 Kentaro Inamura
Kentaro Inamura
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 28 August 2017
Sec. Thoracic Oncology
Volume 7 - 2017 | https://doi.org/10.3389/fonc.2017.00193
Lung cancer is the leading cause of cancer-related death worldwide due to late diagnoses and limited treatment interventions. Recently, comprehensive molecular profiles of lung cancer have been identified. These novel characteristics have enhanced the understanding of the molecular pathology of lung cancer. The identification of driver genetic alterations and potential molecular targets has resulted in molecular-targeted therapies for an increasing number of lung cancer patients. Thus, the histopathological classification of lung cancer was modified in accordance with the increased understanding of molecular profiles. This review focuses on recent developments in the molecular profiling of lung cancer and provides perspectives on updated diagnostic concepts in the new 2015 WHO classification. The WHO classification will require additional revisions to allow for reliable, clinically meaningful tumor diagnoses as we gain a better understanding of the molecular characteristics of lung cancer.
Lung cancer is the leading cause of cancer-related deaths worldwide in both men and women (1). It is categorized into two main histological groups: small cell lung carcinoma (SCLC, 15% of all lung cancers) and non-SCLC (NSCLC, 85% of all lung cancers). NSCLCs are generally subcategorized into adenocarcinoma, squamous cell carcinoma (SqCC), and large cell carcinoma. Accumulating evidence suggests that lung cancer represents a group of histologically and molecularly heterogeneous diseases even within the same histological subtype (2–26).
The histopathological classification of lung cancer has recently been revised and published as the 2015 WHO classification (2). Several major revisions were made in this classification to reflect recent discoveries related to the molecular pathology of lung cancer.
Human comprehensive molecular characterization projects have resulted in the identification of novel molecular characteristics of lung cancer and the different subtypes at levels of DNA alteration, DNA methylation, mRNA expression, microRNA expression, and protein expression. This review introduces and briefly summarizes recent studies on the molecular pathology of lung cancer with a focus on the association between molecular profiles and morphology (2).
The WHO classification was updated based on newly identified molecular profiles and targetable genetic alterations in lung cancer. For lung adenocarcinoma, the 2011 International Association for the Study of Lung Cancer, American Thoracic Society, and European Respiratory Society classification (27) was mostly adopted in the 2015 WHO classification. Table 1 shows the WHO classification of lung tumors (epithelial tumors) (2). The major revisions to the WHO classification are described below.

Table 1. WHO classification of tumors of the lung (epithelial tumors) (2).
Pathologists are required to categorize lung cancer into adenocarcinoma and SqCC due to the targetable driver genetic alterations identified in lung adenocarcinoma and inappropriate drugs for SqCCs due to side effects in patients with SqCC. Before the 2015 WHO classification, adenocarcinoma was defined as carcinoma with an acinar/tubular structure or mucin production, whereas SqCC was defined as carcinoma with keratinization or intercellular bridges. If poorly differentiated carcinoma lacking light microscopic evidence of glandular differentiation (Figure 1A) is proven by immunohistochemistry to express “adenocarcinoma markers,” such as TTF-1 (Figure 1B) and/or Napsin A (Figure 1C), it is diagnosed as a solid adenocarcinoma. If poorly differentiated carcinoma lacking light microscopic evidence of squamous differentiation (Figure 1D) is proven by immunohistochemistry to express “SqCC markers,” (28) such as p40 (Figure 1E), CK5/6 (Figure 1F), and p63, it is diagnosed as non-keratinizing SqCC. Because of this classification, the proportion of large cell carcinoma has been markedly reduced.
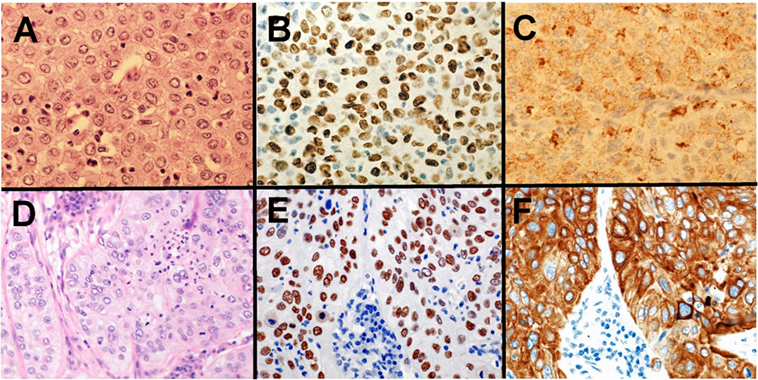
Figure 1. Solid adenocarcinoma (A–C) and non-keratinizing squamous cell carcinoma (SqCC) (D–F). Solid adenocarcinoma [(A) HE staining] is immunohistochemically positive for TTF-1 (B) and Napsin A (C). Non-keratinizing SqCC [(D) HE staining] is immunohistochemically positive for p40 (E) and CK5/6 (F).
