 N. M. Mazure1,2*
N. M. Mazure1,2*
- 1CNRS UMR7284, INSERM U1081, Institute for Research on Cancer and Aging, Nice (IRCAN), University of Nice, Nice, France
- 2CNRS GDR 3697 Micronit (www.micronit.fr)
The voltage-dependent anion channel (VDAC) is the main interface between the cytosol and mitochondria of cells. It plays a crucial role in both mitochondrial metabolism and cell death. The main basic function of this channel is to mediate and gate the flux of small ions, metabolites, and adenosine triphosphate. Changes in its structure, and thus conformation, are expected to affect its activity and modulate the ability of cancer cells to expand. In this review, we describe a novel mechanism by which mitochondria of cells in hypoxia, a low level of oxygen, protects from apoptosis. In hypoxia, some mitochondria become enlarged due to hyperfusion. These mitochondria possess a truncated form of VDAC1 (VDAC1-ΔC), which is linked to the higher metabolic capacity and the greater resistance to cell death of hypoxic cells. However, not all of the VDAC1 protein is truncated, but the amount of the full-length form is diminished compared to the amount in normoxic cells. First, we describe how such a decrease effects cell proliferation, respiration, glycolysis, and other processes. Second, we report on a novel mitochondrial-endolysosomal crosstalk that leads to VDAC1 truncation. By pharmacological targeting of VDAC1-ΔC, the production of energy could be turned off and the sensitivity to cell death restored. This could counteract the favorable microenvironment that gives cancer cells a growth advantage and thereby disrupts the balance between life and death, which is controlled by VDAC1.
Introduction
Mitochondria have evolved over time to take on a symbiotic relationship within eukaryotic cells to produce adenosine triphosphate (ATP) through activation of the electron transport chain (1). The production of ATP is probably the most important function of mitochondria, together with the regulation of apoptosis. Thereby, they are involved in different processes essential to the maintenance of cellular homeostasis. Modifications in metabolism and the redox status, critical steps in tumor cell transformation and progression, make mitochondria attractive targets for therapeutic treatment. Therefore, any modification to cancer cell metabolism and more specifically to mitochondrial metabolism, by increasing reactive oxygen species (ROS) production, or stimulating mitochondrial permeability transition to induce cell death, could be promising new therapeutic strategies (2).
How do mitochondria behave in hypoxia? Hypoxia is a decrease in the oxygen concentration compared to the normal physiological concentration and is a characteristic of the tumor microenvironment. Far from being a disadvantage, hypoxia is an undeniable force for the tumor cell. Functional benefits of hypoxia include epigenetic modifications, tumor vascularization, modified metabolism, signaling of metastasis, invasion and extravasation, cancer stemness, and innate immune activation, all of which are under the control of complex molecular pathways driven by the transcription factor the hypoxia-inducible factor (HIF) (3, 4). These changes help cells to proliferate and resist cell death induced by chemotherapy or radiation. Among the processes activated during tumor growth, metabolism, and more specifically glycolysis, is the one that is the most exacerbated by hypoxia (5, 6). To counterbalance mitochondrial generation of ROS that interfere with cell survival, HIF-1 activates pyruvate dehydrogenase kinase (PDK1) to block the conversion of pyruvate to acetyl CoA resulting in decreased flux through the tricarboxylic acid (TCA) cycle (7, 8). Moreover, HIF-1 upregulates the expression of COX4-2, present in complex IV, and the mitochondrial protease LONP1, which in turn degrades COX4-1. COX4-2 is then more efficient at facilitating electron transfer to O2 and thereby protects the cell from oxidative damage in hypoxia (9). HIF-1-mediated inhibition of MYC (10) and PGC-1 also results in reduced mitochondrial biogenesis (11). However, cancer cells have selectively found new mechanisms that promote their survival.
In this mini review, we highlight a new hypoxic mechanism that modulates the amount (decrease) and structure (a cleaved form lacking its C-terminus) of the most abundant protein of the outer mitochondrial membrane (12), the voltage-dependent anion channel (VDAC), which profoundly impacts cancer cell proliferation and survival.
The Voltage-Dependent Anion Channel: Function, Organization, and Structure
Voltage-dependent anion channel plays a key role in both mitochondrial metabolism and cell death, acting as a convergent point of control (13, 14). The function of VDACs as a pore seems quite clear-cut, but other suspected more complex functions are yet to be elucidated. VDACs play a crucial role as a gatekeeper for the entry and exit of many metabolites. They mediate and gate the flux of small ions (Cl−, K+, Na+) with a preference for anions and metabolites (NADH, citrate, succinate, glutamate, pyruvate, and glucose) and act as channels that constitute the main pathway for passage of ATP/ADP (15–18). In addition to their role in bioenergetics, VDACs act as a scaffold through interactions with numerous proteins. They are anchors for pro- and anti-apoptotic proteins, respectively, of the hexokinase (HK) (19, 20) and Bcl-2 families (21–24) of proteins, which contribute to the balance between survival and cell death.
