progression of cancer but also modulate therapy outcome of treatment with platinum drugs. In this review, we discuss the structural and functional impact of some of the prevalent BRCA mutations in breast and ovarian cancers and their role in platinum therapy response. Understanding the response of platinum drugs in the context of BRCA mutations may contribute toward developing better therapeutics that can improve survival and quality of life of patients.
Introduction
Cancer is one of the leading causes of mortality worldwide. As per WHO estimates, 8.8 million cancer related deaths were reported in 2015, and this number is projected to rise to 13.1 million by the year 2030, with low- and middle-income countries bearing approximately 70% burden of all deaths worldwide. Of these, breast cancer has emerged as the second major cancer type comprising almost 25% of all cancers among women. Breast cancer is the most commonly occurring cancer among women with incidence rates varying widely across the world, having rates ranging from 27 per 100,000 in Middle Africa and Eastern Asia to 92 in Northern America. But the mortality rate is lower in the developed countries as compared with low- and middle-income countries, because of higher survival of breast cancer patients in developed nations (1, 2).
Mutations in breast cancer susceptibility gene type 1 and type 2 (BRCA1 and BRCA2) put women at a higher risk of developing breast and/or ovarian cancer. In individuals harboring mutations in BRCA1, the probability of developing breast cancer over a lifetime is about 57–65% and that of ovarian cancer is about 39–40%. With BRCA2 mutations, the probabilities are at 45–49% for breast cancer and 11–18% for ovarian cancer (3, 4). Women with germline mutations are more prone to develop these cancers at a younger age with more aggressive disease and poorer prognosis as compared to those with somatic mutations. BRCA-mutated tumors exhibit both higher clinical grade and stage disease with greater metastatic potential (5). 70% of breast tumors containing germline mutations in BRCA1 fall in the category of triple-negative breast cancer (TNBC), a highly aggressive, highly metastatic subtype, comprising approximately 15% of all breast cancer cases, characterized by the absence of hormone receptors with no amplification of growth signal receptor (6).
Ovarian cancer patients with BRCA mutations exhibit a higher histological grade disease compared to those with sporadic disease and respond better to platinum therapy, having better prognosis (7, 8). In the absence of functional BRCA proteins, cells fail to repair intra-strand crosslinks formed by DNA cross-linking agents such as platinum drugs, leading to apoptotic cell death. Cisplatin is the most commonly used therapeutic agent for treating gynecological cancers either as a single agent before surgery or in combination with other drugs. Despite the favorable initial response, these cancers eventually develop tolerance to platinum leading to therapy failure. This review examines a few clinically relevant mutations that are common in breast and ovarian cancers. The structural and functional changes resulting from these mutations are explored further, focusing on their implications in modulating response to platinum therapy.
BRCA Mutations in Breast and Ovarian Cancers
BRCA1 and BRCA2 play a crucial role in maintaining genome integrity by repairing double-strand DNA breaks via the homologous recombination repair (HRR) pathway. Any mutations that cause functional disruption of these proteins may prove to be highly deleterious, leading to the development of cancer. In addition, BRCA1 and BRCA2 also play a critical role in cell division where they are transported to the cytosol to participate in regulating various molecular events during mitosis. Mutations impacting these important functions of BRCA1/2 can affect the delicate balance of the tightly regulated cellular processes that may lead to progression of disease.
BRCA mutations show huge diversity in various populations, many of which are functionally neutral or are of unknown pathological significance. However, there are some mutations that are more significant than others (Table 1). One of the most common cancer related mutations found in BRCA1 is the 5382insC, reported to have originated from a common European ancestor about 400–500 years ago. It was first described as the founder mutation in the Ashkenazi Jew population and could be present in other European populations as well. This mutation is also reported to be associated with a higher incidence of ovarian cancer (9.4%) but a lower incidence of breast cancer in Slavic countries (9). 185delAG located in exon 2 of BRCA1 is another common mutation reported in various ethnicities including Ashkenazi Jews and Indian population where it occurs at a high frequency of 16.4%. Missense mutation at the Cys61 (C61G) of BRCA1 is a founder mutation in Polish population and is included as a standard test for diagnosis and treatment of breast and ovarian cancer for Polish women (10, 11). 6174delT mutation is common in BRCA2 in the Ashkenazi Jewish population and other ethnic groups (12). In addition, BRCA1 and BRCA2 contain numerous other mutations that show a more population-specific distribution. This has been summarized in a review by Karami and Mehdipour (9). Screening for specific BRCA1/2 mutations that occur at high frequency in certain populations not only help in better clinical management of breast and ovarian cancers but can also be an invaluable tool in identifying healthy individuals who are currently disease-free but are at an increased risk of developing breast and/or ovarian cancer later in life (13). In addition to the presence of either BRCA1 or BRCA2 mutations in breast and/ovarian cancer patients, there are reports of patients being double heterozygous for both BRCA1 and BRCA2 mutations. These patients develop cancer at a much earlier age and with more severe disease (14, 15).
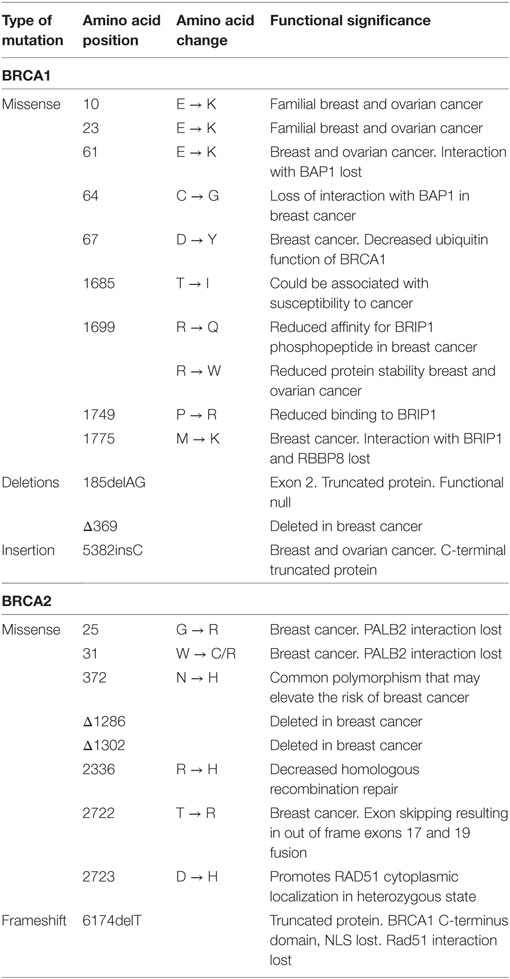
Table 1. Clinically significant BRCA1/2 mutations in breast and ovarian cancers.
