 Maria Livia Sassano
Maria Livia Sassano Alexander R. van Vliet
Alexander R. van Vliet Patrizia Agostinis
Patrizia Agostinis
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 18 August 2017
Sec. Molecular and Cellular Oncology
Volume 7 - 2017 | https://doi.org/10.3389/fonc.2017.00174
This article is part of the Research Topic Inter-Organelle Calcium Communication in Cancer View all 14 articles
The tight cross talk between two essential organelles of the cell, the endoplasmic reticulum (ER) and mitochondria, is spatially and functionally regulated by specific microdomains known as the mitochondria-associated membranes (MAMs). MAMs are hot spots of Ca2+ transfer between the ER and mitochondria, and emerging data indicate their vital role in the regulation of fundamental physiological processes, chief among them mitochondria bioenergetics, proteostasis, cell death, and autophagy. Moreover, and perhaps not surprisingly, it has become clear that signaling events regulated at the ER–mitochondria intersection regulate key processes in oncogenesis and in the response of cancer cells to therapeutics. ER–mitochondria appositions have been shown to dynamically recruit oncogenes and tumor suppressors, modulating their activity and protein complex formation, adapt the bioenergetic demand of cancer cells and to regulate cell death pathways and redox signaling in cancer cells. In this review, we discuss some emerging players of the ER–mitochondria contact sites in mammalian cells, the key processes they regulate and recent evidence highlighting the role of MAMs in shaping cell-autonomous and non-autonomous signals that regulate cancer growth.
The interconnection between mitochondria and the endoplasmic reticulum was the first inter-organelle contact site discovered. This fascinating discovery dates back to 1959, when Copeland and Dalton identified a possible association between ER and mitochondria in cells of the pseudobranch gland of a teleost (1). Ten years later, another research group verified this discovery by using electron microscopy (2). It was only in 1990, however, after several subcellular fractionation studies (3–5), that the mitochondria-associated membranes (MAMs) were isolated as a distinct and purified structure from rat liver. Here, Vance demonstrated that MAMs work as a platform for lipid biosynthesis and transfer, by proposing an alternative non-vesicular mechanism for the inter-organelle transfer of lipid (6–8). Furthermore, eight years later, Rizzuto and co-workers uncovered another crucial functional role of the MAMs (9). They demonstrated by live cell imaging that these regions of tight contact are necessary to maintain Ca2+ homeostasis, suggesting the existence of Ca2+-enriched “hot spots,” or microdomains at the ER, which are exposed to higher concentration of Ca2+ than the bulk cytosol, upon IP3-mediated Ca2+ release. Further studies revealed that the ER and mitochondria interaction could occur at approximately 10–25 nm (9, 10). Importantly, at these sites of organelle interaction, the two membranes do not fuse but rather form a proteinaceous tether (11). It is now estimated that the percentage of the mitochondrial surface that appear to be physically in contact with the ER ranges between 5 and 20%, depending on the cell type (9).
Along with the recognition of the multifaceted roles of the ER and mitochondria in cellular homeostasis and cell death, these landmark discoveries sparked a growing interest in MAM biology. Consistent with this, accumulating evidence indeed indicate that disturbances in this tightly regulated interface can elicit dysfunctions in crucial cellular processes, such as apoptosis, inflammation, and autophagy, which are involved in several pathologies, including—but not limited to—cancer. Several excellent recent reviews have discussed in great details the emerging connections of MAMs with important human diseases (12–16).
However, it should be mentioned that contact sites established by the ER are not only confined to mitochondria and that through specific microdomains the ER may dynamically connect various organelles or structures in the cell. Although the complexity of this phenomenon as well as the molecular composition of these discrete ER regions are not completely solved yet, the ability of the ER to spatially and temporally coordinate interorganellar connections may have a profound physiological meaning (17). For example, it may allow mitochondria to become spatially redistributed in regions or subcellular hot spots, such as the ER–plasma membrane (PM) contacts, in order to couple extracellular cues to rapid changes in mitochondria bioenergetics and/or buffering calcium entry (18) or to the redistribution of endosomes (17). An in depth discussion of the emerging roles of membrane contact sites will not be the focus of this review and is discussed elsewhere (17).
Here, we discuss accumulating evidence demonstrating the essential role of ER–mitochondria contact sites as a molecular platform that regulates fundamental cellular processes, which are harnessed by cancer cells to support their malignant phenotype, such as sustaining ER–mitochondria Ca2+ signaling.
Fundamental knowledge of interorganellar communication accumulated in the past decades has changed the previously accepted “mitochondria-centered” perception that the regulation of key processes, such as metabolism and cell death, could occur solely in a mitochondrion-autonomous way. Indeed, although mitochondria play a pivotal role in various aspects of cellular health, it is now appreciated that cellular processes critical for the cell’s fate, are dictated by the interactions that mitochondria dynamically establish with the ER through the MAMs. Thus, MAMs are no longer considered just a static physical bridge between ER and mitochondria but are now being increasingly considered as a crucial cellular platform for the exchange of molecular signals and protein complex formation, where critical cellular decisions in response to alterations of cellular homeostasis are taken. Several recent reviews have discussed the molecular nature of the ER–mitochondria contact sites and revealed molecular “tags,” such as palmitoylation (19), used to dynamically recruit a variety of proteins to these subdomains (14, 20, 21).
In the next sections, we will discuss some of the vital cellular functions and biological processes regulated by the ER–mitochondria appositions (see Figure 1).
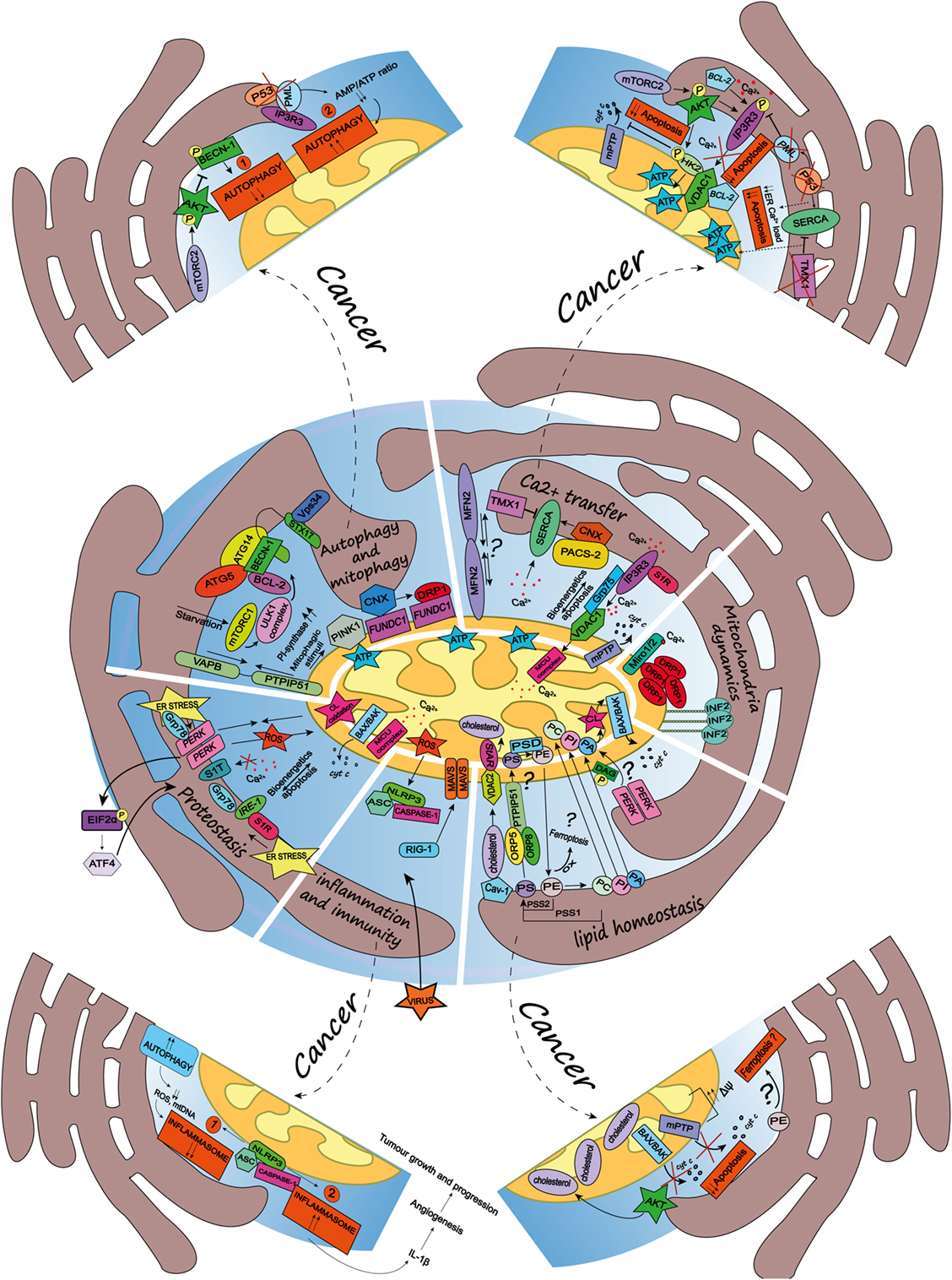
Figure 1. Schematic summary of the multifaceted roles of the endoplasmic reticulum (ER)-mitochondria contact sites and representation of the main pathways they regulate. Here, are represented some of the vital cellular processes regulated by the mitochondria-associated membranes (MAMs), such as autophagy and mitophagy, Ca2+ transfer, mitochondria dynamics, lipid homeostasis, inflammation and immunity, and proteostasis. Dashed arrows link the physiological condition of some of the MAMs-regulated cellular functions to pathways harnessed by cancer (for further details see main text). Autophagy and mitophagy: here is shown the MAMs localization of the autophagy-related gene (ATG5) and ATG14, whose recruitment is mediated by the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein syntaxin 17, upon activation of the mammalian target of rapamycin complex 1- Ulk1 unc-51 like kinase 1 (ULK1) complex, which is located in phosphatidylinositol synthase-enriched domains. Other autophagic key proteins found at MAMs are Beclin-1 and the phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3/Vps34). Mitophagic stimuli lead to the relocalization of the PTEN induced putative kinase 1 (PINK1) at MAMs. Additionally, the hypoxia-stimulated and mitochondria-associated FUN14 domain-containing 1, interacting with calnexin (CNX), relocates at MAMs, thus promoting Drp1 recruitment and hypoxia-induced mitophagy. Furthermore, the tethering complex vesicle-associated membrane protein-associated protein B (VAPB)–protein tyrosine phosphatase-interacting protein 51 (PTPIP51), which by tightening the ER–mitochondria contact sites, impairs rapamycin- and torin1-induced, but not starvation-induced, autophagy. Ca2+ transfer: here is represented the inositol 1,4,5 triphosphate receptor (IP3R) which is stabilized at MAMs by the chaperone Sigma1R (S1R). IP3R is involved in Ca2+ transfer from the ER storage to mitochondria by interacting with the mitochondrial voltage-dependent anion channel 1 (VDAC1), thus allowing Ca2+ transfer into the mitochondria through the mitochondrial calcium uniporter (MCU) and promoting mitochondrial bioenergetics and/or apoptosis, here represented by the opening of the mitochondrial permeability transition pore. Their interaction is allowed by the chaperone glucose-regulated protein 75, enriched at MAMs. Mitofusin 2 is another modulator of the interaction between ER and mitochondria (for details see the main text). The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) residing at the ER–mitochondria contact sites is positively modulated by chaperone CNX, whose localization at MAMs is regulated by the phosphofurin acidic cluster sorting protein 2 and negatively by the thioredoxin-related transmembrane protein 1. Mitochondria dynamics: here is represented the outer mitochondrial membrane (OMM) Rho-GTPase proteins Miro1/2 which, in presence of Ca2+, lose its binding with kinesin 1 (not shown), thereby reducing mitochondria motility and fostering the mitochondrial Ca2+ buffering. Oligomers formed by the dynamin-related guanosine triphosphatase (GTPase) dynamin-related protein 1 (Drp1) at MAMs, facilitate membrane scissions, along with actin filaments and the ER-localized protein inverted formin 2. Lipid