Voltage-dependent anion channels exist as three isoforms: VDAC1, VDAC2, and VDAC3, encoded by three different genes. The three human VDAC genes share the same number of exons for each gene isoform (25). The human VDAC1 gene spans about 30 kb localized on the chromosome 5q31-32 (26), the human VDAC2 has been mapped to chromosome 10q22 and is 16.4 kb in length whereas the human VDAC3 is localized on chromosome 8p11.21 with a length of 14.3 kb. The VDAC2 gene uses several polyadenylation sites, thus giving rise to multiple mRNA, whereas the VDAC3 gene presents an alternative splicing event that corresponds to an additional ATG. At the protein level and in mammals, VDACs share ~70% identity with a very similar molecular mass of 30–35 kDa. They are known to be expressed ubiquitously in mammalian mitochondria, where VDAC1 remains the most abundantly expressed of the three isoforms (27). VDAC1 has also been detected in the plasma membrane of human lymphocytes (27, 28) and in the sarcoplasmic reticulum (29).
Analysis of the structure of VDACs revealed a 19-stranded β-barrel fold (13), yet only 13 of these strands form the wall of the channel. The N-terminal region of VDACs is very dynamic and exposed to the cytoplasm but located inside the pore. It acts as the voltage sensor and maintains the channel in an open or closed status (25). In an open-state configuration, VDACs are capable of passing millions of ATP molecules per second in vitro (17) and up to 100,000 ATP molecules per second under physiological conditions (16, 17), using at least five different trajectories (30). By contrast, very little is known about the function of the C-terminus of VDACs. It possesses NAD+-binding sites, considered essential for glycolysis (31). Finally, VDAC1 can oligomerize and assemble into a dynamic equilibrium of dimers, trimers, tetramers, and higher oligomers (32). These conformational changes could occur upon induction of apoptosis (33). However, the function of VDAC1 oligomers is not known. They may contribute to the stabilization of the protein (34) and may offer a more stable platform to anchor HKs I and II (32).
The Voltage-Dependent Anion Channel: Modifications, Silencing, and Over-Expression
