 Simona Reina
Simona Reina Francesca Guarino
Francesca Guarino Andrea Magrì
Andrea Magrì Vito De Pinto
Vito De Pinto- 1Department of Biomedical and Biotechnological Sciences, University of Catania, Catania, Italy
- 2National Institute of Biostructures and Biosystems (INBB), Rome, Italy
VDAC3 is the least known isoform of the mammalian voltage-dependent anion selective channels of the outer mitochondrial membrane. It has been recently shown that cysteine residues of VDAC3 are found over-oxidized. The VDAC3 cysteine over-oxidation was associated with the oxidizing environment and the abundance of reactive oxygen species (ROS) in the intermembrane space. In this work, we have examined the role of VDAC3 in general pathogenic mechanisms at the basis of mitochondrial dysfunction and involving the mitochondrial quality control. Many of the diseases reported here, including cancer and viral infections, are often associated with significant changes in the intracellular redox state. In this sense, VDAC3 bearing oxidative modifications could become marker of the oxidative load in the mitochondria and part of the ROS signaling pathway.
Introduction
The outer mitochondrial membrane (OMM) is a continuous membrane that separates the cytosol from the inside of the organelle. It has long been imagined as a simple envelope, perforated by plenty of water-filled holes, as it has recently been illustrated in evocative AFM images (1). Nevertheless, the OMM can be considered the border of the mitochondria and any message to be communicated outside the mitochondrion has to be dealt with it. Furthermore, the OMM is involved in fusion–fission, communicates to the cell about the derangements in the bioenergetics processes, and rules the interaction of cytosolic molecules with the mitochondrion: these large or hydrophobic molecules require a specific docking structure. Cytoskeletal components (2, 3), ribosomes (4), and enzymes (5–8) contact the outer surface of mitochondria, or anchor to it, for structural or organizational purposes. The importance of OMM is demonstrated by the catastrophic results of its destruction: when the intactness of the OMM is destroyed, in the so-called permeability transition, the apoptotic cascade begins (9).
The most abundant protein located in the OMM is the family of pore structures called VDACs (for voltage-dependent anion selective channels). They are considered rather unspecific, hollow, water-filled pores (10–12): in contrast with Tom40, another pore-forming protein of the OMM, with similar structure, is devoted to channeling polypeptides to be folded in mitochondria (13). The apparent unspecific function contrasts with many roles, many issues, and many pathologies in which VDACs have been involved. Several important advancements derived by casual bumping of investigations into VDACs: once for all, the very first sequence, at protein level, of human VDAC1 was performed on a protein believed to be something else, a component of the immune system (14). There is thus a need for a deeper study of VDACs, addressing the more profound functions of this group of protein. In this work, we have focused our attention onto the least investigated mammalian VDAC isoform, named VDAC3, since it was the last one to be discovered (15). We report here a survey of its known feature and the information about its role in pathology. In general, however, a defective VDAC should not impact on a single pathology but instead be part of the mitochondrial dysfunction, in turn associated with a myriad of cellular pathological states.
VDAC3 Structure: The Essential Information
The VDACs are a small family of proteins whose primary role is to form an aqueous pore through the OMM that allows the exchange of metabolites and molecules (10–12). In chordates, and in particular in mammals, three distinct VDAC isoforms are coded in the nucleus and targeted to the mitochondria (16). The VDAC sequences are conserved, have a length of about 280 amino acids, and, despite they form a hydrophilic barrel, they behave as integral membrane proteins in the isolation procedures, comparable to the transmembrane carriers (17, 18).
3D structures of mouse and human VDAC1 isoform have been experimentally determined by X-ray crystallography and NMR (19–21). Since the sequences are conserved, a similar structure has been hypothesized for the other two isoforms (22, 23). The VDAC2 structure from zebrafish has been solved, and it is very similar to the VDAC1 (24). Interestingly, the zebrafish VDAC2 has no N-terminal extension typical of this isoform in mammalian and has only one cysteine in its sequence (Cys 127). The β-strands are arranged in an antiparallel bunch strengthened by hydrogen bridges between each couple. Since the number of strands is odd, the first and last strands run in parallel: this is a peculiarity of mitochondrial VDACs in comparison with bacterial porins, which form an even, completely antiparallel β-barrel (25). The β-strands are connected by short turns or loops. As it is more evident for the bacterial porins (25), shorter turns crowd one side of the barrel [toward the intermembrane space (IMS)], and slightly longer loops the other side (the cytosol). The assignment of the turns and loops to a definite side of the membrane was a very important achievement (26) because it allows to investigate the availability of single amino acid residues to one of the two sides of the outer membrane. The last peculiarity of the VDAC structure is the N-terminal tail, a sequence of 25–36 amino acids (36 in VDAC2) predicted to form an exotic α-helix in a whole β-strands structure (27). The sequence has been assigned by structural determinations inside the pore, with a strong tendency to fold as an amphipathic α-helix. However, the exact arrangement of this segment requires further refinement, because there are slight differences in the available structures (28). The importance of the N-terminal tail has been highlighted in many works, since its involvement in the dynamic gating of the pore is possible.
The VDAC3 structure has not yet been obtained. Several bioinformatic predictions have been proposed, based on the large sequence similarity, with a β-barrel core almost identical to the other VDAC isoforms (23). Unfortunately, very little information about this isoform is present in the literature (29).
The puzzling question refers to the presence in the mitochondria of three different isoforms with very similar structure. The three VDACs are encoded by distinct genes located on different chromosomes (22, 30): they share the same exon–intron organization indicating that they are the result of not evolutionary far duplications (22, 30, 31): but the presence, the different expression levels (32–34), the different results of each gene K.O. (35–38) indicate that during evolution three VDAC genes were fixed in the chordate genome, most likely because they improved the cell (or the mitochondria) fitness.
Recent Investigations on the Mammal VDAC3: Over-Oxidation of Cysteines
Our working hypothesis about a specific function of VDAC3 begun from the sequence analysis. It is evident that a striking difference among VDAC isoforms is the number of cysteines in the sequences. In human, VDAC1 has two Cys, VDAC2 has nine Cys, and VDAC3 has six Cys. Furthermore, it is indeed interesting that the three isoforms show different structural localization of their sulfur amino acids (Figure 1).