homeostasis: the figure illustrates the lipid transfer and synthesis occurring at MAMs. The transfer of the phosphatidylserine (PS), which is formed from phosphatidylethanolamine (PE) or phosphatidylcholine (PC) by PS synthases (PSS1, PSS2), is required for the mitochondrial synthesis of PE, which is enabled by PS decarboxylase. Moreover, the ER-anchored oxysterol-binding protein-related protein (ORP) 5 and 8, are localized at MAMs where they could promote the transport of PS. PE, once exported to the ER is converted in PC. Here is also shown a speculative link to ferroptosis regulated at the MAMs, where the oxidation of polyunsaturated lipids, such as PE, occurs. The phosphatidic acid (PA) is transferred from the ER to the mitochondria as well, where it is essential for the synthesis of cardiolipin (CL), pivotal to apoptosis induction. Here is represented an interesting although speculative link involving the lipid activity kinase of the ER stress sensor RNA-dependent protein kinase (PKR)-like ER kinase (PERK), which by phosphorylating diacylglycerol can generate PA at MAMs. Cholesterol transfer into mitochondria is facilitated by the interaction with the cholesterol-binding caveolin-1, a MAM-resident protein. Moreover, the cholesterol transport steroidogenic acute regulatory protein can shuttle cholesterol from the OMM to the inner mitochondrial membrane interacting with the voltage-dependent anion channel 2. Inflammation and immunity: here is illustrated how the inflammasome is assembled at the MAMs after sensing host danger or intracellular damage, like damaged mitochondria. The inflammasome is made up of the pattern recognition receptors and nucleotide-binding domain and leucine-rich repeat-containing (NLR) protein family, NOD-like receptor family 3 and the adaptor protein apoptosis-associated speck-like protein (ASC), which recruits caspase-1 and allows its activation leading to the release of pro-inflammatory cytokines, like IL-1beta. Upon RNA virus infection, the recognition receptor RIG-I moved at MAMs, where it recruits its adaptor mitochondrial antiviral-signaling protein, thus starting an intracellular immune response. Proteostasis: here is reported that key ER stress sensors localize at MAMs, where, they can regulate signaling events linked to loss of ER homeostasis. In particular, the ER stress sensor PERK can modulate the tethering ER–mitochondria thus promoting the transfer of reactive oxygen species from the ER to the mitochondria, CL oxidation and the consequent BAX/BAK-dependent release of cytochrome c (cyt c). The PERK-activating transcription factor 4 axis of the unfolded protein response regulates the expression of S1T (truncated variants of SERCA1), thus leading to apoptosis. Moreover, here is also reported the MAMs localization of the inositol requiring enzyme 1, which interacts with S1R and Grp78, as well present at MAMs.
The importance of the ER–mitochondria platform in the regulation of Ca2+ homeostasis has been firmly established (22, 23). The tight tether between the ER membranes and mitochondria allows Ca2+ to be rapidly transferred through specialized microdomains that overcome the low apparent Ca2+ affinity (Kd ~ 15–20 M) of the mitochondrial Ca2+ uniporter (MCU) (9, 24). Many ER-associated Ca2+ handling proteins have been found enriched at MAMs, thus supporting the close correlation between this organellar intersection and Ca2+ regulation. Chiefly among these are the inositol 1,4,5 triphosphate receptors (IP3Rs), which together with the ryanodine receptors, represent the major ER Ca2+ channels (25). In particular, it has been reported that isoform 3 of the IP3Rs is preferentially involved in transmitting Ca2+ signals to mitochondria and co-localizes most strongly with them (26). The IP3Rs have been identified to be physically linked to the mitochondrial voltage-dependent anion channel 1 (VDAC1), located at the outer mitochondrial membrane (OMM) and associated to the MAMs as well (27). The interaction between IP3Rs and VDAC1 is modulated by the molecular chaperone glucose-regulated protein 75, which acts as a bridge allowing the Ca2+ transfer from the ER to the Ca2+ permeable OMM, from which the ions can easily reach the mitochondrial matrix through the mitochondrial Ca2+ uniporter MCU. Moreover, the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump localized to the ER membrane, is regulated by several proteins residing at the ER–mitochondria contact sites, thereby affecting Ca2+ flux and modulating MAMs interplay. SERCA is the ER “housekeeping” Ca2+ pump, which ensures the refilling of the ER Ca2+ storage by actively pumping this ion from the cytosol to the ER, thus restoring the Ca2+ balance between cytosol and ER creating a high [Ca2+] gradient between these regions (~0.1 μM in the cytosol and ~400 μM in the ER) (28). SERCA2b is the SERCA isoform that is particularly enriched at the ER–mitochondria contact sites (19). Among the SERCA2b interacting partners, the MAMs-resident chaperone calnexin (CNX) functions as a positive regulator while thioredoxin-related transmembrane protein 1 (TMX1) inhibits SERCA2b and thus increases mitochondrial Ca2+ flux (19). The phosphofurin acidic cluster sorting protein 2 (PACS-2), known to regulate the localization of CNX at the MAMs (19, 29), has also been implicated in Ca2+ homeostasis through its interaction with the Ca2+ channel transient receptor potential protein 2 (30). However, whether PACS-2 exerts this role through a direct modulation of ER–mitochondria contact sites is still a matter of debate (31).
A strong link exists between the strength of the connection between the ER and mitochondria and its Ca2+ trafficking role and is often used as experimental evidence in essays exploring MAMs defects. One of the major players in MAMs tethering is vesicle-associated membrane protein-associated protein B (VAPB)-protein tyrosine phosphatase-interacting protein 51 (PTPIP51) complex whose loss affects Ca2+ homeostasis, causing a significant delay in mitochondria Ca2+ uptake and, as a consequence, an increase in [Ca2+]cyt following release from the ER store (32). A recent study indicated that the loss of both these proteins decreases the interaction between the IP3R3 and VDAC1, affecting the mitochondrial Ca2+ uptake (33). In this vein, another important ER–mitochondria tethering protein is mitofusin 2 (MFN2). Together with MFN1, MFN2 belongs to the OMM-associated fusogenic GTPases family of proteins regulating mitochondrial fusion (34). Only MFN2, however, is enriched at the ER–mitochondria contact sites and controls their stability, Ca2+ and lipid transfer and the morphology of both these organelles (35). In line with this, loss of MFN2 has been reported to disrupt ER–mitochondria juxtapositions and cause a decrease in the mitochondrial Ca2+ uptake after IP3-generated stimuli (35). However, this MFN2 model has been challenged recently (36). Some studies showed that MFN2 actually works antagonistically on ER–mitochondria contacts (36–38). These studies also showed that MFN2 ablation leads to an increase in ER–mitochondria coupling, thus boosting—rather than reducing—the efficiency of the IP3-induced Ca2+ transfer to mitochondria and increasing Ca2+ dependent cell death sensitivity (36). However, recent work has since tried to reinforce the original finding of a tethering role for MFN2 at the ER–mitochondria contact sites and its consequence for mitochondrial Ca2+ uptake (39). In other words, the role of MFN2 at the MAMs still remains controversial.
The ER is the major lipid factory in the cell and provides a supply of various lipids to different organelles that are unable to meet all of their own lipid requirements. The environment generated by the ER–mitochondria juxtapositions represents a privileged site for non-vesicular lipid trafficking between these organelles and facilitates transport of lipids via carrier proteins (40).
Although the mitochondria are capable of synthesizing several lipids through their own repertoire of enzymes, they require phosphatidylcholine (PC), phosphatidylinositol (PI), phosphatidylserine (PS), and sterols from the ER (41, 42). Mitochondria import PS through the MAMs where it is synthesized from PC or phosphatidylethanolamine (PE) by PS synthases (PSS1, PSS2). In line with this, MAMs are enriched for several phospholipid-synthesizing enzymes, such as PS and PI synthase (8, 43). After being transferred to mitochondria, PS is decarboxylated by PS decarboxylase in the inner mitochondrial membrane (IMM) to form PE. PE is then rapidly exported from the mitochondria to other organelles, such as the ER, where it is converted to PC (44). Inhibiting the enzymatic activity of PDS leads to a massive accumulation of PS in MAMs, thus supporting and confirming the involvement of the ER–mitochondria contact sites in the transfer of PS (45). The ER–mitochondria interface is also the site of phosphatidic acid (PA) transfer. PA is the precursor of cardiolipin (CL), a phospholipid enriched in the inner membrane of the mitochondria and essential for mitochondrial bioenergetics and apoptosis induction (see further section) (7, 46–48). Although PA can be made by mitochondria as well, most of the PA used for CL synthesis originates from the ER and is thus transported through the MAMs (49, 50). Interestingly, the ER stress sensor and Ser/Thr kinase PERK [RNA-dependent protein kinase (PKR)-like ER kinase], which has been shown to be enriched at the ER–mitochondria contact sites (51) is endowed with a lipid kinase and PA-generating activity (52). This raises the intriguing possibility that the lipid kinase activity of PERK may be regulated at the ER–mitochondria contact sites, a hypothesis urging experimental confirmation.