Post-translational modifications, changes in expression, or even mutation in VDACs profoundly disrupt metabolism and, thus, the balance between cell survival and cell death. The three isoforms of VDAC can be post-translationally modified by phosphorylation and acetylation at multiple sites (35). The role of VDAC1 phosphorylation remains unclear, as it is difficult to study these modifications on highly hydrophobic integral mitochondrial outer membrane proteins. The impact of these modifications has been studied mostly in the context of apoptosis. However, no direct relationship to VDAC function or activity has been demonstrated. The relevance of acetylation remains to be determined. Recently, our studies showed a new form of post-translational modification of VDAC1; C-terminal truncation of the protein to give VDAC1-ΔC (discussed in Section “The Hypoxic Mitochondrial Phenotype and VDAC1-ΔC”) (Figure 1). This modification occurred specifically under hypoxic conditions. This hypoxic form was associated in some cancer cell lines with resistance to chemotherapy-induced apoptosis, a higher output of ATP and was found in late stage tumors of patients with lung cancer (36). A mutation in VDAC1 that resulted in the removal of 60% of the length of the C-terminal region has been described in colorectal and gastric cancers, but the consequence on metabolism and apoptosis is still to be determined (37).
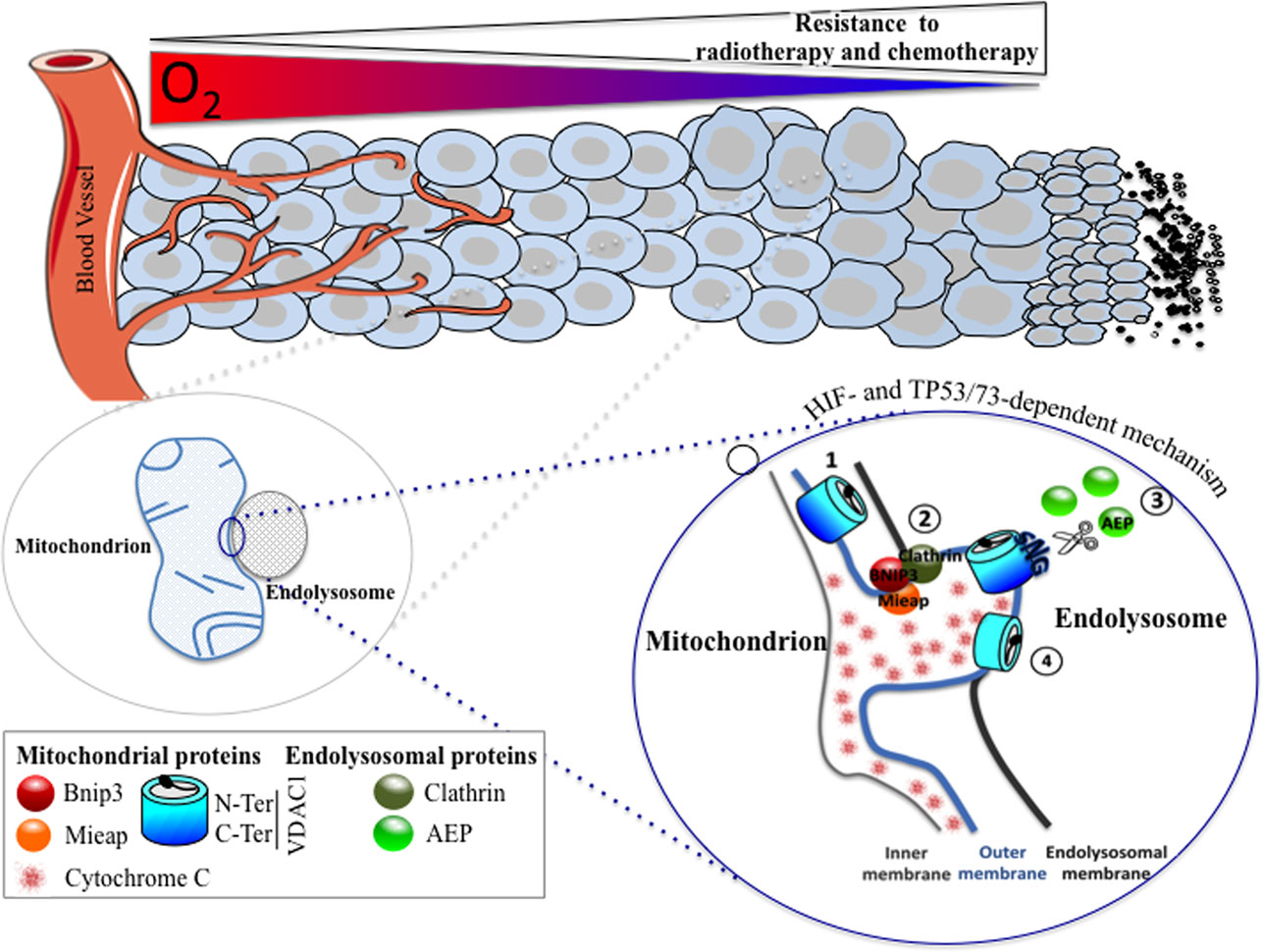
Figure 1. A HIF- and TP53/73-dependent model that potentiates tumor cell survival in hypoxia through the formation of enlarged mitochondria that interact with endolysosomes to modify mitochondrial VDAC1, an ATP channel-regulating metabolism and apoptosis. As oxygen diffuses from a vessel, a decreasing gradient in the oxygen concentration occurs in the adjacent tissue (top). As the level in oxygen decreases, resistance to radiotherapy and chemotherapy increases since the former requires oxygen for DNA damage, and the latter depends on the limits of tissue diffusion of the drug. Hypoxic cells with resistance to chemotherapy show the presence of enlarged hypoxic mitochondria (magnified on the bottom left). Microfusion between the mitochondrial outer membrane and an endolysosomal membrane takes place (magnified on the bottom right), and a cleaved form of VDAC1 is produced, according to the following steps: (1) VDAC1 is exposed to an endolysosome, (2) a complex formed of BNIP3/Mieap (mitochondrial proteins) and clathrin (endolysosomal protein) maintained microfusion of both membranes, (3) the endolysosomal asparagine endopeptidase (AEP), in contact with the mitochondrial outer membrane, specifically cleaves VDAC1, and (4) VDAC1-ΔC promotes resistance to apoptosis and a blockade in cytochrome c release.