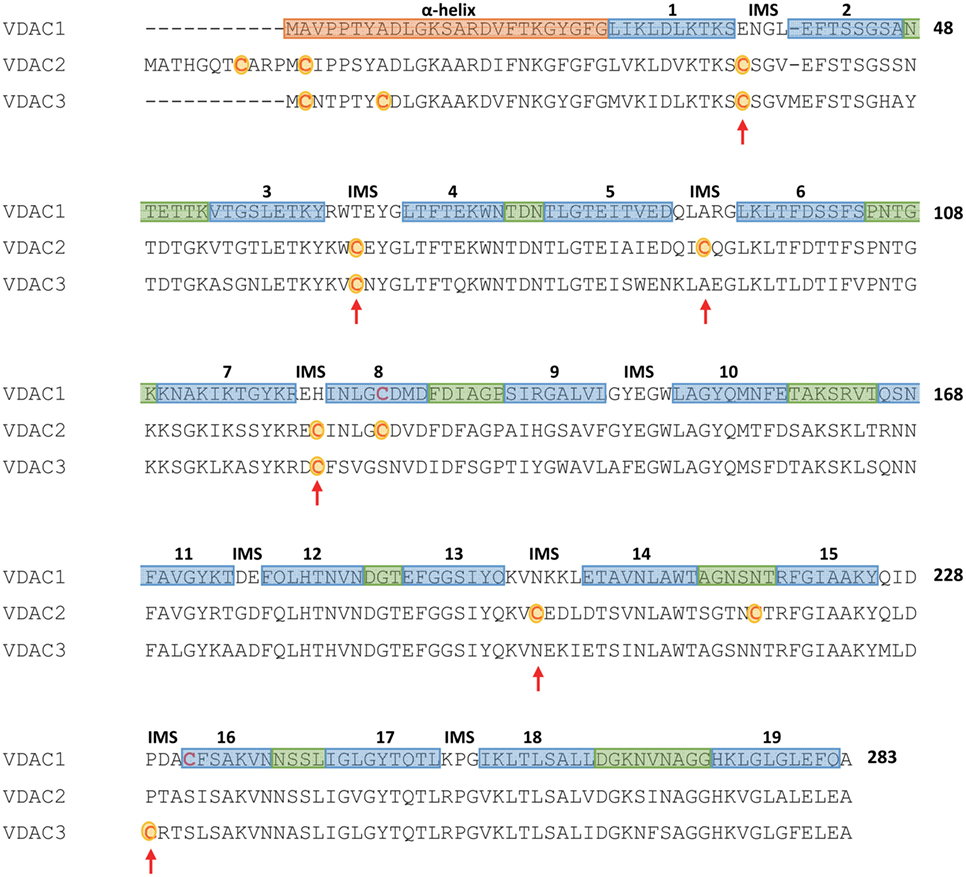
Figure 1. Multi-alignment of human mammalian voltage-dependent anion selective channel (VDAC) isoforms. The multi-alignment, obtained by the Clustal-X software, reports as a pink box the N-terminal sequence of VDAC1, in mauve the sequences forming the 19 β-strands, and in pale green the sequences corresponding to cytosol-exposed loops. The non-colored sequences correspond to the intermembrane-exposed turns (intermembrane space, IMS). The strands localization refers to the structure reported in Ref. (19), while the assignment of IMS or cytosolic loops follows the paper by Tomasello et al. (26). Cysteine residues of VDAC2 and VDAC3 are in red and outlined in yellow, and the red arrows show those cysteine residues protruding in the intermembrane space.
The sideness determination allowed the assignment of the cysteine residues position in interstrands loops or turns. It helped to speculate about their exposition to the water environment. While VDAC1 contains two Cys, located far from each other and interacting, respectively, with the water interior of the pore (Cys 232 in the β-strand 16) and with the hydrophobic, phospholipidic milieu (Cys 127 in the β-strand 8) (19, 39, 40), the other two isoforms’ cysteines mostly protrude toward the IMS (16, 26, 41). In particular, VDAC2 and VDAC3 show conserved cysteine residues at positions 36, 65, and 122; in addition, VDAC3 has Cys 229 exposed to the IMS, while VDAC2 has not only Cys 92 and 199 exposed to IMS but also Cys 127 in the middle of β-strand 8 and Cys 216 in the loop between β-strands 14 and 15. At the end, in VDAC2 and VDAC3, two cysteines are localized in the N-terminus moiety, thus in the water interior but only one of them is conserved at the same position in the two isoforms (Cys 2 in VDAC3 numbering) [the definition of the turns and loops is based on the human VDAC1 structure (19), thus it is predictive for the other isoforms; numbering follows the VDAC1 sequence, for the sake of clarity; please refer to the scheme in Figure 1]. Thus, human VDAC2 has more cysteines than VDAC3 but only in VDAC3 they look all preferentially exposed to the IMS.
The IMS is one of the oxidizing environments in the cell, together with the ER lumen (42). Its oxidizing power is a consequence of the proton unbalance across the inner mitochondrial membrane, due to the electron chain oxidation; the glutathione redox balance is also in favor of an oxidative potential (43, 44) and Complex III and Monoamine Oxidase directly pour reactive oxygen species (ROS) in the IMS (45, 46). It has been observed that VDACs are the conduit for the diffusion of the superoxide anion outside the organelle (47) and of hydroperoxide, produced by the dismutation of superoxide anion by SOD. Thus, we suspected that the VDAC cysteines exposed to the IMS could be a preferential target to discharge the oxidative load of the ROS in the compartment. We focused in particular on VDAC3 and, to get a molecular evidence of this hypothesis, we performed high resolution Mass Spectrometry analysis on VDAC3 derived from rat liver mitochondria (41). The results showed that (i) VDAC3 is electrophoretically heterogeneous (i.e., it is present with different mobility in SDS-PAGE) because the arrangement of oxidized cysteine is different in the various bands; (ii) different molecules contain different extents, in terms of type and quantity, of oxidized cysteine (we like to call them: “redox isomers”); (iii) the oxidation states of the cysteine are not random but some of them (Cysteines 2 and 8) are always found in a reduced (exactly derivatized by iodoacetamide) state, others (Cysteines 36, 65, and 229) in different oxidation levels (+1, +3, and +5); (iv) the oxidation to sulfinic (+3) or sulfonic (+5) acid is biologically irreversible. The only known reductase is thioredoxin/sulforedoxin system that targets sulfinic cysteine (48, 49), while there are no known enzymes able to reverse the sulfonic acid (+5) whose function is unknown and possibly related to either modify the protein electrostatic equilibrium or target it for degradation.
What can be the meaning of VDAC3 oxidations for the mitochondria within the cellular context?
Relevance of Oxidation States of VDAC3 in Physiopathology: Is VDAC3 a Marker of the Mitochondrial Quality Control and Aging?
We have proposed, well before the discovery of over-oxidized cysteines, two explanations for the VDAC3 cysteines exceeding in number those present in VDAC1 (22). In the former hypothesis, the load of oxidations of VDAC3, whose role in the permeability of the OMM is not preeminent, can be considered as a defensive mechanism: the accumulation of damages on an apparently unimportant protein may drain the excess ROS. In the latter hypothesis, the accumulation of ROS modifications on the same protein, in addition to protect other molecules, can change the conformation and/or the docking capability of VDAC3 and this modification, in turn, signals the ROS load of the single mitochondrion to other structures within the cell.
Recently, deletion of cysteines in engineered VDAC3 molecules (41, 50) suggested that the oxidized state of these residues can decrease the VDAC3 pore-conductance activity (41, 50, 51), even though the molecular mechanism of such hindrance to the conductance is not clear yet. Okazaki et al. proposed that the transient formation of a disulfide bridge inside the pore could strongly change the permeability (50). In our hands, we found that a different disulfide bridge can form but does not change much the permeation available diameter of the pore (41, 52). The modifications to the polypeptide chain conformation due to the cys oxidation can also affect the electrical charge disposition on the protein surface: in VDACs, the surfaces inside the barrel or at the mouths of the pore are hydrophilic (53). Furthermore, the insertion of negative charges due to sulfinic and sulfonic oxidation can provoke electric repulsions inside the chain or toward phospholipids, and next modify the conformation of the protein. A similar output has been evidenced in Ref. (52), where a molecular dynamic simulation has shown that, in case of disulfide formation between the closest cysteine residues available, the pore diameter would not change, while the exposition of residues, or of part of the N-terminal moiety, to the extra-membranous environment would change (52, 54). These conformational changes can be “sensed” in the cell as modification of the docking properties of the outer mitochondrial surface. The amounts of ROS in each mitochondrion can indeed cause VDAC cysteine oxidation proportional to the ROS concentration. We thus speculated that the quantitative extent of VDAC oxidative modifications on the mitochondrial surface can be used as a signal of oxidation load, allowing the monitoring of the ROS amounts in each mitochondrion or mitochondrial network section (Figure 2). At the end, oxidation of the cysteines in VDAC could provoke the binding or the release of yet unknown interactors.