Cancer-Associated Mutations Alter the Biology of BRCA Proteins
BRCA1 and BRCA2 are at the intersection of numerous key cellular pathways and perform multiple functions ranging from DNA damage response and DNA repair activity, chromatin remodeling and transcription, and protein ubiquitination (16–19) (Figure 1). Single- or double-strand DNA breaks may arise from various sources like natural metabolic processes or from extraneous sources like chemical agents or irradiation. Left unrepaired, these DNA breaks may lead to the accumulation of deleterious mutations with a potential to cause genomic instability. Efficient repair of double-strand breaks via the HRR pathway requires functional involvement of BRCA1 and BRCA2 via their interaction with numerous other proteins (20, 21). Upon DNA damage, BRCA1 acts as a mediator bringing together components of the DNA repair pathway to the site of damage where it interacts with a large complex called the BRCA1-associated genome surveillance complex (BASC complex), along with the components of the DNA repair machinery (22, 23). Although BRCA1 and BRCA2 function in the DNA damage repair pathway, both have functionally distinct roles (23). BRCA1 functions as a DNA damage checkpoint activator and also in DNA repair, whereas BRCA2 is a core component of the HRR machinery. Homozygous BRCA1 knockout mice are embryonic lethal at age E7.5–E13.5, suggesting that functional loss of BRCA1 cannot be compensated by the presence of wild-type BRCA2 (24).
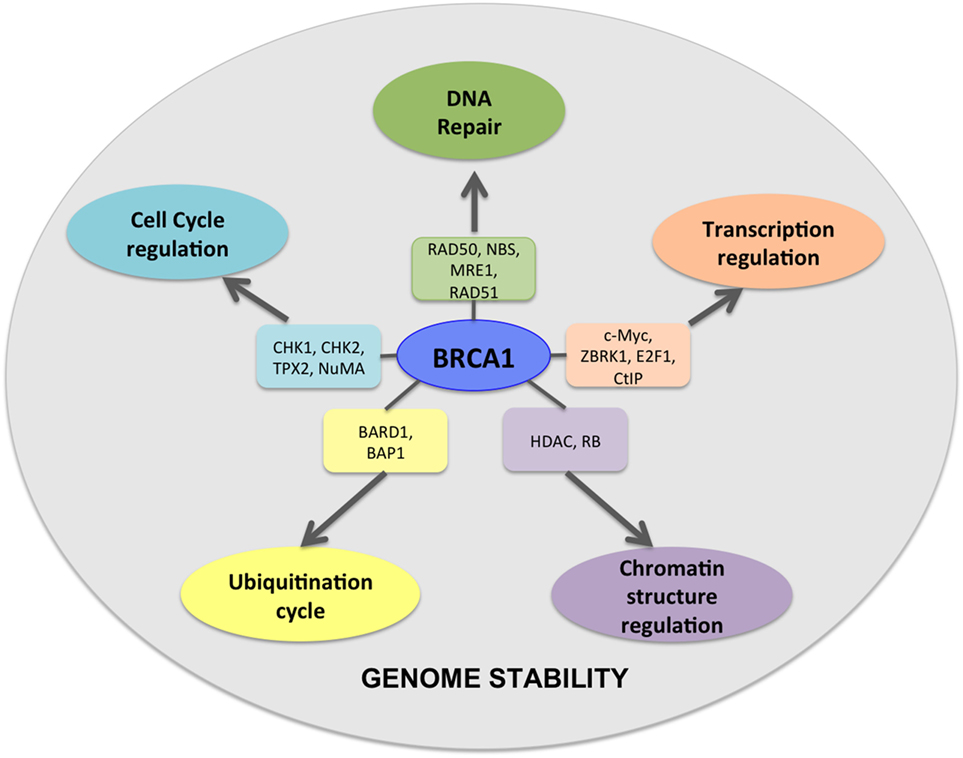
Figure 1. BRCA1 is at the hub of numerous interconnecting cellular pathways. BRCA1 interacts with numerous intermediate proteins in these pathways that contribute toward genomic and cellular stability.
The BRCA proteins are organized into functional domains that enable numerous protein–protein interactions that are vital for their optimal function. BRCA1, a 220 kDa protein, contains an RING domain in the N-terminus that interacts with BRCA1-associated RING domain protein 1 (BARD1), the heterodimerization of which increases BRCA1 ubiquitin ligase activity by many folds. The BRCA1 C-terminus domain is conserved across many proteins that are involved in DNA repair and is the site for numerous phosphoprotein interactions. Limited structural data are available for the region of BRCA1 between exons 11 and 13, despite it being the binding site for a number of proteins that are involved in multiple cellular pathways. Some of the proteins that bind to this region of BRCA1 are retinoblastoma protein (RB), the transcription factor c-Myc, DNA repair proteins RAD50, RAD51, and PALB2 that forms a scaffold for BRCA1 and BRCA2 interaction (25). A common feature shared by many proteins that are at the hub of interconnecting pathways is the intrinsically disordered structure (26). Exons 11–13 of BRCA1 exhibit such disordered structure that perhaps provide a scaffold for multiple interactions and signal integration from various pathways (27). BRCA2 is a large 385 kDa protein with an N-terminus transactivation domain, a long exon 11 containing RAD51-specific binding site and a DNA binding domain toward the C-terminus (28). BRCA2, like BRCA1, contains disordered structure interspersed between more structured motifs, suggesting its participation in multiple cellular pathways (29, 30) (Figure 2). Here, we discuss some key mutations in BRCA1 and BRCA2 that have strong correlation with breast and ovarian cancer and their functional consequences (Figures 3A,B).