Deletion of genes coding Vdac in mice models has provided information concerning the functions of VDACs. Vdac1 and Vdac3 heterozygote embryonic stem cells have been generated to obtain hetero- and homozygote knockout mice (38, 39). Vdac1−/− mice did not meet the normal Mendelian pattern, suggesting partial embryonic lethality at days 10.5–11.5. For the mice that survived, multiple respiratory defects appeared in skeletal and cardiac muscles. The mice were fertile but were retarded in growth. Mitochondria from muscle fibers of skin contained enlarged mitochondria with very compact cristea. Vdac3−/− mice survived with defects in mitochondrial respiration in heart tissue. Males were also infertile with alterations to their sperm. A combination of both Vdac1−/− and Vdac3−/− deletions was not lethal. Finally, the homozygote mice lacking Vdac2 died during development (40). Although it seems surprising not to find VDACs in a tissue, we recently reported, using the Cancer Genome Atlas (TCGA) data sets from 89 cancer studies, that deletion of the VDAC1 gene is found in some cancer types (41). Even if the VDAC1 gene was mainly heterologously lost, some homologous loss was found. Clear cell renal cell carcinoma (ccRCC) and ovarian cancers seemed to be the most affected by the homologous loss of the VDAC1 gene, whereas the heterologous loss was almost ubiquitous. It might be interesting to further examine these data to check if the VDAC2 and VDAC3 genes are also deleted or if compensation has taken place to counterbalance for the homologous loss of VDAC1. New aspects of the function of VDAC1 were highlighted in our recent transcriptome analysis of mouse embryonic fibroblasts (MEFs) knocked out for Vdac1 (41). We characterized the cellular and molecular phenotype of both Vdac1−/− MEF and MEFs transformed with the pBabe-RASv12 vector, Vdac1−/− RAS MEF. Our results pointed to alterations in programs controlling HIF-1, cell death, and survival, as well as cell proliferation and motility. We confirmed the presence of alterations in OXPHOS and glycolysis in knocked out cells, which was accompanied by a higher level of apoptosis. Of note, Vdac1−/− MEF and Vdac1−/− RAS MEF grew better in hypoxia, by maintaining respiration and promoting glycolysis. Vdac1−/− RAS MEF formed tumors faster than Wt RAS MEF in nod-scid mice. Moreover, after dissection of the various mechanisms involved, our results showed a strong impact of VDAC1 on tumor development, through alterations in the inflammatory response as a result of an abnormal vasculature due to ROS production and HIF-1α stabilization. Changes in metabolism were also observed in both Vdac1−/− MEF and Vdac1−/− RAS MEF. The first surprise came from the impact of hypoxia on the behavior of Vdac1−/− cells, in particular on cell proliferation. These initial results lead us to revisit the Mendelian ratio observed in the Vdac1−/− mice. Indeed, a large number of Vdac1−/− embryos did not survive (60%), whereas the Vdac1−/− embryos that survived gave an almost normal phenotype. Is it possible that the non-viable Vdac1−/− embryos were not exposed to low oxygen concentrations during development and, therefore, apoptosis prevailed? Our second hypothesis is that non-viable Vdac1−/− embryos may have developed dysfunctional blood vessels during embryogenesis. The second surprise came from our result that suggested the involvement of VDAC1 in modulating the structure of blood vessels and in enhancing the inflammatory response (41). Indeed, accumulation of ROS in Vdac1−/− RAS MEF-derived tumors triggered HIF-1α stabilization, abnormal vasculature, and leakage of red blood cells, thus generating an inflammatory response that resulted on a strong impact on VDAC1 tumor development. To our knowledge, this is the first time that VDAC1 has been connected to such events. These pathways should not be neglected in the future when considering VDAC1 as a therapeutic target.
Exogenous over-expression of VDAC1 in different cell lines always seems to be linked to apoptosis (24, 25, 42, 43). However, the impact of VDAC1 directly or indirectly on apoptosis is not yet clear. It may depolarize the inner membrane (44) as it could trigger the mitochondrial permeability transition pore (MPTP) (45). However, we showed that VDAC1 was over-expressed in lung adenocarcinomas tumor tissue from 44 patients (36). More recently, after analysis of the same TCGA data sets, as described above, expression of Vdac1 was also gained and amplified (41). The in vitro results contrast with the in vivo ones. It is easy to hypothesize that as VDAC1 regulates metabolism through its association with HK, cancer cells draw a substantial profit from increasing glycolysis. Similarly, because of its association with members of the Bcl-2 family, cancer cells, again, take advantage of such an association by minimizing apoptosis.
The Hypoxic Mitochondrial Phenotype and VDAC1-ΔC
Mitochondria are dynamic organelles that undergo membrane remodeling through cycles of fusion and fission (Figure 1). This balance controls the mitochondrial structure and, thus, mitochondrial activity. The key factors regulating fusion are the dynamin-related GTPases mitofusin 1 (Mfn1) and 2 (Mfn2), and optic atrophy 1 (OPA1) that mediate the OMM, while the dynamin-related protein 1 (DRP1) regulates the opposing process of fission of the inner mitochondrial membrane (IMM) (46). Cells lacking mitochondrial fusion show changes in mitochondrial shape associated with a loss of their membrane potential, a reduced growth rate, and a lower activity of respiratory complexes (47). By contrast, forced expression of Mfns resulted in the formation of clusters with enlarged mitochondria due to the fusion of the OMM (48). In 2010, we reported that a number of human cancer cells, including colon carcinoma cells (LS174) and non-neoplastic CCL39 lung fibroblasts exposed to long-term hypoxia (72 h – 1% O2) showed a change in mitochondrial phenotype from a tubular network to an enlarged morphology (49). The modification of the shape of mitochondria observed in LS174 cells was HIF-1-dependent (36). The formation of these enlarged mitochondria resulted from hyperfusion as expression of Mfn1 was increased in hypoxia and as silencing of the expression of Mfn1 reverted the mitochondrial morphology. Moreover, we reported that Bcl-2/adenovirus E1B 19-kDa interacting protein (BNIP3) and BNIP3 like (BNIP3L), two pro-autophagy proteins from the Bcl-2 family, also participated in the dynamic process of fusion induced in hypoxia. Finally, cytochrome c was retained inside these structurally unusual mitochondria when the cells were treated with staurosporine, a pro-apoptotic drug. We concluded that cells with enlarged mitochondria were more resistant to cell death than normoxic cells and that these cells possessed a selective growth advantage.