A recent study investigating the role of the ER-anchored oxysterol-binding protein-related protein (ORP) 5 and 8, which transport PS from the ER to the PM at the ER–PM contact sites, found that ORP5/8 are localized at the ER–mitochondria contact sites. MAMs localization of ORP5/8 required their interaction with the OMM protein PTPIP51 and their functional lipid transfer domain (53). Depletion of ORP5/8 caused defects in mitochondrial morphology and respiratory function, suggesting that their ability to transfer essential lipids, such as PS, within the MAMs contributes to the maintenance of mitochondrial integrity.
The lipid composition of the MAMs is critical for their functions and biological roles. MAMs are highly enriched in cholesterol, and the altering cholesterol levels itself has been shown to modulate the ER–mitochondrial connections and mitochondrial functions. Cholesterol transport into the mitochondria requires cholesterol-binding proteins. In mouse liver, the absence of the cholesterol-binding caveolin-1 (Cav-1), shown to be a MAM-resident protein, reduced the stability of ER–mitochondria contacts and increased their cholesterol content (54). Moreover, genetic ablation of Cav-1 caused a decreased electron flux through the respiratory chain, increased reactive oxygen species (ROS) generation, and apoptosis sensitization (55). Likewise, disturbed glycosphingolipid content (primarily synthesized in the ER) as caused by accumulation of GM1-ganglioside at the ER–mitochondria contact sites, in a mouse model of human lysosomal storage disease GM1-gangliosidosis, elicited stimulation of IP3R-mediated ER Ca2+ depletion, mitochondrial Ca2+ overload and OMM permeabilization, thus triggering ER stress-induced and mitochondria-mediated apoptosis (56) (see further section on MAMs and ER Proteostasis).
Recently, in steroid-producing cells, the cholesterol transport steroidogenic acute regulatory (StAR) protein, which shuttles cholesterol from the OMM to the IMM was found to be localized to the MAMs where it interacts with the voltage-dependent anion channel 2, enabling StAR to enter in the mitochondrial matrix and regulate steroidogenesis (57, 58). Interestingly, the generation of the intermediate StAR folding state required for the delivery of cholesterol to the mitochondria occurs at the MAMs and necessitates the presence of the ER-chaperone glucose-regulated protein 78 (GRP78) (59). This mechanism, operating at the ER–mitochondria appositions, therefore couples crucial sensors of unfolded proteins and ER proteostasis (see below), like the chaperone GRP78, to critical mitochondrial functions of steroid-producing cells (59).
Finally, the ER–mitochondria lipid connection through the MAMs could potentially modulate crucial steps in the initiation of autophagy and cell death (see next sections). A recent report shows that components of the Ulk1 unc-51 like kinase 1 (ULK1) complex involved in autophagy initiation, are recruited to ER subdomains enriched in PI synthase (60), which prime for the initiation of the autophagosome. Although not formally tested, these subdomains are reminiscent of the ER–mitochondria contact sites which have been shown to be a site for autophagosome formation (see in further section).
Both the ER and mitochondria are highly dynamic organelles, forming networks that may undergo rapid changes in the size, length, and shape, depending on metabolic and Ca2+ buffering needs, or in response to different cellular insults. Beside the MAMs, the mitochondria are able to establish other type of organelle-contact sites, such as the dynamic interactions with the ER and the PM, facilitating their apposition with the PM, to regulate Ca2+ homeostasis (61, 62).
A large body of evidence links MAMs to the regulation of intracellular transport, motility, and morphology of both the mitochondria and ER. In mammalian cells, several proteins involved in the anterograde and retrograde movement of these organelles, through kinesin 1 and cytoplasmic dynein molecular motors, respectively, are tightly regulated by local Ca2+ concentration rises (63). Perhaps, the most important players in this perspective are the OMM Rho-GTPase proteins Miro1/2, Ca2+ sensors connecting the mitochondria to kinesin 1, which have been shown to localize to the ER–mitochondria contact sites (64, 65). In subdomains that are exposed to high Ca2+ concentrations, such as the MAMs, Miro 1/2 lose their connection to kinesin 1. This halts mitochondrial mobility, while tightening MAMs-association, thus allowing buffering of (excessively) high cytosolic Ca2+ levels or stimulating oxidative phosphorylation. Beyond motility, mitochondrial fission events, driven by the dynamin-related guanosine triphosphatase (GTPase) dynamin-related protein 1 (Drp1), which forms oligomers that assemble around mitochondria to facilitate membrane scission, are orchestrated at the interface between the tubular ER and the mitochondria (66). A recent study provided compelling evidence that tubular ER is strongly associated with mitochondrial fission mechanics (67). During mitochondrial fission, the formation of constriction sites has been shown to occur at the ER–mitochondrial tether (67). Guided by the ER-localized protein inverted formin 2, actin filament polymerization in correspondence to the fission foci provides the driving force to constrict the mitochondria thus allowing Drp1 recruitment and mitochondrial fission (68). This physical intersection between tubular ER and mitochondria hinted at the involvement of the MAMs in mitochondrial division. This hypothesis has now been confirmed. An elegant study evidenced that a subset of ER–mitochondria contacts assembles the machinery for mitochondrial DNA synthesis. Thus, along with the coordination of mitochondrial motility and constriction/fission events, the ER–mitochondria contacts mark the site for mitochondrial mtDNA replication and ensure the accurate distribution of newly replicated mtDNA to daughter mitochondria upon division (69).
The rapid exchange of Ca2+-occurring through the MAMs is essential for mitochondrial bioenergetics and, consequently, cell fate decisions (70). The Ca2+-concentration in mitochondria is fundamental for several cellular processes, among these ATP production. Three dehydrogenases of the Krebs cycle are Ca2+-dependent enzymes, the pyruvate dehydrogenase, whose activation is due to Ca2+-dependent dephosphorylation, the α-ketoglutarate, the isocitrate dehydrogenases and FAD-glycerol phosphate dehydrogenase, activated directly by Ca2+ (71–73).
Moreover, several loss or gain-of-function studies have demonstrated that MFN2, one of the most studied modulators of the ER–mitochondria contacts, critically affects mitochondrial bioenergetics, through mechanisms that are independent of its fusogenic function (34). Thus, while MFN2 depletion reduces glucose oxidation, cellular respiration, mitochondrial membrane potential and proton leak, and mitochondrial coenzyme Q levels (74), its overexpression activates mitochondrial metabolism (34, 69). Interestingly, MFN2 also confers transient protection against starvation-induced autophagy and apoptosis by promoting mitochondrial hyperfusion and elongation (75), redox-regulated processes (76, 77) that optimize ATP production while sparing mitochondria from degradation (75).
While regulated ER-to-mitochondrial Ca2+ transport favors bioenergetics, it is well established that massive and prolonged mitochondrial Ca2+ overload triggers the opening of the mitochondrial permeability transition pore, precipitating apoptosis (72). As a result of Ca2+ overload and consequent OMM permeabilization, proapoptotic and caspase-activating factors, including cytochrome c (cyt c) are released in the cytoplasm (78). Once in the cytoplasm cyt c can also exacerbate Ca2+ release from IP3R by binding to it, thus avoiding the Ca2+-dependent inhibition of the receptor and amplifying caspase activation (79). To highlight the importance of ER–mitochondria juxtaposition in controlling cellular fate [for a complete overview please see Ref. (80)], work done in the lab of Hajnócsky and coworkers showed that tightening of the ER–mitochondria interaction leads to apoptotic cell death by increasing mitochondrial Ca2+ uptake, while loosening these links promote mitochondrial respiration and cellular survival (10, 81).
Beyond Ca2+ overload, MAMs could be decisive for the induction of either apoptosis or a newly described form of iron-regulated necrotic cell death, called ferroptosis, mediated by ROS and hallmarked by the accumulation of lipid peroxidation products (51, 82). During apoptosis induced by photo-oxidative ER stress (i.e., hypericin-mediated photodynamic therapy (83)), weakening of the ER–mitochondria contacts by the depletion of PERK protected cancer cells from rapid ROS-induced CL oxidation—preceding the unfolded protein response (UPR)-mediated cell death pathway—and the consequent release of cyt c in the cytosol (51). Indeed, during apoptosis, the interaction of CL with cyt c in the presence of mitochondrial ROS, drives the formation of cyt c/cardiolipin peroxidase complexes that oxidize CL and cause the release of cyt c from mitochondria into the cytosol (48, 84).
But could MAMs also be relevant for the induction of ferropotosis? Recent studies using quantitative redox lipidomics showed that oxidation of polyunsaturated lipids, the molecular executioners of ferroptosis, occurs in ER subdomains and involves oxidation of one class of phospholipids; PE molecules with arachidonoyl (AA) or adrenoyl (AdA) fatty acyls, driven by lipoxygenase (15-LOX) (85). A direct link between ferroptosis and MAMs has not been explored yet. Notably, acyl-CoA synthetase long-chain family member 4 (ACSL4 or FACL4), which catalyzes the synthesis of AA thus promoting its esterification in phospholipids and conferring ferroptosis susceptibility (86), is found enriched at the MAMs (87). Thus, these observations raise the tantalizing possibility that ROS-mediated death signals generated at the ER–mitochondria contact sites could be decisive to trigger either apoptosis of ferroptosis, depending on the type of oxidized phospholipids.
Accumulating evidence indicates the tight connection between the UPR and MAMs components. The UPR is a conserved intracellular pathway engaged by various physiological and stressful cues triggering the accumulation of unfolded proteins in the ER, a condition known as ER stress [extensively reviewed in Ref. (88, 89)]. ER–mitochondria contacts establish an intense and mutual cross talk that facilitates stress responses evoked by the UPR (14, 90, 91). Perhaps, the strongest evidence in support of this link is the finding that key ER stress sensors have been shown to regulate signaling events linked to loss of ER homeostasis by directly localizing at the MAMs and/or by regulating the expression of ER–mitochondria tethers. In particular, our lab discovered that the ER stress sensor, PERK is an integral member of the MAMs and has a tethering role at the ER-mitochondria contact sites (51). PERK-deficient cells display weakened ER–mitochondria contact sites and increased apoptosis resistance against agents that simultaneously mobilize Ca2+ and induce ER stress through ROS (51). Interestingly, one study showed that MFN2 interacts with PERK and prevents its activation (92). In response to ER stress, PERK ablation increases apoptosis in MFN2-ablated cells, but it also partially rescues the aberrant mitochondrial Ca2+ content and the fragmentation of the mitochondrial network, caused by loss of MFN2 (92). While this study would suggest that MFN2 deficiency causes mitochondrial aberrations through sustained activation of the PERK pathway of the UPR, the still controversial aspects around the role of MFN2 in ER–mitochondria tethering and the presence of PERK at the MAMs, ask for further mechanistic analysis of this signaling cross talk.
