 Tyler A. Herek
Tyler A. Herek Christine E. Cutucache
Christine E. Cutucache- Department of Biology, University of Nebraska at Omaha, Omaha, NE, USA
The role of the tumor microenvironment in leukemias and lymphomas is well established, yet the intricacies of how the malignant cells regulate and influence their non-malignant counterparts remain elusive. For example, chronic lymphocytic leukemia (CLL) is an expansion of malignant CD5+CD19+ B cells, yet the non-malignant T cells play just as large of a role in disease presentation and etiology. Herein, we review the dynamic tumor cell to lymphoid repertoire interactions found in two non-Hodgkin’s lymphoma subtypes: CLL and angioimmunoblastic T-cell lymphoma. We aim to highlight the pivot work done in the murine models which recapitulate these diseases and explore the insights that can be gained from studying the immuno-oncological regulation of non-malignant lymphoid counterparts.
Introduction
The cancer immunosurveillance and immunoediting concepts posit that the immune system plays a protective role in tumor development through its ability to recognize and eradicate subclinical tumors (1, 2). These theories suggest an ever-shifting balance of tumor eradication and tumor growth wherein equilibrium can be lost and tumor growth can be promoted (3, 4). The promotion of tumor growth can be viewed as a consequence of the immune system as a function of microenvironmental interactions that have been shown to be vital for tumor development and survival (5). Therefore, with the immune system playing seemingly two separate, yet opposite, roles within immuno-oncology interactions it has garnered the “Janus-faced” moniker (3).
To understand the Janus-faced nature of tumor immunology is to understand the impact that a growing tumor has upon its microenvironment; by which we mean how tumors regulate their non-malignant counterparts. Perhaps it is not surprising the impact a tumor can have on the immune system when one considers all the working parts of the immune system function that function in an antitumor response (6). From NK cells lysing transformed clones, to presentation of tumor-associated antigens by macrophages and dendritic cells to activate T- and therefore B-cells, the immune system plays an integral role in producing and expanding tumor-specific responses (3).
Currently, the most effective method of studying the impact of a tumor microenvironment (TME) on a host immune system is through the use of murine models (7). Specifically, this would extend to transgenic murine models as they allow for the “natural” propagation of a clinical tumor within an immunocompetent host; most closely mirroring what is found in humans. In this review, we summarize and discuss the impact two distinct hematological malignancies, chronic lymphocytic leukemia (CLL) and angioimmunoblastic T-cell lymphoma (AITL), have upon their adaptive immunological microenvironmental counterparts (i.e., T cells and B cells, respectively) (Figure 1). Explicitly, we summarize how murine models play a role in elucidating the impact of a B-cell malignancy (i.e., CLL) on resident T cells, and the impact of a T-cell malignancy (i.e., AITL) on resident B cells. The aforementioned hematological malignancies were chosen as representative malignancies to study immuno-oncological interactions due to their intense microenvironmental components. As CLL is the most common adult leukemia, we are able rely on a wealth of publications and data from both human and murine investigations to summarize and discuss the above-mentioned interactions. AITL on-the-other-hand is a much rarer and less studied disease. Therefore, we will focus on what is currently known concerning AITL immuno-oncological interactions and put forward recommendations concerning AITL murine models and directions for future investigations.
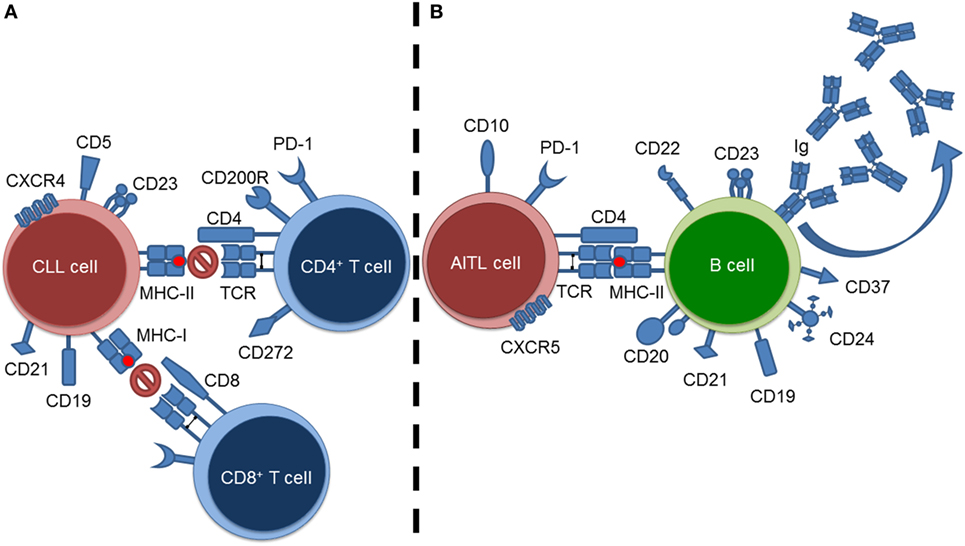
Figure 1. Summary of tumor–lymphoid interactions in chronic lymphocytic leukemia (CLL) and angioimmunoblastic T-cell lymphoma (AITL). (A) CLL cell–resident T cell interactions exemplified by blockade of T-cell-mediated responses against the CLL cells. (B) AITL cell–resident B cell interactions exemplified by stimulation of B cells resulting in hypergammaglobulinemia.
Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia is the most common adult leukemia characterized by the clonal expansion of malignant CD5+CD19+ B cells. The clinical presentation of CLL is heterogeneous, leading to both an indolent and aggressive form demarcated by well-defined prognostic markers (i.e., IGVH status, CD38 and ZAP70 expression, and chromosomal aberrations) (8–10).
Chronic lymphocytic leukemia is a malignancy highly dependent on its microenvironment illustrated by the fact that CLL cells readily undergo apoptosis in vitro without coculture of bone marrow stromal cells (BMSC) (11–13) or monocyte-derived nurse-like cells (NLCs) (13). While both BMSC and NLCs display similar recruitment of CLL cells through CXCL12–CXCR4 signaling (12–14), the mechanisms by which these compulsory stromal cells engage in cross talk with CLL cells differ. In vitro, CLL cells are attracted to BMSC and form a contact-dependent protective niche. While in contact with the BMSC, the CLL cells undergo a processes termed pseudoemperipolesis, which describes the migration of a fraction of the CLL cells beneath the BMSC (14–16). This migration results in the cross talk and bi-directional activation of both the BMSC and CLL cells leading to the upregulation of TCL1 and FOS/JUN in the CLL cells (17). Conversely, NLC-dependent activation of CLL cells is characterized by enhanced CLL cell viability through NF-κB activation and the BAFF-/APRIL-binding pathways resulting in the expression of the anti-apoptotic protein MCL-1 by CLL cells for prolonged survival (18). Additionally, CLL cell activation through the B-cell receptor pathway is associated with increased secretion of CCL3 and CCL4 chemokines, allowing for the enhanced recruitment of accessory cells to the TME (19).