We were, therefore, faced with the challenge of exploring the underlying mechanisms that lead to the protective phenotype of hypoxic cancer cell. We found that VDAC1 was detected in hypoxia as a smaller than usual form (26 kDa rather than 30 kDa on SDS-PAGE). The amount of the 30 kDa form was decreased by around 50% with a parallel increase in the smaller 26 kDa form in hypoxia. Using a VDAC1 antibody directed to the C-terminus of VDAC1 that did not detect this fast migrating form of VDAC1 on immunoblots (36) and after analysis by mass spectrometry (50), we concluded that it was a form truncated in the C-terminal region: VDAC1-ΔC. After reconstitution into a planar lipid bilayer system, VDAC1-ΔC presented a similar but not identical channel activity and voltage dependency as VDAC1. At a higher voltage of −40 mV, the full-length channel showed two major conducting states with higher occupancy at the closed substate, whereas VDAC1-ΔC showed a higher open-state occupancy in comparison to the occupancy of low-conducting substates. Moreover, at the high voltages, VDAC1-ΔC showed a slightly higher conductance than VDAC1 (36). Indeed, we observed an increase in ATP levels in cells in hypoxia when VDAC1-ΔC and enlarged mitochondria were present. We also demonstrated that VDAC1-ΔC associated with the same partners as VDAC1, i.e., HKI/II and Bcl-XL. Anchoring of HKI/II is probably involved in the exacerbated metabolism of tumor cells in hypoxia in the presence of VDAC1-ΔC. Both the association of VDAC1-ΔC with HK and the increase in the expression of HKI/II in hypoxia increased OXPHOS and glycolysis. Thus, VDAC1-ΔC seems to control cell survival in hypoxia by regulating the export of ATP and probably NADH and brings advantage to cancer cells in promoting survival via mitochondria that are probably not as dormant as previously described. We do not know yet whether the conformation and the oligomerization of VDAC1-ΔC are different to that of VDAC1.
The VDAC1–HK complex has been reported to also play an important role in apoptosis (14). Thus, the VDAC1–HK complex already represents a target for cancer therapy, using, for example, specific VDAC1-based peptides that disrupt the connections between these proteins (51). HK provides an apoptosis-suppressive capacity by interfering with the ability of Bax to bind to mitochondria and induce apoptosis (52, 53). In addition, Bcl-XL, which interacts with VDAC1-ΔC (36), was found to protect cells from apoptosis via a block in the Bax–Bak interaction, subsequently preventing cytochrome c release (54, 55). Currently, cytochrome c is considered to play an important role in the resistance to cell death observed in cells with enlarged mitochondria and VDAC1-ΔC. We have previously shown that resistance to chemotherapy was linked to the phenotype of enlarged mitochondria. Subsequently, we demonstrated that silencing of VDAC1/VDAC1-ΔC in hypoxia or re-exposure of cells to normoxia, which inhibited the formation of VDAC1-ΔC, restored the sensitivity of the cells to apoptosis. We also showed that the mitochondrial transmembrane potential was unchanged in hypoxia and that cytochrome c was not released in the presence of staurosporine-induced apoptosis, whereas the transmembrane potential was decreased in normoxic cells under these conditions. We showed that cytochrome c was trapped inside the mitochondrial intermembrane space due to a change in the mitochondrial conformation and a hypothetical modification to VDAC oligomerization. These interactions occur specifically in hypoxia, when cancer cells are known to be highly resistant to anti-cancer treatments, and may, therefore, be exploited for therapy in the future.
Finally, we investigated further the mechanism behind the hypoxic regulation of the truncated form of VDAC1. We found that the cleavage of VDAC1 was dependent on TP53 or p73 as only cells expressing p53 (LS174, A549, or HepG2 cells) or p73 (HeLa cells) contained the hypoxic VDAC1-ΔC. Moreover, silencing of TP53 and/or HIF-1α diminished VDAC1 truncation and, thus, cell survival in the presence of staurosporine. Mieap, a TP53-inducible protein that controls mitochondrial quality was also involved, as silencing of MIEAP diminished also the hypoxic VDAC1-ΔC. We found that bafilomycin A1 and chloroquine, two compounds that increase the lysosomal pH, inhibited the cleavage of VDAC1 to VDAC1-ΔC, suggesting implication of lysosomes. This was confirmed by electron microscopy, which showed microfusion between mitochondria and endolysosomes and by cleavage of VDAC1 by an endolysosomal asparagine endopeptidase (AEP). Analysis by mass spectrometry of VDAC1-ΔC showed cleavage at asparagine 214 and glycine 213. Of note, AEP is also regulated by TP53 (56). Moreover, we reported that BNIP3, already known to be involved in regulating mitochondria morphology in hypoxia (36), acted as a docking site for lysosomes together with clathrin, a protein involved in multiple membrane vesicle trafficking pathways (57). This mechanism was identified not only in vitro but also in vivo in patients with lung cancer. We propose that this novel mechanism is a readout of mitochondrial–endolysosomal microfusion in hypoxia, in vitro and most importantly in vivo, and represents an additional defense mechanism that cancer cells have developed to resist chemotherapy.
