 Emily Curran
Emily Curran Leticia Corrales
Leticia Corrales Justin Kline
Justin Kline
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 09 April 2015
Sec. Cancer Immunity and Immunotherapy
Volume 5 - 2015 | https://doi.org/10.3389/fonc.2015.00083
This article is part of the Research Topic Advances in Combination Tumor Immunotherapy View all 8 articles
Because of its disseminated nature and lack of tumor-draining lymph nodes, acute myeloid leukemia (AML) likely employs unique immune evasion strategies as compared to solid malignancies. Targeting these unique mechanisms may result in improved immunotherapeutic approaches. Emerging data suggest that a specific dendritic cell (DC) subset, CD8α DCs, may be responsible for mediating tolerance in AML and thus targeting the innate immune system may be of benefit in this disease. Promising immune targets include the toll-like receptors, calreticulin/CD47, the stimulator of interferon genes pathway, and signal transducer and activator of transcription 3 (STAT3). However, it is becoming clear that compensatory mechanisms may limit the efficacy of these agents alone and thus rationale combinations of immunotherapies are warranted. This review discusses the potential immune evasion strategies in AML, as well as discussion of the promising innate immune targets, both alone and in combination, for this disease.
Over 18,000 people will be diagnosed with acute myeloid leukemia (AML) in the United States this year (1). While the majority of patients will achieve a complete remission with standard induction chemotherapy (2), the relapse rate is high and over half will ultimately succumb to the disease. Despite an improved understanding of the molecular pathways altered in AML, no significant therapeutic advances have been achieved in many years. Allogeneic stem cell transplantation can be curative for some patients with AML (3), thought to be secondary to therapeutic graft-versus-leukemia effects resulting from donor-derived T cell recognition of minor histocompatibility antigens expressed on host leukemia cells (3–6). Unfortunately, only a minority of patients are candidates for stem cell transplantation, but the success of this procedure suggests that other immune strategies may be beneficial in AML.
It is known that AML cells express leukemia-associated antigens (LAA) that can be recognized by the immune system, including those derived from proteins such as proteinase 3 (PR3), receptor for hyaluronic acid-mediated motility (RHAMM), and Wilm’s tumor-1 (WT1), among others (7, 8). Objective clinical responses have been documented in patients with myeloid malignancies following PR3 and WT1 peptide vaccination (8, 9) and with cellular-based WT1 vaccines (10). These observations argue that vaccine-based immunotherapy for AML may be an effective strategy to reduce the risk of disease relapse, particularly those in a minimal residual disease state after remission induction and consolidation chemotherapy. However, there is limited data at this point to support this hypothesis. Further, many years of experience with cancer vaccine approaches for solid tumors has been ripe with failures, including dozens of therapeutic vaccination studies, which demonstrated minimal clinical efficacy (11).
It is clear that the development of cancer immunotherapies for hematologic malignancies, including AML, has lagged behind that for solid tumors. In part, this may be due to the availability of relatively effective chemotherapies and allogeneic stem cell transplantation for AML patients, thus limiting the interest in cultivating immune-based therapies in the field. Further, recent results of AML exome and genome sequencing has revealed a lower mutational burden in AML compared to most other cancers, such as melanoma and lung carcinoma, which impacts the number of leukemia-specific antigens (LSA) available for discovery and targeting (12, 13). While a number of LAA, including those mentioned above, have been identified in AML, they are also typically expressed in other tissues, including the thymus. Developing thymocytes capable of recognizing LAA with high affinity are likely deleted via central tolerance mechanisms, leaving behind low affinity T cells, which upon antigen encounter would be expected to elicit a weak and ineffective immune response.
Yet another barrier to generating effective immunotherapy for AML is that of immune evasion. Similar to solid malignancies, AML activates mechanisms in the host to avoid its immune-mediated elimination. Several of these negative regulatory mechanisms appear to be shared between solid and hematological cancers, including expression of negative costimulatory ligands, such as programed death-ligand 1 (PD-L1) and galectin 9 (Gal-9) on AML cells, and induced expansion of immunoregulatory cells, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) (14–18). However, recent work from our laboratory has demonstrated that AML may also promote unique immune evasion pathways not previously described in solid tumor settings (described below). Further understanding and exploration of the mechanisms of through which AML regulates the host immune system is expected to culminate in the development of effective immune therapies for this disease, as has been the case in solid tumors (19–21).
