 Jenny He1
Jenny He1 Sunit Das
Sunit Das
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 08 January 2018
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 7 - 2017 | https://doi.org/10.3389/fonc.2017.00322
Gliomas are the most common primary malignant brain tumor in humans. Lower grade gliomas are usually less aggressive but many cases eventually progress to a more aggressive secondary glioblastoma (GBM, WHO Grade IV), which has a universally fatal prognosis despite maximal surgical resection and concurrent chemo-radiation. With the identification of molecular markers, however, there is promise for improving diagnostic and therapeutic strategies. One of the key molecular alterations in gliomas is the alpha thalassemia/mental retardation syndrome X-linked (ATRX) gene, which is frequently mutated. One-third of pediatric GBM cases are also found to have the ATRX mutation and the genetic signatures are different from adult cases. The exact role of ATRX mutations in gliomagenesis, however, is unclear. In this review, we describe the normal cellular function of the ATRX gene product followed by consequences of its dysfunction. Furthermore, its possible association with the alternative lengthening of telomeres (ALT) phenotype is outlined. Lastly, therapeutic options potentiated through a better understanding of ATRX and the ALT phenotype are explored.
The ATRX gene is located on the q arm of the X chromosome. It has been hypothesized that in patients with the ATRX syndrome, both copies are inactivated, one by mutation and the other by X inactivation (1). ATRX mutations result in a truncated protein and, therefore, diminished protein expression. Functional ATRX is one subunit of a chromatin-remodeling complex, forming a heterodimer with death domain-associated protein (DAXX). In oncology, ATRX gene mutations were first discovered in pancreatic neuroendocrine tumors (PanNETs) (2), where mutations of ATRX and DAXX are associated with a better prognosis. Subsequent work has identified these mutations in a range of other cancer subtypes, including gliomas.
Alpha thalassemia/mental retardation syndrome X-linked is a member of the SWI/SNF family of chromatin-remodeling proteins, with two highly conserved domains (3). The N-terminal ATRX-Dnmt3-Dnmt3L (ADD) domain contains a GATA-like zinc finger, a PHD finger, and an alpha helix. The ATPase/helicase domain is located at the C-terminus. SWI/SNF family proteins disrupt DNA–histone interactions in order to facilitate histone octamer sliding, which changes the conformation of the nucleosome and facilitates chromatin remodeling (4).
Many chromatin-remodeling enzymes have been implicated in human disease (4). Mutations in either of the two ATRX domains are known to cause ATRX syndrome, an X-linked intellectual disability syndrome, although the consequences of mutations in the ADD domain are generally more severe (5). The ATRX gene product interacts with the transcription factor DAXX to form a heterodimer that displays ATP-dependent chromatin-remodeling functions, a non-covalent mechanism that helps regulate gene expression in eukaryotes (6). The ADD domain of ATRX binds histone H3 tail peptides, which contain lysine 9 trimethylation (H3K9me3) marks, while DAXX is a specific chaperone for the histone variant H3.3. Together, the heterodimer facilitates the deposition of H3.3 into telomeric and peri-centromeric chromatin which are rich in H3K9me3 marks (7). ATRX and DAXX localize predominantly in the nucleus and are associated with PML nuclear bodies (8).
The histone variant H3.3 has been implicated in the formation and maintenance of chromatin structure. Nucleosomes containing H3.3 are assembled independent of replication by chaperone proteins, such as DAXX (9). The mammalian genome encodes three different H3 variants. H3.1 and H3.2 are primarily expressed during S-phase of the cell cycle, while H3.3 is replication independent and expressed throughout the cell cycle. H3.3 has previously been found to associate with regulatory elements and heterochromatin at telomeres, and the ATRX/DAXX complex is necessary for its incorporation (7, 10).
Alpha thalassemia/mental retardation syndrome X-linked-Dnmt3-Dnmt3L domain mutations disrupt H3K9me3 binding, which is the general pathway of ATRX mutation pathogenicity. Although the complete function of the ADD domain remains unknown, the PHD finger is known to differentially recognize methylated or unmodified lysine residues in histone tails (11, 12). Reduced ADD binding to H3 tail peptides leads to loss of heterochromatic localization of the ATRX/DAXX complex in cells (5).