Targeting VDAC1 for Therapy
Studies into cellular metabolism have lead to the characterization of a number of drugs that have already showed promise in pre-clinical and clinical trials. However, the quest for new therapeutic targets is hampered by the fact that cells show a great degree of plasticity, which already augurs the challenges we face. Studying metabolism per se is important and will allow us to identify new targets. However, metabolism should be studied in the context of a changing microenvironment (hypoxia, pH, changes in concentrations of metabolites, etc.) and in the context of malignant transformation. Thus, VDAC1-ΔC appears to be an interesting therapeutic target. Various compounds have already been identified for their capability to directly interact with and modify the activity of VDAC. Avicins (closes VDAC) (58), acrolein (used in Alzheimer’s disease to carbonylate VDAC) (59), erastine (binds to VDAC2) (60), endostatin (inducing PTP opening) (61), fluoxetine and cisplatin (inhibition of PTP opening and apoptosis) (62, 63), furanonaphthoquinones (induces VDAC-dependent apoptosis) (64), and oblimersen (blocks channel activity) (65, 66) are chemicals that will be tested under hypoxic conditions in the near future. In addition, VDAC-based peptides, novel pro-apoptotic agents that specifically target domains for interaction with HK, Bcl-2, and Bcl-XL, could be an interesting alternative to chemicals to restore the sensitivity to apoptosis in hypoxia (67).
We hope that, in the near future, this hypoxia/VDAC1-ΔC duo will meet the expectations that we have discussed in this review.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Dr M. C. Brahimi-Horn (web: Y2JyYWhpbWlob3JuLmZyZWUuZnI=; Y2Job3JuQG9yYW5nZS5mcg==) for critical reviewing and editorial correction.
Funding
The laboratory is funded by the Ligue Nationale Contre le Cancer, the Association pour la Recherche contre le Cancer, the Institut National du Cancer, the Agence Nationale pour la Recherche, the Centre A. Lacassagne, the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, and the University of Nice.
References
1. Andersson SG, Karlberg O, Canback B, Kurland CG. On the origin of mitochondria: a genomics perspective. Philos Trans R Soc Lond B Biol Sci (2003) 358:165–77;discussion77–9. doi: 10.1098/rstb.2002.1193
2. Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab (2014) 2:17. doi:10.1186/2049-3002-2-17
3. Brahimi-Horn C, Mazure N, Pouyssegur J. Signalling via the hypoxia-inducible factor-1alpha requires multiple posttranslational modifications. Cell Signal (2005) 17:1–9. doi:10.1016/j.cellsig.2004.04.010
4. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci (2012) 33:207–14. doi:10.1016/j.tips.2012.01.005
5. Brahimi-Horn M, Laferrière J, Mazure N, Pouysségur J. Hypoxia and tumour progression. In: Marmé D, Fusenig N, editors. Tumor Angiogenesis. Berlin Heidelberg, NY: Springer (2007). p. 171–94.
6. Brahimi-Horn MC, Bellot G, Pouyssegur J. Hypoxia and energetic tumour metabolism. Curr Opin Genet Dev (2011) 21:67–72. doi:10.1016/j.gde.2010.10.006
7. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3:177–85. doi:10.1016/j.cmet.2006.02.002
8. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3:187–97. doi:10.1016/j.cmet.2006.01.012
9. Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell (2007) 129:111–22. doi:10.1016/j.cell.2007.01.047
10. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell (2007) 11:407–20. doi:10.1016/j.ccr.2007.04.001
11. Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab (2012) 23:459–66. doi:10.1016/j.tem.2012.06.006
12. Linden M, Andersson G, Gellerfors P, Nelson BD. Subcellular distribution of rat liver porin. Biochim Biophys Acta (1984) 770:93–6. doi:10.1016/0005-2736(84)90077-4
13. Colombini M. VDAC structure, selectivity, and dynamics. Biochim Biophys Acta (2012) 1818:1457–65. doi:10.1016/j.bbamem.2011.12.026
14. Shoshan-Barmatz V, Ben-Hail D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion (2012) 12:24–34. doi:10.1016/j.mito.2011.04.001
15. Abu-Hamad S, Sivan S, Shoshan-Barmatz V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc Natl Acad Sci USA (2006) 103:5787–92. doi:10.1073/pnas.0600103103
16. Rostovtseva T, Colombini M. ATP flux is controlled by a voltage-gated channel from the mitochondrial outer membrane. J Biol Chem (1996) 271:28006–8. doi:10.1074/jbc.271.45.28006
17. Rostovtseva T, Colombini M. VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function. Biophys J (1997) 72:1954–62. doi:10.1016/S0006-3495(97)78841-6