The progressive accumulation of CLL clones is primarily attributed to increased apoptotic resistance via exploitation by the above-described microenvironments (20); however, proliferative centers of CLL have been identified (21). These proliferation centers, termed pseudo-follicles, are composed of Ki-67+, Survivin+, p27−, Bcl-2+, CD23Hi CLL cells and CD40L+ T cells (21–24). As a result, CD40 activation and IL-2/IL-10 signaling heighten CLL proliferation and upregulate IRF4 (25).
The Impact of CLL on T Cells
The T cells of CLL patients display distinct expression profiles (26) and are hallmarked by an exhausted phenotype, attenuated immune synapse formation, decreased cytolitic activity, migratory impairments, and dysregulated Rho-GTPase signaling.
T-cell exhaustion is defined by T cells displaying an overexpression of inhibitory receptors, decreased effector function, attenuated cytokine production, and decreased cytolitic activity (27). CLL-T cells have been shown to upregulate the surface expression of PD-1, CD160, and CD244, indicative of an exhausted phenotype (28). Moreover, these markers are known to be highly expressed on effector T cells, indicating a skewing of the T-cell compartment of CLL patients to a more mature, effector differentiation, albeit with attenuated functionality. Further support for a skewed T-cell compartment comes from CD4+ CLL-T cells having decreased gene expression of the JNK and p38 MAPK pathway activators (MINK, NFRKB, PIK3CB) (26) suggesting a defect in Th1 differentiation as the aforementioned genes are crucial in the production of IFN-γ (29–31).
The immune synapse is a localization of receptors, costimulatory molecules, and adhesive anchors that allows for the engagement and recognition of antigen and activation by T cells (32). During the activation process, the cytoskeleton of the T cell undergoes intense rearrangements to better facilitate both contact and signaling through pseudopodia formation and cellular polarization. In essence, the T cell grasps the antigen-presenting cell (APC) forming what is called the immunological synapse (33). Structurally, the immune synapse is described as possessing three concentric rings called supramolecular activation clusters (SMACs) (34). The central supramolecular activation cluster houses the TCR molecules which are relocated from the periphery to be recycled, the peripheral SMAC is vital for maintaining adhesion between the cells and is primarily composed of the integrin, lymphocyte function-associated antigen 1 (LFA-1), and the distal SMAC is a clustering of F-actin. The engagement of the components of these SMACs influences the rearrangement of the cytoskeleton both by signaling relayed via the TCRs and through integrins such as LFA-1. For example, once LAT is phosphorylated via Zap70 following T-cell activation the Rho-family GTPase exchange factor vav guanine nucleotide exchange factor 1 (Vav1) is recruited to the synapse (35, 36). Following Vav1 activation, the small GTPases Ras-related C3 botulinum toxin substrate 1 (Rac1) and cell division control protein 42 homolog (Cdc42) bind GTP and activate actin nucleation promoting factors such as Wiskott–Aldrich syndrome protein (WASp) and Wasp-family verprolin-homologous protein 2 (WAVE2) which coordinate actin-related protein 2/3 (Arp2/3)-dependent polymerization of branched actin filaments (37–39). Following actin polymerization, the immunological synapse functions as a signal specifier acting to focus TCR signaling responses to ensure efficient cross talk with the bound APC. Dysregulation of the polymerization processes could therefore lead to inefficient effector function delivery through poor coordination of T-cell signaling or ineffectual delivery of signals to the APC.
Immune synapse formation, including both antigen presentation by CLL cells and the subsequent response by T cells, is attenuated in CLL patients. Phenotypically, immune synapse malformation of CLL-T cells is displayed as a decrease in T-cell–APC conjugation and F-actin polymerization, with further decrement in T-cell receptor, WASp, Dynamin-2, Lck, Cdc42, and Filamin-A recruitment to the synapse site (40, 41). Additionally, the inhibitory receptors CD200R, CD272, and CD279 are upregulated in CLL-T cells, and further impede immune synapse formation (28, 42). These aforementioned defects also contribute to decreased CD8+ T-cell cytolitic effector function, as granzyme B is inefficiently packaged and localized to the immune synapse in CLL CD8+ T cells (26, 28).
T cells from CLL patients display migratory defects through LFA-1 directed T-cell motility (36). This defect is concurrent with the previously described deficiency in cytoskeletal remodeling in CLL-T cells (26) and has been determined to be dependent upon LFA-1 conformation status. In terms of mRNA expression, there is no difference in gene expression of LFA-1 between healthy T cells and CLL-T cells. Rather, CLL-T cells showed impaired migration due to the inefficiency of the low-affinity conformation of LFA-1 to bind intercellular adhesion molecule 1 causing an impediment in leukocyte rolling, arrest, firm adhesion, and diapedesis (43).
At the center of CLL-mediated T-cell impairments is the dysregulation of Rho-GTPase signaling in CLL-T cells. Rho-GTPases are members of the Ras superfamily of small GTP-binding proteins and play key roles in T-cell development, T-cell migration, and T-cell differentiation (44). This regulation is carried out through Rho-GTPase-mediated cytoskeletal rearrangements, as well as signaling through numerous pathways (45, 46). After coculture with CLL cells, T cells display lower active GTPase signaling for RhoA, Rac1, and Cdc42, as compared with coculture of T cells with healthy B cells (42). The dysregulation of these signaling molecules is also tied to defective LFA-1-mediated migration, where downregulated RhoA, Rac1, and potentiated Cdc42 are noted among CLL-T cells. Importantly, Cdc42 levels differed in CLL-T cell migration and stimulation assays and suggest that CLL-T cells do not lose all Rho-GTPase signaling potentials; however, they do not display identical signaling patterns as healthy T cells.
Evidence from the Eμ-TCL1 Model
T-cell impairment is a hallmark feature of CLL and is evident within the Eμ-TCL1 murine model. Early investigations into the T-cell phenotype in Eμ-TCL1 mice demonstrated decreased antigen-specific activation, suppressed mitogen initiated proliferation, and impaired idiotype specific CD8+ T cell-mediated CLL clone lysis (47). Gene expression profiling experiments supported the phenotypic differences, giving evidence to a dysregulation of the actin-cytoskeleton formation pathways, impaired F-actin polymerization, and immune synapse formation (47). Additionally, decreased expression of CD28, BTLA, Nfrkb, Pi3k, Kfkb, Gadd45b, and Vav3 were observed.