Our laboratory has recently characterized a novel and potent pathway through which the host innate immune system generates a T cell tolerant state in an animal AML model. In animals with AML, leukemia-specific CD8+ T cells underwent abortive proliferation and were deleted from the host. A small number of surviving antigen-specific T cells were partially dysfunctional and produced low levels of effector cytokines upon restimulation ex vivo, which suggested that they may have been anergized following antigen encounter. Deletional T cell tolerance in AML-bearing hosts appeared to be regulated by host antigen presenting cells (APCs) as it could be reversed in vivo following the administration of an agonistic anti-CD40 antibody, which resulted in enhanced anti-leukemia T cell immunity and prolonged survival (22).
More recent observations from our laboratory have suggested that a subset of host dendritic cells (DCs), called CD8α+ DCs, may mediate T cell tolerance in hosts with AML. Experiments in which fluorescently labeled AML cells were inoculated into mice revealed that CD8α+ DCs were uniquely capable of engulfing AML cells in vivo and of cross-presenting AML cell-derived antigens to T cells ex vivo (23). These results support a critical role for CD8α+ DCs in the immune recognition of AML. These data are important because they suggest that immune tolerance to AML may be initiated at the level of the innate immune system.
The ability of DCs to activate T cells is dependent upon their “activation state.” In the absence of inflammatory stimuli (i.e., under steady-state conditions), DCs are quiescent and are important in this context to maintain peripheral tolerance to self-antigens. Conversely, in the solid tumor context, danger-associated molecular patterns (DAMPs) released by dying cancer cells are sensed by DCs, leading to enhanced antigen presentation, as well as increased expression of costimulatory ligands, chemokines, and cytokines. These changes effectively license DCs to prime a functional anti-tumor T cell response. Although speculative, we believe that due to the disseminated nature of AML, as well as the lack of a classical tumor-draining lymph node, DCs, which engulf and cross-present AML-derived antigens, may not be exposed to sufficient “danger signals” from AML cells to mediate their licensing. The net result is the induction of T cell tolerance to AML. If, in fact, innate immune cells are central to tolerance induction in leukemia-bearing hosts, then targeted activation of innate immunity may be sufficient to overcome tolerance and promote clinically meaningful immunity against AML. In the following sections, we will discuss several innate immune pathways that are amenable to targeting in order to enhance immunity in hosts with AML, as well as the potential for combination therapy (see Figure 1).
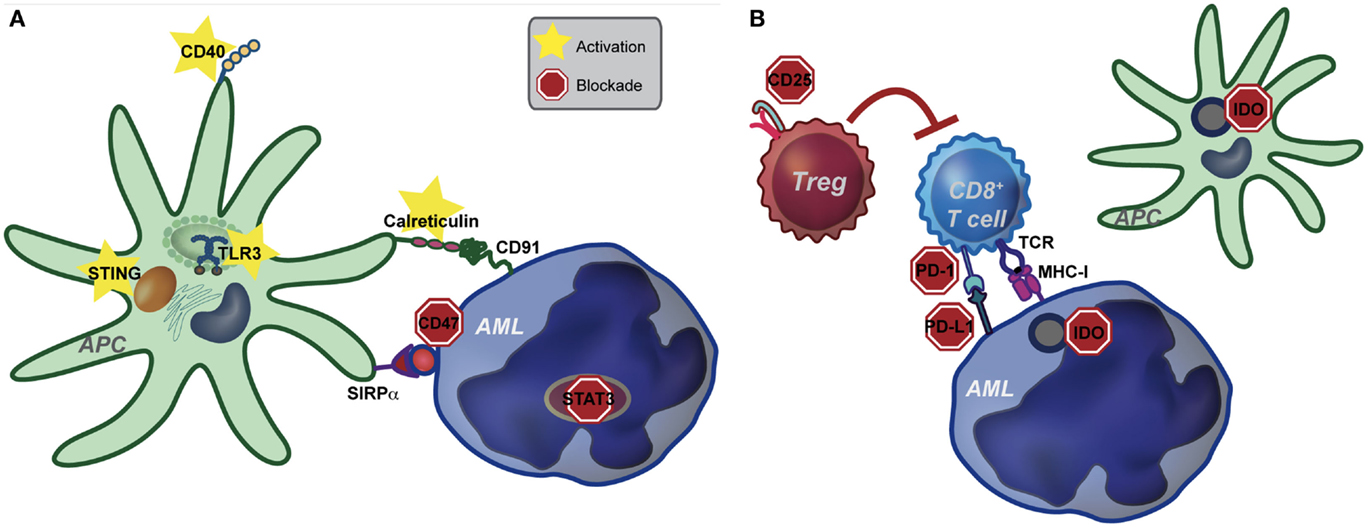