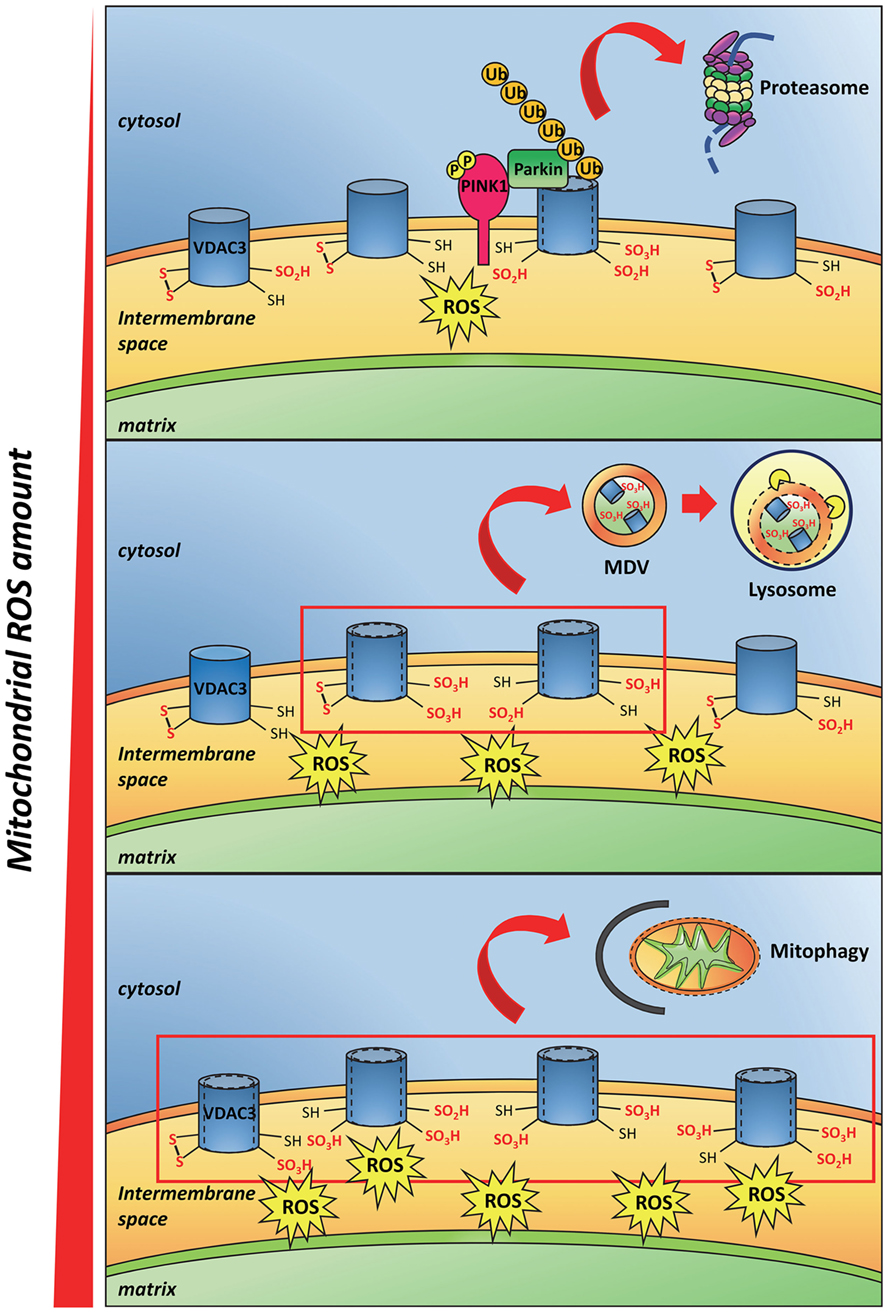
Figure 2. Proposed model of mitochondrial reactive oxygen species (ROS) and VDAC3 cysteine residues interaction. Little amounts of ROS oxidize some VDAC3 cysteines that protrude toward the mitochondrial intermembrane space up to sulfinic and sulfonic oxidation states. In addition to a conformational change in the protein, such irreversible modifications recruit the PINK/Parkin system that in turn ubiquitinates VDAC3. This step, followed by the proteasome degradation of the ubiquitinated protein, is preliminary to the mitochondrial quality control (upper panel). The progressive accumulation of ROS, due to mitochondrial stress, increases the amount of oxidized cysteines in VDAC3. This phenomenon stimulates the incorporation of single damaged proteins, or membrane patches containing damaged proteins, into mitochondria-derived vesicles, subsequently targeted to lysosomes (middle panel). When the ROS level reaches a maximum threshold, almost all VDAC3 proteins of the outer mitochondrial membrane become heavily modified by irreversible oxidations. Conformational changes derived from these modifications signal the redox state of the mitochondria to the rest of the cell. Damaged, reactive oxygen species-producing mitochondria are therefore removed through mitophagy (lower panel).
VDAC2 is also rich in cysteines: it is possible that this isoform could also fulfill a similar function. The differences in number and location of cysteine residues do not support an overall overlapping of the VDAC2 and VDAC3 functions. Unfortunately, the molecular analysis of VDAC2 by mass spectrometry has not been completed, thus it is premature to draw conclusions about VDAC2.
It is well known that mitochondria are subjected to quality control systems that allow the cell to dispose malfunctioning organelles or just part of them. Such a quality control has the role to avoid that the cell is affected by a relevant mitochondrial dysfunction, an event reported in a large number of degenerative pathologies. The single affected organelle is targeted to lysosomes and destroyed. As an alternative, single proteins, or membrane patches are identified as damaged portions of the organelle and destroyed by a complex process involving the formation of mitochondria-derived vesicles (MDVs) and their targeting to lysosomes [reviewed in Ref. (55)].
The production of MDVs is a process stimulated by the mitochondrial stress. In a recent work where the MDV pathway has been dissected and characterized, it was demonstrated that MDVs are enriched in oxidized proteins derived from mitochondria and in particular from the outer membrane (56). The authors suggested indeed that the protein conformational changes subsequent to oxidation can initiate their incorporation into MDVs. The oxidized state of proteins was demonstrated with immunoblots targeting carbonylated amino acids (56). VDAC1 was found among the proteins in MDVs. Unfortunately, the oxidation state of cysteines was not investigated nor the presence of VDAC3 in MDVs.
It is peculiar for VDAC3 that the post-translational modification deletes the starting methionine in the sequence, leaving N-terminal residue the cysteine encoded as second amino acid (Cys 2). Cysteine at the N-terminal position is subjected to the so-called “N-end rule pathway,” a destructive process involving the oxidation of the N-terminal Cys, its selective arginylation and, at the end, its ubiquitination (53, 57). This pathway forwards the modified protein from the mitochondria through a retrotranslocation pathway to the cytosolic proteasome for its recycling, similar to the ER-associated degradation pathway (ERAD). In our studies, Cys 2 in VDAC3 was never found oxidized: the oxidized N-terminal cysteine is the precursor stage for the N-end pathway. Cysteine 2 in VDAC3 was instead always found involved in a disulfide bridge (41). During the MS pre-processing of the samples, Cys 2 was reduced and modified with iodoacetoamide. Iodoacetoamide irreversibly modifies the reduced cysteines, marking them as reduced or reducible in the conditions of analysis. The lack of any evidence of Cys 2 oxidation might just mean that each single protein altered in this way in the cell is immediately destroyed by the retrotranslocation pathway (N-end rule pathway). VDAC3 has been identified as a Parkin interactor and subjected to ubiquitination by Parkin in defective mitochondria (58). The PINK1/Parkin system is responsible for the elimination of defective mitochondria by autophagy (59). PINK1 is a kinase present, at very low levels, in the OMM, that is able to phosphorylate Parkin upon its recruitment from cytosol to the OMM. The phosphorylated Parkin starts ubiquitination of selected targets in the mitochondria and this event is considered preliminary to the mitochondrial quality control (or to mitophagy). Experiments by Sun et al. support a model in which VDACs are part of the machinery that recruits Parkin to the organelle (58). By mass spectrometry, VDAC isoforms have been found to be the most abundant Parkin-associated proteins, indicating their role as mitochondrial docking site for Parkin. Interestingly, while VDAC3 was found ubiquitinated, VDAC2 can bind Parkin but it was not detected ubiquitinated (58). This difference can be due to the lack of the N-terminal cysteine in the mature form of VDAC2. When the Cys 2 is reduced or forms a disulfide bridge (41, 50), the VDAC3 protein can follow the same degradation pathway followed by the other isoforms.
