 Cora A. Ricker
Cora A. Ricker Yuan Pan2†
Yuan Pan2† Charles Keller
Charles Keller
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 20 December 2016
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 6 - 2016 | https://doi.org/10.3389/fonc.2016.00259
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder that results from germline mutations of the NF1 gene, creating a predisposition to low-grade gliomas (LGGs; pilocytic astrocytoma) in young children. Insufficient data and resources represent major challenges to identifying the best possible drug therapies for children with this tumor. Herein, we summarize the currently available cell lines, genetically engineered mouse models, and therapeutic targets for these LGGs. Conspicuously absent are human tumor-derived cell lines or patient-derived xenograft models for NF1-LGG. New collaborative initiatives between patients and their families, research groups, and pharmaceutical companies are needed to create transformative resources and broaden the knowledge base relevant to identifying cooperating genetic drivers and possible drug therapeutics for this common pediatric brain tumor.
Neurofibromatosis type 1 (NF1) predisposes children to the low-grade glioma (LGG), pilocytic astrocytoma (PA). While these brain tumors are slow growing, the locations in which PAs arise make them difficult to surgically excise – often devastating to a child‘s sight or neurological function. Furthermore, no cell lines or xenograft models exist from which to develop new targeted therapies, leading to a sole reliance on transgenic models for mechanistic insights. Increased collaborations with affected patients and their families may hold the key to building the necessary resources to unravel biology and new therapies for this cancer.
Low-grade gliomas account for 30% of primary pediatric central nervous system tumors, with PA predominating in children younger than 15 years of age (1). As a group, LGGs encompass both World Health Organization (WHO) grade I and II gliomas (2). Histologically, these tumors have low proliferative indices (<4%) with rare or absent mitotic figures and no evidence of necrosis. While distinguishing between these two malignancy grades can be challenging in some pediatric LGGs, PAs, unlike their WHO II counterparts, have characteristic eosinophilic granular bodies and Rosenthal fibers. In addition, PAs often contain a cystic component, especially when occurring in the cerebellum, and frequently harbor areas of compacted bipolar cells alternating with loose-textured multipolar cells and microcysts. Many PAs may also enhance, especially in their peripheral rim if a cystic component exists. Enhancement does not denote high-grade glioma (HGG) in this case. Also, intense meningeal enhancement may occur in the absence of a cystic component. Cystic components do not usually exist in optic nerve gliomas, but do exist often in PAs. Similar to other glial malignancies, these pediatric LGGs are immunopositive for expression of glial fibrillary acidic protein (GFAP) and Olig2. Finally, these tumors harbor a rich extracellular matrix with prominent infiltration of monocytes.
In general, children with PA can be divided into distinct subgroups based on their molecular etiologies. First, the majority of children with LGGs lack a genetic predisposition to brain cancer (sporadic PA). Pioneering genomic sequencing efforts by the Pfister laboratory and colleagues identified that the majority of sporadic PA tumors are caused by a somatic genomic rearrangement of the KIAA1549 and BRAF genes to result in a fusion protein containing an unregulated and active BRAF kinase domain (3, 4). BRAFV600E mutations are most likely found in the extracerebellar and diencephalic regions (2). Subsequent studies revealed that this signature genomic alteration predominates in cerebellar and optic pathway PAs (5, 6). In addition to the KIAA1549:BRAF fusion, mutations in the fibroblast growth factor receptor 1 (FGFR1) and neurotrophic tyrosine kinase receptor 2 (NTRK2) genes are (more) common in non-cerebellar PAs (7).
The second group of children with PA includes those who harbor a germline mutation in the Neurofibromin (NF1) tumor suppressor gene and, therefore, have NF1 as the genetic etiology for their brain tumors. Examination of these tumors reveals somatic loss of the remaining NF1 allele, resulting in bi-allelic NF1 inactivation (8). Laboratory-based research over the past 20 years has revealed several potential opportunities for targeted inhibition of the growth control pathways deregulated in sporadic and NF1-associated PAs. The NF1 gene encodes neurofibromin, a protein that primarily functions as a negative regulator of the RAS proto-oncogene. In this regard, loss of NF1 gene expression leads to increased RAS activation and hyperactivation of the downstream RAS effectors, including the RAF/MEK/ERK and the PI3K/AKT (4) pathways, thereafter converging on the mechanistic target of rapamycin (mTOR) complex (9). Similarly, the KIAA1549:BRAF mutation results in increased MEK activation (10), which also operates to control cell growth through the mTOR complex (11). While less is known about the downstream signaling pathways operative in FGFR1- and NTRK2-mutant PAs, these receptor tyrosine kinase molecules are also known to activate RAS and RAS downstream signaling and regulate growth in numerous other cancers (12–16). These insights have resulted in the execution of early-phase clinical trials of MEK and mTOR inhibitors (NCT02285439 and NCT01734512; http://clinicaltrials.org), yet it is clear that additional resources and research will be required in order to develop personalized and effective therapies for these brain tumors.
Clinically, children with cerebellar PA come to medical attention when they present with signs and symptoms that reflect raised intracranial pressure, such as headaches, nausea/vomiting, blurred vision, and balance problems. Children with PAs involving the brainstem may exhibit reduced appetite, cranial neuropathies, and drowsiness, while those with optic pathway gliomas (OPGs) may have reduced visual acuity or early-onset puberty when affecting the hypothalamus. For this reason, accurate age-appropriate visual assessments are critical, especially for children with NF1, who are most at risk for the development of OPGs.
Therapeutically, individuals with sporadic PAs typically undergo surgical resection of the tumor, when feasible. This approach is most often employed for those tumors located within the cerebellum, but not for midline or optic pathway tumors. In these situations, chemotherapy (carboplatin/vincristine) is employed to halt further tumor growth. However, allergic reactions have been reported from 2 to 30% of children with LGG (17). Risk of an allergic reaction from carboplatin and increased peripheral neuropathy from vincristine has led to vincristine being the first- or second-line monotherapy of choice (18–20). Since NF1-associated PAs are most commonly located in the optic pathway (optic nerve, chiasm, tracts, and radiations, Figure 1), surgery is rarely performed, and most children with symptomatic tumors are treated with chemotherapy in the absence of a tissue diagnosis unless patients demonstrate atypical symptoms and a biopsy is deemed unnecessary. Alkylator-based chemotherapy is typically avoided due to the risk of treatment-induced malignancy. Importantly, radiation therapy is rarely ever used due to the elevated risk of radiation-induced vasculopathy (21). With carboplatin/vincristine combination therapy, 56% (44/78) children with LGG exhibit an objective response, with two-year progression-free survival rates approaching 80% (22). In another study examining the combination of vincristine and carboplatin, 52% (12/23) of children with recurrent LGG had an objective response, and seven of these children showed tumor reductions greater than 50% (23). Although outcomes are far from optimal, approximately one-third of patients who have received chemotherapy demonstrated improvement of vision (almost all being carboplatin-based within the study). These responses speak to the pressing need to develop targeted therapeutic agents for these tumors that inhibit the specific growth pathways deregulated in NF1-associated tumors.