Figure 1. Pathways of potential immune targeting in AML. (A) Innate immunity may be targeted in AML through activation of the STING, TLR (specifically TLR-3) and CD40 receptors. Strategies to enhance calreticulin exposure on AML cells, combined with CD47-blocking antibodies may also be effective. Lastly, STAT3 signaling appears to inhibit the innate immune response, including hampering the effect of TLR9, and inhibition of STAT3 signaling in AML cells leads to differentiation to antigen presenting cells (APCs) with an activated DC phenotype. Activation of the innate immune system, either by stimulating activating pathways or blocking inhibitory pathways, ultimately leads to increased IFN-γ production by CD8+ T cells. (B) Increased IFN-γ resulting from activating of the innate immune system may lead to compensatory up-regulation of other immune evasion pathways, such as PD-L1, indoleamine-2,3-dioxygenase (IDO), and regulatory T cells (Treg). While innate immune activation will be critical to induce anti-leukemia T cell priming, combination therapy targeting compensatory pathways will be key in eliciting a clinically significant anti-leukemia immune response.
The toll-like receptors (TLRs) are a family of transmembrane receptors, which recognize highly conserved microbial structures (i.e., bacterial cell wall components, CpG DNA, viral nucleic acids), termed pathogen-associated molecular patterns (PAMPs). Activation of TLR signal transduction pathways leads to induction of inflammatory cytokines, chemokines, MHC, and costimulatory molecules (24). There are at least 10 TLRs in humans and several, including TLR3, have been targeted as immunotherapy for cancer.
The natural TLR3 ligand is double-stranded RNA. TLR3 stimulation results in the activation of the transcription factors interferon regulatory factor 3 (IRF3) and NF-κB through the adaptor molecule TRIF, culminating in interferon (IFN)-β production (25). Unlike the majority of TLRs, TLR3 signals in a MyD88-independent manner (24). Interestingly, TLR3 appears to be preferentially expressed on CD8α+ DCs (26, 27). Because this DC subset appears to be responsible for mediating tolerance to AML, at least in pre-clinical models, TLR3 may be a promising target for immunotherapy in leukemia. Polyriboinosinic polyribocytidylic acid [poly(I:C)] and a related compound, polyinosinic-polycytidylic acid-polylysine-carboxymethylcellulose (poly-ICLC), are TLR3 agonists, which have been utilized to target TLR3 both in pre-clinical and clinical studies. It has been shown that loading leukemia cells with poly(I:C) by electroporation, and thus mimicking a viral infection, leads to enhanced immunogenicity of leukemia cells, as well as DC maturation and activation (28). Thus, targeting TLR3, with the goal of activating host DCs to reverse T cell tolerance is a promising therapeutic strategy.
