
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 30 June 2015
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 5 - 2015 | https://doi.org/10.3389/fonc.2015.00147
This article is part of the Research Topic Advances in Diffuse Intrinsic Pontine Glioma View all 5 articles
 Pawel Buczkowicz1,2*
Pawel Buczkowicz1,2* Cynthia Hawkins1,2,3*
Cynthia Hawkins1,2,3*
Diffuse intrinsic pontine glioma (DIPG) is a devastating pediatric brain cancer with no effective therapy. Histological similarity of DIPG to supratentorial high-grade astrocytomas of adults has led to assumptions that these entities possess similar underlying molecular properties and therefore similar therapeutic responses to standard therapies. The failure of all clinical trials in the last 30 years to improve DIPG patient outcome has suggested otherwise. Recent studies employing next-generation sequencing and microarray technologies have provided a breadth of evidence highlighting the unique molecular genetics and epigenetics of this cancer, distinguishing it from both adult and pediatric cerebral high-grade astrocytomas. This review describes the most common molecular genetic and epigenetic signatures of DIPG in the context of molecular subgroups and histopathological diagnosis, including this tumor entity’s unique mutational landscape, copy number alterations, and structural variants, as well as epigenetic changes on the global DNA and histone levels. The increased knowledge of DIPG biology and histopathology has opened doors to new diagnostic and therapeutic avenues.
Diffuse intrinsic pontine gliomas (DIPG), brainstem tumors that diffusely involve the pons, are the most common type of brainstem gliomas (BSG) (1). The mean age of diagnosis for this devastating pediatric neoplasm is 6–7 years (2, 3). The delicate location of these tumors eliminates surgical intervention as a treatment option. Radiation therapy (RT) is the standard of care, although it offers temporary relief of symptoms rather than a real hope of cure (4). Despite 68 clinical trials using various adjuvant chemotherapeutic agents between 1984 and 2014, there has been no improvement in survival compared to radiation alone, and DIPG are currently the number one cause of brain tumor related death in children (5–18). The median survival of DIPG patients is only 10 months post diagnosis and <10% of patients survive 2 years (19, 20). Since the 1980s, the diagnosis of DIPG was based on clinical findings and diagnostic imaging characteristics on computerized tomography (CT) or magnetic resonance imaging (MRI) (4, 21). The lack of surgical and biopsy material has limited most studies of DIPG biology and histology to post-mortem tissue. Although initial investigation into the safety of incorporating biopsy for BSG showed no surgical mortality and low surgical morbidity (22), the advent of CT and MRI allowed for accurate non-invasive localization of BSG (23). It was noted that patients with DIPG represented the majority of deaths in children with BSG and biopsies, which did not stratify patients into different treatment groups, were abandoned (23). Recently, the role for biopsy in DIPG diagnosis has been substantially strengthened due to several important discoveries related to the biology and histopathology of this universally fatal tumor (3, 24, 25) and successful use of stereotactic biopsy by several centers with limited morbidity (26–29). Biologically, DIPG is a unique tumor entity which possesses properties that are antithetical when compared to other brain tumors, such as pediatric and adult supratentorial high-grade astrocytomas (HGA), which they most often resemble histologically. This review highlights the unique pathology, genetics, and epigenetics of DIPG.
Early molecular profiling of pediatric gliomas was limited to candidate gene approaches (30–33) focusing on mutational and copy number profiling of genes known to be frequently implicated in adult HGA, namely EGFR, TP53, IDH1/2, CDKN2A, MGMT, and PTEN (34–36). All early studies of DIPG were performed on small cohorts or case studies, which limited their usefulness in defining the biology of this deadly pediatric cancer. However, conclusions from these studies still highlighted some differences between DIPG and adult HGA. Unfortunately, these candidate gene approaches were limited to what was known about adult HGA and future studies using whole-genome profiling approaches would be required to discover more robust differences between these entities. Whole genome profiling technologies such as array CGH and SNP-genotyping allowed for the first genome-wide copy number analysis of cancers. Although giving a low resolution perspective at first, newer iterations allowed for greater and greater resolution and began to highlight the unique molecular profiles of DIPG when compared to pediatric and adult supratentorial astrocytomas. Copy number signatures at the whole chromosome arm level revealed differences between DIPG and adult HGA (37). Gains of chromosome 1q and losses of 11p, 13q, and 14q were frequently observed in DIPG and less so in supratentorial HGA (37). Subsequent copy number studies, with larger DIPG patient tumor cohorts, revealed frequently altered genes (Table 1) including PDGFRA, TP53, PARP1, PVT-1/MYC, RB1, and PTEN (3, 38, 39). Most recently, next-generation sequencing approaches, including whole-genome sequencing (WGS), whole-exome sequencing (WES), and RNA-sequencing integrated with histopathology, copy number, gene expression, and methylation profiling, and other molecular techniques have re-defined what we know about DIPG genetics and epigenetics.
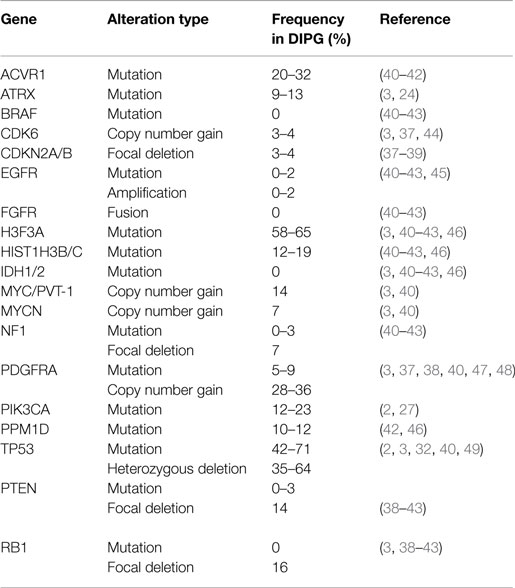
Table 1. Frequencies of most common mutations and copy number alterations in diffuse intrinsic pontine glioma from the literature.
The first major breakthrough in defining the DIPG mutational landscape came in 2012, when studies on pediatric brain tumors using whole-genome and WES reported that 70–84% of DIPG possess mutations in histone H3, and that these mutations were predictive of outcome (3, 24, 25). These recurrent mutations, in H3F3A or HIST1H3B/C/I (Figure 1A), result in a p. Lys27Met (K27M) substitution. A subset of other midline astrocytomas, such as those arising in the thalamus, have also been found to harbor K27M histone H3 mutations although at a lower frequency (50). Conversely, pediatric supratentorial HGA rarely possess these mutations, and more frequently have p. Gly34Arg (G34R) or p. Gly34Val (G34V) substitution in histone H3.3. The G34R/V-H3.3 mutations occur in 10–19% of supratentorial HGA cases (24) and are never found in DIPG (3). Furthermore, among supratentorial GBM, these G34R/V-H3.3 mutations are predominantly found in older children and young adults (3). Mutations affecting these two histone residues are extremely rare in adult HGA.