Hofbauer and colleagues (48) were the first to report on the immunophenotype of Eμ-TCL1 T-cells, finding an increase in the absolute numbers of T cells in the peripheral blood of leukemic mice, with the increased population consisting primarily of CD8+ T cells (~72%). Further, CLL-onset Eμ-TCL1 mice experienced a shift between naïve and antigen experience T cells (as assessed by CD44 expression), presenting with a 48:48 CD4+ naïve:experienced ratio, compared to a 80:20 ratio in wild-type control mice. The CD8+ T cell ratio held a similar shift, with an 86:14 naïve:experienced ratio in Eμ-TCL1 mice compared to a 70:27 ratio in wild-type controls (48). Importantly, this investigation brought a unique insight into the pathogenesis of CLL by establishing the engraftment of CLL spleenocytes within an immunocompetent host. This adoptive transfer model decreased the latency of CLL onset (83 days post-transfer compared to 247 days of Eμ-TCL1 development) and fully recapitulated the abovementioned skewing of the T-cell compartment. These data implicate the transferred CLL clones as being directly responsible for the onset of disease progression and changes to the T-cell compartment (48).
Recently, extensive work by the Gribben lab sought to delineate the mechanism behind the PD-1/PD-L1 signaling axis in T cells from Eμ-TCL1 mice, as well as provide important evidence for use of PD-L1 checkpoint blockade therapy (49, 50). Importantly, these studies lend microenvironmental data to the known role of PD-1/PD-L1 signaling in human CLL (49). As CLL developed in the Eμ-TCL1 mice, there was a significant correlation in the reduction of the percentage of CD3+, CD3+CD4+ T cells, and an expansion of CD3+CD8+ T cells in the spleen. This finding is in agreement with Hofbauer et al. (48) of a decrease in the CD4+/CD8+ ratio in Eμ-TCL1 mice for the spleen, peripheral blood, and lymph node microenvironments. Further, CD3+CD8+CD44−CD62L+ naïve cells were lost with CLL development, with a shift toward CD44+ antigen-experienced T cells in the spleen, peripheral blood, lymph node, and bone marrow microenvironments. These antigen-experienced CD44+ T cells held higher proliferation indices, as evidenced by increased Ki-67+ ratios in Eμ-TCL1 mice and adoptive transfer recipients (spleenocytes transferred into 3-month Eμ-TCL1 mice).
Consistent with aging, both Eμ-TCL1 and wild-type mice developed CD3+CD8+PD-1+ T cells, with higher absolute numbers found in Eμ-TCL1 and adoptive transfer mice (49). Investigation into the differential function of PD-1+ subsets in Eμ-TCL1 mice revealed an enrichment within the PD-1+ population for cytotoxic cells (as assessed by CD107a+) with increased proliferation ascribed to both PD-1high and PD-1low subsets, with the PD-1high population having greater EdU incorporation compared to PD-1low. Finally, immune synapse assays indicated the PD-1high population to form smaller synapses when compared to the PD-1low population in Eμ-TCL1 T cells, suggesting the PD-1high phenotype to contribute more toward T-cell impairment (49).
A causal role for the PD-1/PD-L1 signaling axis impairing clone-specific immune responses in Eμ-TCL1 mice has been supported by the work of both McClanahan and colleagues (50) and Gassner et al. (51). α-PD-L1 blockade administered to Eμ-TCL1 mice resulted in the effective control of CLL development, concurrent with reduction in both spleen sizes and weights (50). Treatment of adoptively transferred CLL tumor cells with either recombinant PD-1 or anti-CD274 resulted in a significant reduction of transferred tumor cells at both 2- and 24-h time points in peripheral blood (both time points), as well as in the spleen, lymph nodes, and lungs (24-h time point) (51). Antibody blockade restored the relative median frequencies of CD3+, CD4+, and CD8+ T cells, while normalizing the CD4+/CD8+ ratio (50). This restoration was observed in conjunction with a reduced shift toward the CD44+ phenotype seen in the peripheral blood, bone marrow, and spleen.
Angioimmunoblastic T-Cell Lymphoma
Angioimmunoblastic T-cell lymphoma is the second most common subtype of mature T-cell lymphoma, known to originate by the pathogenic clonal expansion of T-follicular helper (Tfh) cells and concordant immune dysfunction (52, 53). AITL is an aggressive disease, with a median survival <3 years post-diagnosis and 10–30% of patients alive at 5 years post-diagnosis (54, 55). The poor prognosis is partially due to the highly dysregulated immune system of AITL patients, as most patients succumb to infectious complications rather than the disease itself (54, 55).
The changes AITL imparts upon the lymph node microenvironment are considered a consequence of the cellular origins of the disease. AITL patients display aberrant lymph node architecture, including: hyperplasia of the follicular dendritic meshwork (53), proliferation of high-endothelial venules, thickened extra follicular meshworks, and neoplastic infiltrate (56). The neoplastic cells can be medium sized with round, slightly irregular nuclei featuring abundant clear cytoplasm, with clustering around the high-endothelial venules (57, 58). The neoplastic cells themselves may only account for 5–30% of the tumor (59), and most of the clinical manifestations of the disease (i.e., lymphadenopathy, hepatosplenomegaly, bone marrow involvement) represent complications arising from a compromised immune system (60, 61).
As stated, the cell-of-origin for AITL is the Tfh cell. In healthy conditions, these cells are formed from high affinity interactions with B cells though a T-cell receptor:antigen-dependent manner (62). Tfh cells are located within germinal centers and assist with the production of high affinity antibodies by B cells, as well as plasma cell differentiation, primarily through secretion of IL-21 and IL-4 (63, 64). These cells express high levels of ICOS and CXCR5 and can be immunophenotypically described as CD4+, CD8−, PD-1+, CD10+, BCL6+, and CXCL13+ cells (59, 65, 66). As with many mature T-cell lymphomas, AITL cells also exhibit defective expression of CD5 and CD7 (65), with further clonal aberrations detected in ~90% of cases (58, 67–69). The gene signature of AITL is described as very closely related to that of Tfh cells, with upregulations of VEGF, PDGFRα, B-cell and follicular dendritic cell genes, chemokines, extracellular matrix components, and vascular biology-associated genes (70, 71). Such findings are in accordance with the changes to the lymph node architecture and the common involvement of B cells in AITL disease progression.
The Impact of AITL on B-Cells
Due to a compromised immune system, in conjunction with dysregulation of normal immune effector functions, there is a high proclivity for developing secondary B-cell malignancies among AITL patients (72). These secondary malignancies commonly present as either composite lymphomas of diffuse large B-cell lymphoma + AITL (73, 74) or polyclonal populations of immunoblasts or plasma cells (75, 76). The development of these secondary malignancies suggests that the malignant T cells still hold the effector capacity to stimulate B-cell proliferation and antibody production and indicate a relationship between the developments of the two separate disease phenotypes (77, 78). Evidence supporting the above hypothesis includes: development of hypergammaglobulinemia, gene signature analyses demonstrating an upregulation of genes for B-cell activation and/or receptor signaling (CD22, CD20, CD21, CD23, CD24, CD37) (79), and histological evaluation of germinal centers displaying SH2D1A staining, a protein involved in bi-directional activation of T and B cells (80).