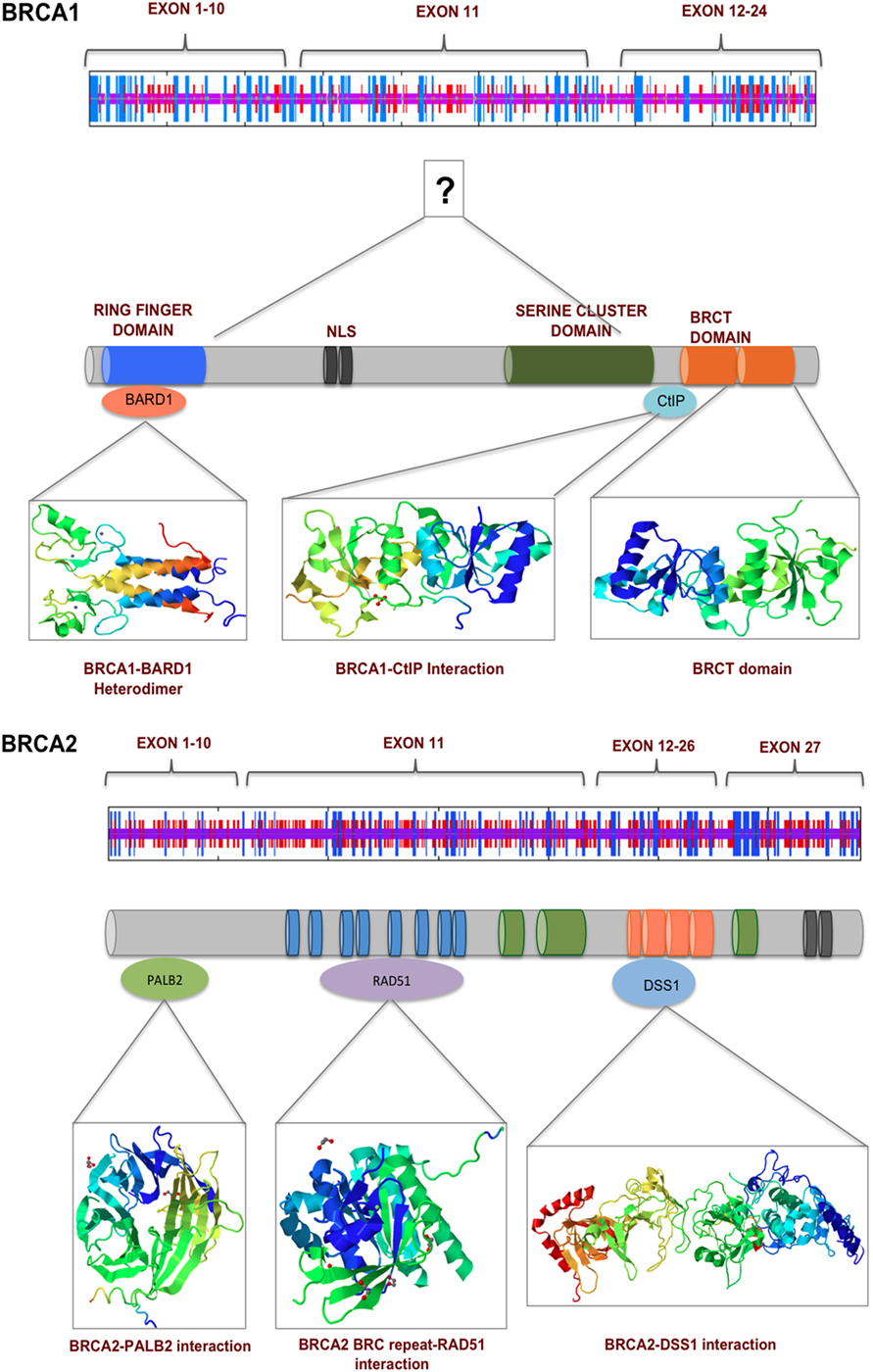
Figure 2. BRCA1 and BRCA2 interact with numerous proteins via their multiple functional domains. The N and C termini of BRCA1 have structural motifs that allow multiple protein–protein interaction. Exons 11–13 in the middle of BRCA1 are more unstructured, which contains two nuclear localization signals. BRCA2 secondary structure prediction indicate a more helical middle region, which contains BRC repeats and binding sited for various proteins of the DNA damage response pathway.
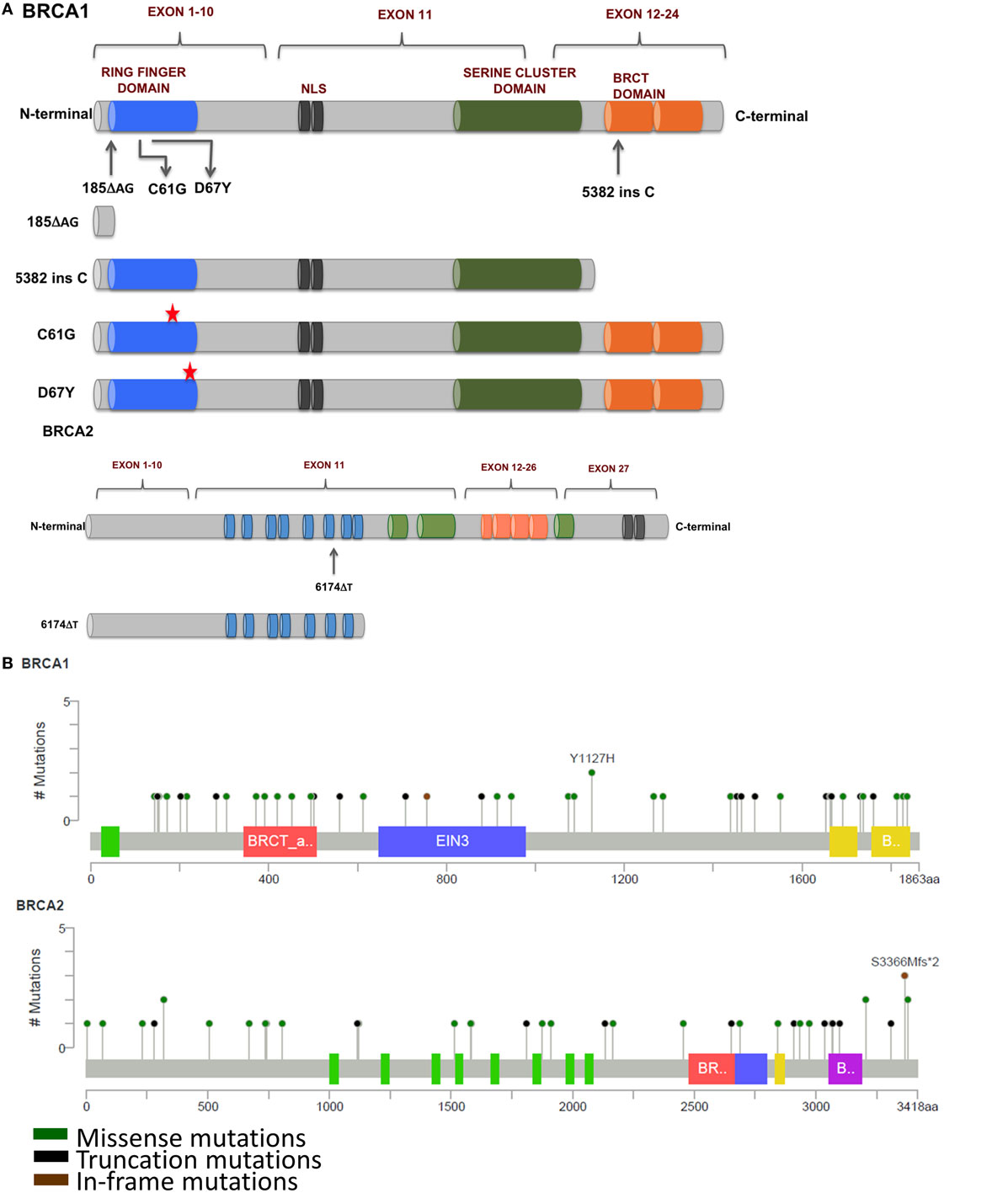
Figure 3. (A) Mutations in BRCA1 and BRCA2 found in breast and ovarian cancers that affect response to platinum therapy. Deletion of multiple functional domains from BRCA1 result in truncated protein that is unable to repair DNA damage efficiently whereas missense mutations that result in amino acid substitutions result in altered response to platinum therapy. (B) Schematic representation of prevalent somatic mutations in BRCA1 and BRCA2 from breast cancer patients showing relative positions of various missense, truncations, and in-frame mutations (Adapted from www.cBioPortal.org) (31).