The 2015 WHO classification divides adenocarcinomas into adenocarcinoma in situ (AIS, preinvasive lesion), minimally invasive adenocarcinoma (MIA), or (overt) invasive adenocarcinoma based on the extent of invasiveness. The disease-free survival rate of AIS and MIA when completely resected is 100% (29).
Adenocarcinoma in situ is defined as an adenocarcinoma comprising a lepidic pattern with a diameter of ≤3 cm. If the tumor diameter exceeds 3 cm, it is defined as “lepidic predominant adenocarcinoma, suspect AIS” because these tumors are rare and lack adequate characterization.
Minimally invasive adenocarcinoma is defined as an adenocarcinoma with a diameter of ≤3 cm and an invasion size of ≤5 mm. Even if the tumor size and invasion size comply with the definition of MIA, the presence of lymphovascular invasion, pleural invasion, or tumor necrosis can be an exclusion factor for an MIA diagnosis. If the tumor size exceeds 3 cm with an invasion size of ≤5 mm, it is defined as “lepidic predominant adenocarcinoma, suspect MIA” because these tumors are rare and lack adequate characterization.
The term “invasive adenocarcinoma, mixed subtype” for invasive adenocarcinoma is no longer used. Invasive adenocarcinoma is now classified using five predominant patterns: lepidic, papillary, acinar, micropapillary, and solid adenocarcinoma.
The term “mucinous bronchioloalveolar carcinoma (BAC)” is no longer used because most mucinous BACs included invasive components. Therefore, the term “invasive mucinous adenocarcinoma (IMA)” replaced mucinous BAC. IMA and mucinous AIS are accurately classified based on invasiveness. Besides IMA, variants of invasive adenocarcinoma comprise enteric, colloid, and fetal adenocarcinoma. Enteric adenocarcinoma is defined as adenocarcinoma with a predominant component that resembles adenocarcinoma arising in the colorectum and often shows CDX2 immunoreactivity (30).
In the 2015 WHO classification, SqCCs are classified into keratinizing SqCC, non-keratinizing SqCC, and basaloid SqCC. Before this classification, basaloid SqCC was categorized as a variant of large cell carcinoma. However, basaloid SqCC immunohistochemically shows “SqCC markers” (e.g., p40, CK5/6, and p63) and is therefore categorized as SqCC.
In the 2015 WHO classification, a new category of “neuroendocrine tumors” was established. Invasive neuroendocrine tumors comprise three subtypes: SCLC, large cell neuroendocrine carcinoma (LCNEC), and carcinoid tumor (typical/atypical). Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia is extremely rare and non-invasive; therefore, its clinical importance is low. On the other hand, the distinction between a high-grade neuroendocrine tumor (HGNET), comprising SCLC and LCNEC, and a carcinoid tumor is very important in both pathological and clinical practice. HGNET is one of the most aggressive subtypes and characterized by a history of heavy smoking in the patient, whereas carcinoid tumors usually carry a benign prognosis and frequently occur in patients with no history of smoking.
With the emergence of high-throughput sequencing techniques, detailed molecular profiles of lung cancer have been identified. The Cancer Genome Atlas (TCGA) research network identified genomic and other molecular alterations among a number of different types of cancer, including lung cancer. In this section, the comprehensive molecular profiles of lung cancer, mainly determined by TCGA, are introduced.
The comprehensive molecular profiling of 230 lung adenocarcinoma by TCGA was published in 2014 (3). The authors reported high rates of somatic mutations (mean: 8.9 mutations per megabase) and identified 18 statistically significant genetic mutations: TP53 (46%), KRAS (33%), KEAP1 (17%), STK11 (17%), EGFR (14%), NF1 (11%), BRAF (10%), SETD2 (9%), RBM10 (8%), MGA (8%), MET (7%), ARID1A (7%), PIK3CA (7%), SMARCA4 (6%), RB1 (4%), CDKN2A (4%), U2AF1 (3%), and RIT1 (2%).