Murine Models That Recapitulate the Defining Characteristics of AITL
Unlike the Eμ-TCL1 mice for CLL, there is no current consensus murine model for AITL. Currently, three genetic models most faithfully recapitulate the defining disease characteristics of AITL and how they impact resident B cells will be discussed below.
Tet2gt/gt Model of AITL
The ten–eleven translocation 2 (TET2) gene has been found to be mutated in multiple myeloid malignancies (81–83) as well as a wide range of AITL and PTCL-NOS cases (84, 85). A loss of function mutation in TET2 is associated with aberrant methylation processes involving the conversion of methylcytosine to hydroxymethylcytosine, a common epigenetic marker in cancer pathogenesis (86).
While previous studies of Tet2 mutations have been conducted (87–89), Muto and colleagues were the first to describe the resultant disease in terms of its relation to the development of a T-cell lymphoma with follicular helper T-cell-like features (90). After backcrossing Tet2 gene trap mice onto the C57BL/6 background and aging for 40–60 weeks, Tet2gt/gt mice had no differences in complete blood counts or proportions of total T- or B-cell populations in comparison to wild-type or heterozygous controls. The Tet2gt/gt mice presented with splenomegaly and preserved follicle structures and an enlargement of germinal centers in some mice. Flow cytometry analyses revealed an increase in the absolute #’s of T- and B-cell populations, despite harboring no differences in the proportions of the overall populations. Further analyses displayed an increase in the proportions of Tfh populations in the Tet2gt/gt mice, namely: CD4+CD44+PD-1+ and CD4+PD-1+Cxcr5+ Tfh cells.
In five of seven mice aged greater than 60 weeks (median age: 67 weeks), Muto and colleagues (90) observed the development of increased splenomegaly, multiple swollen lymph nodes, and liver and lung nodules in Tet2gt/gt mice. Histological analysis demonstrated the follicular structures of the spleen to be no longer apparent due to the infiltration of large polymorphic cells with irregular nuclei and characterized as CD4+PD-1+Cxcr5+ Tfh cells. These T cells were determined to be clonal in nature, with distinct rearrangements of TCR Vβ/Jβ2. Not observed histologically was proliferation of high-endothelial venules, nor eosinophil infiltration. Then, >60-week mice presented with increased proportions of CD4+ T-cell fractions and smaller B220+ B-cell fractions, with the T-cell population mostly composed of CD44+PD-1+ cells. Of note, there was no difference in serum Ig levels found in Tet2gt/gt mice compared to controls.
Consistent with the observation that the malignant cells in Tet2gt/gt mice were of Tfh origin, microarray analysis of isolated splenic CD4+ T cells from Tet2gt/gt mice compared to control mice found the Tet2gt/gt T cells to be enriched for Tfh genes. Notable genes found to be highly expressed in the Tet2gt/gt mice include: Bcl6, cMaf, PD-1, Icos, and Cxcr5. Control CD4+ T cells were enriched for genes involved in Th1/Th2 differentiation by comparison, suggesting the downregulation of these genes is necessary for the adoption of the Tfh phenotype. Sanger sequencing experiments indicated that none of the concurrent mutations identified within human AITL (e.g., Flt3, Npm1, Dmnt3a, Idh2, Rhoa) were found in the Tet2gt/gt mice. Investigation into the differential methylation patterns resulting from reduced Tet2 function revealed disruption of the distribution of methylcytosine and hydroxymethylcytosine in transcriptional start sites, gene body regions, and CpG islands. Specifically, Bcl6 was identified as having aberrant methylation suggesting this as a mechanism for increased Bcl6 expansion and the induction of the outgrowth of Tfh-like cells (90).
Heterozygous RoquinSan Model of AITL
The sanroquin mouse strain bears a homozygous point mutation in the Roquin/Rc3hl gene (91), designated as Roquinsan. This point mutation results in increased binding affinity between ROQUIN and Icos mRNA resulting in the decreased decay rate of the transcript and overall upregulation of gene product (92). The upregulation of Icos in these mice results in the expansion of Tfh cells driving a disease progression equivocal to a systemic lupus-like disease phenotype—a similar phenotype is observed in Roquinsan/+ mice but in the absence of systemic autoimmunity (91). However, mice heterozygous for the Roquinsan allele (Roquinsan/+) as described in Ellyard et al. (93) do not develop systemic autoimmunity but instead present with asymmetric lymphadenopathy. Roquinsan/+ mice had a complete absence of generalized lymphadenopathy seen in homozygous mice by 8 weeks, but instead, 53% developed one to four enlarged lymph nodes. The resultant lymphadenopathy was deemed non-lethal and caused an increase in the cellularity of the affected lymph nodes by 50- to 150-fold. A higher prevalence in females (65%) compared to males (41%) was noted for the development of the tumor-bearing lymph nodes. Histological examination of tumor nodes in Roquinsan/+ mice demonstrated an effacement of nodal architecture and vascularization, as well as infiltration of small-to-medium sized T cells and large PAX5+ B cells, but no expansion of the follicular dendritic cell network. An increase in F4/80+ macrophages was observed with prominent vascularization and sinusoidal dilation. Reactive B blasts were observed in interfollicular areas along with a proliferative and polymorphic T-cell infiltrate, small clusters of mature plasma cells, and some rosette formation between T cells and large blasts. Tumor lymph nodes appeared to have no bearing on un-affected lymph nodes in tumor-bearing Roquinsan/+ mice, as un-affected nodes displayed typical morphologies with the exception of an increase in mature plasma cells found within interfollicular areas. All Roquinsan/+ mice presented with increased spleen weight compared to Roquin+/+ mice regardless of tumor incidence.
Concurrent with the highly affected lymph node environment of the Roquinsan/+ mice, increased serum IgG is observed consistent with the development of hypergammaglobulinemia at 15 weeks of age. While both Roquinsan/+ tumor-bearing and non-tumor-bearing mice show a significant increase in IgG production compared to Roquin+/+ mice, tumor-bearing Roquinsan/+ mice harbor an additional 1.5-fold increase above non-tumor-bearing Roquinsan/+ mice. A finding further corroborated by flow cytometry analysis revealing an increased B-cell fraction in the tumor-bearing lymph nodes of Roquinsan/+ mice in comparison to normal lymph nodes from both Roquin+/+ and Roquinsan/+ mice. Clonality assessments identified a clonal peak of IgH in only 1 of 15 cases, in contrast to clonal arrangements in 12 of 15 cases for TCR-β.