185delAG—A two nucleotide deletion in exon 2 of BRCA1 produces a 39 amino acids long, functionally null truncated protein that has lost most of its functional domains as compared with a 220 kDa wild-type protein. The loss of the ring finger domain abrogates the binding of BARD1 and BAP1, which are important regulators of E3 ubiquitin ligase activity of BRCA1. Therefore, the loss of the ring finger domain of BRCA1 and consequently, loss of E3 ubiquitin ligase activity may be an important event in the development and progression of breast cancer (32, 33). BRCA1 interacts with both upstream and downstream effectors of cell cycle checkpoint kinases—deletion of binding domains result in the loss of these interactions and failure of the S and G2/M phase checkpoints allowing cell cycle progression even in the presence of DNA damage (34–37). Interaction of BRCA1 and BRCA2 is mediated by PALB2, and the complex is critical in RAD51-mediated HRR of damaged DNA. Disruption of this interaction by functional loss of BRCA1 protein results in defective DNA repair and consequently propagation of mutations that accumulate in daughter cells during cell division (38). However, nuclear expression of Maspin (mammary serine protease inhibitor), a member of the serpin superfamily and a target of truncated BRCA1, has been correlated with increased sensitivity to cisplatin with improved prognosis in ovarian cancer (39).
5382insC produces a C-terminal truncated BRCA1 commonly encountered in breast and ovarian tumors. The C-terminus of BRCA1 contains numerous domains for various protein–protein interactions including RNA-helicase binding, HDAC interaction, CtIP binding, and many others proteins (40–42). Thangaraju and colleagues have reported that the deletion of the C-terminus of BRCA1 through the 5382insC mutation resulted in loss of apoptosis in cell lines. It has been speculated that the deletion of the transactivation domains may be responsible for loss of transcriptional activation and/or repression of several genes, which may lead to apoptotic cell death (43).
6174delT—A frameshift mutation in the BRCA2 produces a 224 kDa truncated protein, about 2,002 amino acids long as compared with the 3,418 amino acids long wild-type protein (390 kDa). This truncation leads to the loss of two BRC repeat domains, loss of DNA binding, and the C-terminus RAD51 binding domain along with the nuclear localization signal, resulting in cells’ defective DNA repair machinery that are unable to form RAD51 foci (44). In addition, cells carrying this mutation have a higher sensitivity to inhibitors of poly(ADP-ribose) polymerase (PARP). Edwards et al. reported that deletion of the 6174delT mutation could make the otherwise PARP inhibitor sensitive cells resistant to the drug. This acquired resistance to PARP inhibitors could be reversed upon restoration of the reading frame of BRCA2 (45), providing a window of opportunity for development of newer and effective strategies for clinical management of BRCA2-mutated cancers. C-terminal truncation of BRCA2 results in the loss of DSS1-mediated stabilization and rapid degradation of BRCA2, leaving cells vulnerable to DNA damage (46, 47). Other interacting partners of BRCA2 include EMSY and DMC1. Sporadic breast and ovarian tumors show amplification of EMSY, a binding partner of BRCA2, and have been associated with poor survival. Interaction of EMSY with exon 3 of wild-type BRCA2 (deleted in cancer) functionally inactivates BRCA2, suggesting a regulatory role for EMSY in the HRR pathway (48). The region between amino acids 2386 and 2411 of BRCA2 is highly conserved across species and is the binding site for DMC1, a germ cell counterpart of RAD51. Loss of this interaction due to the 6174delT mutation results in defective meiosis and propagation of chromosomal abnormalities in the germline (49).
Sporadic Cancer and BRCAness
BRCA1/2 mutations, seen most commonly in familial breast and ovarian tumors, impact the DNA repair pathway leading to genomic instability. However, some sporadic tumors that contain wild-type BRCA1 also have defective DNA repair pathway that may have resulted via other mechanisms. These characteristics of sporadic tumors that are similar to familial cancers are collectively called “BRCAness.” Inactivation of BRCA1 in sporadic breast and ovarian tumors may be brought about by non-genetic mechanisms like promoter methylation that result in lowering of gene expression to undetectable levels and loss of heterozygosity. In contrast, BRCA2 inactivation does not occur by promoter hypermethylation—a significant number of sporadic breast and ovarian tumors show amplification of EMSY at the gene level. As discussed earlier, EMSY–BRCA2 interaction may regulate DNA repair via HRR pathway (50). A common feature that both BRCA-mutated cancers and those showing characteristics of BRCAness share is the elevated susceptibility to DNA cross-linking agents like platinum drugs and this has been the rationale for including these as therapeutic agents (8).
BRCA-Mutated Tumors are Sensitive to Platinum Therapy
Ever since its approval as an antineoplastic agent, cisplatin has been effective in BRCA-mutated breast cancer either as a single agent or in combination with other anticancer drugs (51, 52). The involvement of BRCA1 in efficient DNA repair mechanism has been highlighted by in vitro studies that showed that cells containing mutant BRCA1 showed increased sensitivity to platinum drugs as compared with those cells that have elevated BRCA1 levels. This heightened sensitivity could, however be, reversed upon restoration of the full-length functional BRCA1 (53, 54). BRCA1 has been identified as a key gene of the DNA repair machinery not only by siRNA screens but also using BRCA1, TP53 conditional knockout mice where animals after an initial favorable response to cisplatin, doxorubicin, and docetaxel, became resistant to doxorubicin and docetaxel but remained sensitive to cisplatin (55). Breast cancer xenografts using HCC1935 with mutated BRCA1 showed complete inhibition of tumor growth upon treatment with cisplatin whereas only partial response in xenografts that had the wild-type BRCA1 reconstituted. Moreover, significant cell cycle arrest at the S phase and G2/M transition was observed. This not only points to the role of BRCA1 in DNA repair pathway and cell cycle checkpoint activation but also its involvement in modulating response to platinum drugs (56, 57).
Although BRCA-mutated breast tumors exhibit higher histological grade due to increased accumulation of chromosomal aberrations over time and are particularly sensitive to platinum drugs, their therapeutic use is limited to first-line treatment mainly due to development of resistance (58–62). This was demonstrated as early as 1988 when 47% partial response was achieved with a dose of 30 mg/m2/day of cisplatin for 4 days, every 3 weeks, in six cycles as a first-line therapy for metastatic breast cancer (63). In another study, 83% pathologic complete response (pCR) was reported in breast cancer patients treated with cisplatin as a neoadjuvant treatment for TNBC. Also, higher response to platinum and pCR has been shown to be associated with low BRCA1 expression, promoter methylation, p53 frameshift or nonsense mutation, and E2F3 activation (64). Huang et al. reported more than 11 years of remission in a BRCA2-mutated metastatic breast cancer patient who received chemotherapy using high dose of cisplatin along with anthracyclin and alkylating agents (65). BRCA1-mutated metastatic breast cancer that was unresponsive to docetaxel, responded well to cisplatin and gemcitabine combination therapy for more than 6 months (66). In case of ovarian cancer, BRCA1 and BRCA2 patients exhibit a differential age of tumor development—BRCA1 carriers develop tumors earlier than those carrying BRCA2 mutations (48 vs. 57 years). Also, BRCA mutations seem to render the tumors more responsive to platinum drugs with a better survival (91 vs. 54 months) and longer disease-free interval (49 vs. 19 months) as compared with sporadic ovarian cancer (67, 68).