Moreover, the PERK-activating transcription factor 4 (ATF4) axis of the UPR was shown to be required for the induction of S1T, a truncated variants of SERCA1 which localized to the MAMs and led to an increasing number of contact sites, mitochondrial Ca2+ overload and an inhibition of mitochondrial movement, which ultimately triggered apoptosis (93). This suggests that following ER stress, the activation of the PERK-ATF4 pathway may induce a transcriptional program that reinforces ER–mitochondria contact sites. In line with this, the PERK-ATF4 pathway can also lead to the expression of E3 ubiquitin ligase Parkin (94), which has been reported to increase ER–mitochondrial coupling and Ca2+ transfer favoring mitochondrial bioenergetics (95). This highlights that activation of the PERK-ATF4 pathway of the UPR, may contextually regulate cellular fate through the upregulation of MAMs-resident proteins. Other members of the UPR, such as the inositol requiring enzyme 1 (IRE1), appear to localize to or have a signaling role at the ER–mitochondria contact sites during ER stress. Interestingly, during ER stress, IRE1 dimerization is facilitated through the interaction with the MAMs-resident ER-chaperone sigma1 receptor (96), which operates as a Ca2+ sensor and dissociates from GRP78 upon ER Ca2+ depletion to favor IP3R-mediated Ca2+ transfer to mitochondria (97). Although the exact role of IRE1 at the MAMs still needs to be fully defined, the co-presence of key sensors of the UPR and GRP78 (a chaperone involved in their activation) with Ca2+ sensors and channels at the ER–mitochondria contact sites, suggests that MAMs are a critical signaling hub coordinating responses to changes in ER Ca2+ homeostasis.
The mitochondrial contact sites are not the only site of close contact that the ER is able to form with organelles in order to perform its various functions and maintain the ER homeostasis. The ER–PM juxtapositions are one of the most important ER–organellar contact sites, together with MAMs, which are responsible for orchestrating several cellular responses including—but not limited to—Ca2+ homeostasis, lipid-mediated signaling, metabolism, cell death, and exocytosis (98). In particular, ER–PM juxtapositions are enabled by morphological changes in the ER and the expansion of thin ER structures, mainly known as cortical ER, toward the PM, thus allowing the ER to rapidly sense and respond to alterations in ER homeostasis by modifying the composition of proteins and lipid membranes. Recently we revealed that PERK, beyond its ability to maintain the ER–mitochondria appositions (51), enables rapid and efficient formation of ER–PM contact sites upon ER Ca2+ store depletion, largely through a UPR independent mechanism (99). PERK, through its interaction with Filamin A (FLNA), a major F-actin binding protein, and remodeling of the F-actin network allows the expansion of the ER tubular network underneath the PM and the recruitment of the ER–PM tethers Stromal interaction molecule 1 (STIM1) and Extended-Synaptogamin 1 (E-Syt1) at the PM in response to agents eliciting ER Ca2+ depletion and store operated Ca2+ entry (99).
Thus, it is increasingly becoming clear that key ER stress sensors like PERK orchestrate the communication between the ER and other crucial compartments/organelles, like the PM, mitochondria and nucleus, by regulating ER homeostasis through membrane contact sites as well as UPR-mediated transcriptional regulation.
Endoplasmic reticulum–mitochondria contact sites have been linked to autophagy initiation. Autophagy is a tightly regulated lysosomal pathway for the recycling of cytoplasmic material, including proteins and organelles, which serves as a vital quality control mechanism to preserve cellular integrity and homeostasis and as an alternative energy source to survive in nutrient-deprived conditions [for a detailed overview of the molecular pathways regulating autophagy see Ref. (100, 101)]. Interestingly, although the origin of the autophagosomal membrane still remains a matter of debate it has recently been demonstrated that autophagosomes form at the ER–mitochondria contact sites, thus assuming that this interplay could play a critical role in autophagy (102, 103). Recently, it has been reported that the autophagosome-formation marker ATG5 and the pre-autophagosome marker ATG14, whose recruitment is regulated by soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein syntaxin 17 (STX17), move and localize at MAMs (103). Accordingly, other key proteins involved in autophagosome formation have been found recruited at MAMs under starvation, chief among these are the pro-autophagic proteins Beclin-1 (BECN1) and the phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3/Vps34) (103). Additionally, the lipid raft-composition of the MAMs could facilitate initial organelle interaction, which leads to the formation of autophagosomes, since these lipid microdomains have been recently identified as important actors of the autophagic process (104).
Relocalization of key proteins of the mitophagy machinery such as the phosphatase and tensin homolog deleted on chromosome 10 (PTEN) induced putative kinase 1 (PINK1), a mediator of Parkin-dependent mitophagy, to the MAMs has been shown to occur in response to pro-mitophagic stimuli, indicating that the ER–mitochondria contact sites are a privileged site to tag mitochondria for clearance (105). Strengthening the connection with the mitophagy machinery, the hypoxia-stimulated and mitochondria-associated FUN14 domain-containing 1 (FUNDC1), has been found to serve as an adaptor for Drp1 recruitment at the ER–mitochondria interface, thereby inducing hypoxia-mediated mitochondrial fission and mitophagy (106).
However, a new study shows that the tightening of the ER–mitochondria tether inhibits rather than promotes autophagy (14, 33). Although it seems contradictory, this elegant study sheds light on the mechanism regulating autophagy by MAMs, suggesting that the ER–mitochondria contact sites could play different roles in autophagy signaling depending on the stimuli. In fact, the overexpression of the tethering complex VAPB-PTPIP51, tightening the ER–mitochondria coupling, impairs rapamycin- and torin1-induced, but not starvation-induced, autophagy (33). Additionally, the mechanism by which the VAPB-PTPIP51 complex regulates autophagy is due to their ability in modulating the ER–mitochondria Ca2+ signaling, further supporting the role of MAMs as the crucial signaling hub of autophagy regulation.
Given the tight link, as mentioned before, between ER stress and MAMs, it is perhaps not unexpected that ER–mitochondria juxtapositions are emerging as regulators of inflammation and immunity. ER–mitochondria contact sites have been shown to modulate the activity of a member of the pattern recognition receptors (PRRs) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) protein family, NOD-like receptor family 3 (NLRP3). NLR proteins are involved in sensing host “dangers” elicited by foreign pathogens, tissue and intracellular damage, such as molecules released by damaged mitochondria, and in mounting an inflammatory response (107). This response is driven by the assembly of an inflammasome, a multimolecular complex composed of an NLR and the adaptor protein apoptosis-associated speck-like protein (ASC), which recruits caspase-1. Caspase-1 activation in the complex leads to the proteolytic maturation of the pro-inflammatory cytokine interleukin 1β (IL-1β) and interleukin 18 (IL-18) (108). Recently, it has been shown that upon detection of mitochondrial ROS, and release of mitochondrial factors, NLRP3 relocates from the ER to the ER–mitochondria appositions, thus sensing the mitochondrial alteration and assembling the inflammasome (109). Interestingly, upon RNA virus infection, the cytosolic pathogen recognition receptor RIG-I is recruited at the ER–mitochondria contact sites and recruits its adaptor mitochondrial antiviral-signalling protein (MAVS), to drive an intracellular immune synapse that directs antiviral innate immunity (110).
These observations indicate that ER–mitochondria contacts sites can modulate important cell-non-autonomous processes, like inflammation and immunity, that are emerging as key modulators of the tumor microenvironment and therapeutic responses (111).
Given the crucial role of MAMs in cellular homeostasis and cellular fate, it is not surprising that an increasing number of reports have highlighted a close intersection between processes that are altered during carcinogenesis and ER–mitochondria contact sites. In particular, it is intriguing to note that several oncogenes and tumor suppressors have been found to target dynamically these ER microdomains. In line with this, it is increasingly becoming clear that signaling events regulated at the MAMs modulate cancer cell’s capability to adapt to intracellular stress, caused by oncogenic perturbations, thereby decreasing the cellular responsiveness to cell death signaling, as well as to extracellular cues found in the tumor microenvironment. Beyond the expression of oncogenes or loss-of-function of tumor suppressors, cancer cells hijack key homeostatic processes, which, as mentioned before are highly regulated at the intersection between the ER and mitochondria. For example, perturbations of Ca2+-signaling, autophagy and the inflammasome, may favor both cancer cell-autonomous and extrinsic features that enable cancer progression and therapy responses. Here, we discuss some of the emerging traits of the cancer cells that have been linked, or can be associated, to intracellular pathways and processes regulated at the ER–mitochondria appositions (see Figure 1).
It has been reported that the lipid composition of the IMM is altered in cancer cells (112, 113). In particular, several types of tumors show an increase in the content of cholesterol (114, 115) and phospholipids with shorter and less unsaturated acyl chains (116, 117). The mitochondrial inner membrane lipid composition could affect mitochondrial bioenergetics and impact on apoptotic cell death, altering the susceptibility to apoptotic stimuli (113, 114). For example, higher cholesterol content decreases the permeability of the IMM, thus affecting the passive transfer of protons and increasing the Δψ, effects that result in an impaired respiratory chain activity (118). Moreover, increased cholesterol is commonly associated in several solid tumors to the overactivation of the Akt pathway (119, 120). This observation has been linked to an impairment of B-cell lymphoma 2 (Bcl-2)-Associated X Protein (Bax)-driven OMM and cyt c release (115). As discussed above, the lipid composition of the MAMs could modulate different, not overlapping types of cell death, including apoptosis and ferroptosis, a form of regulated necrosis driven by lipid peroxides (121). This may offer an interesting therapeutic possibility given that alternative cell death pathways may be exploited to break down apoptosis resistance in cancer cells, and suggests that lipid manipulations of MAMs could be targeted to favor the cytotoxic activity of ferroptosis inducers.
Emerging cellular and in vivo evidence indicate that perturbation of Ca2+ homeostasis is one of the vital mechanism through which oncogenes and tumor suppressors impact cancer cell fate (81). Accordingly, and as discussed below, oncogenes and tumor suppressors engage in complex interactions at the ER–mitochondria contact sites to directly shape Ca2+ flux from the main cellular storage, the ER, to the cellular energy–supplier, the mitochondria.