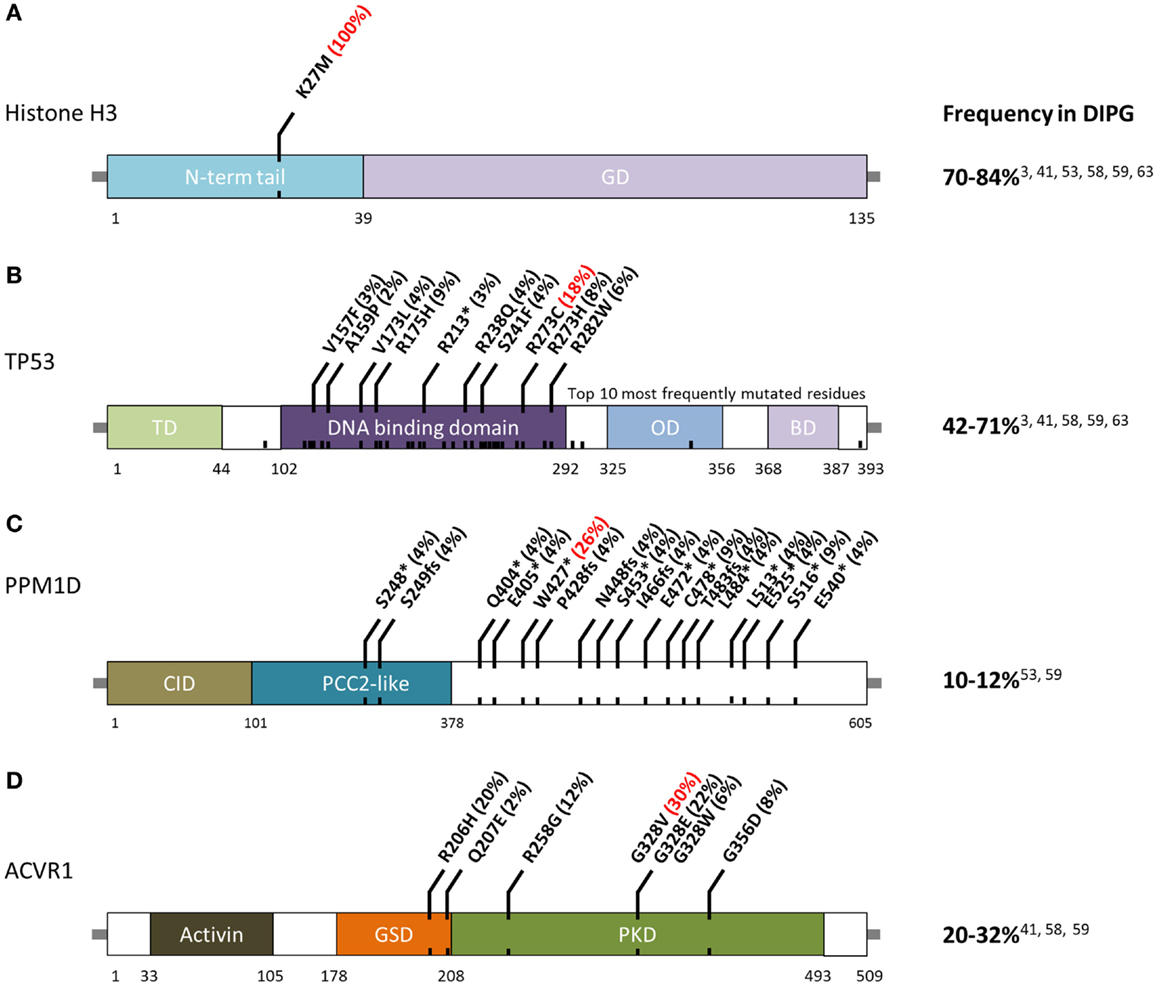
Figure 1. Schematic of residue alterations in (A) histone H3, (B) TP53, (C) PPM1D, and (D) ACVR1 as a result of mutations most frequently identified in DIPG. GD, globular domain; TD, transactivation domain; OD, oligomerization domain; BD, basic domain; CID, CHEK1 interacting domain; GSD, GS domain; PKD, protein Kinase domain. Red indicates most frequently altered amino acid.
Histones are proteins that form octomeric complexes known as nucleosomes around which DNA wraps and condenses into chromatin. The majority of histone mutations in DIPG, 65%, effect histone variant H3.3 (3, 24, 25). Although this histone is coded in two genes, H3F3A and H3F3B, K27M mutations are only found in the former. K27M-H3.1 mutations are found in 12–19% of DIPG and are mutually exclusive from K27M-H3.3 mutations (24, 40). Histone H3.1 is coded by a cluster of 10 genes on chromosome 6 known as the HIST1 cluster (44). Currently, K27M-H3.1 mutations have been detected in HIST1H3B, HIST1H3C, and HISTIH3I.
Histones play a key role in the state of chromatin; however, they themselves do not determine whether chromatin will be in a conformation more permissive to gene expression (euchromatin) or in a conformation less permissive to gene expression (heterochromatin). Several histone marks can be laid on specific residues across the histone tail, including lysine 27, which can be post-translationally modified by either acetylation or mono-, di-, or tri- methylation (51–53). Various cellular machineries are implicated in the reading, writing, and copying of these epigenetic histone marks, including histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), and histone demethylases (54). The methionine substitution at K27 in DIPG cannot be modified and several in vitro and in vivo studies have documented a global decrease in H3K27me3 in the presence of mutant histone (40, 55–58). This global decrease is attributed to polycomb repressive complex 2 (PRC2) inhibition (59); however, other molecular consequences of this mutation, including its effect on histone marks, global DNA methylation, and gene expression, are still under investigation. Histone H3 mutations are undoubtedly important in DIPG tumorigenesis and/or maintenance but in a majority of cases are found to be partnered with other mutations and copy number alterations (CNAs) (3, 40), suggesting that they are not the sole drivers of DIPG tumorigenesis.
TP53 mutations and CNAs occur in 42–71% of DIPG (Figure 1B) (3, 40). Hemizygous deletions are often associated with a mutation and strong protein expression by immunohistochemistry (32, 49). Interestingly, DIPG with low-grade astrocytoma histology (grade II), even those harboring H3-mutations, are not mutated for TP53 (2). Among DIPG with high-grade histology (grade III and IV), both H3-mutant and wild-type DIPG often have mutated TP53 (2). However, a recent exome sequencing study identified PPM1D mutations (Figure 1C) in a subset of H3-mutant but TP53 wild-type DIPG (46). These mutations account of ~50% of TP53 wild-type grade II DIPG. PPM1D gene codes for WIP1 (wild-type p53-induced protein phosphatase 1D) and has been implicated as an oncogene in other cancers (60–62). Mutations of PPM1D have been shown to be functionally equivalent to those of TP53 (46, 63). Taken together, alterations of the TP53 signaling pathway in DIPG are only slightly less common than histone H3 mutations.