Secondary Somatic BRCA Mutations Contribute to the Development of Resistance to Platinum Drugs
28.3% of recurrent ovarian cancer tumors contain secondary mutations as compared with only 3.1% in primary tumors, while 46.2% of platinum resistant tumors have secondary mutations that restored the function of BRCA1/2 as compared with 5.3% that are sensitive to platinum drugs (69). This implies that the secondary mutations in BRCA1/2 may be instrumental in the development of drug resistance to platinum therapeutics. The 39 amino acid long, non-functional BRCA1, produced due to the 185delAG mutation in the exon 2, severely impair its DNA repair function making the cells highly sensitive to platinum. This may get circumvented by the restoration of the reading frame that produces the full-length functional protein resulting in efficient DNA damage repair. Various studies indicate that platinum refractory BRCA1-mutated tumors carry two nucleotide insertions that are otherwise not present in BRCA1-mutated platinum-sensitive tumors. Similar results were demonstrated in case of BRCA2 mutations. In addition, new secondary mutations arise due to selection pressure exerted by prolonged exposure to platinum drugs resulting in resistance to both platinum drugs as well as PARP inhibitors (70). Such accumulation of secondary mutations could contribute to the development of resistance to platinum drugs over time. The presence of secondary mutations restoring the function of BRCA1/2 could be developed as a promising prognostic marker for either screening patients that are likely to respond favorably to platinum therapy or for predicting clinical outcome in patients already receiving platinum drugs or PARP inhibitor as single agent or in combination (71).
Apart from accumulated secondary mutations in BRCA, missense mutations in the ring finger domain affect response to platinum drugs. C61G missense mutation in exon 5 of BRCA1 abrogates BRCA1–BARD1 heterodimerization leading to loss of E3 ubiquitin ligase activity and cytoplasmic mislocalization of BRCA1. This results in diminished availability of BRCA1 at the site of DNA damage and impaired DNA repair, increasing vulnerability of these cells to platinum drugs. Homozygous C61G mice are embryonic lethal with severe developmental delays, mimicking phenotype of BRCA1 null mice. Interestingly, unlike BRCA1 null mice, these mice were reported to have developed resistance to both cisplatin and the PARP inhibitor olaparib, suggesting a hypomorphic activity of C61G mutation in BRCA1 (72). D67Y, another missense mutation that substitutes aspartic acid for a tyrosine residue at the 67th position result in decreased ubiquitin function of the mutated BRCA1 protein. In contrast to the C61G mutation, the D67Y mutation has been reported to increase cisplatin sensitivity in vitro (73).
In addition to mutational inactivation of BRCA function contributing to resistance to platinum drugs, several other mechanisms may exist that result in tumors not responding to platinum therapy (Figure 4). The dynamic balance between influx and efflux of platinum drugs influence its availability in the intracellular milieu to form DNA adducts and ultimately lead to apoptotic cell death. Copper transporters (CTR1 and CTR2) and copper-transporting p-type adenosine triphosphatase 1 and 2 (ATP7A and ATP7B) are key molecules that are responsible for intracellular transport of platinum drugs. In a meta-analysis study, Sun and colleagues reported positive correlation between CTR1 expression levels and response to cisplatin with favorable overall survival, progression, and disease-free survival. However, no such correlation could be made for CTR2, ATP7A, and ATP7B (74). Therefore, how a patient would response to platinum therapy is dependent not only on the occurrence of mutations that functionally inactivate BRCA1/2 but also to a large degree on other cellular mechanisms such as ion transport pathways that influence the dynamics of the drug within the tumor.
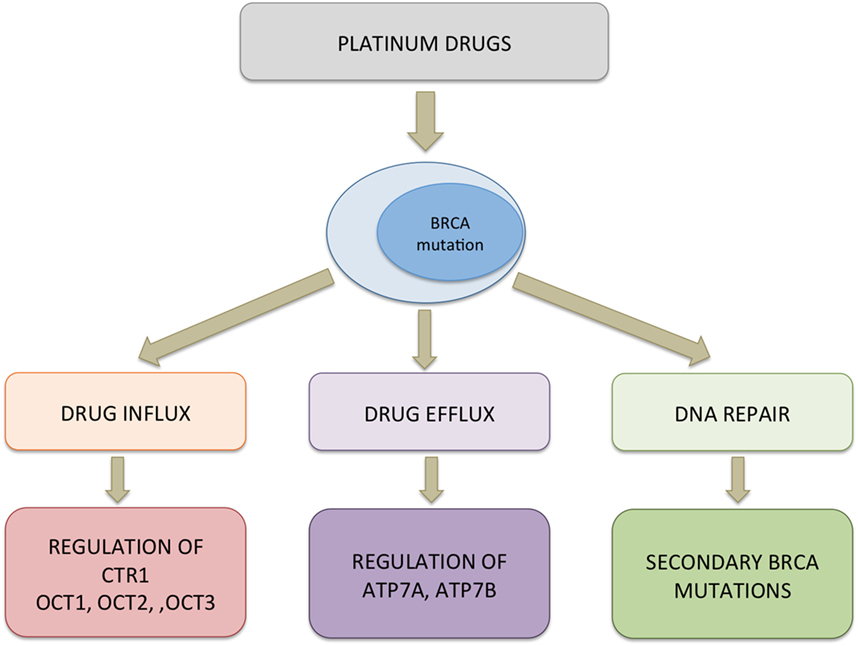
Figure 4. Probable mechanism of resistance to platinum drugs in BRCA-mutated cells. Multiple pathways could be activated simultaneously conferring resistance to platinum drugs in cells. Downregulation of copper transporter (CTR1) and organic cation transporters (OCT1/2/3) can decrease the uptake of platinum drugs whereas upregulation of efflux pumps can bring about increased transport of the drug out of the cells. In the cytosol, platinum drugs can bind to a protein such as chaperons that interfere with the drug reaching the target. Functional restoration of BRCA1/2 and elevated expression of ERCC1 leading to effective removal of platinum–DNA adducts and efficient DNA repair contribute to decreased cytotoxicity of platinum drugs.
Overcoming Platinum Resistance in BRCA-Mutated Cancers
Treatment failure as a consequence of development of resistance to platinum drugs resulting in disease recurrence is a major roadblock in clinical management of cancers that carry BRCA mutations. In such cases, non-platinum-based drugs can achieve improved response and extended survival. PARP-1 and PARP-2, members of the PARP family, recruit the base excision repair (BER) machinery, a parallel, but less efficient mechanism for repairing damaged DNA, to the site of single-strand DNA breaks. In the absence of PARP, these lesions persist resulting in stalling of the replication fork during DNA synthesis and formation of DSBs (75). The concept of synthetic lethality, the underlying premise of which is that functional depletion of two genes singly may not cause deleterious effect but together are lethal, was thus introduced to cancer therapeutics to develop better treatment strategies. This was mainly achieved by blocking the BER pathway by small molecule inhibitors against PARP in BRCA1/2-mutated cancers (76).