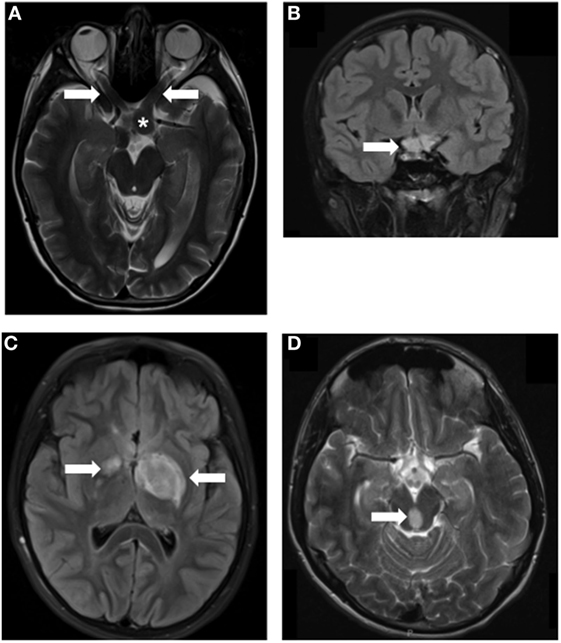
Figure 1. MRI images of Neurofibromatosis type 1 (NF1)-associated low-grade glioma (LGG). LGGs (arrows) developing in children with NF1 at various locations, including optic nerves (A), asterisk indicates the chiasm, chiasm (B), thalamus (C), and brainstem (D). Arrows denote the location of the tumors.
A total of 15–20% of children with NF1 develop LGGs (24–26), affecting 1:2,500 individuals worldwide. Over two-thirds of these PAs arise in the optic pathway (optic nerves, chiasm, tracts, and radiations), with another 15% located in the brainstem (27). Although rare, NF1 is one of the most common genetic disorders in humans, and PAs, when included in the LGG category, are the most common pediatric brain tumors. Thus, developing therapies for NF1-associated PAs could have a major impact on LGGs.
Patient registries provide important information regarding demographic and clinical characteristics. Currently, there are several registries for brain tumors and others, specifically for NF1. Dr. Michael Fisher at the Children‘s Hospital of Philadelphia leads a large international study group for NF1-OPG aimed at defining the natural history and outcomes for these tumors. In 2012, the Children‘s Tumor Foundation (CTF) created the NF Registry. The NF Registry is a tissue repository managed by Precision Bioservices and curated by Patients Crossroads Inc. The NF registry notifies patients of new clinical trials and possible treatments, allows patients to compare medical histories, and enables patients to connect with researchers on pressing issues.
Additionally, the Washington University Neurofibromatosis (NF) Center created (May 2011) and currently hosts the Neurofibromatosis Type 1 (NF1) Patient Registry Initiative (28). This epidemiologic database serves a different purpose than the CTF Registry. It was specifically designed to facilitate clinical association studies that can be mechanistically explored in the laboratory. As such, this registry has been invaluable for assessing how internet-based epidemiological research can be optimally conducted (29, 30) and for identifying unanticipated associations between specific clinical features for future risk assessment analysis (31–33).
Although not specific to NF1, the National Cancer Data Base is a program established by the American College of Surgeons in collaboration with the American Cancer Society to provide oncology results from Commission on Cancer (CoC)-accredited hospital registries. In addition, the Central Brain Tumor Registry of the United States provides an invaluable resource for descriptive statistical data on all primary tumors, while the Swedish Cancer Registry houses similar data on LGG and HGG (34). Several other cancer registries, which track LGGs from around the world, are summarized in Table 1.

Table 1. International Cancer Registries for Low-Grade Astrocytoma.
Finally, a number of institutions and laboratories, in close collaboration with neurosurgical teams, are pioneering the creation of LGG tumor banks. For example, the Pediatric Low-Grade Astrocytoma Program at the Dana Farber Cancer Institute has invested in a tissue banking research effort to abstract data of each tumor and distribute the frozen tissue to investigators. Similar biospecimen repositories also exist at Johns Hopkins University, St. Louis Children‘s Hospital/Washington University, Children‘s National Medical Center, and St. Jude Children‘s Research Hospital. Nevertheless, the general lack of frozen tissue samples has hindered comprehensive analyses of cooperating molecular changes in NF1-associated PA. The lack of surgical specimens reflects the current practice of treating these tumors without a tissue diagnosis because current clinical practices exclude giving patients a biopsy unless they present clear abnormal clinical or radiological symptoms. However, by pooling these rare biopsy materials from a large number of institutions, programs like the NF1 Synodos LGG Initiative, spearheaded by the Children‘s Tumor Foundation, aim to perform comprehensive genetic and genomic sequencing of these tumors.
Cell lines are typically derived from patients or animal models. Establishing cell lines from patient tumors would be ideal for testing preclinical drugs because these lines closely resemble actual human tumors and may more closely mirror the response of a particular patient‘s tumor to treatment. Unfortunately, despite efforts by many research groups, human NF1-PA cell cultures have yet to be successfully established. It is not clear whether this reflects issues with cellular senescence or the need for trophic growth factors from the normal microenvironment. As such, continual passage of sporadic tumors is associated with loss of the signature fusion BRAF alteration (KIAA1549:BRAF) and oncogene-induced senescence (35). To date, only a small number of PA cell lines and their genetic characteristics have been reported, which are summarized in Table 2, including NCH492 (BRAF duplication (4)), IPNT-H (36), and Res186 (PTEN deletion and TP73 promoter hypermethylation (37, 38)). However, none of the LGG cell lines have reported NF1 gene loss/mutation. Further characterization of the existing sporadic LGG cell lines as well as the development of NF1-LGG cell lines are needed.