18. Ferrer I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J Bioenerg Biomembr (2009) 41:425–31. doi:10.1007/s10863-009-9243-5
19. Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem (2008) 283:13482–90. doi:10.1074/jbc.M708216200
20. Rosano C. Molecular model of hexokinase binding to the outer mitochondrial membrane porin (VDAC1): implication for the design of new cancer therapies. Mitochondrion (2011) 11:513–9. doi:10.1016/j.mito.2011.01.012
21. Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem (2010) 285:6053–62. doi:10.1074/jbc.M109.082990
22. Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J (2004) 377:347–55. doi:10.1042/bj20031465
23. Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol (2001) 152:237–50. doi:10.1083/jcb.152.2.237
24. Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ (2005) 12:751–60. doi:10.1038/sj.cdd.4401599
25. Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med (2010) 31:227–85. doi:10.1016/j.mam.2010.03.002
26. Messina A, Oliva M, Rosato C, Huizing M, Ruitenbeek W, van den Heuvel LP, et al. Mapping of the human Voltage-Dependent Anion Channel isoforms 1 and 2 reconsidered. Biochem Biophys Res Commun (1999) 255:707–10. doi:10.1006/bbrc.1998.0136
27. De Pinto V, Messina A, Lane DJ, Lawen A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett (2010) 584:1793–9. doi:10.1016/j.febslet.2010.02.049
28. Bathori G, Parolini I, Szabo I, Tombola F, Messina A, Oliva M, et al. Extramitochondrial porin: facts and hypotheses. J Bioenerg Biomembr (2000) 32:79–89. doi:10.1023/A:1005516513313
29. Shoshan-Barmatz V, Israelson A. The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: characterization, modulation and possible function. J Membr Biol (2005) 204:57–66. doi:10.1007/s00232-005-0749-4
30. Choudhary OP, Paz A, Adelman JL, Colletier JP, Abramson J, Grabe M. Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat Struct Mol Biol (2014) 21:626–32. doi:10.1038/nsmb.2841
31. Brahimi-Horn MC, Mazure NM. Hypoxic VDAC1: a potential mitochondrial marker for cancer therapy. Adv Exp Med Biol (2014) 772:101–10. doi:10.1007/978-1-4614-5915-6_5
32. Zalk R, Israelson A, Garty ES, Azoulay-Zohar H, Shoshan-Barmatz V. Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem J (2005) 386:73–83. doi:10.1042/BJ20041356
33. Shoshan-Barmatz V, Mizrachi D. VDAC1: from structure to cancer therapy. Front Oncol (2012) 2:164. doi:10.3389/fonc.2012.00164
34. Ujwal R, Cascio D, Chaptal V, Ping P, Abramson J. Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation. Channels (Austin) (2009) 3:167–70. doi:10.4161/chan.3.3.9196
35. Kerner J, Lee K, Tandler B, Hoppel CL. VDAC proteomics: post-translation modifications. Biochim Biophys Acta (2012) 1818:1520–5. doi:10.1016/j.bbamem.2011.11.013
36. Brahimi-Horn MC, Ben-Hail D, Ilie M, Gounon P, Rouleau M, Hofman V, et al. Expression of a truncated active form of VDAC1 in lung cancer associates with hypoxic cell survival and correlates with progression to chemotherapy resistance. Cancer Res (2012) 72:2140–50. doi:10.1158/0008-5472.CAN-11-3940
37. Yoo NJ, Park SW, Lee SH. A Frameshift Mutation of the Pro-Apoptotic VDAC1 Gene in Cancers with Microsatellite Instability. Gut Liver (2011) 5:548–9. doi:10.5009/gnl.2011.5.4.548
38. Anflous K, Armstrong DD, Craigen WJ. Altered mitochondrial sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J Biol Chem (2001) 276:1954–60. doi:10.1074/jbc.M006587200
39. Sampson MJ, Decker WK, Beaudet AL, Ruitenbeek W, Armstrong D, Hicks MJ, et al. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J Biol Chem (2001) 276:39206–12. doi:10.1074/jbc.M104724200
40. Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science (2003) 301:513–7. doi:10.1126/science.1083995
41. Brahimi-Horn MC, Giuliano S, Saland E, Lacas-Gervais S, Sheiko T, Pelletier J, et al. Knockout of Vdac1 activates hypoxia-inducible factor through reactive oxygen species generation and induces tumor growth by promoting metabolic reprogramming and inflammation. Cancer Metab (2015) 3:8. doi:10.1186/s40170-015-0133-5
42. Ghosh T, Pandey N, Maitra A, Brahmachari SK, Pillai B. A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS One (2007) 2:e1170. doi:10.1371/journal.pone.0001170
43. Godbole A, Varghese J, Sarin A, Mathew MK. VDAC is a conserved element of death pathways in plant and animal systems. Biochim Biophys Acta (2003) 1642:87–96. doi:10.1016/S0167-4889(03)00102-2