Furthermore, approximately 75% of the lung adenocarcinomas examined harbored genetic alterations that promote the RTK/RAS/RAF signaling pathway. Of all the cases, 62% showed driver genetic alterations that promote the RTK/RAS/RAF pathway. Among them, mutations in KRAS, EGFR, and BRAF comprised 32, 11, and 7.0%, respectively. Other genetic alterations that promote the RTK/RAS/RAF pathway included MET exon 14 skipping (4.3%), ERBB2 (or HER2) mutation (1.7%), ROS1 fusion (1.7%), ALK fusion (1.3%), MAP2K1 mutation (0.9%), RET fusion (0.9%), NRAS mutation (0.4%), and HRAS mutation (0.4%). An examination of the DNA copy number of the remaining 38% cases without driver genetic alterations that promote the RTK/RAS/RAF pathway revealed amplification of oncogenes in the RTK/RAS/RAF pathway: ERBB2 amplification (0.9%) and MET amplification (2.2%). The authors also identified new genetic alterations in this pathway: mutations in NF1 and RIT1. NF1 is a tumor suppressor gene that regulates the RTK/RAS/RAF pathway, and the frequency of NF1 mutations was 8.3%. Similarly, RIT1 constitutes a part of the RTK/RAS/RAF pathway, and the frequency of RIT1 mutations was 2.2%. Consequently, 75% of lung adenocarcinomas have genetic alterations that promote the RTK/RAS/RAF pathway. This study on the comprehensive molecular characterization of lung adenocarcinoma widened the potential therapeutic targets of lung adenocarcinoma.
This study conducted mRNA profiling and provided new transcriptional subtypes: i.e., the terminal respiratory unit (TRU, formerly bronchioid), the proximal-inflammatory (PI, formerly squamoid), and the proximal-proliferative (PP, formerly magnoid) mRNA subtypes. The TRU subtype was enriched for EGFR mutation and kinase fusions. The PI subtype was characterized by solid morphology and co-mutation of NF1 and TP53. The PP subtype was enriched for KRAS mutation and STK11 inactivation.
DNA methylation profiling divided lung adenocarcinomas into three subtypes: i.e., CpG island methylator phenotype (CIMP)-high, CIMP-intermediate, and CIMP-low subtypes. CIMP-high tumors often showed DNA hypermethylation of CDKN2A, GATA2, GATA5, HIC1, HOXA9, HOXD13, RASSF1, SFRP1, SOX17, and WIF1. The CIMP-high subtype was enriched for MYC overexpression as well as for DNA hypermethylation of genes in WNT pathway.
Protein profiling divided lung adenocarcinomas into six subtypes. The top 50 differentially expressed proteins among the 6 subtypes included Cyclin D1, Smad4, p-mTOR, Rad50, beta-Catenin, and HER2. The six subtypes partially overlapped with the mRNA three subtypes.
The comprehensive molecular profiling of 178 cases of SqCC by TCGA was published in 2012 (4). As can be expected from the history of heavy smoking in SqCC patients, SqCCs are characterized by complex genomic alterations. The authors indeed detected a mean of 360 exonic mutations, 165 genomic rearrangements, and 323 segments of copy number alteration per tumor. They identified 11 statistically significant genetic mutations: TP53, CDKN2A, PTEN, PIK3CA, KEAP1, MLL2, HLA-A, NFE2L2, NOTCH1, RB1, and PDYN. The frequency of TP53 mutations was 90%. The authors identified novel loss-of-function mutations in the HLA-A class I major histocompatibility gene. They also conducted pathway analyses and identified pathways related to oxidative damage, including KEAP1 and NFE2L2 in 34% of cases, squamous cell differentiation pathway, including overexpression of SOX2 and TP63 in 44% of cases, PI3K/AKT pathway in 47% of cases, and inactivation of CDKN2A in 72% of SqCC cases. These results provided us multiple potential targets for the treatment of lung SqCC.
The mRNA profiling divided SqCCs into four subtypes: i.e., classical, basal, secretory, and primitive subtypes. The classical subtype was characterized by pronounced hypermethylation, chromosomal instability, and alterations in KEAP1, NFE2L2, and PTEN. The basal subtype showed NF1 alterations. The primitive subtype was enriched for RB1 and PTEN alterations.