Mutations in the ATRX gene lead to changes in methylation patterns of many CpG-rich repetitive sequences, which presents a link between chromatin-remodeling processes and DNA methylation (13). Patients with ATRX syndrome display hypomethylation in subtelomeric regions and rDNA genes, particularly in the CpG-rich regions of the rDNA repeats. Conversely, hypermethylation has been found in the Y-specific repeat (DYZ2) of the Y chromosome. These findings suggest that ATRX may influence CpG-rich regions of the genome and ATRX mutation may have implications on gene expression (13).
Additionally, mutations in SWI/SNF family proteins have been found in approximately 19.6% of human cancers, indicating that SWI/SNF is the most frequently mutated chromatin-regulatory complex in human cancer (14). These proteins have been implicated as tumor suppressors in various tissues; thus, dysfunction may contribute to tumor formation or progression. Various tumor suppression mechanisms have been presented, including the possibility that these complexes contribute to genome stability (15) and influence DNA damage responses (DDRs) (16). One study suggested that SWI/SNF complexes contribute to tumor suppression by activating the DDR and attenuating replicative stress (17). In fact, disruption of both catalytic subunits of SWI/SNF complexes in mouse embryos or D98 tissue-culture cells (cervical carcinoma subline) decreases replication fork efficiency by 50%. Collapsed replication forks highly contribute to genomic instability (18), suggesting the involvement of SWI/SNF complexes in the DDR. SWI/SNF complex deficiency was also found to prevent full activation of DDR kinases such as ATM and ATR, preventing high levels H2AX phosphorylation, an early event in the DDR.
Gliomas represent about 80% of all malignant brain tumors (13) and are classified into four major grades based on their clinical behavior and histology (19). The discovery of ATRX mutations has dramatically influenced the understanding of gliomagenesis.
Data from one study showed that the majority (~75%) of adult lower grade (WHO II/III) gliomas which have p53 and IDH1 mutations also carry ATRX mutations, suggesting an association or a possible driver role in gliomagenesis and progression to secondary glioblastoma (GBM) (20). The incidence of ATRX mutation is higher in diffuse lower grade astrocytomas, compared with oligodendrogliomas and GBMs (2). However, not all oligodendroglial tumors are free of ATRX mutations. Therefore, additional strategies are necessary to delineate whether the cell of origin is oligodendroglial or astrocytic. Given that mutation in the ATRX gene results in a loss of protein expression, immunohistochemical (IHC) analysis should demonstrate a lack of nuclear ATRX expression in tumor cells with mutated/lost ATRX. Reuss et al. have shown a high concordance between retention of nuclear ATRX expression and Grade II/III oligodendrogliomas; a much higher proportion of Grade II/III astrocytomas were found to have loss of nuclear ATRX expression (2). The addition of IHC staining for canonical IDH mutations can help increase the granularity of tumor classification. Tumors with loss of ATRX expression and IDH1/2 mutations can be reliably classified as diffuse astrocytomas while IDH1/2 mutant tumors with retained ATRX expression should undergo testing for 1p/19q co-deletion to help differentiate between a diffuse astrocytoma and oligodendroglioma (21). In younger patients (age <55 years), given the high prevalence of IDH mutations, the absence of mutation detection by standard IHC methods should prompt sequencing for canonical/non-canonical mutations (22).
The majority of human cancer cells upregulate telomerase to avoid cellular senescence or apoptosis. However, about 10–15% of cancers use an alternative approach to achieve immortality (23). The ALT phenotype is a homologous recombination-associated process but the mechanism is not fully understood. The ALT phenotype is characterized by the presence of ALT-associated promyelocytic leukemia bodies (APBs), which differ from common PML bodies, as well as by telomere recombination with the presence of extrachromosomal telomeric repeats (ECTRs) (24), which may serve as a template for extension of telomeres (25). Recently, it has been shown that many ALT-positive tumors, including PanNETs, oligodendrogliomas, and GBM, contain inactivating mutations in ATRX and/or DAXX, implicating the possible role of ATRX/DAXX inactivation in the manifestation of the ALT phenotype (23, 26).