Poly(ADP-ribose) polymerase inhibitor olaparib has recently received FDA approval for treatment of ovarian cancer with mutated BRCA1/2 and EMEA approval for maintenance therapy for platinum-sensitive ovarian cancer (77). Randomized phase II clinical trial for evaluating the efficacy of olaparib, the first clinically approved PARP inhibitor, in combination with either paclitaxel or carboplatin, followed by olaparib monotherapy resulted in improved progression-free survival in BRCA-mutated high grade ovarian cancer patients compared with treatment with paclitaxel and carboplatin (78). PARP inhibitors have also been reported to be effective in other BRCA-mutated cancers. Clinical trials are underway to assess the efficacy of these (79, 80). Other PARP inhibitors such as niraparib, veliparib, and rucaparib are currently being tested in various clinical trials (81). Kim et al. reported that there is no clear correlation between BRCA1 expression and response to docetaxel (82), while Byrski et al. reported that non-BRCA1 mutation carriers showed higher complete or partial response to docetaxel as neoadjuvant therapy when compared with patients who were BRCA1 mutation carriers (83). In vitro studies using breast cancer cells indicate that BRCA1 mutations make the cells non-responsive to taxanes, an observation supported by in vivo experiments where targeted deletion of p53 and BRCA1 in mammary tissue rendered tumors resistant to docetaxel but not cisplatin (52). Clinical correlation between BRCA1 mutation and response to taxane is not clear with several studies reporting data that are largely inconclusive.
Conclusion
Structural organization of BRCA1 and BRCA2 into functionally distinct domains allow for multiple protein–protein interactions with numerous binding partners that facilitate participation in various cellular activities including DNA damage repair pathway. Specific protein–protein interaction such as BRCA1 and BARD1 that is crucial for the E3 ubiquitin ligase activity and the numerous phosphoproteins that bind to the C-terminus of BRCA1 make it an important node in the highly branched intracellular signaling network (29, 30, 84–88). Most common type of mutation observed among breast and ovarian cancer patients is deletion of multiple functional domains of BRCA1/2 that lead to not only impaired DNA repair but also abrogation of cell cycle checkpoints and transcriptional mis-regulation of genes, which eventually lead to global genomic instability. Suboptimal DNA repair mechanism arising from BRCA mutations render cells highly vulnerable to DNA cross-linking agents, such as platinum drugs, making them useful therapeutics in many cancers. In vitro data have provided strong evidence toward better response to platinum drugs in the presence of BRCA mutations and is corroborated by clinical studies where BRCA mutation carriers exhibit better survival and longer disease-free intervals upon treatment with platinum drugs, suggesting beneficial therapy outcome. Along with targeting the suboptimal HRR pathway that results from non-functional BRCA1/2, the blocking of the BER pathway with PARP inhibitors significantly improves survival rates of cancer patients. Although BRCA1/2-mutated tumors are highly susceptible to platinum drugs and PARP inhibitors, development of resistance poses a major challenge in the clinical management of these cancers and secondary mutations significantly contribute toward this.
The inherent susceptibility of BRCA-mutated tumors to platinum drugs makes it an appropriate target for development of newer therapeutic agents. The ongoing efforts to design and develop novel inhibitors for the various components of the DNA repair pathway may yield encouraging results and in combination with platinum drugs could further improve the treatment options available for cancer patients. In addition, molecular signatures that can predict the outcome of a treatment regimen are being evaluated as biomarkers, which may help in identifying a target population that is more likely to respond to therapy. For instance, the C118T and C8092A polymorphisms in ERCC1 have been strongly correlated with objective response rate and overall survival (89). Similarly, the levels of annexin A3 in the peripheral blood may be a potential predictor for platinum resistance in ovarian cancer (90). Although molecular markers have the potential to predict therapy response, rigorous validation in large patient cohorts would truly bring out the benefits of such predictions. The formation of DNA–platinum adducts and subsequent progression toward apoptotic cell death was believed to be the mechanism of action for platinum drugs. Recent advances have brought forth other novel mechanisms by which platinum drugs exert their cytotoxic effect. One such mechanism is the ability of platinum drugs to modulate the host immune system. Treatment regimen that includes platinum drugs in combination with immune checkpoint blockers, such as anti-CTLA4 antibody and ipilimumab, has been approved for metastatic melanoma, whereas anti-PD-L1 antibody, atezolizumab, has been approved for metastatic lung cancer (91).
Platinum drugs, although highly efficacious in patients carrying BRCA1 and BRCA2 mutations, come at a cost in the form of severe side effects—nephrotoxicty and ototoxicity being two of the major unwanted effects of cisplatin treatment. The major challenge is to target the drug specifically to the tumor alone and minimize its accumulation in non-tumor tissue. Development of better drug-delivery vehicles that can ensure targeted delivery at the tumor site will not only maximize availability of the therapeutic agent at the site of the tumor but also reduce accumulation in healthier tissue, thereby minimizing toxicity. Thus, a therapeutic regimen that includes inhibitors for the DNA repair pathway, a tumor-specific platinum drug, together with radiotherapy may prove to be an effective way of treating and managing the cancer burden due to BRCA1 and BRCA2 mutations.
Author Contributions
MR was involved in the conceptualization of the topic and supervised the writing of the manuscript. SM wrote the manuscript and drew the figures. AD critically reviewed the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
SM and MR are employees of Invictus Oncology Pvt. Ltd. and hold equity of IOPL. All remaining authors have no potential conflicts to disclose.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136(5):E359–86. doi:10.1002/ijc.29210
2. WHO Factsheet. (2017). Available from: http://www.who.int/mediacentre/factsheets/fs297/en/
3. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet (2003) 72(5):1117–30. doi:10.1086/375033
4. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol (2007) 25(11):1329–33. doi:10.1200/JCO.2006.09.1066
5. Musolino A, Bella MA, Bortesi B, Michiara M, Naldi N, Zanelli P, et al. BRCA mutations, molecular markers, and clinical variables in early-onset breast cancer: a population-based study. Breast (2007) 16(3):280–92. doi:10.1016/j.breast.2006.12.003
6. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res (2004) 10(16):5367–74. doi:10.1158/1078-0432.CCR-04-0220
7. Pan Z, Xie X. BRCA mutations in the manifestation and treatment of ovarian cancer. Oncotarget (2017) 8(57):97657–70. doi:10.18632/oncotarget.18280
8. Byrski T, Huzarski T, Dent R, Marczyk E, Jasiowka M, Gronwald J, et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat (2014) 147(2):401–5. doi:10.1007/s10549-014-3100-x
9. Karami F, Mehdipour P. A comprehensive focus on global spectrum of BRCA1 and BRCA2 mutations in breast cancer. Biomed Res Int (2013) 2013:928562. doi:10.1155/2013/928562
10. Menkiszak J, Chudecka-Głaz A, Gronwald J, Bedner R, Cymbaluk-Płoska A, Wezowska M, et al. Characteristics of selected clinical features in BRCA1 mutation carriers affected with breast cancer undergoing preventive female genital tract surgeries. Ginekol Pol (2013) 84(9):758–64. doi:10.17772/gp/1636
11. Szwiec M, Jakubowska A, Górski B, Huzarski T, Tomiczek-Szwiec J, Gronwald J, et al. Recurrent mutations of BRCA1 and BRCA2 in Poland: an update. Clin Genet (2015) 87(3):288–92. doi:10.1111/cge.12360
12. Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med (1997) 336(20):1401–8. doi:10.1056/NEJM199705153362001
13. Karakasis K, Burnier JV, Bowering V, Oza AM, Lheureux S. Ovarian cancer and BRCA1/2 testing: opportunities to improve clinical care and disease prevention. Front Oncol (2016) 6:119. doi:10.3389/fonc.2016.00119
14. Heidemann S, Fischer C, Engel C, Fischer B, Harder L, Schlegelberger B, et al. Double heterozygosity for mutations in BRCA1 and BRCA2 in German breast cancer patients: implications on test strategies and clinical management. Breast Cancer Res Treat (2012) 134(3):1229–39. doi:10.1007/s10549-012-2050-4
15. Lavie O, Narod S, Lejbkowicz F, Dishon S, Goldberg Y, Gemer O, et al. Double heterozygosity in the BRCA1 and BRCA2 genes in the Jewish population. Ann Oncol (2011) 22(4):964–6. doi:10.1093/annonc/mdq460
16. Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science (1990) 250(4988):1684–9. doi:10.1126/science.2270482
17. Yarden RI, Papa MZ. BRCA1 at the crossroad of multiple cellular pathways: approaches for therapeutic interventions. Mol Cancer Ther (2006) 5(6):1396–404. doi:10.1158/1535-7163.MCT-05-0471
18. Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science (1994) 265(5181):2088–90. doi:10.1126/science.8091231
19. Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene (2003) 22(37):5784–91. doi:10.1038/sj.onc.1206678
20. Haber JE. Partners and pathways repairing a double-strand break. Trends Genet (2000) 16:259–64. doi:10.1016/S0168-9525(00)02022-9
21. Friedenson B. The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer (2007) 7:152. doi:10.1186/1471-2407-7-152
22. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev (2000) 14(8):927–39.
23. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer (2011) 12(1):68–78. doi:10.1038/nrc3181
24. Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene (2006) 25(43):5885–97. doi:10.1038/sj.onc.1209871
25. Clark SL, Rodriguez AM, Snyder RR, Hankins GD, Boehning D. Structure-function of the tumor suppressor BRCA1. Comput Struct Biotechnol J (2012) 1(1). doi:10.5936/csbj.201204005
26. Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J (2005) 272(20):5129–48. doi:10.1111/j.1742-4658.2005.04948.x
27. Mark WY, Liao JC, Lu Y, Ayed A, Laister R, Szymczyna B, et al. Characterization of segments from the central region of BRCA1: an intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J Mol Biol (2005) 345(2):275–87. doi:10.1016/j.jmb.2004.10.045
28. Chen JJ, Silver D, Cantor S, Livingston DM, Scully R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res (1999) 59(7 Suppl):1752s–6s.
29. Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature (2002) 420(6913):287–93. doi:10.1038/nature01230
30. Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science (2002) 297(5588):1837–48. doi:10.1126/science.297.5588.1837
31. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6(269):1. doi:10.1126/scisignal.2004088
32. Nishikawa H, Wu W, Koike A, Kojima R, Gomi H, Fukuda M, et al. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res (2009) 69(1):111–9. doi:10.1158/0008-5472.CAN-08-3355
33. Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene (1998) 16(9):1097–112. doi:10.1038/sj.onc.1201861
34. Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science (1999) 286(5442):1162–6. doi:10.1126/science.286.5442.1162
35. Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, et al. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev (2000) 14(23):2989–3002. doi:10.1101/gad.851000
36. Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol (2004) 24(2):708–18. doi:10.1128/MCB.24.2.708-718.2004
37. Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet (2002) 30(3):285–9. doi:10.1038/ng837
38. Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res (2009) 7(7):1110–8. doi:10.1158/1541-7786.MCR-09-0123
39. O’Donnell JD, Linger RJ, Kruk PA. BRCA1 185delAG mutant protein, BRAt, up-regulates maspin in ovarian epithelial cells. Gynecol Oncol (2010) 116(2):262–8. doi:10.1016/j.ygyno.2009.10.052
40. Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet (1998) 19(3):254–6. doi:10.1038/930
41. Yarden RI, Brody LC. BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci U S A (1999) 96(9):4983–8. doi:10.1073/pnas.96.9.4983
42. Li S, Chen PL, Subramanian T, Chinnadurai G, Tomlinson G, Osborne CK, et al. Binding of CtIP to the BRCT repeats of BRCA1 involved in the transcription regulation of p21 is disrupted upon DNA damage. J Biol Chem (1999) 274(16):11334–8. doi:10.1074/jbc.274.16.11334
43. Thangaraju M, Kaufmann SH, Couch FJ. BRCA1 facilitates stress-induced apoptosis in breast and ovarian cancer cell lines. J Biol Chem (2000) 275(43):33487–96. doi:10.1074/jbc.M005824200
44. Abaji C, Cousineau I, Belmaaza A. BRCA2 regulates homologous recombination in response to DNA damage: implications for genome stability and carcinogenesis. Cancer Res (2005) 65(10):4117–25. doi:10.1158/0008-5472.CAN-04-3071
45. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature (2008) 451(7182):1111–5. doi:10.1038/nature06548
46. Li J, Zou C, Bai Y, Wazer DE, Band V, Gao Q. DSS1 is required for the stability of BRCA2. Oncogene (2006) 25(8):1186–94. doi:10.1038/sj.onc.1209153
47. Gudmundsdottir K, Lord CJ, Witt E, Tutt AN, Ashworth A. DSS1 is required for RAD51 focus formation and genomic stability in mammalian cells. EMBO Rep (2004) 5(10):989–93. doi:10.1038/sj.embor.7400255
48. Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin SF, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell (2003) 115(5):523–35. doi:10.1016/S0092-8674(03)00930-9
49. Thorslund T, Esashi F, West SC. Interactions between human BRCA2 protein and the meiosis-specific recombinase DMC1. EMBO J (2007) 26(12):2915–22. doi:10.1038/sj.emboj.7601739
50. Muggia F, Safra T. ‘BRCAness’ and its implications for platinum action in gynecologic cancer. Anticancer Res (2014) 34(2):551–6.
51. Hu XC, Zhang J, Xu BH, Cai L, Ragaz J, Wang ZH, et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol (2015) 16(4):436–46. doi:10.1016/S1470-2045(15)70064-1
52. Husain A, He G, Venkatraman ES, Spriggs DR. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diamminedichloroplatinum(II). Cancer Res (1998) 58(6):1120–3.
53. Tassone P, Tagliaferri P, Perricelli A, Blotta S, Quaresima B, Martelli ML, et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br J Cancer (2003) 88(8):1285–91. doi:10.1038/sj.bjc.6600859
54. Bartz SR, Zhang Z, Burchard J, Imakura M, Martin M, Palmieri A, et al. Small interfering RNA screens reveal enhanced cisplatin cytotoxicity in tumor cells having both BRCA network and TP53 disruptions. Mol Cell Biol (2006) 26(24):9377–86. doi:10.1128/MCB.01229-06
55. Rottenberg S, Nygren AO, Pajic M, van Leeuwen FW, van der Heijden I, van de Wetering K, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci U S A (2007) 104(29):12117–22. doi:10.1073/pnas.0702955104
56. Tassone P, Di Martino MT, Ventura M, Pietragalla A, Cucinotto I, Calimeri T, et al. Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol Ther (2009) 8(7):648–53. doi:10.4161/cbt.8.7.7968
57. Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res (1999) 59(15):3547–51.