The third most commonly mutated gene in DIPG codes for the activin A receptor, type I (ACVR1), a member of the bone morphogenic protein (BMP) signaling pathway. Approximately, 20–32% of DIPG harbor mutations in ACVR1 which significantly overlap with K27M mutations in histone H3.1 (40–42). Previously only reported in a congenital autosomal dominant disease of the connective tissue called fibrodysplasia ossificans progressiva (FOP), ACVR1 mutations result in ligand-independent constitutive activation of the BMP signaling pathway (64–66). Although seven different ACVR1 mutations have been reported in DIPG, the most common alteration in this tumor type, p.Gly328Val, has not been reported in FOP patients. Several other residues of ACVR1 are frequently mutated in DIPG (Figure 1D). These mutations have been shown to increase levels of phosphorylated SMAD1/5 (40–43) as well as increased gene expression of downstream BMP signaling targets ID1 and ID2 (40). Research into the cooperation between ACVR1 and histone H3.1 mutations, and their effects on tumorigenesis are still ongoing.
Receptor tyrosine kinases (RTKs) are cell surface receptors which are often dysregulated in cancers. The RTK/RAS/PI-3K signaling pathway is the most commonly dysregulated signaling pathway in adult GBM, with 90% of tumors exhibiting CNAs or mutations in pathway members (34). Mutations and CNAs in the platelet-derived growth factor receptor alpha (PDGFRA) have been implicated in both adult and pediatric HGA. Several array based studies of DIPG revealed amplification of PDGFRA in 28–36% of patient tumors (37, 38). The frequency of these amplifications in pediatric and adult supratentorial HGA was reported in the range of 7–14%, and was preferentially identified in the Proneural subtype of adult GBM (36). In DIPG, PDGFRA amplifications are exclusively found in patients with K27M-H3 mutations and across all astrocytic histologies (grade II-IV) (2, 40). Oncogenic mutations of PDGFRA have also been reported in 5–9% of DIPG (47, 48). Epidermal growth factor receptor (EGFR) copy number gains and mutations are among the most frequent alterations in adult HGA, occurring in 60–85% of GBM (34, 35). In adults, EGFR protein expression correlates with amplifications and EGFRvIII mutations; however in DIPG, EGFR CNAs and mutations are extremely rare, found in 0–2% of patient tumors (45), and do not correlate with immunopositivity (67, 68). The RTK/RAS/PI-3K signaling pathway is further dysregulated in DIPG through hemizygous deletions of PTEN (40), as well as mutations of PIK3CA and PIK3R1 (2, 27). Although this pathway has often been targeted in clinical trials, the majority of the CNAs and mutations in the RTK/RAS/PI-3K pathway are clonal events, as determined by fluorescence in situ hybridization (FISH) for PDGFRA amplifications (37) and mutation allele frequency of PIK3CA, suggesting these events likely arise later in tumor development.
Radiotherapy is the standard of care for DIPG and adjuvant chemotherapeutics have been shown to be ineffective. Perturbations in DNA damage repair pathways in DIPG were first identified in 2010 by Zarghooni et al. Loss of heterozygosity (LOH) was identified in many genes involved in nucleotide excision repair, non-homologous end-joining (NHEJ), homologous recombination (HR), base excision repair (BER), and mismatch repair (MMR) by analysis of both SNP arrays and microsatellite markers (37). Poly (ADP-ribose) polymerase (PARP1), a protein essential for repair of single strand DNA breaks induced by alkylating agents, as well as repair of ionizing radiation induced double strand breaks by HR and NHEJ were found to be gained and/or overexpressed in 54% of DIPG, highlighting it as a potential therapeutic target (37). Furthermore, pathway analysis revealed a subset of patients with LOH or deletions in members of the BRCA DNA damage response pathway, including BRCA1 and BRCA2. Defects in either of these two genes have been implicated in promoting sensitivity to single strand DNA repair via PARP inhibition.
Temozolomide (TMZ) is a frontline DNA alkylating agent most often used in treatment of adult GBM. TMZ causes DNA damage by alkylating O6-guanine, N7-guanine, and N3-adenine residues. In adults, it was identified that a subgroup of patients with MGMT (O6-methylguanine DNA methyltransferase) promoter methylation had improved overall survival when treated with radiotherapy and concomitant TMZ compared to patients without MGMT promoter methylation (69). MGMT repairs TMZ alkylated O6-guanine nucleotides. However, the universal lack of response to TMZ in DIPG patients could not be attributed to this resistance mechanism, as DIPG have not been found to express MGMT (37). It has been recently described that TMZ resistance in the pediatric population can be attributed to an ATM-dependent regulation of 3-methylpurine-DNA glycosylase (MPG), an enzyme responsible for repair of alkylated N7 guanine and N3 adenine residues (70). Further investigation into using radio-sensitizing and chemo-sensitizing agents in DIPG therapy is warranted.
Mutations in the mitochondrial enzyme gene, isocitrate dehydrogenase 1 (IDH1), whose gene product catalyzes the oxidative decarboxylation of isocitrate to α-keto glutarate, are found in approximately 70–80% of adult low-grade astrocytomas (LGA) and anaplastic astrocytomas (AA), as well as secondary glioblastoma (GBM) (71). Furthermore, in adult gliomas, IDH1 mutations were found to be associated with ATRX mutations and alternative lengthening of telomeres (ALT) (72). Isocitrate dehydrogenase 2 (IDH2) mutations are also found in a subset of adult HGA but to date, no mutations in IDH1 or IDH2 have been detected in DIPG.