The ALT phenotype is usually detected using telomere fluorescence in situ hybridization, with large, ultrabright signals indicating the presence of ALT (27). Though the ALT mechanism of telomere maintenance is most characteristically found in cancer cells and immortalized cells, some have shown their presence in non-neoplastic cells as well (28, 29). The latter group of cells tended to have a higher rate of DNA damage. Some studies have suggested that the ALT phenotype is not strictly found in ATRX mutant cells and that alteration of other pathways resulting in DNA damage have also been implicated; these include the absence of MYCN amplification and alterations in the TP53 pathway (30). These findings further strengthen the putative association between DNA damage and the ALT phenotype (29). Whether DNA damage is caused by or selects for the ALT phenotype is not clear.
The predominant view of the ALT mechanism is that it maintains telomeres through homology-directed recombination (HDR). ALT-positive cells display a wide range of telomere repeat lengths, increased levels of telomere sister chromatid exchanges (T-SCEs), ECTRs, and the presence of nuclear APBs (31). APBs are nuclear structures that are known to play a role in DNA repair and recombination processes; therefore, it is hypothesized that ALT-mediated telomere elongation takes place within them. In one study performed on PanNETs, a perfect correlation was found between inactivation of ATRX or DAXX and the ALT phenotype (27). The same study also explored the ALT status of CNS tumors with and without ATRX mutations, and a similarly strong correlation was found. Akin to the higher rate of ATRX mutations in lower grade gliomas and secondary GBM, the ALT phenotype was shown to be more common in these tumor groups as well (32).
Despite the role of testing for ATRX to better refine our ability to characterize astrocytic versus oligodendrocytic glioma lineage, the complete role of ATRX in tumorigenesis remains uncertain. Many studies have implicated ATRX loss in promoting a genetically unstable tumor, with impaired telomere maintenance and disrupted DNA repair, which is more aggressive when left untreated, but also more responsive to double-stranded DNA-breaking agents (20). In fact, recent data have shown that pediatric and adult patients with ATRX mutated GBM have a survival advantage when treated with double-stranded DNA-breaking agents like doxorubicin, irinotecan (SN-38), and topotecan (20). It is possible that the genetic instability is partially a result of ALT activation as the ALT phenotype in other cancer types is associated with genetic instability and altered DNA repair (23).
In addition to binding histone H3 tails, ATRX can also be recruited to G-rich repeats present at telomeres. One study used deep sequencing to show that ATRX preferentially binds tandemly repetitive DNA, especially telomeric G-rich repeats and CpG islands (33). These DNA regions are able to form non-canonical conformations such as G-quadruplexes (G4) in vivo (34) and ATRX preferentially binds G4 structured DNA over unfolded sequences (33). Given the association of ATRX with DAXX, the latter is able to enhance H3.3 deposition at these repetitive sequences. Since ATRX preferentially binds these DNA secondary structures, ATRX loss and the presence of abnormal secondary structures may affect the expression of genes near the tandem repeat DNA. G4 structures form barriers to processes like DNA replication and transcription (35, 36), and ATRX loss has been associated with replication defects such as replication fork stalling, prolonged S-phase, accumulation of p53, and H2AX DNA damage marker (37, 38). These problems in replication can cause disruptions of local gene expression and propagation of histone forks during replication. Additionally, increased replication fork stalling, which occurs when functional ATRX is absent, is a known trigger of homologous recombination and can contribute to the activation of the ALT pathway (36). This lends to the hypothesis that stable formation of G4 structures at telomeres impedes replication and triggers the DDR, and functional ATRX (with DAXX and H3.3) may have a role in suppressing ALT pathway activation by binding to G4 structures and resolving them (36).
Additionally, ATRX loss may impair DNA repair. A recent study by Koschmann et al. used a mouse model of ATRX-deficient GBM and found that ATRX loss accelerated GBM growth rate and reduced median survival (39). The mouse brain tumors appeared in the first few weeks of life, similar to the development of pediatric/young adult GBM in humans, which frequently have ATRX mutations (39). They also found that ATRX loss correlated with the ALT phenotype, along with a decrease in expression of non-homologous end joining (NHEJ) repair pathway proteins. Based on the role of ATRX in histone H3.3 deposition, they hypothesized that ATRX loss may lead to conformational changes in heterochromatin that serve to prevent access of NHEJ proteins to the damaged DNA. NHEJ impairment is a mechanism for genetic instability that has been thought to contribute to gliomagenesis, as NHEJ inhibition induces the accumulation of dsDNA breaks (40). A relative increase in HDR compared with NHEJ in ATRX-deficient glioma may play a role in ALT activation.