As mentioned above the IP3Rs are the most important Ca2+-transport systems involved in maintaining Ca2+ homeostasis between ER and mitochondria. Given that these channels are directly responsible for mitochondrial Ca2+ uptake, and considering the role of Ca2+ signaling in cancer cells (122), IP3R activity has been shown to be directly or indirectly regulated by several oncogenes. It is now firmly established that the functionality of the IP3Rs is modulated by post-transcriptional modifications, including phosphorylation. Several oncogenes and tumor suppressors have been shown to act by modulating IP3R phosphorylation state in order to modify the rate of Ca2+ efflux from intracellular stores and, as a consequence, altering the cellular response to apoptosis (123, 124). The proto-oncogene serine/threonine kinase Akt (also known as protein kinase B), is able to phosphorylate all the isoforms of the IP3R in a conserved consensus sequence found in the cytosol-exposed C-terminal tail (125, 126). Accordingly, in cancer cells in which Akt is upregulated, IP3Rs are hyper-phosphorylated (126) and as a result, Ca2+ efflux from ER to mitochondria is blunted, thus protecting cells from apoptotic stimuli (127, 128). Interestingly, Akt mainly phosphorylates the isoform 3 IP3R (IP3R3), which is enriched at the ER–mitochondria interface, thus suggesting that this antiapoptotic function of Akt requires its compartmentalization at the MAMs (128). Interestingly, the mechanistic target of the Rapamycin complex 2 (mTORc2), which activates Akt, is also found at the ER–mitochondria appositions (129). Along with regulation of the phosphorylation state of IP3R and Ca2+ flux, the mTORc2/Akt complex controls mitochondrial metabolism and physiology, through the phosphorylation of the glycolytic enzyme hexokinase 2, thus promoting cancer cell’s aerobic glycolysis (Warburg effect) and preventing mitochondrial apoptosis (130, 131).
On the other side of the coin, the phosphorylation state of IP3R3 is, directly or indirectly, modulated by the recruitment of different tumor suppressors, including the lipid phosphatase and negative regulator of Akt, the PTEN (132, 133), p53 (134) and the promyelocytic leukemia (PML) protein (135). The fraction of PML localized at the MAMs (135) is essential to maintain a normal Ca2+ flux between ER and mitochondria since the absence of PML leads to a decrease in the amplitude of Ca2+ signals after induction of IP3-mediated Ca2+ release. At the MAMs, PML resides in a multi-protein complex, consisting of IP3R3, Akt and the protein phosphatase 2A (PP2A). PML appeared to be vital for the binding to IP3R of PP2A, which, by de-phosphorylating Akt, inhibits its activation (135, 136). The consequent reduced IP3R3 phosphorylation, ultimately drives Ca2+ flux from ER to mitochondria, thus preserving cellular susceptibility to Ca2+-dependent apoptotic signals. In addition, recent data showed that PML recruitment at the ER–mitochondria contact sites is modulated by the tumor suppressor p53 (134), thus suggesting that key tumor suppressors cooperate to hinder oncogenic signaling, by physically localizing to this strategic subcellular compartment.
The modulation of the SERCA pump functional activity is another common mechanism through which MAMs-located oncogenes and tumor suppressors, by remodeling the ER-Ca2+ steady state, can modulate cancer cell fate (81, 137, 138). For example, the non-nuclear proapoptotic function of the tumor suppressor p53 at MAMs involves its interaction with the SERCA pumps (139). Recent work showed that the pool of MAMs-associated p53 keeps the oxidation state of SERCA in check, thereby favoring its activity and filling of the ER Ca2+ store. Upon an apoptotic stress or treatment with an anticancer agent, this p53 mediated mechanism at the ER–mitochondria interface allows rapid mitochondrial Ca2+ overload, thus precipitating apoptotic cell death (139). Opposite to this, the redox-sensitive oxidoreductase TMX1 binds and inhibits the SERCA2b pump at the ER–mitochondria appositions, under oxidizing conditions, thus decreasing ER Ca2+ load and impairing mitochondrial respiration (19). This is in line with the notion that low-level constitutive IP3-mediated Ca2+ release supports oxidative phosphorylation of cancer cells, in the absence of which low ATP levels may trigger autophagy (70). Accordingly, cancer cells displaying lower levels of TMX1 show an increased ER Ca2+ content and an accelerated cytosolic Ca2+ clearance, along with a reduced ability of the ER to direct Ca2+ toward mitochondria. Interestingly, human cancer cells with impaired expression of TMX1 levels, when xenografted in nude mice exhibited faster tumor growth, which was explained by a metabolic reprogramming and induction of a Warburg phenotype in these cancer cells (19).
Beyond Akt and various antiapoptotic Bcl-2 family members including Bcl-2 and B-cell lymphoma-extra large (Bcl-XL), which are commonly overexpressed in a variety of cancers (140), have been proposed to suppress the IP3-mediated Ca2+-flux from ER to mitochondria by targeting the IP3Rs at the ER-mitochondria contact sites (141, 142). Although a full understanding of the antiapoptotic function of the Bcl-2 oncogene at the ER–mitochondria contact site is still elusive, Bcl-2 is thought to either lower the ER Ca2+-store content, or act directly on Ca2+-discharge from ER to the mitochondria [extensively reviewed in Ref. (143)]. Recently, the specific BH4 domain of Bcl-2 was shown to directly target all the three isoforms of IP3Rs and to inhibit their activity, hence suppressing ER Ca2+ release and reducing the cellular sensitivity to apoptosis (141, 144, 145). Likewise, the binding between Bcl-XL the IP3Rs has recently been determined to be fundamental for exerting its antiapoptotic functions (146–148). Both the antiapoptotic proteins Bcl-2 and Bcl-XL inhibit the MAMs-resident voltage-dependent anion channel VDAC1, by controlling its permeability to Ca2+ and thereby preventing cyt c release and apoptotic cell death (149–152). Although both Bcl-2 and Bcl-XL directly bind to the N-terminal region of VDAC1 (153, 154), the regions involved in this binding are different. In fact, whereas the BH4 domain of Bcl-XL is able to selectively target and inhibit the N-terminal domain of VDAC1, the BH4 domain of Bcl-2 is uniquely involved in inhibiting the IP3Rs (151). However, the functional role of the Bcl-XL-VDAC1 is still a matter of debate since a different impact of Bcl-XL interaction on VDAC1’s proprieties has been reported (155).
Emerging evidence indicates that autophagy has a highly dynamic role during tumorigenesis and can act either as a tumor-promoting or as a tumor-suppressing process, depending on the different stages of the tumor and the tight cross talk between cancer cells and stromal cells in the tumor microenvironment. Mechanistically, the tumor suppressor role of autophagy has been ascribed to the vital cell-autonomous functions of autophagy in maintaining cellular integrity and mitigating cellular damage under conditions of metabolic stress, including (but not limited to) the removal of damaged mitochondria and ROS [reviewed in Ref. (156)]. On the other hand, during tumor development, heightened autophagy becomes vitally required to maintain cancer cell’s elevated energy balance, through the recycling of intracellular components into biosynthetic pathways or ATP synthesis, and to regulate secretion of pro-tumorigenic factors. Thus in established tumors, autophagy allows cancer cells to plastically adapt to the increasingly hostile tumor microenvironment, characterized by poor nutrient availability, and foster tumor growth by establishing an intense interface with stromal cells (157, 158).
As mentioned before, several oncogenes and tumor suppressors that have been found localized at MAMs are also critical modulators of autophagy (159–162). In particular, the MAMs-associated mTORc2/Akt complex could inhibit autophagy by phosphorylating and inhibiting BECN1, an interacting partner of IP3R3 and Bcl-2 (162–164). Furthermore, autophagy induction in cancer cells is under the control of MAMs-associated p53 and PML. The absence of p53 or mislocalization of PML away from MAMs activates autophagy in response to cellular stress, suggesting that p53 via PML suppresses cytoprotective autophagy (134). These data indicate that the p53-PML interaction at the ER-mitochondria appositions, by regulating the transfer of Ca2+ from the ER to the mitochondria, on one hand, favors Ca2+-dependent apoptosis, while on the other, suppresses autophagy. Indeed the loss of PML, by decreasing constitutive Ca2+-release reduces mitochondrial respiration and ATP production, which results in the stimulation of autophagy via AMPK activation. This may explain why the loss of PML from MAMs promoted tumor growth and increased chemoresistance by simultaneously increasing resistance to apoptotic stimuli and, through the stimulation of autophagy, adaptation to metabolic stress and anticancer therapy-mediated cellular damage (134). Interestingly, in cancer cells with constitutively low level of ER-to-mitochondrial Ca2+ transfer, AMPK-dependent autophagy activation is not sufficient to maintain cancer cell survival. Inhibition of IP3R attenuates the growth of B16 melanomas, implanted in immunocompromised mice, due to a severe bioenergetics failure that triggers necrotic cancer cell death (165). This study further unravels a cancer cell vulnerability, which is imparted by the dependency on constitutive Ca2+ transfer to mitochondria for viability, at least in cancer cells still relying on mitochondrial respiration to supply their energy needs. It moreover suggests that targeting the ER–mitochondria interface, and breaking down the “mitochondrial Ca2+ addiction,” may provide a novel therapeutic strategy to halt tumor growth. A hypothesis that should be further tested in immunocompetent mice, to assess the impact of inhibition of cancer cell-associated ER–mitochondria Ca2+ transfer and perturbations of ER–mitochondria appositions, on a fully competent tumor microenvironment.
As mentioned above, recent research has highlighted an important role for MAMs in the regulation of the NLRP3 inflammasome. The role of the inflammasomes is best appreciated in innate immune cells, such as myeloid cells, but certain aggressive cancer cells, such as melanoma cells, are able to produce autonomously high levels of pro-inflammatory cytokines, such as IL-1 β through the activation of the inflammasome (166). In tumors, activation of the inflammasome has contextual pro-tumorigenic and anti-tumorigenic roles, reflecting the double-edged role played by inflammation in cancer progression (167, 168). Indeed, the main cytokines produced by the assembly of the NLRP3 inflammasome, IL-1β and IL-18, exert pleiotropic effects in inflammation, immunosurveillance and therapy responses and have both pro- and anti-tumorigenic functions depending on the context and type of cancer, as well as in response to anticancer therapies and their ability to induce immunogenic cell death (169, 170). Interestingly, MAMs could have a central role in priming the activation of the inflammasome and at the same time preventing excessive inflammation through the recruitment of the autophagy machinery. In line with this, by the removal of aged and malfunctioning mitochondria, autophagy has been shown to downregulate the activation of the NLRP3 inflammasome at the ER-mitochondria contact sites, thereby preventing the release of crucial NLRP3 activators, including mtDNA and mitochondrial ROS (171). Thus although speculative, this connection deserves to be further tested in the near future.