Swiss Jim Lambert (SJL) Model of AITL
Swiss Jim Lambert mice have been studied extensively as a model of lymphoproliferative malignancies. In these mice, the germinal center B cells express an endogenous mouse mammary tumor virus (mtv-29) superantigen (vSAg) known stimulate and activate CD4+ T cells which in turn drive B cell proliferation (94, 95). Through this dual activation paradigm, heterogeneous lymphoproliferative disorders develop in >90% of mice (96). Recent studies have characterized the heterogeneity of SJL lymphomas finding similarities to non-Hodgkin lymphomas and moving away from the classical reticulum cell sarcoma diagnosis (97, 98).
Jain and colleagues (99) are the first to describe the similarities between RCS and AITL. In an analysis of 80 SJL mice, ~44% developed classical SJL disease [i.e., Non-Hodgkin lymphoma and/or reticulum cell sarcoma (100)]. In SJL disease-diagnosed mice ranging from 2 to 24 months of age, flow cytometry demonstrated an expansion of Tfh cell frequencies (CD4+CXCR5+ICOS+PD-1+) in the spleens of SJL mice compared to C57BL/6 controls. Additionally, increased frequencies of splenic GC B cells (B220+FAS+GL-7+) were also found in SJL mice compared to C57BL/6 controls. Microarray analysis of a time course of SJL spleens (mice aged 6 weeks, 6 months, 12 months) revealed a progressive upregulation of genes involved in Tfh signaling, namely: Il21, Il10, Ccl8, and Ccl12. Knockout experiments investigating the impact of IL-21-mediated signaling on the SJL mice established that Il21r−/− SJL mice showed significant reductions in frequencies of total CD4+ T cells and Tfh cells in 9.5- to 12-month mice. A finding concurrent with reduced frequencies of GC B cells seen in 3- to 12-month Il21r−/− SJL compared to SJL WT controls (99). This finding verifies the IL-21 signaling as the etiology of SJL-diseased mice, and re-affirms SJL disease as Tfh-based as IL-21 is considered the major effector cytokine for Tfh cells (101).
Diagnosed SJL-diseased mice underwent age-related changes in splenic and lymph node architecture as assessed by peanut agglutinin (PNA) reactivity. Younger mice had well preserved follicular structures, but by 12 months had uniformly enlarged structures with uneven PNA reactivity. Some mice were noted as having highly enlarged follicles that did not contain germinal centers. The uncontrolled expansion of these follicles leads to the white pulp compressing the red pulp in the spleen.
Higher sera IgG2b protein levels were found in SJL mice compared to C57BL/6 controls. Il21r−/− mice exhibited lower sera IgG2b sera levels compared to wild-type SJL mice, implicating the expanded Tfh cell population as a driver of the development of hypergammaglobulinemia. Igh and Tcrb genes from spleens of primary lymphoma or SJL-diagnosed mice were sequenced to assess the presence or absence of clonal populations within the diseased mice. All sequenced Tcrb clones contained identical V gene sequences, albeit with different J gene sequences. For Igh sequences, high somatic hypermutation was evident across all malignant samples with overall identities <95%.
Conclusion
The Eμ-TCL1 mouse is a powerful model for CLL with a highly analogous microenvironment to that of the human disease (102). Reviewed herein, we see the powerful effect a B-cell malignancy can have on its T-cell counterparts. In CLL, there is a skewing of the T-cell compartment toward antigen-experienced CD8+ cytotoxic T cells. These cells further express exhaustion markers (e.g., PD-1) that demonstrate their inability to effectively control malignant clones. In this scenario, we see that a tumor-specific immune response has been stymied by the malignancy itself, allowing for continued tumor growth and dissemination. Support for this observation is seen in that adoption of the exhausted and skewed T-cell phenotype is dependent on the presence of CLL clones (48), and α-PD-1 blockade therapies are successful in restoring the protective capability of the immune system (50, 51). These results demonstrate the potential of treatments aimed at restoring immune equilibrium to help combat tumors in vivo and give further credence to the necessity and power of murine models to support bench-to-bedside investigations.
While there is no current consensus murine model for AITL, we have reviewed the three most prominent proposals to-date. Of these, both the heterozygous Roquinsan and SJL models appear most suited to be used in future studies modeling AITL microenvironmental interactions. Importantly, both of these models faithfully recapitulate defining disease characteristics of AITL, including: Tfh expansion, hypergammaglobulinemia, effacement of lymphoid architecture, clonal populations, and increased high-endothelial vascularization. It stands to reason that future investigations should be aimed not only at additional model validations and Tfh-based characterization but also focus on the B-cell involvement of AITL that could be key to disease pathogenesis.
Taken together, both murine models for CLL and AITL effectively recapitulate the in vivo expansion of key immune cells. Studies on CLL that have included both cell types in their investigations have provided exceptional clinical insight to modulate immune competence and provide treatment for CLL (including through immunomodulation therapy). Consequently, as seen in CLL, therapies targeted at the lymphoid counterpart of AITL (i.e., B cells) could hold high efficacy and aide in the understanding of tumor microenvironmental manipulation of resident B cells.
Author Contributions
TH and CC cowrote the paper in its entirety.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewers, CS and RJ, and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
The authors acknowledge the University of Nebraska Foundation, the National Science Foundation, and the UNO College of Arts and Sciences for the support of this work.
Funding
This work was supported by the University of Nebraska Foundation, Dr. George Haddix (CC), and the National Science Foundation Graduate Research Fellowship Program (TH).
References
1. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3:991–8. doi:10.1038/ni1102-991
2. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi:10.1146/annurev.immunol.22.012703.104803
3. Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol (2012) 23(Suppl 8):viii6–9. doi:10.1093/annonc/mds256
4. Harris TJ, Drake CG. Primer on tumor immunology and cancer immunotherapy. J Immunother Cancer (2013) 1:12. doi:10.1186/2051-1426-1-12
5. Goubran HA, Kotb RR, Stakiw J, Emara ME, Burnouf T. Regulation of tumor growth and metastasis: the role of tumor microenvironment. Cancer Growth Metastasis (2014) 7:9. doi:10.4137/CGM.S11285
6. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer (2004) 4:11–22. doi:10.1038/nrc1252
7. Pattengale PK, Taylor CR. Experimental models of lymphoproliferative disease. The mouse as a model for human non-Hodgkin’s lymphomas and related leukemias. Am J Pathol (1983) 113:237–65.
8. Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med (2004) 351:893–901. doi:10.1056/NEJMoa040857
9. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med (2005) 352:804–15. doi:10.1056/NEJMra041720
10. Smoley SA, Van Dyke DL, Kay NE, Heerema NA, Dell’Aquila ML, Dal Cin P, et al. Standardization of fluorescence in situ hybridization studies on chronic lymphocytic leukemia (CLL) blood and marrow cells by the CLL research consortium. Cancer Genet Cytogenet (2010) 203:141–8. doi:10.1016/j.cancergencyto.2010.08.009
11. Panayiotidis P, Jones D, Ganeshaguru K, Foroni L, Hoffbrand A. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br J Haematol (1996) 92:97–103. doi:10.1046/j.1365-2141.1996.00305.x
12. Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood (1998) 91:2387–96.
13. Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood (2000) 96:2655–63.
14. Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood (1999) 94:3658–67.
15. Hiai H, Shisa H, Nishi Y, Inoue Y, Ikawa Y, Matsudaira Y, et al. Symbiotic culture of mouse leukaemias: regulation of cell interaction by an activity of serum. Virchows Arch B Cell Pathol Incl Mol Pathol (1980) 32:261–79. doi:10.1007/BF02889031
16. Miyake K, Hasunuma Y, Yagita H, Kimoto M. Requirement for VLA-4 and VLA-5 integrins in lymphoma cells binding to and migration beneath stromal cells in culture. J Cell Biol (1992) 119:653–62. doi:10.1083/jcb.119.3.653
17. Fecteau JF, Messmer D, Zhang S, Cui B, Chen L, Kipps TJ. Impact of oxygen concentration on growth of mesenchymal stromal cells from the marrow of patients with chronic lymphocytic leukemia. Blood (2013) 121:971–4. doi:10.1182/blood-2012-08-447813
18. Nishio M, Endo T, Tsukada N, Ohata J, Kitada S, Reed JC, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood (2005) 106:1012–20. doi:10.1182/blood-2004-03-0889
19. Burger JA, Quiroga MP, Hartmann E, Burkle A, Wierda WG, Keating MJ, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood (2009) 113:3050–8. doi:10.1182/blood-2008-07-170415
20. Bhattacharya N, Diener S, Idler IS, Rauen J, Häbe S, Busch H, et al. Nurse-like cells show deregulated expression of genes involved in immunocompetence. Br J Haematol (2011) 154:349–56. doi:10.1111/j.1365-2141.2011.08747.x
21. Schmid C, Isaacson P. Proliferation centres in B-cell malignant lymphoma, lymphocytic (B-CLL): an immunophenotypic study. Histopathology (1994) 24:445–51. doi:10.1111/j.1365-2559.1994.tb00553.x
22. Lampert IA, Wotherspoon A, Van Noorden S, Hasserjian RP. High expression of CD23 in the proliferation centers of chronic lymphocytic leukemia in lymph nodes and spleen. Hum Pathol (1999) 30:648–54. doi:10.1016/S0046-8177(99)90089-8
23. Granziero L, Ghia P, Circosta P, Gottardi D, Strola G, Geuna M, et al. Survivin is expressed on CD40 stimulation and interfaces proliferation and apoptosis in B-cell chronic lymphocytic leukemia. Blood (2001) 97:2777–83. doi:10.1182/blood.V97.9.2777
24. Ghia P, Strola G, Granziero L, Geuna M, Guida G, Sallusto F, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4, CD40L T cells by producing CCL22. Eur J Immunol (2002) 32:1403–13. doi:10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y
25. Fluckiger AC, Garrone P, Durand I, Galizzi JP, Banchereau J. Interleukin 10 (IL-10) upregulates functional high affinity IL-2 receptors on normal and leukemic B lymphocytes. J Exp Med (1993) 178:1473–81. doi:10.1084/jem.178.5.1473
26. Gorgun G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest (2005) 115:1797–805. doi:10.1172/JCI24176
27. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis (2015) 6:e1792. doi:10.1038/cddis.2015.162
28. Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, et al. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood (2013) 121:1612–21. doi:10.1182/blood-2012-09-457531
29. Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincon M, et al. Differentiation of CD4 T cells to Th1 cells requires MAP kinase JNK2. Immunity (1998) 9:575–85. doi:10.1016/S1074-7613(00)80640-8
30. Rincon M, Enslen H, Raingeaud J, Recht M, Zapton T, Su MS, et al. Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J (1998) 17:2817–29. doi:10.1093/emboj/17.10.2817
31. Yamashita M, Kimura M, Kubo M, Shimizu C, Tada T, Perlmutter RM, et al. T cell antigen receptor-mediated activation of the Ras/mitogen-activated protein kinase pathway controls interleukin 4 receptor function and type-2 helper T cell differentiation. Proc Natl Acad Sci U S A (1999) 96:1024–9. doi:10.1073/pnas.96.3.1024
32. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285:221–7. doi:10.1126/science.285.5425.221
33. Dustin ML, Chakraborty AK, Shaw AS. Understanding the structure and function of the immunological synapse. Cold Spring Harb Perspect Biol (2010) 2:a002311. doi:10.1101/cshperspect.a002311
34. Le Floc’h A, Huse M. Molecular mechanisms and functional implications of polarized actin remodeling at the T cell immunological synapse. Cell Mol Life Sci (2015) 72:537–56. doi:10.1007/s00018-014-1760-7
35. Ku GM, Yablonski D, Manser E, Lim L, Weiss A. A PAK1-PIX-PKL complex is activated by the T-cell receptor independent of Nck, Slp-76 and LAT. EMBO J (2001) 20:457–65. doi:10.1093/emboj/20.3.457
36. Missy K, Hu B, Schilling K, Harenberg A, Sakk V, Kuchenbecker K, et al. AlphaPIX Rho GTPase guanine nucleotide exchange factor regulates lymphocyte functions and antigen receptor signaling. Mol Cell Biol (2008) 28:3776–89. doi:10.1128/MCB.00507-07
37. Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, Nolz JC, et al. HS1 functions as an essential actin-regulatory adaptor protein at the immune synapse. Immunity (2006) 24:741–52. doi:10.1016/j.immuni.2006.03.022
38. Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, Shimizu Y, et al. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol (2006) 16:24–34. doi:10.1016/j.cub.2005.11.036
39. Zhang J, Shehabeldin A, da Cruz LA, Butler J, Somani AK, McGavin M, et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J Exp Med (1999) 190:1329–42. doi:10.1084/jem.190.9.1329
40. Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest (2008) 118:2427–37. doi:10.1172/JCI35017
41. Gilling CE, Mittal AK, Chaturvedi NK, Iqbal J, Aoun P, Bierman PJ, et al. Lymph node-induced immune tolerance in chronic lymphocytic leukaemia: a role for caveolin-1. Br J Haematol (2012) 158:216–31. doi:10.1111/j.1365-2141.2012.09148.x
42. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood (2012) 120:1412–21. doi:10.1182/blood-2012-02-411678