Mutations of chromatin remodeling genes are less common in DIPG than in supratentorial HGA (3, 24). ATRX, which codes for the α-thalassemia/mental retardation syndrome X-linked gene, was found to be mutated in a subset of DIPG (~9%) but in contrast to pediatric supratentorial HGA, had no clear overlap with histone H3 mutations (3). Pediatric supratentorial HGA showed high overlap with ATRX or DAXX mutations (15–25%) and G34R/V-H3.3 alterations, as well as mutual exclusivity of IDH1/2 mutations (24, 73). Irrespective of tumor location, in the pediatric population ATRX mutations significantly overlap with TP53 mutations and are predominantly found in older children (3). ATRX and DAXX (death-domain associated protein) are genes encoding subunits of a chromatin remodeling complex required for histone H3.3 incorporation at telomeric regions. Telomeres are repetitive regions of DNA found on the ends of chromosomes and shorten during every cell division due to incomplete DNA replication (74). Over many cell divisions, telomeres may reach a critically short length, which results in cellular senescence (75). This fate can be avoided by expressing telomerase, an enzyme that can extend telomeres. Although telomerase is not expressed in most mature, terminally differentiated cells (76, 77), its expression and activity has been implicated in several brain cancers as a poor prognostic marker and potential therapeutic target (78–80). Extension of telomeres in certain cancers can be attained by a telomerase independent mechanism known as alternative lengthening of telomeres (ALT). Twenty percent of DIPG test positive for ALT by either TRF (telomere restriction fragment) assay or C-circles assay and ALT positive DIPG are exclusive carriers of the K27M-H3.3 mutation (40). ALT phenotype is also associated with an older age of diagnosis in DIPG and to date has not been detected in patients with low-grade astrocytoma histology (2, 40).
Diffuse intrinsic pontine glioma is a heterogeneous disease, and represents a varied histological spectrum. These tumors are very diffuse and often involve adjacent brain structures beyond the pons. Several studies report leptomeningeal dissemination and subventricular spread as a common occurrence seen in as many as one-third of DIPG, with tumor cells found as far rostrally as the frontal lobe (2, 81, 82). A review by Jansen et al. reported World Health Organization (WHO) central nervous system tumor classification on 108 biopsies from 13 studies, including 37 AA (WHO grade III), 27 GBM(WHO grade IV), 22 LGA (WHO grade II), 3 anaplastic oligoastrocytomas (WHO grade III), and 19 tumors with “not further specified” or undefined characterization (9). Data from this biopsy series would suggest that WHO grade III AA are the most common histological entity in brainstem glioma; however, autopsy based histopathological studies report WHO grade IV GBM to be the most common histology, potentially highlighting the caveat of limited tissue sampling during biopsy or anaplastic progress and/or treatment effect seen in late stage disease at autopsy. Autopsy based studies revealed that GBM histology was most common in DIPG. Of the 33 pediatric patients examined by Yoshimura et al., 29 were reported to be GBM and 4 with anaplastic astrocytoma histology (83). A larger autopsy based study of DIPG histology reported 42 GBM, 18 anaplastic astrocytoma, 8 low-grade astrocytoma, and 2 with features of primitive neuroectodermal tumor (PNET, WHO grade IV) (13). Previous investigations into DIPG histology have also observed this rare PNET histology in the brainstem (84). Importantly, regional differences within one DIPG specimen can bias biopsy based diagnoses. Autopsy studies allow extensive tissue sampling and have highlighted intratumoral histopathologic heterogeneity (2). Areas within or around a grade IV astrocytoma may present with features of grade II or grade III histology that could be inadvertently targeted at biopsy (Figure 2).
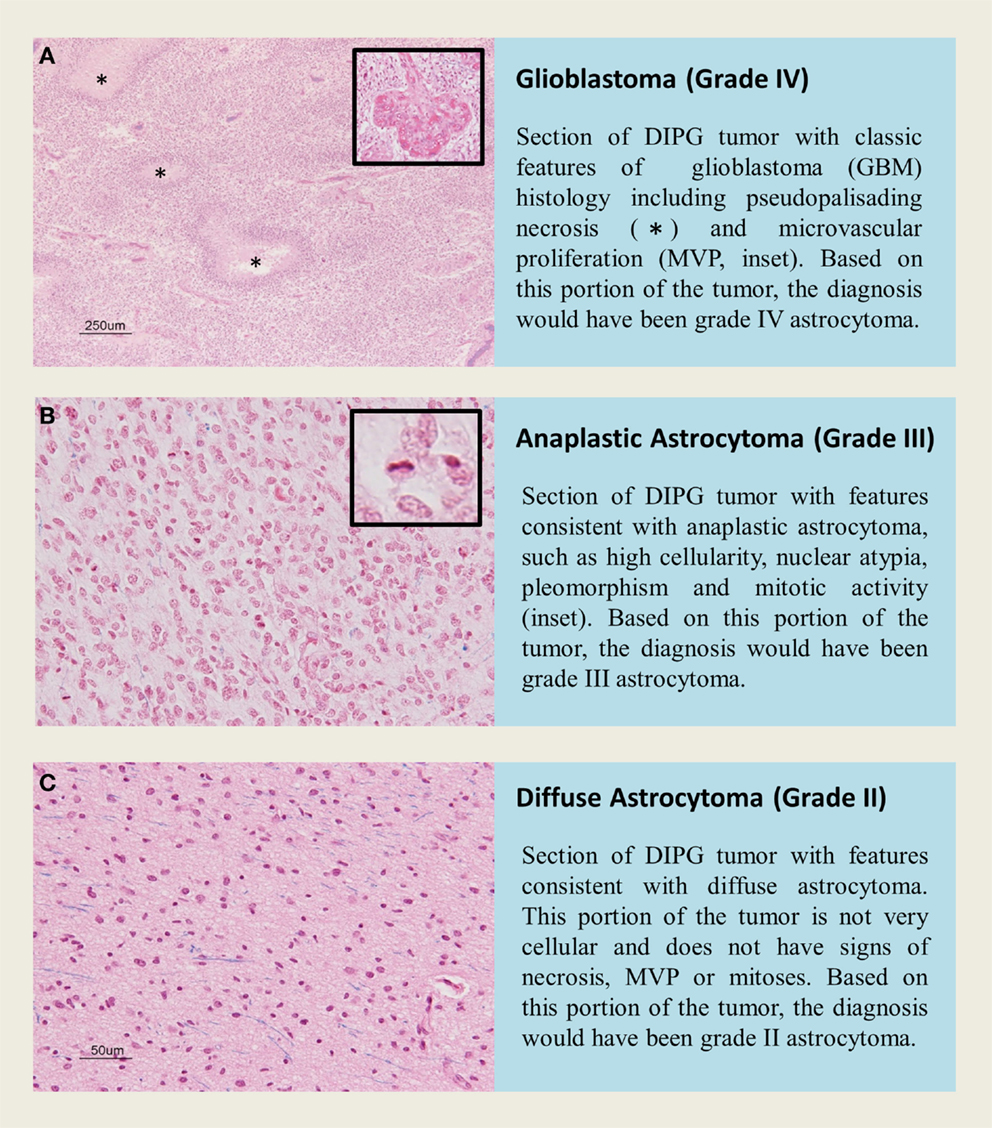
Figure 2. H&E staining of tissue sections from an autopsy of a K27M-H3.1 mutant diffuse intrinsic pontine glioma patient highlights vast regional differences that histologically resemble (A,B) high grade astrocytoma (WHO grade III–IV) or (C) low grade astrocytoma (WHO grade II). Inadvertently targeting these regions on biopsy could lead to misdiagnosis. Histone H3 mutation predicts overall survival better than histologic grade for DIPG. Mutational testing at the time of stereotactic biopsy should be implemented into clinical practice for DIPG patients.