Finally, a recent study proposes an intriguing link between ER-mitochondria contacts sites and immunosurveillance (172). In glioblastoma, a highly aggressive and heterogeneous tumor, glioma stem-like cells (GSCs) displayed an increased susceptibility to killing by cytotoxic T lymphocytes and natural killer cells—the two major players of the immunosurveillance process—than their differentiated glioma cells (GDCs). This demonstrated increased susceptibility of GSCs to T cell killing both in vitro and in vivo. This also correlated to the reduced presence of surface sialylated glycans, which in turn extended the duration of immune synapses formed with cytotoxic T cells when compared to GDCs. Remarkably, GSCs had more fragmented mitochondria compared to GDCs and reduced ER-mitochondria tethering along with reduced expression of Mfn2. Importantly, the causative link between reduced MAMs and low levels of sialylated glycans in GSCs was evidenced by artificially forcing ER–mitochondria tethering, which resulted in increased cell surface glycans and reduced killing by CTLs (172). Although it is still elusive how mitochondrial morphology and ER–mitochondria appositions regulate the composition and expression of surface glycans, this study provides the first evidence that MAMs can modify the cell surface glycoprotein glycocalyx, a key determinant of immune modulation harnessed by cancer cells to evade control from both innate and adaptive immune destruction (173).
The increasingly important and pleiotropic role of ER–mitochondria tethers in the coordination of key cellular functions has been elucidated by the growing interest in this fundamental cell biology process sparked in the last decades. Along with the complex and dynamic molecular composition of the MAMs, several studies have highlighted the fundamental signaling role of these ER subdomains and their relevance for cancer biology and treatment. However, several outstanding questions still need to be answered before reaching a complete mechanistic and functional understanding of the ER–mitochondria interface. For example, how do MAMs regulate the strength and outcome of signaling pathways, such as those engaged by the loss of ER homeostasis, which are crucial for cancer cell survival, secretion, inflammation, and therapy responses? Which are the key mediators of this process and can they be harnessed for therapeutic intervention in cancer therapy? Given their role in lipid trafficking between the ER and mitochondria, do MAMs strategically control the type of cancer cell death induced by metabolic alterations or cellular stress? How do ER–mitochondria contact sites regulate immunosurveillance mechanisms through changes in the surface composition of the cancer cells and what is the role of MAMs in the release of mitochondria-derived danger signals, such as mtDNA and formyl peptides?
In this context, the use of modulators of ER–mitochondria tethers, such as the recently developed MFN2 inhibitors (174), could provide a relevant approach to further study cancer cell-intrinsic (such as the ER-to-mitochondria Ca2+ dependence for survival) and extrinsic (immunosurveillance mechanisms, inflammation and release of mitochondria-derived danger signals) processes that are modulated by ER–mitochondria contact sites. This further urges clarity about the role of MFN2 in ER–mitochondria appositions in both normal and cancerous cells. Finally, it is still not clear whether ER–mitochondria contact sites modulate the mitochondrial UPR, an emerging process modulating mitochondrial proteostasis (175). All these are outstanding questions that await future studies.
MS and PA conceived and wrote the manuscript. AV helped in improving and editing the manuscript. PA supervised and critically revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
MS is funded by the EU’s Horizon 2020 Marie Sklodowska-Curie grant 675448. AV is funded by the FWO. This work was supported by grants from the Fund for Scientific Research Flanders (FWO Vlaanderen; G.0498.17N) and KU Leuven (C16/15/073) to PA.
AA, arachidonoyl; ATF4, activating transcription factor 4; ATG, autophagy-related gene; Bcl-2, B-cell lymphoma 2; Bcl-XL, B-cell lymphoma-extra large; BECN1, Beclin-1; Ca2 +, calcium; Cav-1, caveolin-1; CL, cardiolipin; CNX, calnexin; cyt c, cytochrome c; Drp1, dynamin-related protein 1; ER, endoplasmic reticulum; Grp78, glucose-regulated protein 78; HK2, hexokinase 2; IL-1β, interleukin 1β; IL-18, interleukin 18; IP3, inositol trisphosphate; IP3R, inositol 1, 4, 5-trisphosphate receptor; IP3R3, inositol 1, 4, 5-trisphosphate receptor 3; IRE1, inositol requiring enzyme 1; MAMs, mitochondria-associated membranes; MAVS, mitochondrial antiviral-signaling protein; MCU, mitochondrial calcium uniporter; MFN2, mitofusin 2; mTORC2, mammalian target of rapamycin 2; NLRP3, NOD-like receptor family 3; OMM, outer mitochondrial membrane; ORP5, oxysterol-binding protein (OSBP)–related protein (ORP) 5; ORP8, oxysterol-binding protein (OSBP)–related protein (ORP) 8; PA, phosphatidic acid; PACS-2, phosphofurin acidic cluster sorting protein 2; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PERK, double stranded RNA-activated protein kinase (PKR)-like ER kinase; PI, phosphatidylinositol; PM, plasma membrane; PML, promyelocytic leukemia; PP2A, protein phosphatase 2A; PS, phosphatidylserine; PTEN, phosphatase and tensin homolog deleted on chromosome 10; PTPIP51, protein tyrosine phosphatase-interacting protein 51; ROS, reactive oxygen species; SERCA, sarco/endoplasmic reticulum Ca2+ ATPase; SERCA2b, sarco/endoplasmic reticulum Ca2 + ATPase isoform 2b; Sigma1 receptor, S1R; StAR, cholesterol transport steroidogenic acute regulatory protein; TMX, thioredoxin-related transmembrane protein; VAPB, vesicle-associated membrane protein-associated protein B; VDAC1, voltage-dependent anion channel 1; VDAC2, voltage-dependent anion channel 2.
1. Copeland D, Dalton A. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Cell Biol (1959) 5(3):393–6. doi:10.1083/jcb.5.3.393
2. Ruby JR, Dyer RF, Skalko RG. Continuities between mitochondria and endoplasmic reticulum in the mammalian ovary. Cell Tissue Res (1969) 97(1):30–7.
3. Wanson JC, Drochmans P, May C, Penasse W, Popowski A. Isolation of centrolobular and perilobular hepatocytes after phenobarbital treatment. J Cell Biol (1975) 66(1):23–41. doi:10.1083/jcb.66.1.23
4. Pickett CB, Montisano D, Eisner D, Cascarano J. The physical association between rat liver mitochondria and rough endoplasmic reticulum: I. Isolation, electron microscopic examination and sedimentation equilibrium centrifugation analyses of rough endoplasmic reticulum-mitochondrial complexes. Exp Cell Res (1980) 128(2):343–52. doi:10.1016/0014-4827(80)90070-1
5. Katz J, Wals PA, Golden S, Raijman L. Mitochondrial-reticular cytostructure in liver cells. Biochem J (1983) 214(3):795–813. doi:10.1042/bj2140795
6. Vance JE. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J Biol Chem (1991) 266(1):89–97.
7. Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta (2014) 1841(4):595–609. doi:10.1016/j.bbalip.2013.11.014
8. Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem (1990) 265(13):7248–56.
9. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science (1998) 280(5370):1763–6. doi:10.1126/science.280.5370.1763
10. Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol (2006) 174(7):915–21. doi:10.1083/jcb.200604016
11. Rowland AA, Voeltz GK. Endoplasmic reticulum–mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol (2012) 13(10):607–25. doi:10.1038/nrm3440
12. Tubbs E, Rieusset J. Metabolic signaling functions of ER-mitochondria contact sites: role in metabolic diseases. J Mol Endocrinol (2017) 58(2):R87–106. doi:10.1530/JME-16-0189
13. Calì T, Ottolini D, Brini M. Calcium and endoplasmic reticulum-mitochondria tethering in neurodegeneration. DNA Cell Biol (2013) 32(4):140–6. doi:10.1089/dna.2013.2011
14. van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta (2014) 1843(10):2253–62. doi:10.1016/j.bbamcr.2014.03.009
15. Filadi R, Theurey P, Pizzo P. The endoplasmic reticulum-mitochondria coupling in heath and disease: molecules, functions and significance. Cell Calcium (2017) 62:1–15. doi:10.1016/j.ceca.2017.01.003
16. Rodríguez-Arribas M, Yakhine-Diop SM, Pedro JM, Gómez-Suaga P, Gómez-Sánchez R, Martínez-Chacón G, et al. Mitochondria-associated membranes (MAMs): overview and its role in Parkinson’s disease. Mol Neurobiol (2016) 1–17. doi:10.1007/s12035-016-0140-8
17. Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol (2016) 17(2):69. doi:10.1038/nrm.2015.8
18. Kozieł K, Lebiedzinska M, Szabadkai G, Onopiuk M, Brutkowski W, Wierzbicka K, et al. Plasma membrane associated membranes (PAM) from Jurkat cells contain STIM1 protein: is PAM involved in the capacitative calcium entry? Int J Biochem Cell Biol (2009) 41(12):2440–9. doi:10.1016/j.biocel.2009.07.003
19. Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, et al. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J (2012) 31(2):457–70. doi:10.1038/emboj.2011.384
20. Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol (2009) 19(2):81–8. doi:10.1016/j.tcb.2008.12.002
21. Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal (2015) 22(12):995–1019. doi:10.1089/ars.2014.6223
22. Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev (2006) 86(1):369–408. doi:10.1152/physrev.00004.2005
23. Giacomello M, Pellegrini L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ (2016) 23(9):1417–27. doi:10.1038/cdd.2016.52
24. Filippin L, Magalhães PJ, Di Benedetto G, Colella M, Pozzan T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J Biol Chem (2003) 278(40):39224–34. doi:10.1074/jbc.M302301200
25. Parys JB, De Smedt H. Inositol 1, 4, 5-trisphosphate and its receptors. In: Islam M, editor. Calcium Signaling. Advances in Experimental Medicine and Biology, Vol. 740. Dordrecht: Springer (2012). p. 255–79.
26. Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, et al. The type III inositol 1, 4, 5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem (2005) 280(49):40892–900. doi:10.1074/jbc.M506623200
27. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol (2006) 175(6):901–11. doi:10.1083/jcb.200608073
28. Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol (2011) 3(5):a004184. doi:10.1101/cshperspect.a004184
29. Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, et al. The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell (2008) 19(7):2777–88. doi:10.1091/mbc.E07-10-0995
30. Köttgen M, Benzing T, Simmen T, Tauber R, Buchholz B, Feliciangeli S, et al. Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J (2005) 24(4):705–16. doi:10.1038/sj.emboj.7600566
31. Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, et al. PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. EMBO J (2005) 24(4):717–29. doi:10.1038/sj.emboj.7600559
32. De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet (2012) 21(6):1299–311. doi:10.1093/hmg/ddr559
33. Gomez-Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CC. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr Biol (2017) 27(3):371–85. doi:10.1016/j.cub.2016.12.038
34. Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell (2016) 61(5):683–94. doi:10.1016/j.molcel.2016.02.022
35. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature (2008) 456(7222):605–10. doi:10.1038/nature07534
36. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc Natl Acad Sci U S A (2015) 112(17):E2174–81. doi:10.1073/pnas.1504880112
37. Wang PT, Garcin PO, Fu M, Masoudi M, St-Pierre P, Panté N, et al. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J Cell Sci (2015) 128(15):2759–65. doi:10.1242/jcs.171132
38. Cosson P, Marchetti A, Ravazzola M, Orci L. Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS One (2012) 7(9):e46293. doi:10.1371/journal.pone.0046293
39. Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc Natl Acad Sci U S A (2016) 113(40):11249–54. doi:10.1073/pnas.1606786113
40. Holthuis JC, Levine TP. Lipid traffic: floppy drives and a superhighway. Nat Rev Mol Cell Biol (2005) 6(3):209–20. doi:10.1038/nrm1591
41. Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol (2008) 9(2):112–24. doi:10.1038/nrm2330
42. Futerman AH. Intracellular trafficking of sphingolipids: relationship to biosynthesis. Biochim Biophys Acta (2006) 1758(12):1885–92. doi:10.1016/j.bbamem.2006.08.004
43. Kuchler K, Daum G, Paltauf F. Subcellular and submitochondrial localization of phospholipid-synthesizing enzymes in Saccharomyces cerevisiae. J Bacteriol (1986) 165(3):901–10. doi:10.1128/jb.165.3.901-910.1986
44. Ardail D, Lerme F, Louisot P. Involvement of contact sites in phosphatidylserine import into liver mitochondria. J Biol Chem (1991) 266(13):7978–81.
45. Voelker DR. Phosphatidylserine functions as the major precursor of phosphatidylethanolamine in cultured BHK-21 cells. Proc Natl Acad Sci U S A (1984) 81(9):2669–73. doi:10.1073/pnas.81.9.2669
46. Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta (2009) 1788(10):2022–31. doi:10.1016/j.bbamem.2009.05.004
47. Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol (2008) 183(4):681–96. doi:10.1083/jcb.200803129
48. Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, et al. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med (2009) 46(11):1439–53. doi:10.1016/j.freeradbiomed.2009.03.004
49. Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol (2011) 192(1):7–16. doi:10.1083/jcb.201006159
50. Potting C, Tatsuta T, König T, Haag M, Wai T, Aaltonen MJ, et al. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab (2013) 18(2):287–95. doi:10.1016/j.cmet.2013.07.008
51. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ (2012) 19(11):1880–91. doi:10.1038/cdd.2012.74
52. Bobrovnikova-Marjon E, Pytel D, Riese MJ, Vaites LP, Singh N, Koretzky GA, et al. PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate AKT activation and promote adipocyte differentiation. Mol Cell Biol (2012) 32:2268–78. doi:10.1128/MCB.00063-12
53. Galmes R, Houcine A, Van Vliet AR, Agostinis P, Jackson CL, Giordano F. ORP5/ORP8 localize to endoplasmic reticulum–mitochondria contacts and are involved in mitochondrial function. EMBO Rep (2016) 17(6):800–10. doi:10.15252/embr.201541108
54. Sala-Vila A, Navarro-Lérida I, Sánchez-Alvarez M, Bosch M, Calvo C, López JA, et al. Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci Rep (2016) 6:27351. doi:10.1038/srep27351
55. Bosch M, Marí M, Herms A, Fernández A, Fajardo A, Kassan A, et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr Biol (2011) 21(8):681–6. doi:10.1016/j.cub.2011.03.030
56. Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, et al. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca2+-dependent mitochondrial apoptosis. Mol Cell (2009) 36(3):500–11. doi:10.1016/j.molcel.2009.10.021
57. Miller WL. Steroid hormone synthesis in mitochondria. Mol Cell Endocrinol (2013) 379(1):62–73. doi:10.1016/j.mce.2013.04.014
58. Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, Bose HS. Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. J Biol Chem (2015) 290(5):2604–16. doi:10.1074/jbc.M114.605808
59. Prasad M, Pawlak KJ, Burak WE, Perry EE, Marshall B, Whittal RM, et al. Mitochondrial metabolic regulation by GRP78. Sci Adv (2017) 3(2):e1602038. doi:10.1126/sciadv.1602038
60. Nishimura T, Tamura N, Kono N, Shimanaka Y, Arai H, Yamamoto H, et al. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J (2017) 36:1719–35. doi:10.15252/embj.201695189
61. Frieden M, Arnaudeau S, Castelbou C, Demaurex N. Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2+-ATPases. J Biol Chem (2005) 280(52):43198–208. doi:10.1074/jbc.M510279200
62. García-Sancho J. The coupling of plasma membrane calcium entry to calcium uptake by endoplasmic reticulum and mitochondria. J Physiol (2014) 592(2):261–8. doi:10.1113/jphysiol.2013.255661
63. Yi M, Weaver D, Hajnóczky G. Control of mitochondrial motility and distribution by the calcium signal. J Cell Biol (2004) 167(4):661–72. doi:10.1083/jcb.200406038
64. Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A (2008) 105(52):20728–33. doi:10.1073/pnas.0808953105
65. Fransson Å, Ruusala A, Aspenström P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun (2006) 344(2):500–10. doi:10.1016/j.bbrc.2006.03.163
66. Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol (2005) 170(7):1021–7. doi:10.1083/jcb.200506078
67. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science (2011) 334(6054):358–62. doi:10.1126/science.1207385
68. Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science (2013) 339(6118):464–7. doi:10.1126/science.1228360
69. Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science (2016) 353(6296):aaf5549. doi:10.1126/science.aaf5549
70. Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, et al. Essential regulation of cell bioenergetics by constitutive InsP 3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142(2):270–83. doi:10.1016/j.cell.2010.06.007
71. Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE (2004) 2004(215):re1. doi:10.1126/stke.2152004re1
72. Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (2008) 23(2):84–94. doi:10.1152/physiol.00046.2007
73. Hansford R, Chappell J. The effect of Ca2+ on the oxidation of glycerol phosphate by blowfly flight-muscle mitochondria. Biochem Biophy Res Commun (1967) 27(6):686–92. doi:10.1016/S0006-291X(67)80090-1
74. Mourier A, Motori E, Brandt T, Lagouge M, Atanassov I, Galinier A, et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J Cell Biol (2015) 208(4):429–42. doi:10.1083/jcb.201411100
75. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol (2011) 13(5):589–98. doi:10.1038/ncb2220
76. Shutt T, Geoffrion M, Milne R, McBride HM. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep (2012) 13(10):909–15. doi:10.1038/embor.2012.128
77. Thaher O, Wolf C, Dey PN, Pouya A, Wüllner V, Tenzer S, et al. The thiol switch C684 in Mitofusin-2 mediates redox-induced alterations of mitochondrial shape and respiration. Neurochem Int (2017). doi:10.1016/j.neuint.2017.05.009
78. Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A (1999) 96(24):13807–12. doi:10.1073/pnas.96.24.13807
79. Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1, 4, 5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol (2003) 5(12):1051–61. doi:10.1038/ncb1063
80. Grimm S. The ER-mitochondria interface: the social network of cell death. Biochim Biophys Acta (2012) 1823(2):327–34. doi:10.1016/j.bbamcr.2011.11.018
81. Marchi S, Giorgi C, Oparka M, Duszynski J, Wieckowski MR, Pinton P. Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol Cell Oncol (2014) 1(2):e956469. doi:10.4161/23723548.2014.956469
82. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1):317–31. doi:10.1016/j.cell.2013.12.010
83. Buytaert E, Dewaele M, Agostinis P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim Biophys Acta (2007) 1776(1):86–107. doi:10.1016/j.bbcan.2007.07.001
84. Basova LV, Kurnikov IV, Wang L, Ritov VB, Belikova NA, Vlasova II, et al. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry (2007) 46(11):3423–34. doi:10.1021/bi061854k
85. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13:81–90. doi:10.1038/nchembio.2238
86. Golej DL, Askari B, Kramer F, Barnhart S, Vivekanandan-Giri A, Pennathur S, et al. Long-chain acyl-CoA synthetase 4 modulates prostaglandin E2 release from human arterial smooth muscle cells. J Lipid Res (2011) 52(4):782–93. doi:10.1194/jlr.M013292
87. Lewin TM, Van Horn CG, Krisans SK, Coleman RA. Rat liver acyl-CoA synthetase 4 is a peripheral-membrane protein located in two distinct subcellular organelles, peroxisomes, and mitochondrial-associated membrane. Arch Biochem Biophys (2002) 404(2):263–70. doi:10.1016/S0003-9861(02)00247-3
88. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 13(2):89–102. doi:10.1038/nrm3270
89. Maurel M, McGrath EP, Mnich K, Healy S, Chevet E, Samali A. Controlling the unfolded protein response-mediated life and death decisions in cancer. Semin Cancer Biol (2015) 33:57–66. doi:10.1016/j.semcancer.2015.03.003
90. van Vliet AR, Agostinis P. When under pressure, get closer: PERKing up membrane contact sites during ER stress. Biochem Soc Trans (2016) 44(2):499–504. doi:10.1042/BST20150272
91. Carreras-Sureda A, Pihán P, Hetz C. The unfolded protein response: at the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front Oncol (2017) 7:55. doi:10.3389/fonc.2017.00055
92. Muñoz JP, Ivanova S, Sánchez-Wandelmer J, Martínez-Cristóbal P, Noguera E, Sancho A, et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J (2013) 32(17):2348–61. doi:10.1038/emboj.2013.168
93. Chami M, Oulès B, Szabadkai G, Tacine R, Rizzuto R, Paterlini-Bréchot P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell (2008) 32(5):641–51. doi:10.1016/j.molcel.2008.11.014
94. Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ (2011) 18(5):769–82. doi:10.1038/cdd.2010.142
95. Calì T, Ottolini D, Negro A, Brini M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca2+ transfer to sustain cell bioenergetics. Biochim Biophys Acta (2013) 1832(4):495–508. doi:10.1016/j.bbadis.2013.01.004
96. Mori T, Hayashi T, Hayashi E, Su TP. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One (2013) 8(10):e76941. doi:10.1371/journal.pone.0076941
97. Hayashi T, Su T-P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell (2007) 131(3):596–610. doi:10.1016/j.cell.2007.08.036
98. van Vliet AR, Garg AD, Agostinis P. Coordination of stress, Ca2+, and immunogenic signaling pathways by PERK at the endoplasmic reticulum. Biol Chem (2016) 397(7):649–56. doi:10.1515/hsz-2016-0108
99. van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, et al. The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol Cell (2017) 65(5):885–99.e6. doi:10.1016/j.molcel.2017.01.020
100. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24(1):24–41. doi:10.1038/cr.2013.168
101. Klionsky DJ, Codogno P. The mechanism and physiological function of macroautophagy. J Innate Immun (2013) 5(5):427–33. doi:10.1159/000351979
102. Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol (2010) 12(9):831–5. doi:10.1038/ncb0910-831
103. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature (2013) 495(7441):389–93. doi:10.1038/nature11910
104. Garofalo T, Matarrese P, Manganelli V, Marconi M, Tinari A, Gambardella L, et al. Evidence for the involvement of lipid rafts localized at the ER-mitochondria associated membranes in autophagosome formation. Autophagy (2016) 12(6):917–35. doi:10.1080/15548627.2016.1160971
105. Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, et al. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy (2017) 13(4):654–69. doi:10.1080/15548627.2016.1277309
106. Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J (2016) 35:1368–84. doi:10.15252/embj.201593102
107. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi:10.1038/nri.2016.58
108. Kim JK, Jin HS, Suh H-W, Jo E-K. Negative regulators and their mechanisms in NLRP3 inflammasome activation and signaling. Immunol Cell Biol (2017). doi:10.1038/icb.2017.23
109. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi:10.1038/nature09663
110. Horner SM, Liu HM, Park HS, Briley J, Gale M Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A (2011) 108(35):14590–5. doi:10.1073/pnas.1110133108
111. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest (2015) 125(9):3347–55. doi:10.1172/JCI80007
112. Kiebish MA, Han X, Cheng H, Chuang JH, Seyfried TN. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the Warburg theory of cancer. J Lipid Res (2008) 49(12):2545–56. doi:10.1194/jlr.M800319-JLR200
113. Monteiro JP, Oliveira PJ, Jurado AS. Mitochondrial membrane lipid remodeling in pathophysiology: a new target for diet and therapeutic interventions. Prog Lipid Res (2013) 52(4):513–28. doi:10.1016/j.plipres.2013.06.002
114. Baggetto LG, Testa-Parussini R. Role of acetoin on the regulation of intermediate metabolism of Ehrlich ascites tumor mitochondria: its contribution to membrane cholesterol enrichment modifying passive proton permeability. Arch Biochem Biophys (1990) 283(2):241–8. doi:10.1016/0003-9861(90)90638-F
115. Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basañez G, et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res (2008) 68(13):5246–56. doi:10.1158/0008-5472.CAN-07-6161
116. Kiebish MA, Han X, Cheng H, Seyfried TN. In vitro growth environment produces lipidomic and electron transport chain abnormalities in mitochondria from non-tumorigenic astrocytes and brain tumours. ASN Neuro (2009) 1(3):e00011. doi:10.1042/AN20090011
117. Morton R, Cunningham C, Jester R, Waite M, Miller N, Morris HP. Alteration of mitochondrial function and lipid composition in Morris 7777 hepatoma. Cancer Res (1976) 36(9 Pt 1):3246–54.