58. Foulkes WD. BRCA1 and BRCA2: chemosensitivity, treatment outcomes and prognosis. Fam Cancer (2006) 5(2):135–42. doi:10.1007/s10689-005-2832-5
59. Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer (2004) 4(10):814–9. doi:10.1038/nrc1457
60. Stefansson OA, Jonasson JG, Johannsson OT, Olafsdottir K, Steinarsdottir M, Valgeirsdottir S, et al. Genomic profiling of breast tumours in relation to BRCA abnormalities and phenotypes. Breast Cancer Res (2009) 11(4):R47. doi:10.1186/bcr2334
61. Shamseddine AI, Farhat FS. Platinum-based compounds for the treatment of metastatic breast cancer. Chemotherapy (2011) 57(6):468–87. doi:10.1159/000334093
62. Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res (2008) 14(5):1291–5. doi:10.1158/1078-0432.CCR-07-2238
63. Sledge GW Jr, Loehrer PJ Sr, Roth BJ, Einhorn LH. Cisplatin as first-line therapy for metastatic breast cancer. J Clin Oncol (1988) 6(12):1811–4. doi:10.1200/JCO.1988.6.12.1811
64. Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol (2010) 28(7):1145–53. doi:10.1200/JCO.2009.22.4725
65. Huang F, Kushner YB, Langleben A, Foulkes WD. Eleven years disease-free: role of chemotherapy in metastatic BRCA2-related breast cancer. Nat Rev Clin Oncol (2009) 6(8):488–92. doi:10.1038/nrclinonc.2009.90
66. Rhiem K, Wappenschmidt B, Bosse K, Köppler H, Tutt AN, Schmutzler RK. Platinum sensitivity in a BRCA1 mutation carrier with advanced breast cancer. Clin Oncol (R Coll Radiol) (2009) 21(6):448–50. doi:10.1016/j.clon.2009.01.006
67. Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer (2003) 97(9):2187–95. doi:10.1002/cncr.11310
68. Tan DS, Rothermundt C, Thomas K, Bancroft E, Eeles R, Shanley S, et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol (2008) 26(34):5530–6. doi:10.1200/JCO.2008.16.1703
69. Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol (2011) 29(22):3008–15. doi:10.1200/JCO.2010.34.2980
70. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res (2008) 68(8):2581–6. doi:10.1158/0008-5472.CAN-08-0088
71. Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature (2008) 451(7182):1116–20. doi:10.1038/nature06633
72. Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell (2011) 20(6):797–809. doi:10.1016/j.ccr.2011.11.014
73. Atipairin A, Ratanaphan A. In vitro enhanced sensitivity to cisplatin in D67Y BRCA1 RING domain protein. Breast Cancer (Auckl) (2011) 5:201–8. doi:10.4137/BCBCR.S8184
74. Sun S, Cai J, Yang Q, Zhao S, Wang Z. The association between copper transporters and the prognosis of cancer patients undergoing chemotherapy: a meta-analysis of literatures and datasets. Oncotarget (2017) 8(9):16036–51. doi:10.18632/oncotarget.13917
75. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434(7035):917–21. doi:10.1038/nature03445
76. Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer (2005) 5(9):689–98. doi:10.1038/nrc1691
77. Cerrato A, Morra F, Celetti A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: the rationale for their inclusion in the clinic. J Exp Clin Cancer Res (2016) 35(1):179. doi:10.1186/s13046-016-0456-2
78. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol (2015) 16(1):87–97. doi:10.1016/S1470-2045(14)71135-0
79. Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta (2014) 1846(1):201–15. doi:10.1016/j.bbcan.2014.07.004
80. Basourakos SP, Li L, Aparicio AM, Corn PG, Kim J, Thompson TC. Combination platinum-based and DNA damage response-targeting cancer therapy: evolution and future directions. Curr Med Chem (2017) 24(15):1586–606. doi:10.2174/0929867323666161214114948
81. Konecny GE, Kristeleit RS. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions. Br J Cancer (2016) 115(10):1157–73. doi:10.1038/bjc.2016.311
82. Kim SJ, Miyoshi Y, Taguchi T, Tamaki Y, Nakamura H, Yodoi J, et al. High thioredoxin expression is associated with resistance to docetaxel in primary breast cancer. Clin Cancer Res (2005) 11(23):8425–30. doi:10.1158/1078-0432.CCR-05-0449
83. Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, et al. Response to neo-adjuvant chemotherapy in women with BRCA1-positive breast cancers. Breast Cancer Res Treat (2008) 108(2):289–96. doi:10.1007/s10549-007-9600-1
84. De Ligio JT, Velkova A, Zorio DA, Monteiro AN. Can the status of the breast and ovarian cancer susceptibility gene 1 product (BRCA1) predict response to taxane-based cancer therapy? Anticancer Agents Med Chem (2009) 9(5):543–9. doi:10.2174/187152009788451798
85. Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol (2001) 8(10):833–7. doi:10.1038/nsb1001-833
86. Varma AK, Brown RS, Birrane G, Ladias JA. Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry (2005) 44(33):10941–6. doi:10.1021/bi0509651
87. Williams RS, Green R, Glover JN. Crystal structure of the BRCT repeat region from the breast cancer-associated protein BRCA1. Nat Struct Biol (2001) 8(10):838–42. doi:10.1038/nsb1001-838
88. Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep (2009) 10(9):990–6. doi:10.1038/embor.2009.126
89. Han Y, Liu J, Sun M, Zhang Z, Liu C, Sun Y. A significant statistical advancement on the predictive values of ERCC1 polymorphisms for clinical outcomes of platinum-based chemotherapy in non-small cell lung cancer: an updated meta-analysis. Dis Markers (2016) 2016:7643981. doi:10.1155/2016/7643981
90. Jin Y, Feng LP, Jiang X, Wang YX, Yin J, Yang ZP, et al. Annexin A3 is a potential predictor of platinum resistance in epithelial ovarian cancer patients in a prospective cohort. J Cancer (2015) 6(7):678–85. doi:10.7150/jca
Keywords: breast cancer, ovarian cancer, BRCA1/2 mutations, platinum drugs, response, resistance
Citation: Mylavarapu S, Das A and Roy M (2018) Role of BRCA Mutations in the Modulation of Response to Platinum Therapy. Front. Oncol. 8:16. doi: 10.3389/fonc.2018.00016
Received: 25 October 2017; Accepted: 17 January 2018;
Published: 05 February 2018
Edited by:
José - Bines, Instituto Nacional de Câncer (INCA), BrazilReviewed by:
Dayanidhi Raman, University of Toledo, United StatesLuigi Formisano, Vanderbilt University Medical Center, United States
Marcio Debiasi, Latin American Cooperative Group (LACOG), Brazil
Copyright: © 2018 Mylavarapu, Das and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monideepa Roy, bXJveUBpbnZpY3R1c29uY29sb2d5LmNvbQ==