43. Ramsay AG, Evans R, Kiaii S, Svensson L, Hogg N, Gribben JG. Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by altering Rho GTPase signaling that is reversible with lenalidomide. Blood (2013) 121:2704–14. doi:10.1182/blood-2012-08-448332
44. Saoudi A, Kassem S, Dejean AS, Gaud G. Rho-GTPases as key regulators of T lymphocyte biology. Small GTPases (2014) 5:e983862. doi:10.4161/sgtp.28208
45. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature (2002) 420:629–35. doi:10.1038/nature01148
46. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol (2005) 21:247–69. doi:10.1146/annurev.cellbio.21.020604.150721
47. Gorgun G, Ramsay AG, Holderried TA, Zahrieh D, Le Dieu R, Liu F, et al. E(mu)-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc Natl Acad Sci U S A (2009) 106:6250–5. doi:10.1073/pnas.0901166106
48. Hofbauer JP, Heyder C, Denk U, Kocher T, Holler C, Trapin D, et al. Development of CLL in the TCL1 transgenic mouse model is associated with severe skewing of the T-cell compartment homologous to human CLL. Leukemia (2011) 25:1452–8. doi:10.1038/leu.2011.111
49. McClanahan F, Riches JC, Miller S, Day WP, Kotsiou E, Neuberg D, et al. Mechanisms of PD-L1/PD-1-mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Emicro-TCL1 CLL mouse model. Blood (2015) 126:212–21. doi:10.1182/blood-2015-02-626754
50. McClanahan F, Hanna B, Miller S, Clear AJ, Lichter P, Gribben JG, et al. PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood (2015) 126:203–11. doi:10.1182/blood-2015-01-622936
51. Gassner FJ, Zaborsky N, Catakovic K, Rebhandl S, Huemer M, Egle A, et al. Chronic lymphocytic leukaemia induces an exhausted T cell phenotype in the TCL1 transgenic mouse model. Br J Haematol (2015) 170:515–22. doi:10.1111/bjh.13467
52. O’Connor NT, Crick JA, Wainscoat JS, Gatter KC, Stein H, Falini B, et al. Evidence for monoclonal T lymphocyte proliferation in angioimmunoblastic lymphadenopathy. J Clin Pathol (1986) 39:1229–32. doi:10.1136/jcp.39.11.1229
53. Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H, et al. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. 4th ed. Lyon, France (2008).
54. Nathwani BN, Rappaport H, Moran EM, Pangalis GA, Kim H. Malignant lymphoma arising in angio-immunoblastic lymphadenopathy. Cancer (1978) 41:578–606. doi:10.1002/1097-0142(197802)41:2<578::AID-CNCR2820410226>3.0.CO;2-8
55. Vose JM. Peripheral T-cell non-Hodgkin’s lymphoma. Hematol Oncol Clin North Am (2008) 22:997–1005. doi:10.1016/j.hoc.2008.07.010
56. Iannitto E, Ferreri AJ, Minardi V, Tripodo C, Kreipe HH. Angioimmunoblastic T-cell lymphoma. Crit Rev Oncol Hematol (2008) 68:264–71. doi:10.1016/j.critrevonc.2008.06.012
57. Merchant SH, Amin MB, Viswanatha DS. Morphologic and immunophenotypic analysis of angioimmunoblastic T-cell lymphoma: emphasis on phenotypic aberrancies for early diagnosis. Am J Clin Pathol (2006) 126:29–38. doi:10.1309/28YP0DELGKEJGRXG
58. De Leval L, Gisselbrecht C, Gaulard P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br J Haematol (2010) 148:673–89. doi:10.1111/j.1365-2141.2009.08003.x
59. Grogg KL, Attygalle AD, Macon WR, Remstein ED, Kurtin PJ, Dogan A. Angioimmunoblastic T-cell lymphoma: a neoplasm of germinal-center T-helper cells? Blood (2005) 106:1501–2. doi:10.1182/blood-2005-03-1083
60. Mourad N, Mounier N, Briere J, Raffoux E, Delmer A, Feller A, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood (2008) 111:4463–70. doi:10.1182/blood-2007-08-105759
61. Luminari S, Bellei M, Biasoli I, Federico M. Follicular lymphoma: treatment and prognostic factors. Rev Bras Hematol Hemoter (2012) 34:54–9. doi:10.5581/1516-8484.20120015
62. Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol (2009) 10:375–84. doi:10.1038/ni.1704
63. Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol (2004) 173:68–78. doi:10.4049/jimmunol.173.1.68
64. Kim JR, Lim HW, Kang SG, Hillsamer P, Kim CH. Human CD57+ germinal center-T cells are the major helpers for GC-B cells and induce class switch recombination. BMC Immunol (2005) 6:3. doi:10.1186/1471-2172-6-3
65. Went P, Agostinelli C, Gallamini A, Piccaluga PP, Ascani S, Sabattini E, et al. Marker expression in peripheral T-cell lymphoma: a proposed clinical-pathologic prognostic score. J Clin Oncol (2006) 24:2472–9. doi:10.1200/JCO.2005.03.6327
66. Laurent C, Fazilleau N, Brousset P. A novel subset of T-helper cells: follicular T-helper cells and their markers. Haematologica (2010) 95:356–8. doi:10.3324/haematol.2009.019133
67. Kaneko Y, Maseki N, Sakurai M, Takayama S, Nanba K, Kikuchi M, et al. Characteristic karyotypic pattern in T-cell lymphoproliferative disorders with reactive “angioimmunoblastic lymphadenopathy with dysproteinemia-type” features. Blood (1988) 72:413–21.
68. Dogan A, Attygalle AD, Kyriakou C. Angioimmunoblastic T-cell lymphoma. Br J Haematol (2003) 121:681–91. doi:10.1046/j.1365-2141.2003.04335.x
69. Nelson M, Horsman DE, Weisenburger DD, Gascoyne RD, Dave BJ, Loberiza FR, et al. Cytogenetic abnormalities and clinical correlations in peripheral T-cell lymphoma. Br J Haematol (2008) 141:461–9. doi:10.1111/j.1365-2141.2008.07042.x
70. de Leval L, Rickman DS, Thielen C, Reynies A, Huang YL, Delsol G, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood (2007) 109:4952–63. doi:10.1182/blood-2006-10-055145
71. Piccaluga PP, Agostinelli C, Califano A, Carbone A, Fantoni L, Ferrari S, et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res (2007) 67:10703–10. doi:10.1158/0008-5472.CAN-07-1708
72. Willenbrock K, Bräuninger A, Hansmann M. Frequent occurrence of B-cell lymphomas in angioimmunoblastic T-cell lymphoma and proliferation of Epstein-Barr virus-infected cells in early cases. Br J Haematol (2007) 138:733–9. doi:10.1111/j.1365-2141.2007.06725.x
73. Suefuji N, Niino D, Arakawa F, Karube K, Kimura Y, Kiyasu J, et al. Clinicopathological analysis of a composite lymphoma containing both T- and B-cell lymphomas. Pathol Int (2012) 62:690–8. doi:10.1111/j.1440-1827.2012.02858.x
74. Hoffmann JC, Chisholm KM, Cherry A, Chen J, Arber DA, Natkunam Y, et al. An analysis of MYC and EBV in diffuse large B-cell lymphomas associated with angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma not otherwise specified. Hum Pathol (2016) 48:9–17. doi:10.1016/j.humpath.2015.09.033
75. Ahsanuddin AN, Brynes RK, Li S. Peripheral blood polyclonal plasmacytosis mimicking plasma cell leukemia in patients with angioimmunoblastic T-cell lymphoma: report of 3 cases and review of the literature. Int J Clin Exp Pathol (2011) 4:416–20.