118. Campbell AM, Chan SH. Mitochondrial membrane cholesterol, the voltage dependent anion channel (VDAC), and the Warburg effect. J Bioenerg Biomembr (2008) 40(3):193–7. doi:10.1007/s10863-008-9138-x
119. Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest (2005) 115(4):959–68. doi:10.1172/JCI200519935
120. Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol (2006) 168(4):1107–18. doi:10.2353/ajpath.2006.050959
121. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ (2016) 23(3):369–79. doi:10.1038/cdd.2015.158
122. Ivanova H, Kerkhofs M, La Rovere RM, Bultynck G. Endoplasmic reticulum-mitochondrial Ca2+ fluxes underlying cancer cell survival. Front Oncol (2017) 7:70. doi:10.3389/fonc.2017.00070
123. Mikoshiba K. IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem (2007) 102(5):1426–46. doi:10.1111/j.1471-4159.2007.04825.x
124. Vanderheyden V, Devogelaere B, Missiaen L, De Smedt H, Bultynck G, Parys JB. Regulation of inositol 1, 4, 5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta (2009) 1793(6):959–70. doi:10.1016/j.bbamcr.2008.12.003
125. Khan MT, Wagner L, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1, 4, 5-trisphosphate receptors. J Biol Chem (2006) 281(6):3731–7. doi:10.1074/jbc.M509262200
126. Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, et al. Phosphorylation of inositol 1, 4, 5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A (2008) 105(7):2427–32. doi:10.1073/pnas.0711324105
127. Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun (2008) 375(4):501–5. doi:10.1016/j.bbrc.2008.07.153
128. Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, et al. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis (2012) 3(5):e304. doi:10.1038/cddis.2012.45
129. Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A (2013) 110(31):12526–34. doi:10.1073/pnas.1302455110
130. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev (2001) 15(11):1406–18. doi:10.1101/gad.889901
131. Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell (2004) 16(5):819–30. doi:10.1016/j.molcel.2004.11.014
132. Dahia P. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer (2000) 7(2):115–29. doi:10.1677/erc.0.0070115
133. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol (2012) 13(5):283–96. doi:10.1038/nrm3330
134. Missiroli S, Bonora M, Patergnani S, Poletti F, Perrone M, Gafà R, et al. PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell Rep (2016) 16(9):2415–27. doi:10.1016/j.celrep.2016.07.082
135. Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science (2010) 330(6008):1247–51. doi:10.1126/science.1189157
136. Pinton P, Giorgi C, Pandolfi P. The role of PML in the control of apoptotic cell fate: a new key player at ER–mitochondria sites. Cell Death Differ (2011) 18(9):1450–6. doi:10.1038/cdd.2011.31
137. Bittremieux M, Parys JB, Pinton P, Bultynck G. ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca2+ signaling. Biochim Biophys Acta (2016) 1863(6):1364–78. doi:10.1016/j.bbamcr.2016.01.002
138. Bittremieux M, Bultynck G. p53 and Ca2+ signaling from the endoplasmic reticulum: partners in anti-cancer therapies. Oncoscience (2015) 2(3):233. doi:10.18632/oncoscience.139
139. Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A (2015) 112(6):1779–84. doi:10.1073/pnas.1410723112
140. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer (2016) 16(2):99–109. doi:10.1038/nrc.2015.17
141. Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ (2012) 19(2):295–309. doi:10.1038/cdd.2011.97
142. Meunier J, Hayashi T. Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor κB. J Pharmacol Exp Ther (2010) 332(2):388–97. doi:10.1124/jpet.109.160960
143. Akl H, Bultynck G. Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP 3 receptors. Biochim Biophys Acta (2013) 1835(2):180–93. doi:10.1016/j.bbcan.2012.12.001
144. Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A (2009) 106(34):14397–402. doi:10.1073/pnas.0907555106
145. Rong Y-P, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell (2008) 31(2):255–65. doi:10.1016/j.molcel.2008.06.014
146. Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1, 4, 5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem (2010) 285(18):13678–84. doi:10.1074/jbc.M109.096040
147. White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, et al. The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nat Cell Biol (2005) 7(10):1021–8. doi:10.1038/ncb1302
148. Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C. Apoptosis regulation by Bcl-xL modulation of mammalian inositol 1, 4, 5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci U S A (2007) 104(30):12565–70. doi:10.1073/pnas.0702489104
149. Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J Biol Chem (2012) 287(27):23152–61. doi:10.1074/jbc.M112.345918
150. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature (1999) 399(6735):483–7. doi:10.1038/20959
151. Monaco G, Decrock E, Arbel N, van Vliet AR, La Rovere RM, De Smedt H, et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J Biol Chem (2015) 290(14):9150–61. doi:10.1074/jbc.M114.622514
152. Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem (2010) 285(9):6053–62. doi:10.1074/jbc.M109.082990
153. Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, et al. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci (2009) 122(11):1906–16. doi:10.1242/jcs.040188
154. Mertins B, Psakis G, Grosse W, Back KC, Salisowski A, Reiss P, et al. Flexibility of the N-terminal mVDAC1 segment controls the channel’s gating behavior. PLoS One (2012) 7(10):e47938. doi:10.1371/journal.pone.0047938
155. Huang H, Hu X, Eno CO, Zhao G, Li C, White C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J Biol Chem (2013) 288(27):19870–81. doi:10.1074/jbc.M112.448290
156. Galluzzi L, Pietrocola F, Bravo‐San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34(7):856–80. doi:10.15252/embj.201490784
157. Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis (2011) 32(7):955–63. doi:10.1093/carcin/bgr031
158. Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med (2013) 19(7):428–46. doi:10.1016/j.molmed.2013.04.005
159. Gross A, Katz SG. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ (2017) 24:1348–58. doi:10.1038/cdd.2017.22
160. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J (2007) 26(10):2527–39. doi:10.1038/sj.emboj.7601689
161. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell (2005) 122(6):927–39. doi:10.1016/j.cell.2005.07.002
162. Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science (2012) 338(6109):956–9. doi:10.1126/science.1225967
163. Bustamante V, Miguel J. The inositol 1, 4, 5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Paris 11 (2009).
164. Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol (1998) 72(11):8586–96.
165. Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep (2016) 14(10):2313–24. doi:10.1016/j.celrep.2016.02.030
166. Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1β. J Biol Chem (2010) 285(9):6477–88. doi:10.1074/jbc.M109.064907
167. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi:10.1038/nm.3893
168. Drexler SK, Yazdi AS. Complex roles of inflammasomes in carcinogenesis. Cancer J (2013) 19(6):468–72. doi:10.1097/PPO.0000000000000004
169. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat Med (2009) 15(10):1170–8. doi:10.1038/nm.2028
170. Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol (2015) 6:588. doi:10.3389/fimmu.2015.00588
171. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell (2016) 164(5):896–910. doi:10.1016/j.cell.2015.12.057
172. Bassoy EY, Kasahara A, Chiusolo V, Jacquemin G, Boydell E, Zamorano S, et al. ER-mitochondria contacts control surface glycan expression and sensitivity to killer lymphocytes in glioma stem-like cells. EMBO J (2017) 36:1493–512. doi:10.15252/embj.201695429
173. Varki A. Letter to the glyco-forum: since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology (2011) 21(9):1121–4. doi:10.1093/glycob/cwr087
174. Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature (2016) 540(7631):74–9. doi:10.1038/nature20156
Keywords: endoplasmic reticulum, mitochondria, mitochondria-associated membranes, Ca2+ signaling, ER stress, autophagy, inflammasome, cancer cell
Citation: Sassano ML, van Vliet AR and Agostinis P (2017) Mitochondria-Associated Membranes As Networking Platforms and Regulators of Cancer Cell Fate. Front. Oncol. 7:174. doi: 10.3389/fonc.2017.00174
Received: 29 June 2017; Accepted: 31 July 2017;
Published: 18 August 2017
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Carlos Villalobos, Consejo Superior de Investigaciones Científicas (CSIC), SpainCopyright: © 2017 Sassano, van Vliet and Agostinis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrizia Agostinis, cGF0cml6aWEuYWdvc3RpbmlzQGt1bGV1dmVuLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.