MicroRNA profiling divided SqCCs into four subtypes. These four subtypes roughly overlapped with the mRNA four subtypes. DNA methylation profiling also divided SqCCs into four subtypes (methylation clusters 1–4). Methylation cluster 4 showed little DNA hypermethylation and included most of the primitive mRNA subtype. Cluster 3 was predominantly made up of the classical mRNA subtype and enriched for NFE2L2 mutations. Cluster 3 and Cluster 2 showed the highest levels of DNA hypermethylation. Cluster 1 showed intermediate DNA hypermethylation levels.
In 2012, comprehensive genomic analyses were reported by two groups. Rudin et al. identified SOX2 as a frequently amplified gene in SCLC (6). In vitro suppression of SOX2 blocked the proliferation of SOX2-amplified SCLC cell lines. Furthermore, they identified a recurrent RLF–MYCL1 fusion in SCLC with RNA sequencing. In vitro silencing of MYCL1 in SCLC cell lines with an RLF–MYCL1 fusion decreased cell proliferation. On the other hand, Peifer et al. reported recurrent mutations in genes that encode histone modifiers, including CREBBP, EP300, and MLL, suggesting histone modification as a major feature of SCLC (7).
In 2015, George et al. sequenced the genome of 110 SCLCs and found comprehensive genomic profiles (5). SCLC is characterized by highly complex genomic alterations, and C:G>A:T transversions were found in 28% of all mutations on average, which is a characteristic pattern of heavy smoking. Almost all examined SCLCs showed bi-allelic inactivation of TP53 and RB1. Genomic alterations of tumor suppressor gene TP73 was observed in 13% of SCLCs. Among TP73 genomic alterations, genomic rearrangement of TP73Δex2/3 was identified. Because TP73Δex2/3 promotes carcinogenesis, TP73Δex2/3-targeted strategy is a promising treatment for SCLC. The authors also observed inactivating mutations of NOTCH family genes (31), which suppressed neuroendocrine differentiation via the regulation of ASCL1 expression in 25% of SCLCs.
The mRNA profiling divided SCLCs into two groups (5). Most of SCLCs (83%) were categorized into group 2, which was characterized by higher expressions of CHGA, GRP, ASCL1, and DLK1. Group 1, comprising 17% of SCLCs, showed lower expressions of these four genes.
Close associations exist between histology/morphology and genetic profiles. In this section, several of these associations are introduced.
EGFR mutation is one of the most common driver mutations in lung adenocarcinoma, and EGFR-mutated adenocarcinoma is characterized by East-Asian ethnicity, female gender, and non/light-smoking history (32). Pathologically, EGFR-mutated lung adenocarcinomas typically show nuclear TTF-1 (NKX2-1) immunostaining and a hobnail cell type. Adenocarcinomas with a micropapillary pattern have a higher frequency of EGFR mutations than adenocarcinomas without this pattern (33, 34).
Fusion genes were recently identified as oncogenic drivers. In lung adenocarcinoma, rearranged genes, including ALK, ROS1, RET, NTRK1, and NRG1, have been reported. ALK-rearranged adenocarcinoma comprises 4–5% of adenocarcinomas (35). ROS1- and RET-rearranged adenocarcinoma each comprises approximately 1% (35, 36). These rearranged adenocarcinomas show a good clinical response to molecular-targeted drugs. ALK-rearranged adenocarcinoma is characterized by a TTF-1 cell lineage, an acinar structure with mucin/signet-ring cell pattern, non-/light-smoking history, and young onset (37–39). ROS1- and RET-rearranged adenocarcinomas have a similar histology to ALK-rearranged adenocarcinoma, such as mucinous cribriform pattern or solid signet-ring cell pattern (35, 40, 41).
NTRK1 fusion in lung adenocarcinoma was identified by Vaishnavi et al. (42) A NTRK1-rearranged adenocarcinoma identified by Shim et al. belonged to IMA subtype (43). NTRK1 is a TRKA kinase; thus, TRKA kinase inhibitors have the potential to be used in the treatments of NTRK1-rearranged adenocarcinomas. NRG1-rearranged adenocarcinoma is also characterized by IMA (44, 45). IMAs are frequently KRAS-mutated; therefore, Nakaoku et al. examined 34 IMA cases without KRAS mutations, partly by whole-transcriptome sequencing (45). They identified five oncogenic fusions: CD74–NRG1, SLC3A2–NRG1, EZR–ERBB4, TRIM24–BRAF, and KIAA1468–RET. These fusion genes were mutually exclusive from KRAS mutations. NRG1 fusions were present in 17.6% (6/34) of KRAS-wild-type IMAs. Because fusions of NRG1, ERBB4, BRAF, and RET are potential molecular targets, their clinical applications are promising.