44. De Pinto V, Tomasello F, Messina A, Guarino F, Benz R, La Mendola D, et al. Determination of the conformation of the human VDAC1 N-terminal peptide, a protein moiety essential for the functional properties of the pore. Chembiochem (2007) 8:744–56. doi:10.1002/cbic.200700009
45. Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina S, et al. Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res (2009) 19:1363–76. doi:10.1038/cr.2009.98
46. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol (2014) 15:634–46. doi:10.1038/nrm3877
47. Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet (2005) 14(Spec No. 2):R283–9. doi:10.1093/hmg/ddi270
48. Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci (2003) 116:2763–74. doi:10.1242/jcs.00479
49. Chiche J, Rouleau M, Gounon P, Brahimi-Horn MC, Pouyssegur J, Mazure NM. Hypoxic enlarged mitochondria protect cancer cells from apoptotic stimuli. J Cell Physiol (2010) 222:648–57. doi:10.1002/jcp.21984
50. Brahimi-Horn MC, Lacas-Gervais S, Adaixo R, Ilc K, Rouleau M, Notte A, et al. Local mitochondrial-endolysosomal microfusion cleaves the voltage-dependent anion channel 1 to promote survival in hypoxia. Mol Cell Biol (2015) 35(9):1491–505. doi:10.1128/MCB.01402-14
51. Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J Biol Chem (2009) 284:3946–55. doi:10.1074/jbc.M803614200
52. Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr (2008) 40:171–82. doi:10.1007/s10863-008-9148-8
53. Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem (2002) 277:7610–8. doi:10.1074/jbc.M109950200
54. Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, Saikumar P. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J Biol Chem (2003) 278:5367–76. doi:10.1074/jbc.M203392200
55. Dlugosz PJ, Billen LP, Annis MG, Zhu W, Zhang Z, Lin J, et al. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J (2006) 25:2287–96. doi:10.1038/sj.emboj.7601126
56. Yamane T, Murao S, Kato-Ose I, Kashima L, Yuguchi M, Kozuka M, et al. Transcriptional regulation of the legumain gene by p53 in HCT116 cells. Biochem Biophys Res Commun (2013) 438:613–8. doi:10.1016/j.bbrc.2013.08.007
57. Young A. Structural insights into the clathrin coat. Semin Cell Dev Biol (2007) 18:448–58. doi:10.1016/j.semcdb.2007.07.006
58. Haridas V, Li X, Mizumachi T, Higuchi M, Lemeshko VV, Colombini M, et al. Avicins, a novel plant-derived metabolite lowers energy metabolism in tumor cells by targeting the outer mitochondrial membrane. Mitochondrion (2007) 7:234–40. doi:10.1016/j.mito.2006.12.005
59. Mello CF, Sultana R, Piroddi M, Cai J, Pierce WM, Klein JB, et al. Acrolein induces selective protein carbonylation in synaptosomes. Neuroscience (2007) 147:674–9. doi:10.1016/j.neuroscience.2007.04.003
60. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447:864–8. doi:10.1038/nature05859
61. Yuan S, Fu Y, Wang X, Shi H, Huang Y, Song X, et al. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J (2008) 22:2809–20. doi:10.1096/fj.08-107417
62. Nahon E, Israelson A, Abu-Hamad S, Varda SB. Fluoxetine (Prozac) interaction with the mitochondrial voltage-dependent anion channel and protection against apoptotic cell death. FEBS Lett (2005) 579:5105–10. doi:10.1016/j.febslet.2005.08.020
63. Castagna A, Antonioli P, Astner H, Hamdan M, Righetti SC, Perego P, et al. A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431. Proteomics (2004) 4:3246–67. doi:10.1002/pmic.200400835
64. Simamura E, Shimada H, Hatta T, Hirai K. Mitochondrial voltage-dependent anion channels (VDACs) as novel pharmacological targets for anti-cancer agents. J Bioenerg Biomembr (2008) 40:213–7. doi:10.1007/s10863-008-9158-6
65. Tan W, Lai JC, Miller P, Stein CA, Colombini M. Phosphorothioate oligonucleotides reduce mitochondrial outer membrane permeability to ADP. Am J Physiol Cell Physiol (2007) 292:C1388–97. doi:10.1152/ajpcell.00490.2006
66. Tan W, Loke YH, Stein CA, Miller P, Colombini M. Phosphorothioate oligonucleotides block the VDAC channel. Biophys J (2007) 93:1184–91. doi:10.1529/biophysj.107.105379
Keywords: cancer, mitochondria dysfunction, VDAC1, hypoxia-inducible factor 1, resistance to cell death
Citation: Mazure NM (2016) News about VDAC1 in Hypoxia. Front. Oncol. 6:193. doi: 10.3389/fonc.2016.00193
Received: 18 January 2016; Accepted: 15 August 2016;
Published: 30 August 2016
Edited by:
Christian Gomez, University of Mississippi Medical Center School of Dentistry, USAReviewed by:
Varda Shoshan-Barmatz, Ben-Gurion University of the Negev, IsraelEverardo Macias, Cedars-Sinai Medical Center, USA
Copyright: © 2016 Mazure. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: N. M. Mazure, bmF0aGFsaWUubWF6dXJlQHVuaWNlLmZy