76. Papadi B, Polski JM, Clarkson DR, Liu-Dumlao TO. Atypical angioimmunoblastic T-cell lymphomas masquerading as systemic polyclonal B-immunoblastic proliferation. Virchows Arch (2012) 461:323–31. doi:10.1007/s00428-012-1280-5
77. Feller AC, Griesser H, Schilling CV, Wacker HH, Dallenbach F, Bartels H, et al. Clonal gene rearrangement patterns correlate with immunophenotype and clinical parameters in patients with angioimmunoblastic lymphadenopathy. Am J Pathol (1988) 133:549–56.
78. Smith JL, Hodges E, Quin CT, McCarthy KP, Wright DH. Frequent T and B cell oligoclones in histologically and immunophenotypically characterized angioimmunoblastic lymphadenopathy. Am J Pathol (2000) 156:661–9. doi:10.1016/S0002-9440(10)64770-0
79. Iqbal J, Weisenburger DD, Greiner TC, Vose JM, McKeithan T, Kucuk C, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood (2010) 115:1026–36. doi:10.1182/blood-2009-06-227579
80. Roncador G, Garcia Verdes-Montenegro JF, Tedoldi S, Paterson JC, Klapper W, Ballabio E, et al. Expression of two markers of germinal center T cells (SAP and PD-1) in angioimmunoblastic T-cell lymphoma. Haematologica (2007) 92:1059–66. doi:10.3324/921059
81. Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood (2009) 114:144–7. doi:10.1182/blood-2009-03-210039
82. Delhommeau F, Dupont S, Valle VD, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med (2009) 360:2289–301. doi:10.1056/NEJMoa0810069
83. Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood (2009) 113:6403–10. doi:10.1182/blood-2009-02-205690
84. Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell (2011) 20:25–38. doi:10.1016/j.ccr.2011.06.003
85. Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood (2012) 120:1466–9. doi:10.1182/blood-2012-02-408542
86. Vasanthakumar A, Godley LA. 5-Hydroxymethylcytosine in cancer: significance in diagnosis and therapy. Cancer Genet (2015) 208:167–77. doi:10.1016/j.cancergen.2015.02.009
87. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell (2011) 20:11–24. doi:10.1016/j.ccr.2011.06.001
88. Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood (2011) 118:4509–18. doi:10.1182/blood-2010-12-325241
89. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A (2011) 108:14566–71. doi:10.1073/pnas.1112317108
90. Muto H, Sakata-Yanagimoto M, Nagae G, Shiozawa Y, Miyake Y, Yoshida K, et al. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J (2014) 4:e264. doi:10.1038/bcj.2014.83
91. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi:10.1038/nature03555
92. Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C, et al. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol (2010) 11:725–33. doi:10.1038/ni.1902
93. Ellyard JI, Chia T, Rodriguez-Pinilla SM, Martin JL, Hu X, Navarro-Gonzalez M, et al. Heterozygosity for Roquinsan leads to angioimmunoblastic T-cell lymphoma-like tumors in mice. Blood (2012) 120:812–21. doi:10.1182/blood-2011-07-365130
94. Tsiagbe VK, Asakawa J, Miranda A, Sutherland RM, Paterson Y, Thorbecke GJ. Syngeneic response to SJL follicular center B cell lymphoma (reticular cell sarcoma) cells is primarily in V beta 16+ CD4+ T cells. J Immunol (1993) 150:5519–28.
95. Tsiagbe VK, Yoshimoto T, Asakawa J, Cho SY, Meruelo D, Thorbecke GJ. Linkage of superantigen-like stimulation of syngeneic T cells in a mouse model of follicular center B cell lymphoma to transcription of endogenous mammary tumor virus. EMBO J (1993) 12:2313–20.
96. Zhang DJ, Tsiagbe VK, Huang C, Thorbecke GJ. Control of endogenous mouse mammary tumor virus superantigen expression in SJL lymphomas by a promoter within the env region. J Immunol (1996) 157:3510–7.
97. Stavnezer J, Lasky JL, Ponzio NM, Scheid MP, Jeanette Thorbecke G. Reticulum cell sarcomas of SJL mice have rearranged immunoglobulin heavy and light chain genes. Eur J Immunol (1989) 19:1063–9. doi:10.1002/eji.1830190616
98. Tang JC, Ho FC, Chan AC, Srivastava G. Clonality of lymphomas at multiple sites in SJL mice. Lab Invest (1998) 78:205–12.
99. Jain S, Chen J, Nicolae A, Wang H, Shin D, Adkins EB, et al. IL-21-driven neoplasms in SJL mice mimic some key features of human angioimmunoblastic T-cell lymphoma. Am J Pathol (2015) 185:3102–14. doi:10.1016/j.ajpath.2015.07.021
101. Crotty S. Follicular helper CD4 T cells (Tfh). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
Keywords: lymphoma, chronic lymphocytic leukemia, angioimmunoblastic T-cell lymphoma, immune surveillance, tumor-induced immunosuppression
Citation: Herek TA and Cutucache CE (2017) Using Murine Models to Investigate Tumor–Lymphoid Interactions: Spotlight on Chronic Lymphocytic Leukemia and Angioimmunoblastic T-Cell Lymphoma. Front. Oncol. 7:86. doi: 10.3389/fonc.2017.00086
Received: 13 May 2016; Accepted: 18 April 2017;
Published: 02 May 2017
Edited by:
Gautam Borthakur, University of Texas MD Anderson Cancer Center, USAReviewed by:
Rodrigo Jacamo, University of Texas MD Anderson Cancer Center, USAChristine Marie Stellrecht, University of Texas MD Anderson Cancer Center, USA
Elias Hallack Atta, Brazilian National Cancer Institute, Brazil
Copyright: © 2017 Herek and Cutucache. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine E. Cutucache, Y2N1dHVjYWNoZUB1bm9tYWhhLmVkdQ==