During aging, stem cells sustain damages until they become exhausted (60). To reduce and slow the accumulation of such damages, stem cell might segregate ruined subcellular components away from the new stem cell. This trick would be extremely useful, excluding damaged mitochondria from the new, daughter stem cells. Katajisto et al. (61) were able to monitor the fate of aged mitochondria by using a photoactivable GFP tag linked to an outer membrane protein, the Omp25. The conclusion of this study was that apportioning of mitochondria to daughter cells from a stem cell is not symmetric: old, aged mitochondria are confined to the somatic cell and not to the daughter stem cell (61). This interesting work showed that, in stem cell, a modification marking old mitochondria must exist, to permit the asymmetric apportioning that is useful to stem cells.
Can the over-oxidation of cysteines in VDAC3 be one of the markers of such aging? It is not straightforward to imagine that the cysteines, exposed to the IMS, can direct a signal on the other side of the membrane (cytosol). We hypothesize that the over-oxidation of cysteines and/or other modification-like formation or break of disulfide bridges (41, 50, 54) can modify the structure of the pore or the electrostatic properties of the water-exposed surfaces and the mobility of the N-terminus. This conformational change could be the real switch (see Figure 2 for a cartoon depicting this hypothesis). A further protein, whose interaction with VDAC3 is modified upon the pore conformational change, should thus be hypothesized as an intermediate messenger.
Interactomic Analysis Confirms the Role of VDAC3 in Mitochondrial Protein Quality Control
The relationship of VDAC3 with cellular systems was highlighted in the catalog of proteins found to interact with this isoform in vivo by a TAP-Tag immunoprecipitation strategy and mass spectrometry identification (62). Proteins from the endoplasmic reticulum were found to be well represented: Grp75, Hsp70, GRP, and calreticulin. They are also markers of the MAM, the mitochondria-associated membranes, considered as contact point and exchange site between mitochondria and ER (63). Other crucial pathways were clearly correlated to VDAC3 based on the presence of interacting proteins involved in it. Proteins correlated to oxidative stress (GSTO-1, PRDX, and GSTK-1), proteins involved in the response to misfolded or unfolded proteins (YWHAQ, KCIP1, or SFN), proteasomal components and chaperons (PDI and Erp5) (64, 65), and proteins related to ribosome contact and control, were identified (62).
Isoforms of protein disulfide isomerases, peroxiredoxins, glutathione transferases, involved in maintaining the redox status, or glutathione S-transferase kinase 1 (GSTK-1) were among VDAC3 interactors (66, 67). Mammalian unfolded protein response utilizes two main mechanisms able to eliminate misfolded proteins: upregulation of chaperons and proteolytic degradation by ubiquitin–proteasome and autophagy–lysosome system (68). Proteins like the interesting but poorly known 14-3-3 protein theta, the protein kinase C-inhibitor, or the stratifin, are components of these pathways and were indeed among the interactors. Also, the ERAD (69, 70), where the endoplasmic reticulum faces accumulation of mis-folded or unfolded proteins, was found well represented with proteins like chaperones, oxidases, and thiol-isomerases (i.e., protein disulfide isomerases, calreticulin) (62).
A very interesting interactor of VDAC3 found in Ref. (62) is VCP (TER-ATPase). VCP is a member of the ERAD pathway and is involved in the “extraction” of protein from the OMM and other membranes to be directed to ubiquitination and degradation through ER, with or without stress conditions (64). VCP mutants have been correlated to the onset of some type of myopathies, of frontotemporal dementia and of Alzheimer disease, Parkinson’s disease, or amyotrophic lateral sclerosis, all pathologies where VDAC involvement has been already reported (71–74). VCP could be in charge of extracting the modified version of VDAC3, addressing it toward microtubules through cytoplasmic granules’ traffic (62) and enriching near the centrosome. VDAC3 has been detected at the centriole and its role in ciliogenesis has been proposed (75, 76). To draw a more careful and comparative comparison of the VDAC isoforms’ roles, the interactomic analysis of VDAC2, presently lacking, would be essential.
VDAC in Pathologies
At dawning of VDAC research, when there were only suspects of more VDAC isoforms and the human genome sequence was not completed, it raised a substantial interest that the claim of VDAC1 lack in a human patient, suffering of an undiagnosed encephalomyopathy (77). The evidence come out from Western blot of the skeletal muscle biopsy of a 3-year-old child, suffering of dysmorphism, hypotonia, respiration and feeding problems, and seizures. The underlying idea was that suspected, but not diagnosed, mitochondrial pathologies could be due to the absence of a transport protein, located in the mitochondrial membranes. This strategy gave raise to a screening with anti-VDAC1 antibodies run on muscle mitochondria membranes. About 100 suspected patients were investigated for VDAC1 deficiency in the Netherlands and Italy (78). As reported in Ref. (71), the patients to be examined were selected on a biochemical basis, having to show (a) diminished substrate oxidation rates not caused by a disturbance in the respiratory chain, citric acid cycle, or pyruvate dehydrogenase complex or (b) normal activities of the respiratory chain enzymes and the pyruvate dehydrogenase complex, together with strong suspicion for mitochondrial disorder based on both clinical and clinical–chemical hallmarks (79). The search for VDAC deficiency at a protein level was nevertheless later abandoned because the paucity of putative patients and their spread. In addition, the lack of reliable antibodies, working specifically against each singular VDAC isoform, hindered the possibility to study different pores. Most of the information about VDACs and in particular VDAC3 involvement in pathologies stemmed, thus, by wide-scale analysis of gene expression or general proteomic surveys. In many of them, evidence of the presence of altered levels of VDAC isoforms and in particular of VDAC3 was often found (80, 81).
Evidence of VDAC3 Involvement in Cancer
In a recent survey with the Ingenuity Pathway Analysis software on VDAC3 interacting proteins, aimed to discover the pathological processes underneath this group of proteins, it was noticed that the processes with the highest score, by far highest than the threshold, were cancer and reproductive system diseases (62).
Evidence for a VDAC3 responsibility in specific mechanisms connected to cancer come from studies with erastin. The anti-tumor agent erastin induces oxidative, non-apoptotic death in human tumor cells with mutations in the oncogenes HRAS, KRAS, or BRAF. Using affinity-based target identification and MS/MS analysis, Yagoda et al. (82) identified VDAC2 and VDAC3 as the docking sites for erastin binding to mitochondria. In response to oncogenic HRAS, the total amount of VDAC protein is increased suggesting that erastin acts by a gain-of-function mechanism and that cells with more VDAC protein are more sensitive to erastin. Following erastin treatment, however, VDAC3 first and subsequently VDAC2 become undetectable. Hence, it is possible to hypothesize that a cellular response to erastin is the downregulation of VDAC2/3 after the generation of lethal oxidative species (Figure 3). On the contrary, VDAC1 is not altered after erastin exposure suggesting that the loss of VDAC2/3 is not simply caused by loss of mitochondria. The K.O. of VDAC3 significantly increases cell resistance to erastin and similar results are reported for VDAC2 K.O. In contrast, overexpression of VDAC3 alone does not increase sensitivity to erastin, suggesting that VDAC3, and partially VDAC2, is necessary but not sufficient to raise sensitivity to erastin, and that other downstream features of RAS–RAF–MEK signaling are necessary. In 2013, Maldonado et al. (83) discovered that erastin prevents and reverses tubulin-induced VDAC blockage to promote mitochondrial metabolism. As already reported, dimeric αβ-tubulin is able to block the conductance of VDAC and suppress mitochondrial respiration (84, 85). Single and double K.O. of VDAC isoforms in HepG2 cells showed that endogenous tubulin inhibits VDAC1 and VDAC2 conductance more than VDAC3 conductance (86). It has been speculated that the interaction between VDAC and tubulin may underlie modulation of energy metabolism in proliferating cells. During interphase, high free tubulin inhibits VDAC fluxes of ATP/ADP and other respiratory substrates across OMM, thereby suppressing mitochondrial respiration and promoting aerobic glycolysis in order to increase biomass formation. The decrease of free tubulin accompanying the cell division, instead, leads to the VDAC opening and the activation of oxidative phosphorylation that in turn generates the energy needed for chromosome movement and cytoplasmic division (86). In the light of the above results, the interaction between free tubulin and VDAC in cancer cells, which frequently show numerous alterations in the microtubule network (87), possibly contributes to the establishment of the Warburg metabolism (86).