Driver genetic alterations in lung adenocarcinomas differ between Caucasians and Asians, and between smokers and non-smokers. For example, KRAS mutations are frequently detected in lung adenocarcinomas in smokers, whereas genetic alterations in EGFR, ALK, ROS1, and RET are frequently detected in lung adenocarcinomas in non-smokers. The frequencies of driver genetic alterations in Caucasians were determined in TCGA study (3), whereas lung adenocarcinoma in Asians is characterized by a high frequency of EGFR mutations (approximately 50%) and low frequency of KRAS mutations (approximately 10%).
MicroRNAs are small single-stranded non-coding RNAs (19–22 nucleotides in length) that play important regulatory roles, including lung carcinogenesis (46–50). Nadal et al. performed microRNA profiling of adenocarcinomas subclassified by the 2015 WHO classification (51). They demonstrated that different histological subtypes of lung adenocarcinoma have distinct microRNA expression profiles. Unsupervised hierarchical clustering divided adenocarcinomas into three major clusters, which correlated with the histological subtypes of the 2015 WHO classification. Cluster 1 included fewer acinar and solid adenocarcinomas, and nearly all the tumors in cluster 1 were categorized as lepidic adenocarcinomas or IMAs. In contrast, clusters 2 and 3 included more acinar and solid adenocarcinomas and fewer lepidic adenocarcinomas and IMAs. Solid adenocarcinoma was characterized by the overexpression of miR-27a, miR-212, and miR-132 (51).
Enteric adenocarcinoma is one of the new variants of lung adenocarcinoma in the 2015 WHO classification. It is defined as an adenocarcinoma with a predominant component that shows enteric differentiation (30). Garajová et al. found that the microRNA signature of enteric adenocarcinomas shows similarities with NSCLCs and pancreatic adenocarcinomas, but not with colorectal adenocarcinomas. Enteric adenocarcinomas share oncogenic microRNAs (miR-31*, miR-126*, miR-506, miR-508-3p, and miR-514) with pancreatic adenocarcinomas (52).
Small cell lung carcinoma, which is categorized into neuroendocrine tumor, shows high expression of ASCL1, which is a transcription factor that promotes neuroendocrine differentiation. A study reported that miR-375 expression was promoted by ASCL1 in lung neuroendocrine carcinoma (53). This study suggested that miR-375 might reduce the YAP1-associated proliferative arrest by inhibiting YAP1. Another study examined microRNAs from 50 SCLC patients and 30 healthy individuals, and suggested that level of miR-92a-2 in plasma could be a potential non-invasive method for the SCLC diagnosis (54). Recent evidence suggests that miR-21 expression may be higher in HGNET (i.e., SCLC and LCNEC) than in typical/atypical carcinoid, and that high expression of miR-34a may be associated with atypical carcinoids (55).
Molecular alterations, which have been recently elucidated in lung cancer, are used in clinical practice or potentially useful therapeutic tools in the treatment of lung cancer.
In lung adenocarcinoma, fusions of ALK, RET, and ROS1 have been shown to be targetable genetic alterations (35, 36). In IMA, new fusions of NTRK1, NRG1, ERBB4, BRAF, and RET were identified as targetable genetic alterations (43, 45). The comprehensive molecular profiling in lung adenocarcinoma by TCGA research network has shown that 75% of lung adenocarcinomas have genetic alterations that promote RTK/RAS/RAF pathway; these genetic alterations included newly identified NF1 and RIT1 mutations, both of which are potentially targetable (3).
For lung SqCC, no effective targetable agents have been developed specifically. The comprehensive molecular profiling in lung SqCC by TCGA research network has identified a potential therapeutic target in the most lung SqCCs that they investigated (4). The alterations in targetable oncogenic pathways in lung SqCCs included RTK pathway (26%), RAS pathway (24%), and PI(3)K pathway (47%) (4). Targeting these pathways are potential ways of the treatment for lung SqCC.