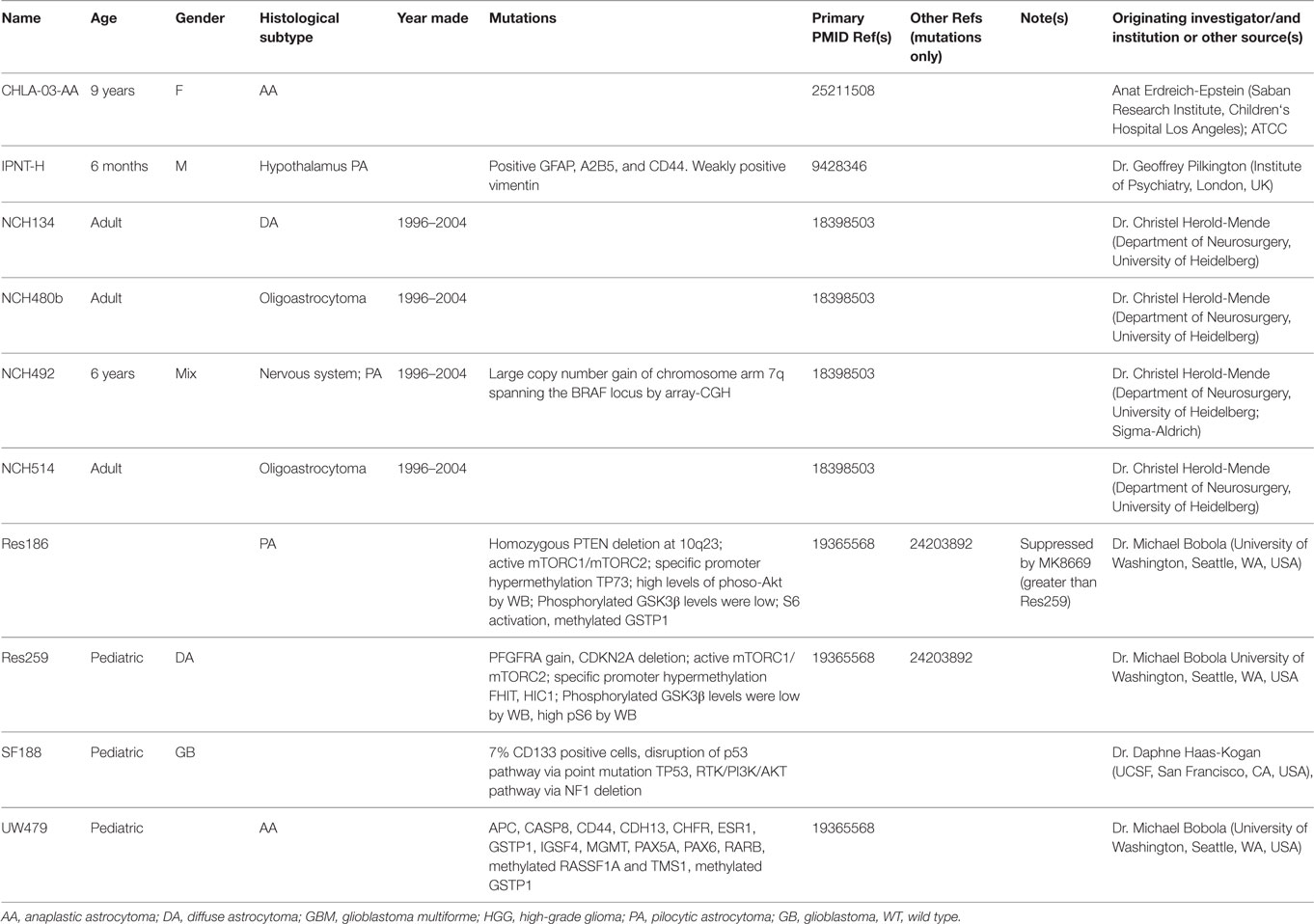
Table 2. Demographic and biological features of glioma cell lines.
At the Washington University NF Center, investigators launched the NF1 Brain Trust Project in October 2012 to create a repository of NF1 patient-induced pluripotent stem cell (iPSC) lines. While these are not glioma cells, they provide a renewable source of human cells for cellular reprograming. Importantly, these iPSC lines were instrumental in demonstrating that different germline NF1 gene mutations result in different levels of neurofibromin expression (39). Current work is underway to use these lines as cellular substrates for glioma modeling, as part of the CTF NF1 Synodos Initiative.
In the absence of patient-derived cell lines or xenografts, genetically engineered mice (GEM), listed in Table 3, have emerged as instructive models for elucidating the cellular and molecular determinants critical for glioma formation and growth. While mice with a germline Nf1 gene mutation (Nf1+/− mice) do not develop gliomas, neither do mice in which both copies of the Nf1 gene are inactivated in neuroglial progenitors (40), suggesting that bi-allelic Nf1 gene inactivation in the proper cell of origin is not sufficient for glioma formation. To better model NF1-PA arising in children with a germline NF1 gene mutation, Nf1+/− mice were generated in which somatic loss of the remaining functional Nf1 allele occurs in neuroglial progenitors (40–42). Analysis of the resulting mice revealed that gliomas arise between 10 and 12 weeks of age in the prechiasmatic optic nerves and chiasm with >90% penetrance. While these LGGs lack the characteristic eosinophilic granule bodies and Rosenthal fibers commonly found in NF1-PA, the murine optic glioma tumors share several similarities with their human counterparts. These tumors display frank tissue distortion evident on MRI, low proliferative indices, increased microglia infiltration, and axonal damage, which culminate in retinal ganglion cell (RGC) loss and reduced visual acuity (40, 43–45).
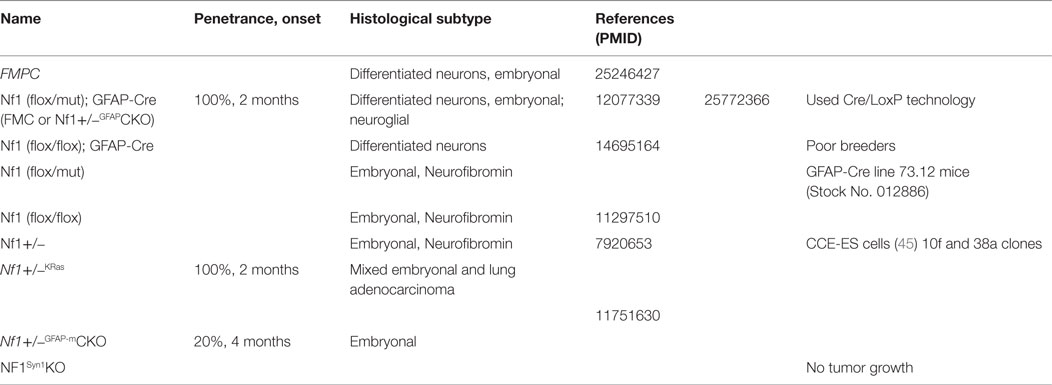
Table 3. Transgenic Mouse Models.