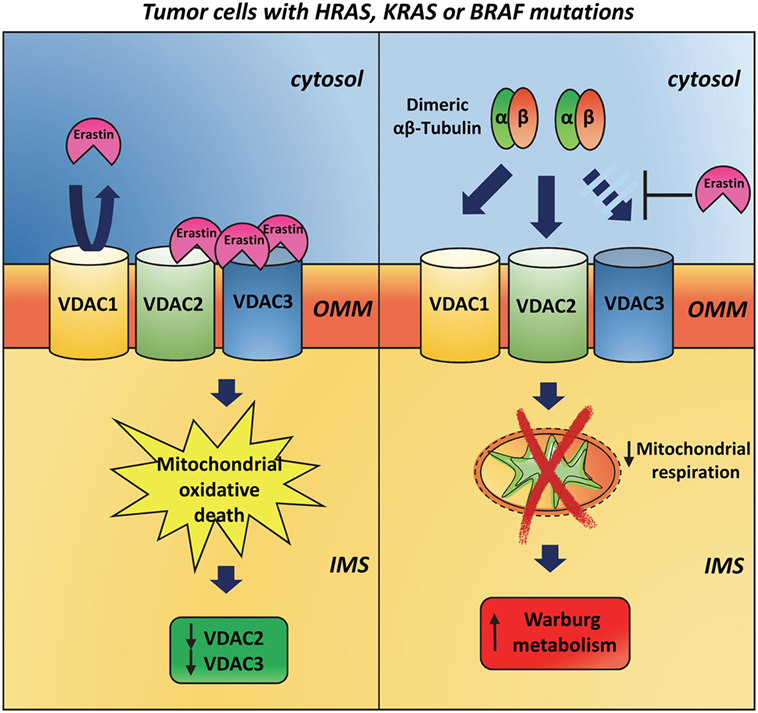
Figure 3. Involvement of VDAC3 in cancer. The anti-tumor agent erastin induces rapid, oxidative, non-apoptotic death in human tumor cells that have mutations in the oncogenes HRAS, KRAS, or BRAF. On the outer mitochondrial membrane, erastin binds VDAC2 and VDAC3 but is not able to interact with VDAC1. After erastin treatment, VDAC2 and VDAC3 expression is strongly reduced, while VDAC1 is still present at later time points (left panel). Dimeric αβ-tubulin is able to inhibit VDAC1 and VDAC2 conductance more than VDAC3 ones. This interaction suppresses mitochondrial respiration and contributes to the establishment of the Warburg metabolism in tumor cells. Erastin prevents and reverses tubulin-induced voltage-dependent anion selective channel blockage and promotes mitochondrial metabolism (right panel).
VDAC3 in Diseases Different from Cancer
Some reports in the literature show the involvement of VDAC3 in pathologies and diseases different from cancer. Chronic unpredictable stress (CUS), one of the most clinically relevant stress paradigms in rodents, mimics several behavioral characteristics of patients affected by depression, anxiety, and mood disorders. In a zebrafish model of CUS, Chakravarty et al. identified VDAC3 as a differentially regulated gene among other proteins involved in mitochondrial function (80). The authors hypothesized that the increased level of VDAC3 in stressed fish brain might increase the efficiency of bioenergetic metabolism and/or protection against ROS (80).
Alterations in the expression levels of VDAC3 have also been reported in the brain of mice infected by Plasmodium berghei, a well-studied mouse model of cerebral malaria (81). Cerebral malaria is the most severe neurological complication of infections by Plasmodium falciparum. This disease, mostly diffused in sub-Saharian Africa, has a high mortality and surviving patients generally manifest long-term neurocognitive impairment. The mechanisms underlying the brain injury are still unclear, and the upregulation of vdac3 is accompanied by alterations of many other genes whose dysfunction is associated with neurological disorders (81).
The Interactions between VDAC3 and Cytoskeletal Proteins are the Basis for Disorganization of Endocellular Functions?
The term “ciliopathies” refers to a series of genetic disorders associated with abnormal formation or function of cilia. The ciliopathies cause multisystem pathologies that can manifest with a plethora of features including retinal degeneration, cerebral anomalies, skeletal dysplasia, and fibrocystic diseases of the liver. It has been reported that VDAC3 is targeted to centrosome where it is mainly associated to the mother centriole/basal body (75). Here, it recruits Mps1, a protein kinase essential for the spindle assembly checkpoint and the modulation of centriole assembly in vertebrates. Depletion of VDAC3 cause defects in centriole assembly and in cell cycle enter. Further evidences revealed that VDAC3 and Mps1 cooperate to promote ciliary disassembly, while VDAC3 might have the additional function to inhibit cilia assembly in cycling cells (76). Interestingly, an earlier study demonstrated that, in VDAC3 K.O. mice, structural defects in the microtubular organization of the sperm tail cause markedly reduced sperm motility (36). As a consequence, male mice lacking VDAC3 are infertile. The molecular relationship between this defect and the VDAC deficiency is not known. It was found that VDAC3 is transcribed at specifically high levels in testis (33). Histochemical staining of bovine testis showed that VDAC1 is present in Sertoli cells and VDAC2 (but not VDAC1) in spermatocytes (88). By means of isoform-specific antibodies and biochemical evidence, it has been found that VDAC2 and VDAC3 are abundant in mature spermatozoa and appear to be associated with outer dense fibers of the sperm flagellum (89). Interestingly, also an association of VDAC isoforms with spermatozoa and ovarian tissue organization was found in Drosophila melanogaster (90, 91). In contrast, the work by Liu et al. assessed that no difference in VDAC3 expression levels exists between normozoospermic fertile donors and infertile patients with idiopathic asthenozoospermia; they instead found a high expression of VDAC2 (92).
It is important to note that human spermatozoa undergo extensive redox regulated signaling since nitric oxide (NO) is involved in sperm motility, capacitation, acrosome reaction, and enhancement of sperm binding to zona pellucida. NO induces S-nitrosylation, a post-translational protein modification that regulates cellular signaling. VDAC3 was identified as a target of S-nitrosylation in spermatozoa (93). Okazaki et al. found that S-nytrosilation activated VDAC3 channel activity by breaking S–S bonds (50).