Small cell lung carcinoma is the deadliest subtype of lung cancer, and no established molecular-targeted therapy for SCLC exists. For SCLC, recent studies discovered potentially targetable genetic alterations, including SOX2 amplification, RLF-MYCL1 fusion, and TP73Δex2/3 (5, 6).
In the era that we have comprehensive genomic data of lung cancer, we should try to treat lung cancer thorough more efficacious targeted therapeutic interventions.
This review focuses on newly identified molecular pathology and the 2015 WHO classification of lung cancer, which was revised based on the better understanding of the molecular pathology of lung cancer and recent advancements in newly developed molecular-targeted drugs. However, in an era of precision medicine, these classification changes remain inadequate. In the near feature, the WHO classification will need to be further revised to allow for reliable, clinically meaningful tumor diagnoses that reflect our better understanding of the molecular characteristics of lung cancer.
KI conceived the review, wrote the text, created the table and figure, and approved the final draft.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by JSPS KAKENHI Grant Number JP16K08679 and the Ministry of the Environment, Japan.
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin (2015) 65(2):87–108. doi:10.3322/caac.21262
2. Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. 4th ed. Lyon: IARC (2015).
3. The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature (2014) 511(7511):543–50. doi:10.1038/nature13385
4. The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature (2012) 489(7417):519–25. doi:10.1038/nature11404
5. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature (2015) 524(7563):47–53. doi:10.1038/nature14664
6. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet (2012) 44(10):1111–6. doi:10.1038/ng.2405
7. Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet (2012) 44(10):1104–10. doi:10.1038/ng.2396
8. Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med (2002) 8(8):816–24. doi:10.1038/nm733
9. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science (2004) 304(5676):1497–500. doi:10.1126/science.1099314
10. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med (2004) 350(21):2129–39. doi:10.1056/NEJMoa040938
11. Inamura K, Fujiwara T, Hoshida Y, Isagawa T, Jones MH, Virtanen C, et al. Two subclasses of lung squamous cell carcinoma with different gene expression profiles and prognosis identified by hierarchical clustering and non-negative matrix factorization. Oncogene (2005) 24(47):7105–13. doi:10.1038/sj.onc.1208858
12. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell (2006) 9(3):189–98. doi:10.1016/j.ccr.2006.01.025
13. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature (2007) 448(7153):561–6. doi:10.1038/nature05945
14. Inamura K, Togashi Y, Nomura K, Ninomiya H, Hiramatsu M, Satoh Y, et al. Let-7 microRNA expression is reduced in bronchioloalveolar carcinoma, a non-invasive carcinoma, and is not correlated with prognosis. Lung Cancer (2007) 58(3):392–6. doi:10.1016/j.lungcan.2007.07.013
15. Bansal P, Osman D, Gan GN, Simon GR, Boumber Y. Recent advances in immunotherapy in metastatic NSCLC. Front Oncol (2015) 6:239. doi:10.3389/fonc.2016.00239
16. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi:10.1126/science.aaa1348
17. Inamura K, Yokouchi Y, Sakakibara R, Kobayashi M, Subat S, Ninomiya H, et al. Relationship of tumor PD-L1 expression with EGFR wild-type status and poor prognosis in lung adenocarcinoma. Jpn J Clin Oncol (2016) 46(10):935–41. doi:10.1093/jjco/hyw087
18. Mirzapoiazova T, Mambetsariev N, Lennon FE, Mambetsariev B, Berlind JE, Salgia R, et al. HABP2 is a novel regulator of hyaluronan-mediated human lung cancer progression. Front Oncol (2015) 5:164. doi:10.3389/fonc.2015.00164