Although ATRX loss has been found to promote some features of the ALT phenotype, and is generally regarded as a hallmark of ALT-immortalized cell lines, it has been shown that ATRX loss is not sufficient for ALT activation, and additional genetic changes are required (39). One study showed that ATRX/DAXX loss and associated lack of H3.3 deposition into telomeric chromatin is crucial and required for ALT but not sufficient, and the ALT phenotype does not arise from a single mutation (40). For example, G2/M checkpoint dysfunction is also required. It is likely that G2/M checkpoint dysfunction is necessary for proliferation despite considerable DNA damage at telomeres (10). Therefore, while there is strong evidence supporting the role of ATRX loss in promoting the ALT phenotype, additional rigorous studies are necessary to establish its contribution to the phenotype and the mechanism(s) through which this is supported. This has implications for diagnostic and therapeutic strategies as well. A schematic diagram, aiming to summarize some of the possible roles of ATRX loss/mutation has been provided in Figure 1.
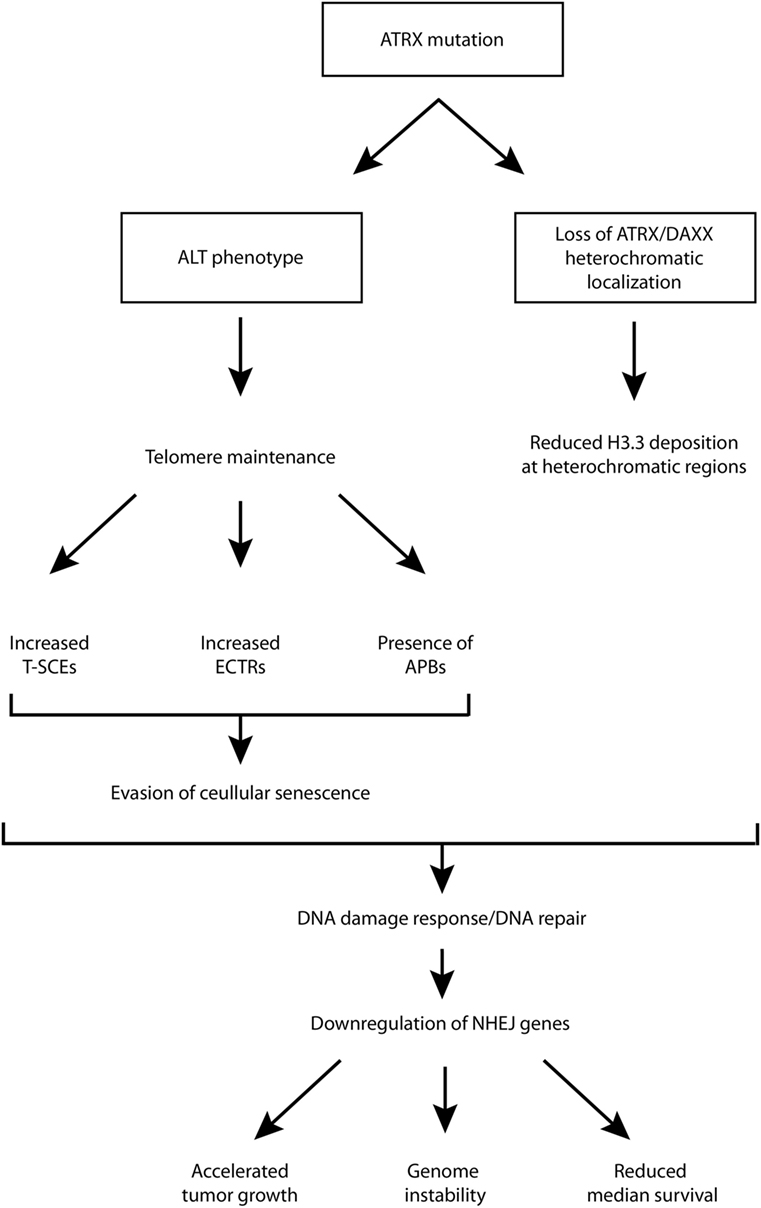
Figure 1. Schematic diagram of putative downstream molecular and cellular events following ATRX mutation. ALT, alternative lengthening of telomeres; APB, ALT-associated promyelocytic leukemia bodies; ATRX, alpha thalassemia/mental retardation syndrome X-linked; DAXX, death domain-associated protein; ECTR, extrachromosomal telomeric repeat; NHEJ, non-homologous end joining; T-SCE, telomere sister chromatid exchanges.