Implications of VDAC3 in Viral Infections
Many viruses encode proteins targeting to mitochondria. These proteins are able to modulate apoptosis in infected cells. In general, all the anti-apoptotic viral proteins contain mitochondrial targeting sequences that are responsible for inserting the protein in the OMM. In contrast, pro-apoptotic proteins like the HBx antigen from hepatitis B virus (HBV) contain amphipathic α-helices. HBV is a small hepatotropic DNA virus responsible for acute and chronic liver disease worldwide. The chronic viral infection constitutes a risk factor for hepatocellular carcinoma. It can indeed not only evolve to cirrhosis but also impact on cell cycle regulation and tumor suppressor genes alterations. It was reported that in HepG2 and HuH7, two hepatocarcinoma cells lines, HBx colocalizes with VDAC3 in the OMM (94, 95) and causes alterations in mitochondrial membrane potential (ΔΨm) leading to the activation of the transcription factors STAT-3 and NF-κB (96). The activation of these factors is sensitive to antioxidants and to the overexpression of Mn-superoxide dismutase II, suggesting a potential role of ROS in the pathogenesis of the disease. The release of cytochrome c in response to the HBx-induced mitochondrial membrane potential variation is instead a late event, indicating that during the stage of chronic HBV infection, HBx expression may sensitize apoptosis in infected hepatocytes, generating HBV pathogenesis and favoring propagation of the viral particles. Since Bcl-2 family proteins bind to VDAC to regulate the release of cytochrome c, it has been proposed that VDAC3–HBx interaction might induce conformational changes that abolish the modulation of channel activity by Bcl-2. VDAC3 seems to be involved also in HHV-8 infection, an oncogenic human herpes virus that has been identified in all types of Kaposi’s sarcoma. Wang et al. (97) reported indeed that HHV-8 K7 protein binds to mitochondrial VDAC3.
Conclusion
In mammals, VDAC3 has been demonstrated to be subjected to over-oxidation of its exposed cysteines. The oxidative modifications, likely due to the concentration of ROS in the IMS, let us propose that it can be candidate to participate in the molecular mechanisms of identification of damaged mitochondria. Damaged mitochondria characterize the mitochondrial dysfunction, a state of the organelle where the oxidative phosphorylation is not able to produce enough ATP and ROS accumulation is a hallmark. This state must be signaled as soon as possible, to remove the damaged organelle. The VDAC3 cysteine oxidations, which were experimentally proved (41), produce changes of the protein electrostatic map, and consequently changes in its presentation to the other compartments. The peculiar modifications of cysteines thus make VDAC3 a potential counter of ROS load in the IMS. The quest for a messenger able to specifically interact with the oxidized VDAC3 and transfer the message to proper responding apparatus will be the next step of this story.
Author Contributions
AM and FG contributed to the bibliographic survey; SR and VP conceived the review and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by MIUR-PRIN project 2015795S5W_005 to VP and by FIR-UNICT project 2014.
References
1. Gonçalves RP, Buzhynskyy N, Prima V, Sturgis JN, Scheuring S. Supramolecular assembly of VDAC in native mitochondrial outer membranes. J Mol Biol (2007) 369:413–8. doi:10.1016/j.jmb.2007.03.063
2. Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, et al. Tubulin binding blocks mitochondrial voltage dependent anion channel and regulates respiration. Proc Natl Acad Sci U S A (2008) 105:18746–51. doi:10.1073/pnas.0806303105
3. Kusano H, Shimizu S, Koya RC, Fujita H, Kamada S, Kuzumaki N, et al. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene (2000) 19:4807–14. doi:10.1038/sj.onc.1203868
4. Lesnik C, Golani-Armon A, Arava Y. Localized translation near the mitochondrial outer membrane: an update. RNA Biol (2015) 12:801–9. doi:10.1080/15476286.2015.1058686
5. Beutner G, Ruck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett (1996) 396:189–95. doi:10.1016/0014-5793(96)01092-7
6. Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem (2003) 10:1535–51. doi:10.2174/0929867033457269
7. Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J (2004) 377:347–55. doi:10.1042/bj20031465
8. Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem (2008) 19:13482–90. doi:10.1074/jbc.M708216200
9. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev (2007) 87:99–163. doi:10.1152/physrev.00013.2006
10. Benz R. Permeation of hydrophilic solutes through mitochondrial outer membranes: review on mitochondrial porins. Biochim Biophys Acta (1994) 1197:167–96. doi:10.1016/0304-4157(94)90004-3
11. Colombini M, Blachly-Dyson E, Forte M. VDAC, a channel in the outer mitochondrial membrane. Ion Channels (1996) 4:169–202. doi:10.1007/978-1-4899-1775-1_5
12. Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med (2010) 31:227–85. doi:10.1016/j.mam.2010.03.002
13. Bay DC, Hafez M, Young MJ, Court DA. Phylogenetic and coevolutionary analysis of the β-barrel protein family comprised of mitochondrial porin (VDAC) and Tom40. Biochim Biophys Acta (2012) 1818:1502–19. doi:10.1016/j.bbamem.2011.11.027
14. Kayser H, Kratzin HD, Thinnes FP, Götz H, Schmidt WE, Eckart K, et al. Identification of human porins. II. Characterization and primary structure of a 31-lDa porin from human B lymphocytes (Porin 31HL). Biol Chem Hoppe Seyler (1989) 370:1265–78.
15. Rahmani Z, Maunoury C, Siddiqui A. Isolation of a novel human voltage-dependent anion channel gene. J Hum Genet (1998) 6:337–40. doi:10.1038/sj.ejhg.5200198
16. Messina A, Reina S, Guarino F, De Pinto V. VDAC isoforms in mammals. Biochim Biophys Acta (2012) 1818:1466–76. doi:10.1016/j.bbamem.2011.10.005
17. Palmieri F, De Pinto V. Purification and properties of the voltage-dependent anion channel of the outer mitochondrial membrane. J Bioenerg Biomembr (1989) 21:417–25. doi:10.1007/BF00762514
18. Menzel VA, Cassarà MC, Benz R, De Pinto V, Messina A, Cunsolo V, et al. Molecular and functional characterization of VDAC2 purified from mammal spermatozoa. Biosci Rep (2009) 29:351–62. doi:10.1042/BSR20080123
19. Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science (2008) 321:1206–10. doi:10.1126/science.1161302
20. Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, et al. Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci U S A (2008) 105:15370–5. doi:10.1073/pnas.0808115105
21. Ujwal R, Cascio D, Colletierc JP, Fahama S, Zhanga J, Torod L, et al. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci U S A (2008) 105:17742–7. doi:10.1073/pnas.0809634105
22. De Pinto V, Guarino F, Guarnera A, Messina A, Reina S, Tomasello FM, et al. Characterization of human VDAC isoforms: a peculiar function for VDAC3? Biochim Biophys Acta (2010) 1797:1268–75. doi:10.1016/j.bbabio.2010.01.031
23. Amodeo GF, Scorciapino MA, Messina A, De Pinto V, Ceccarelli M. Charged residues distribution modulates selectivity of the open state of human isoforms of the voltage dependent anion-selective channel. PLoS One (2014) 9:e103879. doi:10.1371/journal.pone.0103879
24. Schredelseker J, Paz A, López CJ, Altenbach C, Leung CS, Drexler MK, et al. High resolution structure and double electron-electron resonance of the zebrafish voltage-dependent anion channel 2 reveal an oligomeric population. J Biol Chem (2014) 289:12566–77. doi:10.1074/jbc.M113.497438
25. Schulz GE. Beta-barrel membrane proteins. Curr Opin Struct Biol (2004) 10:443–7. doi:10.1016/S0959-440X(00)00120-2
26. Tomasello FM, Guarino FM, Reina S, Messina A, De Pinto V. The voltage-dependent anion selective channel 1 (VDAC1) topography in the mitochondrial outer membrane as detected in intact cell. PLoS One (2013) 8:e81522. doi:10.1371/journal.pone.0081522
27. De Pinto V, Tomasello F, Messina A, Guarino F, Benz R, La Mendola D, et al. Determination of the conformation of the human VDAC1 N-terminal peptide, a protein moiety essential for the functional properties of the pore. Chembiochem (2007) 8:744–56. doi:10.1002/cbic.200700009
28. Guardiani C, Scorciapino MA, Amodeo GF, Grdadolnik J, Pappalardo G, De Pinto V, et al. The N-terminal peptides of the three human isoforms of the mitochondrial voltage-dependent anion channel have different helical propensities. Biochemistry (2015) 54:5646–56. doi:10.1021/acs.biochem.5b00469
29. Reina S, Palermo V, Guarnera A, Guarino F, Messina A, Mazzoni C, et al. Swapping of the N-terminus of VDAC1 with VDAC3 restores full activity of the channel and confers anti-aging features to the cell. FEBS Lett (2010) 584:2837–44. doi:10.1016/j.febslet.2010.04.066
30. Sampson MJ, Lovell RS, Craigen WJ. The murine voltage-dependent anion channel gene family. Conserved structure and function. J Biol Chem (1997) 272:18966–73.