Perhaps most importantly, DIPG histology is not a predictor of survival. Patients with low-grade histology do just as poorly as patients with high-grade histology. On multivariate Cox regression analysis, only histone H3 mutation is a predictor of worse overall survival, irrespective of histological grade, age of diagnosis, and sex (2, 3). Furthermore, DIPG patients, which at autopsy had grade II histology, but were mutated for histone H3, had clinical outcomes similar to what would be expected of high-grade tumors (2, 3). This important finding suggests brainstem glioma require their own WHO grading scheme, which needs to incorporate H3-mutation testing at biopsy.
Several studies have embarked on molecular subgrouping of DIPG based on various molecular signatures, including gene expression profiling, copy number analysis, proteomics, mutational profiles, and methylation profiling (Figure 3). A study by Paugh et al. in 2011 profiled 27 DIPG by gene expression arrays and classified them into three subgroups based on hierarchical clustering and compared the subgroup specific enrichment scores to previously identified subgroups of adult GBM, highlighting significant enrichment in either mesenchymal, proliferative, or pronerual markers (38). These data were also compared to genomic copy number abnormalities determined by single-nucleotide polymorphism arrays which highlighted frequent focal amplifications of PDGFRA and RB1. However, these CNAs were not restricted to any of the three identified subgroups. A subsequent study of gene expression profiles from 23 DIPG matched with copy number data from array CGH identified two subgroups using K-means clustering (48). These two subgroups were enriched for mesenchymal and proneural markers, respectively, with the proneural subgroup displaying oligodendroglial features and alterations of PDGFRA (48). With the discovery of histone mutations in ~80% of DIPG in 2012 and their clinical relevance in patient survival, supervised analysis of CNAs in wild-type and histone mutant subgroups by single-nucleotide polymorphism arrays highlighted PDGFRA and PVT-1/MYC gains and amplifications to be present at high frequency in K27M-H3 mutant DIPG, whereas wild-type DIPG were enriched for MYCN amplification as determined by GISTIC2.0 analysis (3). A study from 2013 by Saratsis et al. utilized mRNA and methylation profiles with protein profiling of 14 DIPG specimens. This study also reported two subgroups, which were characterized by upregulation of N-Myc or Hedgehog signaling through mRNA expression and DNA hypomethylation (85).
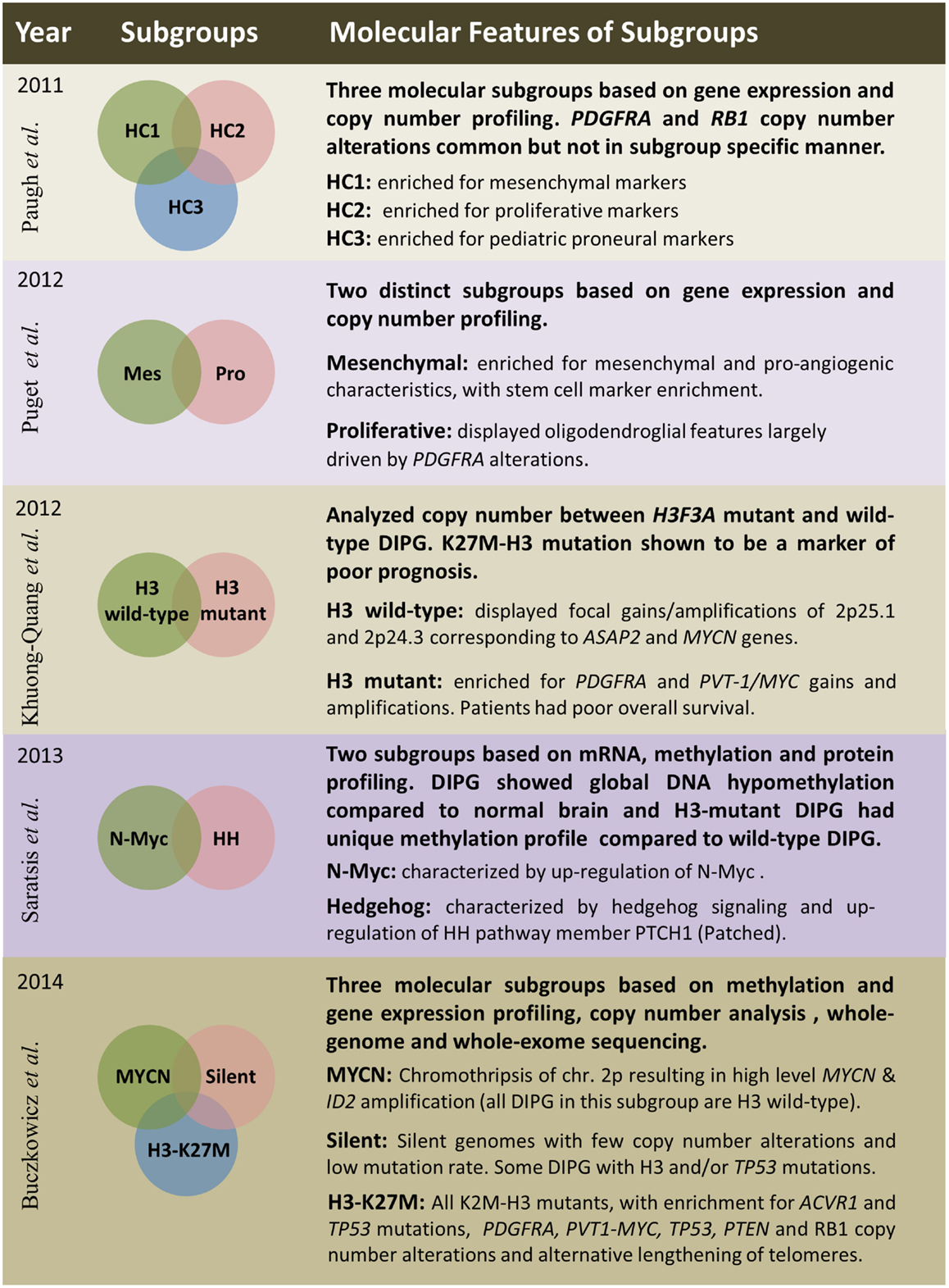
Figure 3. Summary of five publications between 2011 and 2014 outlining clinical and molecular features of diffuse intrinsic pontine gliomas based on various platforms and sample sizes; Paugh et al. (38), three subgroups – n = 27; Puget et al. (48), two subgroups – n = 23; Khuong-Quang et al. (3), two subgroups – n = 42; Saratsis et al. (85), two subgroups – n = 14; Buczkowicz et al. (40), three subgroups – n = 48.