19. Hwang DH, Sholl LM, Rojas-Rudilla V, Hall DL, Shivdasani P, Garcia EP, et al. KRAS and NKX2-1 mutations in invasive mucinous adenocarcinoma of the lung. J Thorac Oncol (2016) 11(4):496–503. doi:10.1016/j.jtho.2016.01.010
20. Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, et al. Small cell lung cancer screen of oncology drugs, investigational agents, and gene and microRNA expression. J Natl Cancer Inst (2016) 108(10):djw122. doi:10.1093/jnci/djw122
21. Bansal P, Osman D, Gan GN, Simon GR, Boumber Y. Recent advances in targetable therapeutics in metastatic non-squamous NSCLC. Front Oncol (2016) 6:112. doi:10.3389/fonc.2016.00112
22. Inamura K, Yokouchi Y, Kobayashi M, Sakakibara R, Ninomiya H, Subat S, et al. Tumor B7-H3 (CD276) expression and smoking history in relation to lung adenocarcinoma prognosis. Lung Cancer (2017) 103:44–51. doi:10.1016/j.lungcan.2016.11.013
23. Kim H, Yang JM, Jin Y, Jheon S, Kim K, Lee CT, et al. MicroRNA expression profiles and clinicopathological implications in lung adenocarcinoma according to EGFR, KRAS, and ALK status. Oncotarget (2017) 8(5):8484–98. doi:10.18632/oncotarget.14298
24. Saruwatari K, Ikemura S, Sekihara K, Kuwata T, Fujii S, Umemura S, et al. Aggressive tumor microenvironment of solid predominant lung adenocarcinoma subtype harboring with epidermal growth factor receptor mutations. Lung Cancer (2016) 91:7–14. doi:10.1016/j.lungcan.2015.11.012
25. Inamura K, Yokouchi Y, Kobayashi M, Ninomiya H, Sakakibara R, Subat S, et al. Association of tumor TROP2 expression with prognosis varies among lung cancer subtypes. Oncotarget (2017) 8(17):28725–35. doi:10.18632/oncotarget.5647
26. Zhang Y, Li ZY, Hou XX, Wang X, Luo YH, Ying YP, et al. Clinical significance and effect of AEG-1 on the proliferation, invasion, and migration of NSCLC: a study based on immunohistochemistry, TCGA, bioinformatics, in vitro and in vivo verification. Oncotarget (2017) 8(10):16531–52. doi:10.18632/oncotarget.4972
27. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society: international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol (2011) 6(2):244–85. doi:10.1097/JTO.0b013e318206a221
28. Bishop JA, Teruya-Feldstein J, Westra WH, Pelosi G, Travis WD, Rekhtman N. p40 (DeltaNp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma. Mod Pathol (2012) 25(3):405–15. doi:10.1038/modpathol.2011.173
29. Noguchi M, Morikawa A, Kawasaki M, Matsuno Y, Yamada T, Hirohashi S, et al. Small adenocarcinoma of the lung. Histologic characteristics and prognosis. Cancer (1995) 75(12):2844–52. doi:10.1002/1097-0142(19950615)75:12<2844::AID-CNCR2820751209>3.0.CO;2-#
30. Inamura K, Satoh Y, Okumura S, Nakagawa K, Tsuchiya E, Fukayama M, et al. Pulmonary adenocarcinomas with enteric differentiation: histologic and immunohistochemical characteristics compared with metastatic colorectal cancers and usual pulmonary adenocarcinomas. Am J Surg Pathol (2005) 29(5):660–5. doi:10.1097/01.pas.0000160438.00652.8b
31. Crabtree JS, Singleton CS, Miele L. Notch signaling in neuroendocrine tumors. Front Oncol (2016) 6:94. doi:10.3389/fonc.2016.00094
32. Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst (2005) 97(5):339–46. doi:10.1093/jnci/dji055
33. Ninomiya H, Hiramatsu M, Inamura K, Nomura K, Okui M, Miyoshi T, et al. Correlation between morphology and EGFR mutations in lung adenocarcinomas significance of the micropapillary pattern and the hobnail cell type. Lung Cancer (2009) 63(2):235–40. doi:10.1016/j.lungcan.2008.04.017
34. Inamura K, Ninomiya H, Ishikawa Y, Matsubara O. Is the epidermal growth factor receptor status in lung cancers reflected in clinicopathologic features? Arch Pathol Lab Med (2010) 134(1):66–72. doi:10.1043/2008-0586-RAR1.1
35. Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med (2012) 18(3):378–81. doi:10.1038/nm.2658
36. Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med (2012) 18(3):375–7. doi:10.1038/nm.2644