Recent studies have also discovered co-existing genetic changes in NF1-PA in addition to NF1 gene mutation, including heterozygous PTEN deletion and KIAA1549:BRAF duplication (46). Based on these observations, two Nf1 optic glioma GEM models were generated that additionally harbored either a heterozygous Pten deletion or KIAA1549:BRAF overexpression. While KIAA1549:BRAF overexpression did not provide an additional growth advantage, tumors from mice with a co-existing heterozygous Pten deletion had larger volumes, increased proliferation indices, and more microglia infiltration. Furthermore, inhibition of Nf1+/− non-neoplastic stromal cells (microglia) significantly decreases optic glioma growth and maintenance (47). These preclinical mouse models suggest that additional genetic changes in NF1-associated PA patients differentially influence tumor growth relevant to the design of future therapeutic strategies.
Whereas xenografts of human tumors maintained in immunocompromised mice have been regularly employed to predict the efficacy of candidate drugs, most of these xenografts derive from high-grade (malignant) cancers. In contrast, generating neoplastic cells from PA tumors has been challenging due to their low clonogenic nature and the requirement for a permissive tumor microenvironment. As presented in Table 4, only a few patient-derived xenograft (PDX) models of sporadic PA have been reported. Two such models, BT-35 and BT-40, have been used in a preclinical study of the selumetinib MEK inhibitor (48). BT-35 and BT-40 express WT and mutant BRAF (BRAFV600E; constitutively active BRAF), respectively. While BT-35 xenografts continued to grow in the presence of selumetinib, BT-40 xenografts are highly sensitive to MEK inhibition. To date, no PDX models exist for NF1-PA.

Table 4. Xenografts.
To develop robust human HGG PDX models, several groups have derived cancer stem cells (CSCs) as in vitro and transplantable in vivo platforms for drug discovery and evaluation (49–51). Similar to their non-neoplastic counterparts, CSCs are capable of self-renewal and multilineage differentiation (52–54). Unfortunately, the CSCs generated from PA tumors have clonogenic frequencies between 0.3 and 1.5% and often undergo oncogene-induced senescence (55). As such, no PA CSC models have been developed from human specimens.
Using GEM, the process of establishing and maintaining human NF1-PA CSC xenografts can be optimized. Using Nf1 optic glioma GEM models, LGG CSCs have been isolated, which were capable of self-renewal, long-term passage, and multilineage differentiation (56). Importantly, these CSCs formed LGG-like lesions when injected into the brainstems of immunocompetent, but not immunocompromised (athymic), mice. This latter finding underscores the obligate role of the tumor microenvironment in Nf1 murine low-grade gliomagenesis. Further work using these CSCs may reveal the critical components of the tumor microenvironment required for CSC engraftment, which might be accurately recapitulated in rodents to enable xenografting of human NF1-PA tumors in the future.
Neurofibromin functions primarily as a suppressor of RAS and its downstream signaling pathways (Figure 2) (57–62). Neurofibromin contains a GAP (GTPase-activating protein)-related domain (GRD) that accelerates the GTP hydrolysis of RAS, leading to RAS inactivation (63). PI3K/AKT and RAF/MEK/ERK are two common pathways downstream of RAS. Hyperactivation of the PI3K/AKT and MEK/ERK pathways are observed in Nf1-deficient neural stem cells (NSCs) (64) and astrocytes (65). While each RAS effector pathway has different functions in NSCs relative to proliferation and glial differentiation, both the PI3K/AKT and MEK/ERK pathways are hyperactivated in Nf1−/− astrocytes relative to their wild-type counterparts (62, 66–69). Importantly, these downstream effectors converge on the mTOR complex to regulate astrocyte growth in vitro and Nf1 optic glioma proliferation in vivo (9). Future laboratory investigations will be required to define the RAS effector signaling networks required for optic glioma maintenance relevant to adaptive and potential escape mechanisms from targeted therapies that might be operative in these tumors (56).
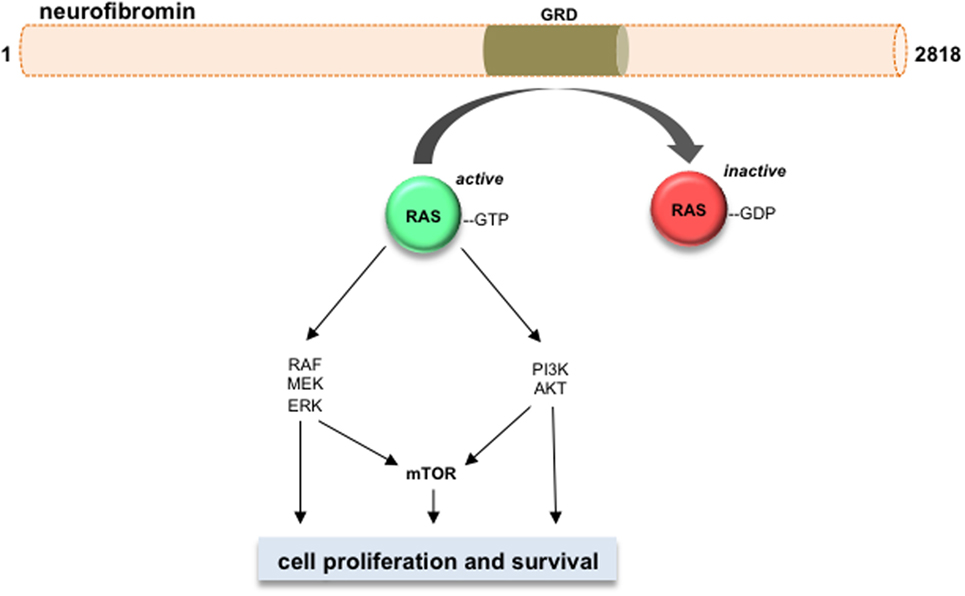
Figure 2. Neurofibromin is a negative regulator of RAS and its downstream effectors. Neurofibromin protein (2818 amino acids) contains a GAP-related domain (GRD) domain, which functions to inhibit RAS activity through accelerating RAS-GTP to RAS-GDP hydrolysis. The RAS downstream effector pathways are shown, including RAF/MEK/ERK, PI3K/AKT, and mTOR. Activation of these pathways increases cell growth and/or cell survival in Nf1-deficient neoplastic cells.