Poly(I:C) was initially investigated as monotherapy for hematologic and solid malignancies several decades ago, with disappointing results. Early clinical trials demonstrated that poly-ICLC was a potent inducer of type I interferon and, among 19 adult and pediatric patients with refractory solid tumors or acute leukemia treated, 1 child with acute lymphoblastic leukemia (ALL) had a complete remission (29). Adverse reactions in these trials included fever, nausea, hypotension, thrombocytopenia, leukopenia, erythema, polyarthralgia, and myalgia (29). In a phase II trial of children with acute leukemia and neuroblastoma, including 28 children with ALL, no complete responses were achieved and significant toxicity was observed, leading the authors to halt further study of this agent in other childhood tumors (30). Similarly, phase I studies of poly(I:C) and poly-ICLC in adults with melanoma, ovarian cancer and other advanced cancers demonstrated significant toxicity with few objective clinical responses (31–33). Newer formulations, lower doses, and intramuscular injections of poly(I:C) may decrease its toxicity (34, 35). Nevertheless, the systemic delivery of poly(I:C) as a single agent is not likely to have a major impact as an immunotherapy for cancer.
More recently, poly(I:C) has re-emerged as a viable cancer immunotherapeutic as a cancer vaccine adjuvant. Pre-clinical studies have demonstrated that, when administered as a single adjuvant to various tumor antigen vaccine formulations (including cell-based, peptide, protein, exome, or viral), poly(I:C), and poly-ICLC resulted in enhanced tumor-associated antigen specific and functional T cells, with reduced tumor growth (36). To date, clinical trials of poly(I:C) and poly-ICLC as cancer vaccine adjuvants have mainly been conducted in solid malignancies, specifically gliomas, but have shown promising results (36). A phase I/II trial of DCs loaded glioma-associated antigens and poly-ICLC adjuvant in 22 patients with recurrent malignant glioma showed progression-free survival of at least 12 months in over one-third of patients, as well as one complete response (37). Recent studies have also investigated the use of poly(I:C) as a vaccine adjuvant in combination with other immune stimulators, such as Monatide-ISA-51, granulocyte–macrophage colony-stimulating factor, or a TLR7/8 agonist (resquimod) (36). These combinations appear to be well-tolerated, even in pediatric patients (38).
Poly(I:C) is currently being investigated as an adjuvant to vaccine therapy in an early phase clinical trial in patients with AML in complete remission following chemotherapy or allogeneic stem cell transplantation (Table 1). This trial combines a WT1 peptide vaccine with basiliximab and either poly(I:C) or montanide ISA 51, with the goal of determining the side effects and best method of administering vaccine therapy. Another ongoing phase I trial combines poly(I:C) with the DEC-205/NY-ESO-1 fusion protein CDX-1401 and decitabine for treating patients with MDS or AML (Table 1). DEC-205 is an endocytic receptor expressed primarily by DCs, as well as thymic epithelial cells (39) and NY-ESO-1 is an immunogenic protein that is usually very low or absent in myeloid leukemias, but is expressed following hypomethylating agents (40). The DEC-205-NY-ESO-1 fusion protein, called CDX-1401, is a full length NY-ESO-1 protein sequence fused to a monoclonal antibody against DEC-205. A previous phase I trial of this agent in solid malignancies, combined with adjuvant TLR agonists, such as poly(I:C), demonstrated disease stabilization and regression in a subset of patients, with no dose-limiting toxicities (41). Thus, while poly(I:C) treatment may be of limited benefit as monotherapy, it has demonstrated moderate effectiveness when coupled with other immunotherapeutic approaches.
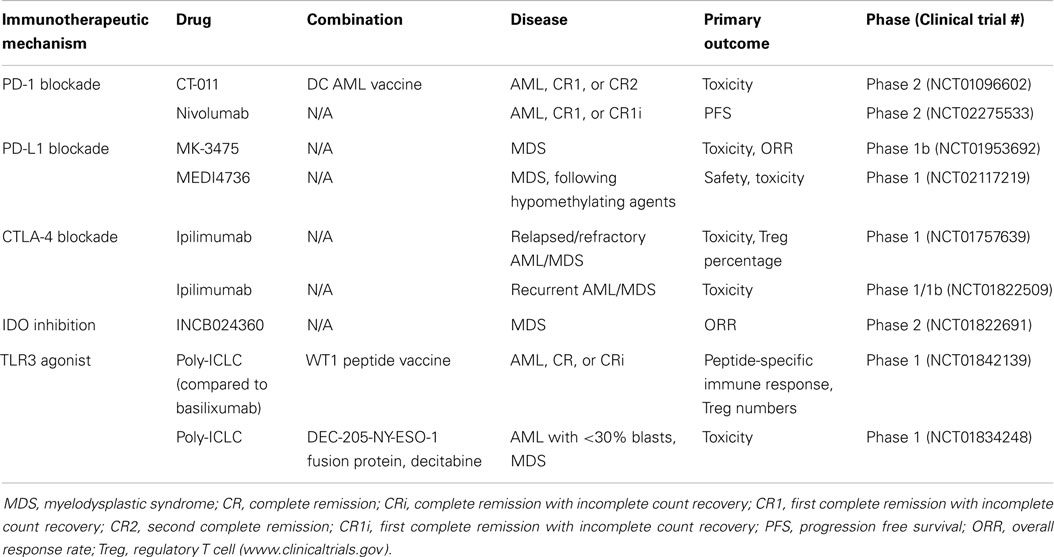
Table 1. Select ongoing trials in AML/MDS.