37. Inamura K, Takeuchi K, Togashi Y, Nomura K, Ninomiya H, Okui M, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol (2008) 3(1):13–7. doi:10.1097/JTO.0b013e31815e8b60
38. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol (2009) 27(26):4247–53. doi:10.1200/JCO.2009.22.6993
39. Inamura K, Takeuchi K, Togashi Y, Hatano S, Ninomiya H, Motoi N, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol (2009) 22(4):508–15. doi:10.1038/modpathol.2009.2
40. Yoshida A, Kohno T, Tsuta K, Wakai S, Arai Y, Shimada Y, et al. ROS1-rearranged lung cancer: a clinicopathologic and molecular study of 15 surgical cases. Am J Surg Pathol (2013) 37(4):554–62. doi:10.1097/PAS.0b013e3182758fe6
41. Tsuta K, Kohno T, Yoshida A, Shimada Y, Asamura H, Furuta K, et al. RET-rearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br J Cancer (2014) 110(6):1571–8. doi:10.1038/bjc.2014.36
42. Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med (2013) 19(11):1469–72. doi:10.1038/nm.3352
43. Shim HS, Kenudson M, Zheng Z, Liebers M, Cha YJ, Hoang Ho Q, et al. Unique genetic and survival characteristics of invasive mucinous adenocarcinoma of the lung. J Thorac Oncol (2015) 10(8):1156–62. doi:10.1097/JTO.0000000000000579
44. Fernandez-Cuesta L, Plenker D, Osada H, Sun R, Menon R, Leenders F, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov (2014) 4(4):415–22. doi:10.1158/2159-8290.CD-13-0633
45. Nakaoku T, Tsuta K, Ichikawa H, Shiraishi K, Sakamoto H, Enari M, et al. Druggable oncogene fusions in invasive mucinous lung adenocarcinoma. Clin Cancer Res (2014) 20(12):3087–93. doi:10.1158/1078-0432.CCR-14-0107
46. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 116(2):281–97. doi:10.1016/S0092-8674(04)00045-5
47. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature (2005) 435(7043):834–8. doi:10.1038/nature03702
48. Inamura K, Ishikawa Y. MicroRNA in lung cancer: novel biomarkers and potential tools for treatment. J Clin Med (2016) 5(3):36. doi:10.3390/jcm5030036
49. Kuninty PR, Schnittert J, Storm G, Prakash J. MicroRNA targeting to modulate tumor microenvironment. Front Oncol (2016) 6:3. doi:10.3389/fonc.2016.00003
50. Jahagirdar D, Purohit S, Jain A, Sharma NK. Export of microRNAs: a bridge between breast carcinoma and their neighboring cells. Front Oncol (2016) 6:147. doi:10.3389/fonc.2016.00147
51. Nadal E, Zhong J, Lin J, Reddy RM, Ramnath N, Orringer MB, et al. A microRNA cluster at 14q32 drives aggressive lung adenocarcinoma. Clin Cancer Res (2014) 20(12):3107–17. doi:10.1158/1078-0432.CCR-13-3348
52. Garajová I, Funel N, Fiorentino M, Agostini V, Ferracin M, Negrini M, et al. MicroRNA profiling of primary pulmonary enteric adenocarcinoma in members from the same family reveals some similarities to pancreatic adenocarcinoma – a step towards personalized therapy. Clin Epigenetics (2015) 7:129. doi:10.1186/s13148-015-0162-5
53. Nishikawa E, Osada H, Okazaki Y, Arima C, Tomida S, Tatematsu Y, et al. miR-375 is activated by ASH1 and inhibits YAP1 in a lineage-dependent manner in lung cancer. Cancer Res (2011) 71(19):6165–73. doi:10.1158/0008-5472.CAN-11-1020
54. Yu Y, Zuo J, Tan Q, Zar Thin K, Li P, Zhu M, et al. Plasma miR-92a-2 as a biomarker for small cell lung cancer. Cancer Biomark (2017) 18(3):319–27. doi:10.3233/CBM-160254
Keywords: adenocarcinoma, driver mutation, genetic alteration, histology, molecular pathology, lung cancer
Citation: Inamura K (2017) Lung Cancer: Understanding Its Molecular Pathology and the 2015 WHO Classification. Front. Oncol. 7:193. doi: 10.3389/fonc.2017.00193
Received: 29 March 2017; Accepted: 11 August 2017;
Published: 28 August 2017
Edited by:
Giulia Veronesi, Humanitas Research Hospital, ItalyReviewed by:
Lorenzo Spaggiari, Istituto Europeo di Oncologia, ItalyCopyright: © 2017 Inamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kentaro Inamura, a2VudGFyby5pbmFtdXJhQGpmY3Iub3IuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.