Evidence from several different experimental studies has revealed a critical role for the tumor microenvironment in murine Nf1 optic glioma formation and growth. Similar to other LGG systems, the tumor microenvironment of Nf1 optic glioma consists of neoplastic cells (glioma cells and CSCs) and non-neoplastic stromal cells (neurons, endothelial cells, and monocytes) (Figure 3). Increasing evidence suggests that neoplastic cells secrete paracrine factors (e.g., chemokines) to recruit stromal cells and stimulate growth factor/cytokine production from these cells. The growth factors then interact with their cognate receptors on the cell surface of neoplastic cells to increase cell proliferation. The roles and interactions of these cells within the microenvironment of NF1-PA remain to be completely elucidated. To date, the importance of microglia to PA pathogenesis is underscored by the finding that 35–50% of the cells in these low-grade tumors are Iba1+ tissue macrophages (monocytes) (70). Monocytes in the brain can represent either resident tissue macrophages (microglia) that enter the brain during mid-embryonic development or peripheral macrophages from the bone marrow (71). In mouse Nf1 optic gliomas, the monocytes are Cd11b+, Cd45low, Cx3cr1+ microglia (70). These microglia are critical for gliomagenesis, such that impairing their directional migration into the tumor by genetic reduction of the Cx3cl1 receptor (Cx3cr1) delays optic glioma formation (72). Moreover, pharmacologic inhibition of microglia function with minocycline (73) or c-Jun-NH2-kinase (JNK) inhibitors (74) reduces optic glioma proliferation. In addition, genetic elimination of microglia using transgenic mice expressing the CD11b-thymidine kinase (TK) transgene results in dramatic reduction in optic glioma proliferation (70). Collectively, these observations establish that microglia in the tumor microenvironment produce critical growth factors that increase optic glioma growth.
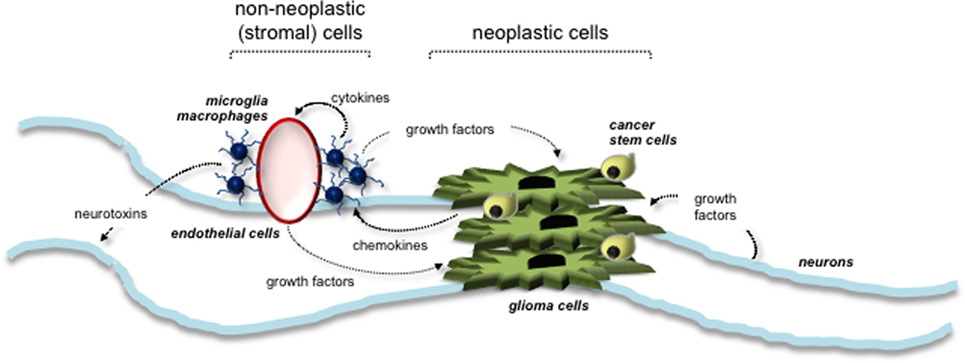
Figure 3. Tumor microenvironment stimulates the growth of NF1-associated PA. Interactions between neoplastic cells (glioma cells and cancer stem cells) and non-neoplastic (stromal) cells are depicted. Stromal cells within the tumor environment include neurons, microglia, macrophages, and endothelial cells. Arrows indicate the paracrine factors (e.g., chemokines and growth factors) secreted by each cell type and their responsive cells.
To identify potential growth factors elaborated by glioma-associated microglia, RNA-sequencing of murine optic glioma-associated microglia has revealed that the Ccl5 chemokine produced by tumor-associated microglia is a potent stromal factor important for optic glioma maintenance in mice (47). In this regard, Ccl5 was sufficient to increase Nf1-deficient astrocytes growth in vitro, and its inhibition using neutralizing antibodies reduced optic glioma proliferation in vivo. Current studies are underway to identify additional chemokines and growth factors for potential stroma-directed glioma therapies.
While 15–20% of NF1 patients develop OPGs, 30–50% will experience impaired visual acuity. Since surgery is rarely employed and radiation is associated with the development of secondary malignancy (75, 76), the standard of care entails the use of chemotherapy. One of the applications of mouse Nf1 optic glioma models is to define the mechanisms underlying tumor-associated visual decline. As such, Nf1 optic glioma mice develop a time-dependent sequence of events, beginning with axonal injury at the tumor site, followed by axonal degeneration, RGC loss (apoptosis), retinal nerve fiber layer thinning, and reduced visual acuity (44, 77).
Neurofibromin plays critical roles in protecting neurons from degeneration and cell death. As such, RGCs harboring a germline Nf1 gene mutation (Nf1+/−) have smaller growth cone areas, shorter neurite lengths, and increased cell death compared to wild-type RGCs (78). These abnormal cellular phenotypes are due to impaired neurofibromin regulation of cyclic AMP (cAMP) generation. In these neurons, reduced neurofibromin expression results in lower cAMP levels, such that elevating cAMP levels using pharmacological methods increases the growth cone areas and neurite lengths of Nf1+/− RGCs in vitro. In addition, elevating cAMP levels greatly attenuates RGC cell death in Nf1 optic glioma mice in vivo (78). These observations raise the potential of future neuroprotective strategies aimed at reducing RGC death.
With respect to NF1-LGG, selumetinib, a MEK inhibitor that inhibits the growth of a BRAFV600E-expressing PA xenograft (48) and blocks growth in both Nf1 optic glioma (9) and KIAA1549:BRAF-driven NSC glioma (11) models, has been evaluated in a phase I clinical trial (http://clinicaltrials.org #NCT01089101) for pediatric LGG. Since the PI3K/AKT and MEK/ERK pathways converge on mTOR, treatments that target mTOR may still have utility. Several mTOR inhibitors other than sirolimus have been investigated for treating NF1- and sporadic LGG. Everolimus (RAD001), a rapamycin analog that targets mTOR, is currently being explored as a potential treatment in two separate trials (http://clinicaltrials.org #NCT01158651 and #NCT01734512, recruiting).
Especially relevant to NF1-LGG, proper communication and collaboration among patient families, clinicians, and researchers are highly recommended in order to preserve valuable LGG specimens for molecular characterization. The importance of donating tumor samples for research purposes should be emphasized to medical groups and families – especially for patients affected by NF1-PA, for which surgical biopsy samples are sometimes sparingly small. Legacy gifts of autopsy tumor tissue and selfless donation can similarly propel the field forward, while simultaneously providing hope for families for whom treatments are not currently available. Most pediatric cancer families (93%) would agree to autopsies if given the option (79); however, there has been a decrease over the past several decades in the acquisition of autopsy specimens for research (79). Patients, advocates of patients, clinicians, and researchers should aim to spread awareness, enhance subject recruitment, and increase funding for the future storage of cryo-viable frozen tissue samples at one or multiple sites, with cataloging by way of a HIPAA-compliant online database such as the CTF NF1 Registry.
While Nf1 mutant mouse models provide unprecedented opportunities for drug discovery and evaluation, the current collection of these strains does not fully capture the full spectrum of human clinical heterogeneity. Laboratory investigations have begun to identify the various factors that might underlie this inherent clinical heterogeneity. First, genotype–phenotype correlation studies in people with NF1 have revealed intriguing associations (80–84); the most striking of which is the finding that individuals with p1809 codon mutations do not develop dermal neurofibromas (85). Relevant to brain tumors, NF1 patients with 5′ NF1 gene mutations have higher chance developing optic gliomas compared to patients with mutations at other locations of the NF1 gene (82). Based on these types of clinical observations, human iPSCs have been used to demonstrate that different NF1 mutations lead to different levels of neurofibromin protein loss (39, 86, 87). These results suggest that not all NF1 gene mutations are equivalent.