Calreticulin (CRT) is a chaperone protein that normally resides in the endoplasmic reticulum (ER), where it functions to ensure that misfolded proteins are not exported to the Golgi apparatus. However, more recent studies have demonstrated that CRT translocates from the ER to the cell-surface following cell stress or apoptosis (42). Upon translocation to the cell surface, CRT appears to stimulate phagocytosis by macrophages via their expression of low-density lipoprotein-receptor related protein (LRP), also known as CD91 (43).
Some chemotherapeutic agents (anthracyclines and oxaliplatin) and radiation have been shown to induce ER stress and promote CRT surface translocation in cancer cells, resulting in their recognition and phagocytosis by innate immune cells, such as macrophages and DCs (44). In turn, these innate immune cells become capable of priming antigen-specific T cell responses directed against malignant cells, termed “immunologic cell death.” Elevated cell surface CRT expression has also been observed on viable malignant cells – including leukemia blasts (45, 46), suggesting that cell death may not be a prerequisite for CRT translocation. However, the mechanistic link between CRT expression and its downstream effects on adaptive immunity has not been elucidated. Furthermore, using publicly available gene profiling data sets, Chao analyzed CRT mRNA levels in tumors from patients with a variety of cancers, including neuroblastoma, bladder cancer, and mantle cell lymphoma and found, perhaps counterintuitively, that elevated CRT expression correlated inversely with event-free and/or overall survival (45).
To directly investigate the role of CRT on immunity to AML, our lab has recently generated AML cells engineered to express high-levels of cell-surface CRT. Consistent with previous studies, CRT-expressing AML cells promoted enhanced anti-leukemia T cell responses and prolonged survival in mice. This effect was dependent on adaptive immunity, as it did not occur in Rag−/− hosts, or following in vivo T cell depletion. Although a clear effect of CRT expression on in vivo phagocytosis by DCs or macrophages was not observed in our experiments, further studies are needed to clarify the mechanism through which CRT promotes anti-leukemia immunity.
Calreticulin-mediated phagocytosis of cancer cells can be inhibited by expression of anti-phagocytic proteins, such as integrin-associated protein (IAP), also known as CD47 (43, 45). CD47 is broadly expressed on all cells and transmits a “do not eat me” signal to phagocytes expressing its receptor, signal regulatory protein-α (SIRP-α) (47). Increased CD47 expression on human AML cells promotes their survival through evasion of phagocytosis (48), and blocking CD47 on human cancer cells in xenotransplantation models promotes their phagocytosis and elimination by innate immune cells (48–50). CD47 is more highly expressed in a subset of human acute lymphocytic leukemia (ALL) samples and is an independent predictor of survival and disease refractoriness (51). Pre-clinical studies of a blocking monoclonal antibody against CD47 enabled phagocytosis of ALL cells by macrophages in vitro, inhibited tumor engraftment in vivo, and eliminated ALL in mice engrafted with primary human ALL (51). In a non-Hodgkin lymphoma model, anti-CD47 antibodies were found to be synergistic with rituximab, leading to disease “cure” through Fc receptor-dependent and -independent stimulation of phagocytosis (52). This data suggest that calreticulin/CD47 targeting may be a beneficial immunotherapeutic strategy for AML as well. Phase I clinical trials utilizing CD47 antibodies are currently being developed (http://stemcell.stanford.edu/CD47/). Thus, CD47 over-expression on leukemia cells could represent yet another immune evasion mechanism, which is targetable through receptor blockade.