The most comprehensive subgrouping of DIPG integrated CpG island methylation with WGS and WES, gene expression, and copy number profiling to find that DIPG are three molecularly distinct subgroups; MYCN, Silent, and H3-K27M (40). The MYCN subgroup did not contain any DIPG with histone mutations, but was characterized by high-level amplifications of MYCN and ID2 genes caused by chromothripsis on chromosome 2p. Furthermore, these MYCN amplified DIPG did not contain PVT-1/MYC, PDGFRA, or RB1 CNAs, which were previously described to be common in these brainstem neoplasms (40). The Silent subgroup did not contain many CNAs or mutations. In fact, this subgroup was characterized by stable genomes when compared to both MYCN and H3-K27M subgroup DIPG. Some DIPG in this subgroup contained histone H3, TP53, and ACVR1 mutations; however, they were found at a lower frequency. All patients within the largest subgroup, H3-K27M, had histone H3-mutations. This subgroup was further characterized by hypomethylated genomes and also enriched in ACVR1 mutations (which significantly co-occurred with K27M-H3.1 mutations), TP53/PPM1D mutations/homozygous deletions, alternative lengthening of telomeres, and CNAs of PVT-1/MYC, PTEN, PDGFRA, and RB1 (40).
In recent years, the combined increase in knowledge of DIPG histology, genetics, and epigenetics has been substantial. Mutations in previously undiscovered oncogenes, histone H3, and ACVR1, molecular subtypes of DIPG, and the rediscovered role for biopsy for this tumor entity are redefining what is clinically possible in treating these patients. The major challenges ahead are developing molecularly accurate pre-clinical models for testing of new therapeutics and integrating new genetic/epigenetic and histopathological knowledge for DIPG patient diagnosis, treatment, and future clinical trial design. In the near future, DIPG patients will benefit from mutational testing and molecular subgrouping at biopsy, which will provide more accurate prognosis and actionable tumor-specific genomic targets for these affected children.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The Guest Associate Editor David D. Eisenstat declares that, despite having collaborated with author Cynthia Hawkins, the review process was handled objectively and no conflict of interest exists.
1. Ramos A, Hilario A, Lagares A, Salvador E, Perez-Nunez A, Sepulveda J. Brainstem gliomas. Semin Ultrasound CT MR (2013) 34(2):104–12. doi:10.1053/j.sult.2013.01.001
2. Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol (2014) 128(4):573–81. doi:10.1007/s00401-014-1319-6
3. Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol (2012) 124(3):439–47. doi:10.1007/s00401-012-0998-0
4. Robison NJ, Kieran MW. Diffuse intrinsic pontine glioma: a reassessment. J Neurooncol (2014) 119(1):7–15. doi:10.1007/s11060-014-1448-8
5. Bailey S, Howman A, Wheatley K, Wherton D, Boota N, Pizer B, et al. Diffuse intrinsic pontine glioma treated with prolonged temozolomide and radiotherapy – results of a United Kingdom phase II trial (CNS 2007 04). Eur J Cancer (2013) 49(18):3856–62. doi:10.1016/j.ejca.2013.08.006
6. Bartels U, Wolff J, Gore L, Dunkel I, Gilheeney S, Allen J, et al. Phase 2 study of safety and efficacy of nimotuzumab in pediatric patients with progressive diffuse intrinsic pontine glioma. Neuro Oncol (2014) 16(11):1554–9. doi:10.1093/neuonc/nou091
7. Broniscer A, Baker SD, Wetmore C, Pai Panandiker AS, Huang J, Davidoff AM, et al. Phase I trial, pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clin Cancer Res (2013) 19(11):3050–8. doi:10.1158/1078-0432.CCR-13-0306
8. Cohen KJ, Gibbs IC, Fisher PG, Hayashi RJ, Macy ME, Gore L. A phase I trial of arsenic trioxide chemoradiotherapy for infiltrating astrocytomas of childhood. Neuro Oncol (2013) 15(6):783–7. doi:10.1093/neuonc/not021
9. Jansen MH, van Vuurden DG, Vandertop WP, Kaspers GJ. Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer Treat Rev (2012) 38(1):27–35. doi:10.1016/j.ctrv.2011.06.007
10. Janssens GO, Jansen MH, Lauwers SJ, Nowak PJ, Oldenburger FR, Bouffet E, et al. Hypofractionation vs conventional radiation therapy for newly diagnosed diffuse intrinsic pontine glioma: a matched-cohort analysis. Int J Radiat Oncol Biol Phys (2013) 85(2):315–20. doi:10.1016/j.ijrobp.2012.04.006
11. Muller K, Schlamann A, Guckenberger M, Warmuth-Metz M, Gluck A, Pietschmann S, et al. Craniospinal irradiation with concurrent temozolomide for primary metastatic pediatric high-grade or diffuse intrinsic pontine gliomas. A first report from the GPOH-HIT-HGG Study Group. Strahlenther Onkol (2014) 190(4):377–81. doi:10.1007/s00066-013-0513-0
12. Okada K, Yamasaki K, Tanaka C, Fujisaki H, Osugi Y, Hara J. Phase I study of bevacizumab plus irinotecan in pediatric patients with recurrent/refractory solid tumors. Jpn J Clin Oncol (2013) 43(11):1073–9. doi:10.1093/jjco/hyt124
13. Porkholm M, Valanne L, Lonnqvist T, Holm S, Lannering B, Riikonen P, et al. Radiation therapy and concurrent topotecan followed by maintenance triple anti-angiogenic therapy with thalidomide, etoposide, and celecoxib for pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer (2014) 61(9):1603–9. doi:10.1002/pbc.25045
14. Rizzo D, Scalzone M, Ruggiero A, Maurizi P, Attina G, Mastrangelo S, et al. Temozolomide in the treatment of newly diagnosed diffuse brainstem glioma in children: a broken promise? J Chemother (2015) 27(2):106–10. doi:10.1179/1973947814Y.0000000228
15. Roos DE, Smith JG. Randomized trials on radioactive iodine ablation of thyroid remnants for thyroid carcinoma – a critique. Int J Radiat Oncol Biol Phys (1999) 44(3):493–5. doi:10.1016/S0360-3016(98)00553-7
16. Veldhuijzen van Zanten SE, Jansen MH, Sanchez Aliaga E, van Vuurden DG, Vandertop WP, Kaspers GJ. A twenty-year review of diagnosing and treating children with diffuse intrinsic pontine glioma in The Netherlands. Expert Rev Anticancer Ther (2015) 15(2):157–64. doi:10.1586/14737140.2015.974563