To explore this further, GEM have recently been engineered to harbor specific Nf1 mutations seen in people with NF1. This is in contrast to the current Nf1 optic glioma mouse model in which the Nf1 gene is inactivated by the insertion of a neomycin targeting cassette, a genetic alteration not seen in individuals with NF1 (88, 89). Strikingly, in these proof-of-principle studies, Nf1 mice harboring a representative missense mutation did not develop optic gliomas, whereas those with a representative nonsense mutation harbored optic gliomas with higher proliferative indices than the conventional Nf1 optic glioma strain (90). Future preclinical studies should be designed to incorporate mice with different germline Nf1 gene mutations and cooperating genetic changes (heterozygous Pten loss; KIAA1549:BRAF expression) as a way of more accurately representing the full spectrum of disease in this patient population.
As mentioned above, the growth of LGG PDXs likely requires trophic support from the tumor microenvironment. Current xenograft methodologies developed using HGG cells may not be suitable for generating LGG PDX due to the relative stromal independence of HGG cells. The methodologies for future PA PDX studies can be tested and optimized using xenografts developed from mouse NF1-LGG cells (56). Current mouse xenograft methodologies involve the isolation of single cells (CD133+) from the optic nerves of 3-month-old mice with NF1-LGG (Nf1+/−GFAPCKO mice) (56). The resulting glioma stem cells express several stem cell markers and a response to therapeutic treatments similar to that observed in humans. This model lays the groundwork for further investigation into potential PDXs.
Since the discovery of the NF1 gene in 1990, great advances have been made to understand the cellular and molecular mechanisms underlying NF1-PA pathogenesis and maintenance. With the establishment of biobanking and registry initiatives, NF1-LGG specimens can be better characterized for actionable mutations. The resulting mutations and genomic alterations can next be evaluated in preclinical model systems as a means of identifying their contributions to tumor growth and targets for therapeutic intervention. Moreover, the incorporation of these cooperating genetic/genomic changes in combination with other factors, like the germline NF1 gene mutation, create a more representative spectrum of models that capture the innate clinical heterogeneity of these tumors for preclinical drug assessment and biomarker discovery. The implementation of these strategies offers the greatest opportunities to discover effective treatments for these common brain tumors, both for children with NF1 and those with non-syndromic LGG.
All authors designed the study and wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding for this internship project was provided by crowdfunding, “The Alexa Project.” We are grateful to Dr. Kellie Nazemi for helpful discussions on the topics of this review. We also acknowledge funding from the Children‘s Tumor Foundation Synodos Low-Grade Glioma Initiative that aims to comprehensively characterize the genetic and genomic changes in NF1-LGG as a means to develop more accurate preclinical models of these common tumors.
cAMP, cyclic adenosine monophosphate; CSC, cancer stem cell; ERK, extracellular signal-regulated kinase; GAP, GTPase-activating protein; GEM, genetically engineered mice; GRD, GAP-related domain; HGG, high-grade glioma; iPSC, induced pluripotent stem cell; JNK, c-Jun-NH2-kinase; PA, pilocytic astrocytoma; LGG, low-grade glioma; MAPK, mitogen-activating protein kinase; MEK, mitogen-activated protein kinase kinase; mTOR, mechanistic target of rapamycin; NF1, neurofibromatosis type 1; NSCs, neural stem cells; PDX, patient-derived xenograft; PI3K, phosphoinositide 3-kinase; PKCγ, protein kinase C gamma; RAF, rapidly accelerated fibrosarcoma; RAS, rat sarcoma; RGC, retinal ganglion cells.
1. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol (2015) 17(Suppl 4):iv1–62. doi: 10.1093/neuonc/nov189
2. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (2007) 114:97–109. doi:10.1007/s00401-007-0243-4
3. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res (2008) 68:8673–7. doi:10.1158/0008-5472.CAN-08-2097
4. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest (2008) 118:1739–49. doi:10.1172/JCI33656
5. Jacob K, Albrecht S, Sollier C, Faury D, Sader E, Montpetit A, et al. Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br J Cancer (2009) 101:722–33. doi:10.1038/sj.bjc.6605179
6. Yu J, Deshmukh H, Gutmann RJ, Emnett RJ, Rodriguez FJ, Watson MA, et al. Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology (2009) 73:1526–31. doi:10.1212/WNL.0b013e3181c0664a
7. Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet (2013) 45:927–32. doi:10.1038/ng.2682
8. Gutmann DH, Mclellan MD, Hussain I, Wallis JW, Fulton LL, Fulton RS, et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res (2013) 23:431–9. doi:10.1101/gr.142604.112
9. Kaul A, Toonen JA, Cimino PJ, Gianino SM, Gutmann DH. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol (2015) 17:843–53. doi:10.1093/neuonc/nou329
10. Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene (2009) 28:2119–23. doi:10.1038/onc.2009.73
11. Kaul A, Chen YH, Emnett RJ, Dahiya S, Gutmann DH. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev (2012) 26:2561–6. doi:10.1101/gad.200907.112
12. Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, et al. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell (1997) 89:693–702. doi:10.1016/S0092-8674(00)80252-4
13. Eggert A, Grotzer MA, Ikegaki N, Liu XG, Evans AE, Brodeur GM. Expression of the neurotrophin receptor TrkA down-regulates expression and function of angiogenic stimulators in SH-SY5Y neuroblastoma cells. Cancer Res (2002) 62:1802–8.
14. Manns M, Gunturkun O, Heumann R, Blochl A. Photic inhibition of TrkB/Ras activity in the pigeon’s tectum during development: impact on brain asymmetry formation. Eur J Neurosci (2005) 22:2180–6. doi:10.1111/j.1460-9568.2005.04410.x
15. Pearse RN, Swendeman SL, Li Y, Rafii D, Hempstead BL. A neurotrophin axis in myeloma: TrkB and BDNF promote tumor-cell survival. Blood (2005) 105:4429–36. doi:10.1182/blood-2004-08-3096
16. Tomlinson DC, Lamont FR, Shnyder SD, Knowles MA. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen-activated protein kinase pathway in bladder cancer. Cancer Res (2009) 69:4613–20. doi:10.1158/0008-5472.CAN-08-2816
17. Yu DY, Dahl GV, Sames RS, Fisher PG. Weekly dosing of carboplatin increases risk of allergy in children. J Pediatr Hematol Oncol (2001) 23:349–52. doi:10.1097/00043426-200108000-00005
18. Lassaletta A, Scheinemann K, Zelcer SM, Hukin J, Wilson BA, Jabado N, et al. Phase II weekly vinblastine for chemotherapy-naive children with progressive low-grade glioma: a Canadian Pediatric Brain Tumor Consortium Study. J Clin Oncol (2016) 29:3537–43. doi:10.1200/JCO.2016.68.1585
19. Jakacki RI, Bouffet E, Adamson PC, Pollack IG, Ingle AM, Voss SD, et al. A phase 1 study of vinblastine in combination with carboplatin for children with low-grade gliomas: a Children’s Oncology Group phase 1 consortium study. Neuro Oncol (2011) 13:910–5. doi:10.1093/neuonc/nor090
20. Bouffet E, Jakacki R, Goldman S, Hargrave D, Hawkins C, Shroff M, et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol (2012) 30:1358–63. doi:10.1200/JCO.2011.34.5843
21. Evans DG, Birch JM, Ramsden RT, Sharif S, Baser ME. Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J Med Genet (2006) 43:289–94. doi:10.1136/jmg.2005.036319
22. Packer RJ, Ater J, Allen J, Phillips P, Geyer R, Nicholson HS, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg (1997) 86:747–54. doi:10.3171/jns.1997.86.5.0747
23. Packer RJ, Lange B, Ater J, Nicholson HS, Allen J, Walker R, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol (1993) 11:850–6.