Alpha thalassemia/mental retardation syndrome X-linked is mutated in nearly a third of pediatric GBM (26), rarely found in adult primary GBM but common in lower grade adult glioma (WHO 2/3) and secondary GBM (41). However, there currently have been minimal efforts to target ATRX deficiency to treat glioma, mainly due to a limited understanding of the exact role of ATRX in gliomagenesis and disease progression. There are currently no actively enrolling or completed clinical trials based on ATRX status. The development of appropriate animal models would be a necessary first step to investigating this. Furthermore, as the possibility of personalized medicine seems to be more in sight, a comprehensive understanding of ATRX along with its regulators, and downstream targets would enable a broader set of management strategies in children and young adults with gliomas.
Alpha thalassemia/mental retardation syndrome X-linked loss can promote tumor cell susceptibility to DNA damaging agents that induce double-stranded DNA breaks, so the use of agents that induce double-stranded breaks may be more effective in ATRX negative cells (20). Additionally, future work could be aimed at exploring possible mechanisms for selectively targeting the molecular features unique to the ALT phenotype, including the presence of APBs, ECTRs, or increased T-SCE levels.
JH and AM performed literature review and analysis and were involved in the writing of the manuscript. SD performed critical analysis and editing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science (2011) 331:1199–203. doi:10.1126/science.1200609
2. Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol (2015) 129:133–46. doi:10.1007/s00401-014-1370-3
3. Dhayalan A, Tamas R, Bock I, Tattermusch A, Dimitrova E, Kudithipudi S, et al. The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum Mol Genet (2011) 20:2195–203. doi:10.1093/hmg/ddr107
4. Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell (2013) 154:490–503. doi:10.1016/j.cell.2013.07.011
5. Badens C, Lacoste C, Philip N, Martini N, Courrier S, Giuliano F, et al. Mutations in PHD-like domain of the ATRX gene correlate with severe psychomotor impairment and severe urogenital abnormalities in patients with ATRX syndrome. Clin Genet (2006) 70:57–62. doi:10.1111/j.1399-0004.2006.00641.x
6. Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with DAXX and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci U S A (2003) 100:10635–40. doi:10.1073/pnas.1937626100
7. Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell (2010) 140:678–91. doi:10.1016/j.cell.2010.01.003
8. Salomoni P, Khelifi AF. DAXX: death or survival protein? Trends Cell Biol (2006) 16:97–104. doi:10.1016/j.tcb.2005.12.002
9. Lewis PW, Elsaesser SJ, Noh K-M, Stadler SC, Allis CD. DAXX is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A (2010) 107:14075–80. doi:10.1073/pnas.1008850107
10. Wong LH, Ren H, Williams E, McGhie J, Ahn S, Sim M, et al. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res (2009) 19:404–14. doi:10.1101/gr.084947.108
11. Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat Res (2008) 647:3–12. doi:10.1016/j.mrfmmm.2008.07.004
12. Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol (2007) 14:1025–40. doi:10.1038/nsmb1338
13. Gibbons RJ, McDowell TL, Raman S, O’Rourke DM, Garrick D, Ayyub H, et al. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet (2000) 24:368–71. doi:10.1038/74191
14. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatics analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet (2013) 45:592–601. doi:10.1038/ng.2628
15. Garraway LA, Lander ES. Lessons from the cancer genome. Cell (2013) 153:17–37. doi:10.1016/j.cell.2013.03.002
16. Watanabe R, Ui A, Kanno S, Ogiwara H, Nagase T, Kohno T, et al. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res (2014) 74:2465–75. doi:10.1158/0008-5472.CAN-13-3608
17. Smith-Roe SL, Nakamura J, Holley D, Chastain PD, Rosson GB, Simpson DA, et al. SWI/SNF complexes are required for full activation of the DNA-damage response. Oncotarget (2015) 6:732–45. doi:10.18632/oncotarget.2715
18. Lambert S, Carr AM. Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma (2013) 122:33–45. doi:10.1007/s00412-013-0398-9
19. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumors of the central nervous system. Acta Neuropathol (2007) 114:97–109. doi:10.1007/s00401-007-0243-4
20. Koschmann C, Calinescu A-A, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med (2016) 8:328ra28. doi:10.1126/scitranslmed.aac8228
21. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (2016) 131:803–20. doi:10.1007/s00401-016-1545-1
22. Chen L, Voronovich Z, Clark K, Hands I, Mannas J, Walsh M, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol (2014) 16:1478–83. doi:10.1093/neuonc/nou097
23. Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet (2012) 8:e1002772. doi:10.1371/journal.pgen.1002772
24. Cesare AJ, Griffith JD. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol (2004) 24:9948–57. doi:10.1128/MCB.24.22.9948-9957.2004
25. Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene (2002) 21:598–610. doi:10.1038/sj.onc.1205058
26. Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature (2012) 482:226–31. doi:10.1038/nature10833
27. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science (2011) 333:425–425. doi:10.1126/science.1207313
28. Chung I, Osterwald S, Deeg KI, Rippe K. PML body meets telomere: the beginning of an ALTernate ending? Nucleus (2012) 3:263–75. doi:10.4161/nucl.20326
29. Slatter TL, Tan X, Yuen YC, Gunningham S, Ma SS, Daly E, et al. The alternative lengthening of telomeres pathway may operate in non-neoplastic human cells. J Pathol (2012) 226:509–18. doi:10.1002/path.2981
30. Farooqi AS, Dagg RA, Choi LM, Shay JW, Reynolds CP, Lau LM. Alternative lengthening of telomeres in neuroblastoma cell lines is associated with a lack of MYCN genomic amplification and with p53 pathway aberrations. J Neurooncol (2014) 119:17–26. doi:10.1007/s11060-014-1456-8
31. Chung I, Leonhardt H, Rippe K. De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. J Cell Sci (2011) 124:3603–18. doi:10.1242/jcs.084681
32. Nonoguchi N, Ohta T, Oh J-E, Kim Y-H, Kleihues P, Ohgaki H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol (2013) 126:931–7. doi:10.1007/s00401-013-1163-0
33. Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell (2010) 143:367–78. doi:10.1016/j.cell.2010.09.023
34. Lipps HJ, Rhodes D. G-quadruplex structures: in vivo evidence and function. Trends Cell Biol (2009) 19:414–22. doi:10.1016/j.tcb.2009.05.002
35. Paeschke K, Capra JA, Zakian VA. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell (2011) 145:678–91. doi:10.1016/j.cell.2011.04.015
36. Rizzo A, Salvati E, Porru M, D’Angelo C, Stevens MF, D’Incalci M, et al. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res (2009) 37:5353–64. doi:10.1093/nar/gkp582
37. Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res (2010) 20:351–60. doi:10.1101/gr.101477.109
38. Leung JW-C, Ghosal G, Wang W, Shen X, Wang J, Li L, et al. Alpha thalassemia/mental retardation syndrome X-linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J Biol Chem (2013) 288:6342–50. doi:10.1074/jbc.M112.411603
39. Koschmann C, Lowenstein PR, Castro MG. ATRX mutations and glioblastoma: impaired DNA damage repair, alternative lengthening of telomeres, and genetic instability. Mol Cell Oncol (2016) 3:e1167158. doi:10.1080/23723556.2016.1167158
40. Azad A, Jackson S, Cullinane C, Natoli A, Neilsen PM, Callen DF, et al. Inhibition of DNA-dependent protein kinase induces accelerated senescence in irradiated human cancer cells. Mol Cancer Res (2011) 9:1696–707. doi:10.1158/1541-7786.MCR-11-0312
Keywords: alternative lengthening of telomeres, alpha thalassemia/mental retardation syndrome X-linked, diagnostics, glioma, therapeutics
Citation: He J, Mansouri A and Das S (2018) Alpha Thalassemia/Mental Retardation Syndrome X-Linked, the Alternative Lengthening of Telomere Phenotype, and Gliomagenesis: Current Understandings and Future Potential. Front. Oncol. 7:322. doi: 10.3389/fonc.2017.00322
Received: 04 October 2017; Accepted: 12 December 2017;
Published: 08 January 2018
Edited by:
Kerrie Leanne McDonald, University of New South Wales, AustraliaReviewed by:
Janice Anne Royds, University of Otago, New ZealandCopyright: © 2018 He, Mansouri and Das. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alireza Mansouri, YWxpcmV6YS5tYW5zb3VyaUB1dG9yb250by5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.