31. Young MJ, Bay DC, Hausner G, Court DA. The evolutionary history of mitochondrial porins. BMC Evol Biol (2007) 7:31. doi:10.1186/1471-2148-7-31
32. Sampson MJ, Lovell RS, Craigen WJ. Isolation, characterization, and mapping of two mouse mitochondrial voltage-dependent anion channel isoforms. Genomics (1996) 33:283–8. doi:10.1006/geno.1996.0193
33. Sampson MJ, Ross L, Decker WK, Craigen WJ. A novel isoform of the mitochondrial outer membrane protein VDAC3 via alternative splicing of a 3-base exon. Functional characteristics and subcellular localization. J Biol Chem (1998) 273:30482–6.
34. Cesar Mde C, Wilson JE. All three isoforms of the voltage-dependent anion channel (VDAC1, VDAC2, and VDAC3) are present in mitochondria from bovine, rabbit, and rat brain. Arch Biochem Biophys (2004) 422:191–6. doi:10.1016/j.abb.2003.12.030
35. Anflous K, Armstrong DD, Craigen WJ. Altered mitochondrial sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J Biol Chem (2001) 276:1954–60. doi:10.1074/jbc.M006587200
36. Sampson MJ, Decker WK, Beaudet AL, Ruitenbeek W, Armstrong D, Hicks MJ, et al. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J Biol Chem (2001) 276:39206–12. doi:10.1074/jbc.M104724200
37. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol (2007) 9:550–5. doi:10.1038/ncb1575
38. Wu S, Sampson MJ, Decker WK, Craigen WJ. Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration. Biochim Biophys Acta (1999) 1452:68–78. doi:10.1016/S0167-4889(99)00120-2
39. Aram L, Geula S, Arbel N, Shoshan-Barmatz V. VDAC1 cysteine residues: topology and function in channel activity and apoptosis. Biochem J (2010) 427:445–54. doi:10.1042/BJ20091690
40. De Pinto V, Al Jamal JA, Benz R, Palmieri F. Characterization of SH-groups in porin of bovine heart mitochondria: porin cysteines are localized in the channel walls. Eur J Biochem (1991) 202:903–11. doi:10.1111/j.1432-1033.1991.tb16450.x
41. Reina S, Checchetto V, Saletti R, Gupta A, Chaturvedi D, Guardiani C, et al. VDAC3 as a sensor of oxidative state of the intermembrane space of mitochondria: the putative role of cysteine residue modifications. Oncotarget (2016) 7:2249–68. doi:10.18632/oncotarget.6850
42. Riemer J, Bulleid N, Herrmann JM. Disulfide formation in the ER and mitochondria: two solutions to a common process. Science (2009) 324:1284–7. doi:10.1126/science.1170653
43. Hu J, Dong L, Outten CE. The redox environment in the mitochondrial intermembrane space is maintained separately from the cytosol and matrix. J Biol Chem (2008) 283:29126–34. doi:10.1074/jbc.M803028200
44. O-Uchi J, Ryu SY, Jhun BS, Hurst S, Sheu SS. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid Redox Signal (2014) 21:987–1006. doi:10.1089/ars.2013.5681
45. Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol (2014) 73:34–42. doi:10.1016/j.yjmcc.2013.12.032
46. Bleier L, Wittig I, Heide H, Steger M, Brandt U, Dröse S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic Biol Med (2015) 78:1–10. doi:10.1016/j.freeradbiomed.2014.10.511
47. Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem (2003) 278:5557–63. doi:10.1074/jbc.M210269200
48. Sevilla F, Camejo D, Ortiz-Espín A, Calderón A, Lázaro JJ, Jiménez A. The thioredoxin/peroxiredoxin/sulfiredoxin system: current overview on its redox function in plants and regulation by reactive oxygen and nitrogen species. J Exp Bot (2015) 66:2945–55. doi:10.1093/jxb/erv146
49. Iglesias-Baena I, Barranco-Medina S, Sevilla F, Làzaro JJ. The dual-targeted plant sulfiredoxin retroreduces the sulfinic form of atypical mitochondrial peroxiredoxin. Plant Physiol (2011) 155:944–55. doi:10.1104/pp.110.166504
50. Okazaki M, Kurabayashi K, Asanuma M, Saito Y, Dodo K, Sodeoka M. VDAC3 gating is activated by suppression of disulfide-bond formation between the N-terminal region and the bottom of the pore. Biochim Biophys Acta (2015) 1848:3188–96. doi:10.1016/j.bbamem.2015.09.017
51. Checchetto V, Reina S, Magrì A, Szabo I, De Pinto V. Recombinant human voltage dependent anion selective channel isoform 3 (hVDAC3) forms pores with a very small conductance. Cell Physiol Biochem (2014) 34:842–53. doi:10.1159/000363047
52. Guardiani C, Leggio L, Scorciapino MA, De Pinto V, Ceccarelli M. A computational study of ion current modulation in hVDAC3 induced by disulfide bonds. Biochim Biophys Acta (2016) 1858:813–23. doi:10.1016/j.bbamem.2016.01.013
53. Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol (2008) 12:746–54. doi:10.1016/j.cbpa.2008.07.028
54. De Pinto V, Reina S, Gupta A, Messina A, Mahalakshmi R. Role of cysteines in mammalian VDAC isoforms’ function. Biochim Biophys Acta (2016) 1857:1219–27. doi:10.1016/j.bbabio.2016.02.020
55. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J (2014) 33:2142–56. doi:10.15252/embj.201488104
56. Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One (2012) 7:e52830. doi:10.1371/journal.pone.0052830
57. Eldeeb M, Fahlman R. The-N-end rule: the beginning determines the end. Protein Pept Lett (2016) 23:343–8. doi:10.2174/0929866523666160108115809
58. Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem (2012) 287:40652–60. doi:10.1074/jbc.M112.419721
59. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron (2015) 85:257–73. doi:10.1016/j.neuron.2014.12.007
60. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature (2007) 447:725–9. doi:10.1038/nature05862
61. Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, et al. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science (2015) 348:340–3. doi:10.1126/science.1260384
62. Messina A, Reina S, Guarino F, Magrì A, Tomasello F, Clark RE, et al. Live cell interactome of the human voltage dependent anion channel 3 (VDAC3) revealed in HeLa cells by affinity purification tag technique. Mol Biosyst (2014) 10:2134–45. doi:10.1039/c4mb00237g
63. Raturiand A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria associated membrane (MAM). Biochim Biophys Acta (2013) 1833:213–24. doi:10.1016/j.bbamcr.2012.04.013
64. Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol (2011) 23:476–82. doi:10.1016/j.ceb.2011.05.007
65. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahleand PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol (2010) 12:119–31. doi:10.1038/ncb2012
66. Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol (2007) 3:89. doi:10.1038/msb4100134
67. Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol (2014) 2:123–39. doi:10.1016/j.redox.2013.12.011