24. Lewis RA, Gerson LP, Axelson KA, Riccardi VM, Whitford RP. von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology (1984) 91:929–35. doi:10.1016/S0161-6420(84)34217-8
25. Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr (1994) 125:63–6. doi:10.1016/S0022-3476(94)70122-9
26. Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet (1999) 89:1–6. doi:10.1002/(SICI)1096-8628(19990326)89:1<1::AID-AJMG3>3.0.CO;2-8
27. Guillamo JS, Creange A, Kalifa C, Grill J, Rodriguez D, Doz F, et al. Prognostic factors of CNS tumours in neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain (2003) 126:152–60. doi:10.1093/brain/awg016
28. Johnson KJ, Hussain I, Williams K, Santens R, Mueller NL, Gutmann DH. Development of an international internet-based neurofibromatosis type 1 patient registry. Contemp Clin Trials (2013) 34:305–11. doi:10.1016/j.cct.2012.12.002
29. Johnson KJ, Mueller NL, Williams K, Gutmann DH. Evaluation of participant recruitment methods to a rare disease online registry. Am J Med Genet A (2014) 164A:1686–94. doi:10.1002/ajmg.a.36530
30. Sharkey EK, Zoellner NL, Abadin S, Gutmann DH, Johnson KJ. Validity of participant-reported diagnoses in an online patient registry: a report from the NF1 Patient Registry Initiative. Contemp Clin Trials (2015) 40:212–7. doi:10.1016/j.cct.2014.12.006
31. Johnson KJ, Fisher MJ, Listernick RL, North KN, Schorry EK, Viskochil D, et al. Parent-of-origin in individuals with familial neurofibromatosis type 1 and optic pathway gliomas. Fam Cancer (2012) 11:653–6. doi:10.1007/s10689-012-9549-z
32. Abadin SS, Zoellner NL, Schaeffer M, Porcelli B, Gutmann DH, Johnson KJ. Racial/ethnic differences in pediatric brain tumor diagnoses in patients with neurofibromatosis type 1. J Pediatr (2015) 167(613–620):e611–2. doi:10.1016/j.jpeds.2015.04.076
33. Porcelli B, Zoellner NL, Abadin SS, Gutmann DH, Johnson KJ. Associations between allergic conditions and pediatric brain tumors in neurofibromatosis type 1. Fam Cancer (2016) 15(2):301–8. doi:10.1007/s10689-015-9855-3
34. Mathiesen T, Peredo I, Lonn S. Two-year survival of low-grade and high-grade glioma patients using data from the Swedish Cancer Registry. Acta Neurochir (Wien) (2011) 153:467–71. doi:10.1007/s00701-010-0894-0
35. Rodriguez FJ, Lim KS, Bowers D, Eberhart CG. Pathological and molecular advances in pediatric low-grade astrocytoma. Annu Rev Pathol (2013) 8:361–79. doi:10.1146/annurev-pathol-020712-164009
36. Rooprai HK, Merzak A, Bullock P, Pilkington GJ. Establishment and characterization of two paediatric brain tumour cell lines in vitro. Anticancer Res (1997) 17:4127–34.
37. Bax DA, Little SE, Gaspar N, Perryman L, Marshall L, Viana-Pereira M, et al. Molecular and phenotypic characterisation of paediatric glioma cell lines as models for preclinical drug development. PLoS One (2009) 4:e5209. doi:10.1371/journal.pone.0005209
38. Hutt-Cabezas M, Karajannis MA, Zagzag D, Shah S, Horkayne-Szakaly I, Rushing EJ, et al. Activation of mTORC1/mTORC2 signaling in pediatric low-grade glioma and pilocytic astrocytoma reveals mTOR as a therapeutic target. Neuro Oncol (2013) 15:1604–14. doi:10.1093/neuonc/not132
39. Anastasaki C, Woo AS, Messiaen LM, Gutmann DH. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum Mol Genet (2015) 24:3518–28. doi:10.1093/hmg/ddv103
40. Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res (2003) 63:8573–7.
41. Gutmann DH, Donahoe J, Brown T, James CD, Perry A. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol (2000) 26:361–7. doi:10.1046/j.1365-2990.2000.00258.x
42. Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development (2005) 132:5577–88. doi:10.1242/dev.02162
43. Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH. Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann Neurol (2005) 57:119–27. doi:10.1002/ana.20337
44. Kim KY, Ju WK, Hegedus B, Gutmann DH, Ellisman MH. Ultrastructural characterization of the optic pathway in a mouse model of neurofibromatosis-1 optic glioma. Neuroscience (2010) 170:178–88. doi:10.1016/j.neuroscience.2010.06.017
45. Robertson R, Jan JE, Wong PK. Electroencephalograms of children with permanent cortical visual impairment. Can J Neurol Sci (1986) 13:256–61. doi:10.1017/S0317167100036386
46. Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B, et al. PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol (2011) 121:407–20. doi:10.1007/s00401-010-0784-9
47. Solga AC, Pong WW, Kim KY, Cimino PJ, Toonen JA, Walker J, et al. RNA sequencing of tumor-associated microglia reveals Ccl5 as a stromal chemokine critical for neurofibromatosis-1 glioma growth. Neoplasia (2015) 17:776–88. doi:10.1016/j.neo.2015.10.002
48. Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST, et al. Initial testing (stage 1) of AZD6244 (ARRY-142886) by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer (2010) 55:668–77. doi:10.1002/pbc.22576
49. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res (2004) 64:7011–21. doi:10.1158/0008-5472.CAN-04-1364
50. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature (2004) 432:396–401. doi:10.1038/nature03128
51. Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell (2005) 8:323–35. doi:10.1016/j.ccr.2005.09.001
52. Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol (2004) 5:738–43. doi:10.1038/ni1080
53. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell (2005) 121:823–35. doi:10.1016/j.cell.2005.03.032
54. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature (2007) 445:111–5. doi:10.1038/nature05384
55. Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res (2011) 17:3590–9. doi:10.1158/1078-0432.CCR-10-3349
56. Chen YH, Mcgowan LD, Cimino PJ, Dahiya S, Leonard JR, Lee Da Y, et al. Mouse low-grade gliomas contain cancer stem cells with unique molecular and functional properties. Cell Rep (2015) 10:1899–912. doi:10.1016/j.celrep.2015.02.041
57. Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature (1992) 356:713–5. doi:10.1038/356713a0
58. DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell (1992) 69:265–73. doi:10.1016/0092-8674(92)90407-4
59. Kim HA, Rosenbaum T, Marchionni MA, Ratner N, Declue JE. Schwann cells from neurofibromin deficient mice exhibit activation of p21ras, inhibition of cell proliferation and morphological changes. Oncogene (1995) 11:325–35.
60. Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet (1996) 12:144–8. doi:10.1038/ng0296-144
61. Cichowski K, Santiago S, Jardim M, Johnson BW, Jacks T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev (2003) 17:449–54. doi:10.1101/gad.1054703
62. Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A (2005) 102:8573–8. doi:10.1073/pnas.0503224102
63. Martin GA, Viskochil D, Bollag G, Mccabe PC, Crosier WJ, Haubruck H, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell (1990) 63:843–9. doi:10.1016/0092-8674(90)90150-D
64. Chen YH, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulation of neural stem cell proliferation and multilineage differentiation operates through distinct RAS effector pathways. Genes Dev (2015) 29:1677–82. doi:10.1101/gad.261677.115
65. Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci (2005) 25:5584–94. doi:10.1523/JNEUROSCI.4693-04.2005
66. Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res (2005) 65:2755–60. doi:10.1158/0008-5472.CAN-04-4058
67. Chang T, Krisman K, Theobald EH, Xu J, Akutagawa J, Lauchle JO, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest (2013) 123:335–9. doi:10.1172/JCI63193
68. Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest (2013) 123:340–7. doi:10.1172/JCI60578
69. Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov (2014) 4:452–65. doi:10.1158/2159-8290.CD-13-0646
70. Simmons GW, Pong WW, Emnett RJ, White CR, Gianino SM, Rodriguez FJ, et al. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol (2011) 70:51–62. doi:10.1097/NEN.0b013e3182032d37
71. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci (2015) 19:20–7. doi:10.1038/nn.4185
72. Pong WW, Higer SB, Gianino SM, Emnett RJ, Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol (2013) 73:303–8. doi:10.1002/ana.23813
73. Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet (2007) 16:1098–112. doi:10.1093/hmg/ddm059
74. Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res (2008) 68:10358–66. doi:10.1158/0008-5472.CAN-08-2506
75. Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol (2006) 24:2570–5. doi:10.1200/JCO.2005.03.8349
76. Madden JR, Rush SZ, Stence N, Foreman NK, Liu AK. Radiation-induced gliomas in 2 pediatric patients with neurofibromatosis type 1: case study and summary of the literature. J Pediatr Hematol Oncol (2014) 36:e105–8. doi:10.1097/MPH.0000000000000006
77. Hegedus B, Hughes FW, Garbow JR, Gianino S, Banerjee D, Kim K, et al. Optic nerve dysfunction in a mouse model of neurofibromatosis-1 optic glioma. J Neuropathol Exp Neurol (2009) 68:542–51. doi:10.1097/NEN.0b013e3181a3240b
78. Brown JA, Gianino SM, Gutmann DH. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J Neurosci (2010) 30:5579–89. doi:10.1523/JNEUROSCI.3994-09.2010
79. Alabran JL, Hooper JE, Hill M, Smith SE, Spady KK, Davis LE, et al. Overcoming autopsy barriers in pediatric cancer research. Pediatr Blood Cancer (2013) 60:204–9. doi:10.1002/pbc.24320
80. De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet (2003) 72:1288–92. doi:10.1086/374821
81. Upadhyaya M, Huson SM, Davies M, Thomas N, Chuzhanova N, Giovannini S, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet (2007) 80:140–51. doi:10.1086/510781
82. Sharif S, Upadhyaya M, Ferner R, Majounie E, Shenton A, Baser M, et al. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J Med Genet (2011) 48:256–60. doi:10.1136/jmg.2010.081760
83. Kehrer-Sawatzki H, Vogt J, Mussotter T, Kluwe L, Cooper DN, Mautner VF. Dissecting the clinical phenotype associated with mosaic type-2 NF1 microdeletions. Neurogenetics (2012) 13:229–36. doi:10.1007/s10048-012-0332-y
84. Pinna V, Lanari V, Daniele P, Consoli F, Agolini E, Margiotti K, et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur J Hum Genet (2015) 23:1068–71. doi:10.1038/ejhg.2014.243
85. Rojnueangnit K, Xie J, Gomes A, Sharp A, Callens T, Chen Y, et al. High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum Mutat (2015) 36:1052–63. doi:10.1002/humu.22832
86. Hoffmeyer S, Assum G. An RsaI polymorphism in the transcribed region of the neurofibromatosis (NF1)-gene. Hum Genet (1994) 93:481–2. doi:10.1007/BF00201684
87. Hoffmeyer S, Assum G, Griesser J, Kaufmann D, Nurnberg P, Krone W. On unequal allelic expression of the neurofibromin gene in neurofibromatosis type 1. Hum Mol Genet (1995) 4:1267–72. doi:10.1093/hmg/4.8.1267
88. Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev (1994) 8:1019–29. doi:10.1101/gad.8.9.1019
89. Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science (2002) 296:920–2. doi:10.1126/science.1068452
Keywords: pilocytic astrocytoma, neurofibromatosis type 1, drug therapy, low-grade glioma, pediatric neuro-oncology
Citation: Ricker CA, Pan Y, Gutmann DH and Keller C (2016) Challenges in Drug Discovery for Neurofibromatosis Type 1-Associated Low-Grade Glioma. Front. Oncol. 6:259. doi: 10.3389/fonc.2016.00259
Received: 22 September 2016; Accepted: 05 December 2016;
Published: 20 December 2016
Edited by:
André O. Von Bueren, University of Göttingen, GermanyReviewed by:
David D. Eisenstat, University of Alberta, CanadaCopyright: © 2016 Ricker, Pan, Gutmann and Keller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles Keller, Y2hhcmxlc0BjYy10ZGkub3Jn
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.