17. Zaghloul MS, Eldebawy E, Ahmed S, Mousa AG, Amin A, Refaat A, et al. Hypofractionated conformal radiotherapy for pediatric diffuse intrinsic pontine glioma (DIPG): a randomized controlled trial. Radiother Oncol (2014) 111(1):35–40. doi:10.1016/j.radonc.2014.01.013
18. Zaky W, Wellner M, Brown RJ, Bluml S, Finlay JL, Dhall G. Treatment of children with diffuse intrinsic pontine gliomas with chemoradiotherapy followed by a combination of temozolomide, irinotecan, and bevacizumab. Pediatr Hematol Oncol (2013) 30(7):623–32. doi:10.3109/08880018.2013.829895
19. Freeman CR, Perilongo G. Chemotherapy for brain stem gliomas. Childs Nerv Syst (1999) 15(10):545–53. doi:10.1007/s003810050542
20. Maria BL, Rehder K, Eskin TA, Hamed LM, Fennell EB, Quisling RG, et al. Brainstem glioma: I. Pathology, clinical features, and therapy. J Child Neurol (1993) 8(2):112–28. doi:10.1177/088307389300800203
21. Donaldson SS, Laningham F, Fisher PG. Advances toward an understanding of brainstem gliomas. J Clin Oncol (2006) 24(8):1266–72. doi:10.1200/JCO.2005.04.6599
22. Albright AL, Price RA, Guthkelch AN. Brain stem gliomas of children. A clinicopathological study. Cancer (1983) 52(12):2313–9. doi:10.1002/1097-0142(19831215)52:12<2313::AID-CNCR2820521226>3.0.CO;2-I
23. Albright AL, Packer RJ, Zimmerman R, Rorke LB, Boyett J, Hammond GD. Magnetic resonance scans should replace biopsies for the diagnosis of diffuse brain stem gliomas: a report from the children’s cancer group. Neurosurgery (1993) 33(6):1026–9. doi:10.1227/00006123-199312000-00010
24. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature (2012) 482(7384):226–31. doi:10.1038/nature10833
25. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet (2012) 44(3):251–3. doi:10.1038/ng.1102
26. Cage TA, Samagh SP, Mueller S, Nicolaides T, Haas-Kogan D, Prados M, et al. Feasibility, safety, and indications for surgical biopsy of intrinsic brainstem tumors in children. Childs Nerv Syst (2013) 29(8):1313–9. doi:10.1007/s00381-013-2101-0
27. Grill J, Puget S, Andreiuolo F, Philippe C, MacConaill L, Kieran MW. Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer (2012) 58(4):489–91. doi:10.1002/pbc.24060
28. Kieran MW. Time to rethink the unthinkable: upfront biopsy of children with newly diagnosed diffuse intrinsic pontine glioma (DIPG). Pediatr Blood Cancer (2015) 62(1):3–4. doi:10.1002/pbc.25266
29. MacDonald TJ. Diffuse intrinsic pontine glioma (DIPG): time to biopsy again? Pediatr Blood Cancer (2012) 58(4):487–8. doi:10.1002/pbc.24090
30. Cheng Y, Ng HK, Zhang SF, Ding M, Pang JC, Zheng J, et al. Genetic alterations in pediatric high-grade astrocytomas. Hum Pathol (1999) 30(11):1284–90. doi:10.1016/S0046-8177(99)90057-6
31. Louis DN, Rubio MP, Correa KM, Gusella JF, von Deimling A. Molecular genetics of pediatric brain stem gliomas. Application of PCR techniques to small and archival brain tumor specimens. J Neuropathol Exp Neurol (1993) 52(5):507–15. doi:10.1097/00005072-199309000-00009
32. Pollack IF, Hamilton RL, Finkelstein SD, Campbell JW, Martinez AJ, Sherwin RN, et al. The relationship between TP53 mutations and overexpression of p53 and prognosis in malignant gliomas of childhood. Cancer Res (1997) 57(2):304–9.
33. Sure U, Ruedi D, Tachibana O, Yonekawa Y, Ohgaki H, Kleihues P, et al. Determination of p53 mutations, EGFR overexpression, and loss of p16 expression in pediatric glioblastomas. J Neuropathol Exp Neurol (1997) 56(7):782–9. doi:10.1097/00005072-199707000-00004
34. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi:10.1016/j.cell.2013.09.034
35. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455(7216):1061–8. doi:10.1038/nature07385
36. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi:10.1016/j.ccr.2009.12.020
37. Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol (2010) 28(8):1337–44. doi:10.1200/JCO.2009.25.5463
38. Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol (2011) 29(30):3999–4006. doi:10.1200/JCO.2011.35.5677
39. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol (2010) 28(18):3061–8. doi:10.1200/JCO.2009.26.7252
40. Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet (2014) 46(5):451–6. doi:10.1038/ng.2936
41. Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet (2014) 46(5):457–61. doi:10.1038/ng.2925
42. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet (2014) 46(5):444–50. doi:10.1038/ng.2938
43. Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet (2014) 46(5):462–6. doi:10.1038/ng.2950
44. Ederveen TH, Mandemaker IK, Logie C. The human histone H3 complement anno 2011. Biochim Biophys Acta (2011) 1809(10):577–86. doi:10.1016/j.bbagrm.2011.07.002
45. Liang ML, Ma J, Ho M, Solomon L, Bouffet E, Rutka JT, et al. Tyrosine kinase expression in pediatric high grade astrocytoma. J Neurooncol (2008) 87(3):247–53. doi:10.1007/s11060-007-9513-1
46. Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J, et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet (2014) 46(7):726–30. doi:10.1038/ng.2995
47. Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res (2013) 73(20):6219–29. doi:10.1158/0008-5472.CAN-13-1491
48. Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One (2012) 7(2):e30313. doi:10.1371/journal.pone.0030313
49. Pollack IF, Finkelstein SD, Woods J, Burnham J, Holmes EJ, Hamilton RL, et al. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med (2002) 346(6):420–7. doi:10.1056/NEJMoa012224
50. Aihara K, Mukasa A, Gotoh K, Saito K, Nagae G, Tsuji S, et al. H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro Oncol (2014) 16(1):140–6. doi:10.1093/neuonc/not144
51. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell (2006) 125(2):315–26. doi:10.1016/j.cell.2006.02.041
52. Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature (2008) 454(7205):766–70. doi:10.1038/nature07107
53. Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, et al. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J (2012) 31(3):593–605. doi:10.1038/emboj.2011.431
54. Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer (2014) 14(2):92–107. doi:10.1038/nrc3655
55. Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell (2013) 24(5):660–72. doi:10.1016/j.ccr.2013.10.006
56. Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev (2013) 27(9):985–90. doi:10.1101/gad.217778.113
57. Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol (2013) 23(5):558–64. doi:10.1111/bpa.12042
58. Venneti S, Santi M, Felicella MM, Yarilin D, Phillips JJ, Sullivan LM, et al. A sensitive and specific histopathologic prognostic marker for H3F3A K27M mutant pediatric glioblastomas. Acta Neuropathol (2014) 128(5):743–53. doi:10.1007/s00401-014-1338-3
59. Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science (2013) 340(6134):857–61. doi:10.1126/science.1232245
60. Akbari MR, Lepage P, Rosen B, McLaughlin J, Risch H, Minden M, et al. PPM1D mutations in circulating white blood cells and the risk for ovarian cancer. J Natl Cancer Inst (2014) 106(1):djt323. doi:10.1093/jnci/djt323
61. Richter M, Dayaram T, Gilmartin AG, Ganji G, Pemmasani SK, Van Der Key H, et al. WIP1 phosphatase as a potential therapeutic target in neuroblastoma. PLoS One (2015) 10(2):e0115635. doi:10.1371/journal.pone.0115635
62. Zajkowicz A, Butkiewicz D, Drosik A, Giglok M, Suwinski R, Rusin M. Truncating mutations of PPM1D are found in blood DNA samples of lung cancer patients. Br J Cancer (2015) 112:1114–20. doi:10.1038/bjc.2015.79
63. Kleiblova P, Shaltiel IA, Benada J, Sevcik J, Pechackova S, Pohlreich P, et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol (2013) 201(4):511–21. doi:10.1083/jcb.201210031
64. Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat (2009) 30(3):379–90. doi:10.1002/humu.20868
65. Petrie KA, Lee WH, Bullock AN, Pointon JJ, Smith R, Russell RG, et al. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS One (2009) 4(3):e5005. doi:10.1371/journal.pone.0005005
66. Song GA, Kim HJ, Woo KM, Baek JH, Kim GS, Choi JY, et al. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem (2010) 285(29):22542–53. doi:10.1074/jbc.M109.094557
67. Kleihues P, Ohgaki H. Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro Oncol (1999) 1(1):44–51. doi:10.1215/15228517-1-1-44
68. Rood BR, MacDonald TJ. Pediatric high-grade glioma: molecular genetic clues for innovative therapeutic approaches. J Neurooncol (2005) 75(3):267–72. doi:10.1007/s11060-005-6749-5
69. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med (2005) 352(10):997–1003. doi:10.1056/NEJMoa043331
70. Agnihotri S, Burrell K, Buczkowicz P, Remke M, Golbourn B, Chornenkyy Y, et al. ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov (2014) 4(10):1198–213. doi:10.1158/2159-8290.CD-14-0157
71. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360(8):765–73. doi:10.1056/NEJMoa0808710
72. Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol (2012) 124(5):615–25. doi:10.1007/s00401-012-1031-3
73. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell (2012) 22(4):425–37. doi:10.1016/j.ccr.2012.08.024
74. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature (1990) 345(6274):458–60. doi:10.1038/345458a0
75. Greider CW. Telomeres, telomerase and senescence. Bioessays (1990) 12(8):363–9. doi:10.1002/bies.950120803
76. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science (1994) 266(5193):2011–5. doi:10.1126/science.7605428
77. Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet (1996) 18(2):173–9. doi:10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3
78. Barszczyk M, Buczkowicz P, Castelo-Branco P, Mack SC, Ramaswamy V, Mangerel J, et al. Telomerase inhibition abolishes the tumorigenicity of pediatric ependymoma tumor-initiating cells. Acta Neuropathol (2014) 128(6):863–77. doi:10.1007/s00401-014-1327-6
79. Mangerel J, Price A, Castelo-Branco P, Brzezinski J, Buczkowicz P, Rakopoulos P, et al. Alternative lengthening of telomeres is enriched in, and impacts survival of TP53 mutant pediatric malignant brain tumors. Acta Neuropathol (2014) 128(6):853–62. doi:10.1007/s00401-014-1348-1
80. Remke M, Ramaswamy V, Peacock J, Shih DJ, Koelsche C, Northcott PA, et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol (2013) 126(6):917–29. doi:10.1007/s00401-013-1198-2
81. Caretti V, Bugiani M, Freret M, Schellen P, Jansen M, van Vuurden D, et al. Subventricular spread of diffuse intrinsic pontine glioma. Acta Neuropathol (2014) 128(4):605–7. doi:10.1007/s00401-014-1307-x
82. Sethi R, Allen J, Donahue B, Karajannis M, Gardner S, Wisoff J, et al. Prospective neuraxis MRI surveillance reveals a high risk of leptomeningeal dissemination in diffuse intrinsic pontine glioma. J Neurooncol (2011) 102(1):121–7. doi:10.1007/s11060-010-0301-y
83. Yoshimura J, Onda K, Tanaka R, Takahashi H. Clinicopathological study of diffuse type brainstem gliomas: analysis of 40 autopsy cases. Neurol Med Chir (2003) 43(8):375–82. doi:10.2176/nmc.43.375
84. Sufit A, Donson AM, Birks DK, Knipstein JA, Fenton LZ, Jedlicka P, et al. Diffuse intrinsic pontine tumors: a study of primitive neuroectodermal tumors versus the more common diffuse intrinsic pontine gliomas. J Neurosurg Pediatr (2012) 10(2):81–8. doi:10.3171/2012.3.PEDS11316
Keywords: DIPG, glioma, glioblastoma, pediatric, ACVR1, K27M, histone, H3F3A
Citation: Buczkowicz P and Hawkins C (2015) Pathology, molecular genetics, and epigenetics of diffuse intrinsic pontine glioma. Front. Oncol. 5:147. doi: 10.3389/fonc.2015.00147
Received: 26 March 2015; Accepted: 16 June 2015;
Published: 30 June 2015
Edited by:
David D. Eisenstat, University of Alberta, CanadaReviewed by:
Keith Giles, New York University School of Medicine, USACopyright: © 2015 Buczkowicz and Hawkins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pawel Buczkowicz, The Arthur and Sonia Labatt Brain Tumour Research Centre, The Hospital for Sick Children, 686 Bay Street, Toronto, ON M5G 0A4, Canada,cGF3ZWwuYnVjemtvd2ljekBzaWNra2lkcy5jYQ==;
Cynthia Hawkins, Department of Paediatric Laboratory Medicine, The Arthur and Sonia Labatt Brain Tumour Research Centre, The Hospital for Sick Children, University of Toronto, 555 University Avenue, Toronto, ON M5G 1X8, Canada,Y3ludGhpYS5oYXdraW5zQHNpY2traWRzLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.