68. Bernales B, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during unfolded protein response. PLoS Biol (2006) 4:e423. doi:10.1371/journal.pbio.0040423
69. Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harbor Perspect Biol (2013) 5:a013185. doi:10.1101/cshperspect.a013185
70. Xu S, Peng G, Wang Y, Fang S, Karbowski M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol Biol Cell (2011) 22:291–300. doi:10.1091/mbc.E10-09-0748
71. Huizing M, Ruitenbeek W, Thinnes F, De Pinto V, Wendel U, Trijbels JMF, et al. Deficiency of the voltage-dependent anion channel: clinical and biochemical aspects of a new mitochondriopathy. Ped Research (1996) 39:1–6. doi:10.1203/00006450-199605000-00003
72. Smilansky A, Dangoor L, Nakdimon I, Ben-Hail D, Mizrachi D, Shoshan-Barmatz V. The voltage-dependent anion channel 1 mediates amyloid β toxicity and represents a potential target for alzheimer disease therapy. J Biol Chem (2015) 290:30670–83. doi:10.1074/jbc.M115.691493
73. Rostovtseva TK, Gurnev PA, Protchenko O, Hoogerheide DP, Yap TL, Philpott CC, et al. α-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J Biol Chem (2015) 290:18467–77. doi:10.1074/jbc.M115.641746
74. Magrì A, Belfiore R, Reina S, Tomasello MF, Di Rosa MC, Guarino F, et al. Hexokinase I N-terminal based peptide prevents the VDAC1-SOD1 G93A interaction and re-establishes ALS cell viability. Sci Rep (2016) 6:34802. doi:10.1038/srep34802
75. Majumder S, Slabodnick M, Pike A, Marquardt J, Fisk HA. VDAC3 regulates centriole assembly by targeting Mps1 to centrosomes. Cell Cycle (2012) 11:3666–78. doi:10.4161/cc.21927
76. Majumder S, Fisk HA. VDAC3 and Mps1 negatively regulate ciliogenesis. Cell Cycle (2013) 12:849–58. doi:10.4161/cc.23824
77. Huizing M, Ruitenbeek W, Thinnes FP, De Pinto V. Lack of voltage-dependent anion channel in human mitochondrial myopathies. Lancet (1994) 344:762. doi:10.1016/S0140-6736(94)92257-8
78. De Pinto V, Messina A, Schmid A, Simonetti S, Carnevale F, Benz R. Characterization of channel-forming activity in muscle biopsy from a porin-deficient human patient. J Bioenerg Biomembr (2000) 32:585–93. doi:10.1023/A:1005622611410
79. Huizing M, De Pinto V, Ruitenbeek W, Trijbels JMF, van den Heuvel LP, Wendel U. Importance of mitochondrial transmembrane processes in human mitochondriopathies. J Bioenerg Biomem (1996) 28:107–12.
80. Chakravarty S, Reddy BR, Sudhakar SR, Saxena S, Das T, Meghah V, et al. Chronic unpredictable stress (CUS)-induced anxiety and related mood disorders in a zebrafish model: altered brain proteome profile implicates mitochondrial dysfunction. PLoS One (2013) 8:e63302. doi:10.1371/journal.pone.0063302
81. Desruisseaux MS, Iacobas DA, Iacobas S, Mukherjee S, Weiss LM, Tanowitz HB, et al. Alteration in the brain transcriptome in Plasmodium berghei ANKA infected mice. J Neuroparasitol (2010) 1:N100803. doi:10.4303/jnp/N100803
82. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447:864–8. doi:10.1038/nature05859
83. Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM, et al. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem (2013) 288:11920–9. doi:10.1074/jbc.M112.433847
84. Gurnev PA, Rostovtseva TK, Bezrukov SM. Tubulin-blocked state of VDAC studied by polymer and ATP partitioning. FEBS Lett (2011) 585:2363–6. doi:10.1016/j.febslet.2011.06.008
85. Rostovtseva TK, Bezrukov SM. VDAC inhibition by tubulin and its physiological implications. Biochim Biophys Acta (2012) 1818:1526–35. doi:10.1016/j.bbamem.2011.11.004
86. Maldonado EN, Lemasters JJ. Warburg revisited: regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J Pharmacol Exp Ther (2012) 342:637–41. doi:10.1124/jpet.112.192153
87. Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front Oncol (2014) 4:153. doi:10.3389/fonc.2014.00153
88. Hinsch KD, Asmarinah, Hinsch E, Konrad L. VDAC2 (porin-2) expression pattern and localization in the bovine testis. Biochim Biophys Acta (2001) 1518:329–33. doi:10.1016/S0167-4781(01)00199-3
89. Hinsch KD, De Pinto V, Aires VA, Schneider X, Messina A, Hinsch E. Voltage-dependent anion-selective channels VDAC2 and VDAC3 are abundant proteins in bovine outer dense fibers, a cytoskeletal component of the sperm flagellum. J Biol Chem (2004) 279:15281–8. doi:10.1074/jbc.M313433200
90. De Pinto V, Benz R, Caggese C, Palmieri F. Characterization of the mitochondrial porin from Drosophila melanogaster. Biochim Biophys Acta (1989) 987:1–7. doi:10.1016/0005-2736(89)90447-1
91. Guarino F, Specchia V, Zapparoli G, Messina A, Aiello R, Bozzetti MP, et al. Expression and localization in spermatozoa of the mitochondrial porin isoform 2 in Drosophila melanogaster. Biochem Biophys Res Commun (2006) 346:665–70. doi:10.1016/j.bbrc.2006.05.172
92. Liu B, Wang P, Wang Z, Jia Y, Niu X, Wang W, et al. Analysis and difference of voltage-dependent anion channel mRNA in ejaculated spermatozoa from normozoospermic fertile donors and infertile patients with idiopathic asthenozoospermia. J Assist Reprod Genet (2010) 27:719–24. doi:10.1007/s10815-010-9466-8
93. Lefièvre L, Chen Y, Conner SJ, Scott JL, Publicover SJ, Ford WC, et al. Human spermatozoa contain multiple targets for protein S-nitrosylation: an alternative mechanism of the modulation of sperm function by nitric oxide? Proteomics (2007) 7:3066–84. doi:10.1002/pmic.200700254
94. Avantaggiati ML, Natoli G, Balsano C, Chirillo P, Artini M, De Marzio E, et al. The hepatitis B virus (HBV) pX transactivates the c-fos promoter through multiple cis-acting elements. Oncogene (1993) 8:1567–74.
95. Rahmani Z, Huh KW, Lasher R, Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol (2000) 74:2840–6.
96. Gulam W, Huh K, Siddiqui A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-κB via oxidative stress. Mol Cell Biol (2001) 21:7721–30. doi:10.1128/MCB.21.22.7721-7730.2001
Keywords: VDAC3, mitochondria quality control, cancer, cysteine over-oxidation, aging, mitophagy, mitochondrial dysfunction
Citation: Reina S, Guarino F, Magrì A and De Pinto V (2016) VDAC3 As a Potential Marker of Mitochondrial Status Is Involved in Cancer and Pathology. Front. Oncol. 6:264. doi: 10.3389/fonc.2016.00264
Received: 27 October 2016; Accepted: 09 December 2016;
Published: 23 December 2016
Edited by:
Hanna Kmita, Adam Mickiewicz University in Poznań, PolandReviewed by:
Catherine Brenner Jan, INSERM, FranceFabrice Homblé, Université Libre de Bruxelles, Belgium
Copyright: © 2016 Reina, Guarino, Magrì and De Pinto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Reina, c2ltb25hcmVpbmFAeWFob28uaXQ=;
Vito De Pinto, dmRwYmlvZmFAdW5pY3QuaXQ=