The importance of type I IFN signaling in the development of functional anti-tumor immunity has been well-characterized (53). Studies in bone marrow chimeric mice harboring autochthonous, carcinogen-induced sarcomas have revealed that type I IFN produced in response to a developing cancer must be “sensed” by host hematopoietic cells, rather than by malignant cells, in order for immune-mediated tumor rejection to ensue (54, 55). Type I IFN signaling, particularly by DCs, is critical to generate functional T cell responses that ultimately mediate tumor elimination (54, 55), and mice lacking the IFNα/β receptor (IFNAR) in DCs fail to reject immunogenic solid tumors (54). Within the DC compartment, CD8α+ DCs were required to respond to type I IFN and to prime anti-tumor T cell responses in a pre-clinical melanoma model, as Batf3−/− mice, in which CD8α+ DCs fail to develop, generate very poor spontaneous anti-tumor T cell responses (55). Collectively, these data suggest a critical role for type I IFN in the host immune response to cancer and may be mediated by CD8α+ DCs.
Until recently, the tumor-derived signals that regulate type I IFN production in the tumor-bearing host were unknown. However, it has been recently demonstrated that cytosolic DNA from dying tumor cells may be a potent inducer of the type I IFN response, through the activation of a cytosolic DNA-sensing receptor called stimulator of interferon genes (STING) (56). STING was originally discovered to stimulate of production of type I interferon using expression screening of human and mouse cDNA transfected into T cells harboring a luciferase reporter under control of the IFNβ promoter (57). Subsequently, STING was found to be required for the induction of innate immune responses to cytosolic, (non-CpG) pathogenic DNA following viral infection (58). Recently, the ligands for STING have been identified as cyclic dinucleotides, known as cyclic-GMP-AMPs (cGAMPS), which are generated from GTP and ATP by the cytosolic enzyme cGMP-AMP synthase (cGAS). cGAMP binding to STING induces its homodimerization and trafficking from the ER to the Golgi (59, 60). In the Golgi, STING recruits tank binding kinase 1 (TBK1), resulting in its phosphorylation and activation of the transcription factor interferon regulatory factor-3 (IRF-3). IRF-3 then translocates to the nucleus and activates transcription of type I IFN (57, 58).
Administration of STING agonists to C1498 AML-bearing mice bearing results in significant expansion of functional leukemia antigen-specific T cells, as well as significantly improved survival (61). While further understanding of the mechanisms underlying the efficacy of STING agonists are warranted prior to human translation, these data suggest that the STING pathway, by stimulating IFN-β production in the host, may represent an effective therapeutic target for AML.
Signal transducer and activator of transcription 3 (STAT3) belongs to a family of seven cytoplasmic transcription factors, which mediate cellular growth, differentiation and apoptosis (62, 63). While a number of cytokines and growth factor receptors can activate STAT3, IL-6 signaling represents one key pathway. IL-6 signaling through GP130 and Janus kinases (JAKs) results in STAT3 phosphorylation, dimerization, and translocation to the nucleus, leading to further production of IL-6 (creating a feed-forward loop), and up-regulation of anti-apoptotic genes (63, 64). Other activators of STAT3 include IL-10, IL-23, and LPS activation of TLR4 and TLR9 (64).
Signal transducer and activator of transcription 3 has been shown to be aberrantly activated in a majority of cancers, most commonly secondary to activation of upstream kinases, such as epidermal growth factor receptor and platelet-derived growth factor receptor (64). Constitutive activation of STAT3 in malignant cells results from the loss of function of negative regulators, such as the suppressor of cytokine signaling (SOCS) family of proteins, protein tyrosine phosphatases (PTPs), the protein inhibitors of activated STATs (PIAS) family, and the ubiquitin-proteasome degradation pathway (62). Constitutive STAT3 activation in malignant cells promotes cell-cycle progression and prevents apoptosis through a variety of mechanisms, including p53 inhibition and up-regulation of genes such as cyclin D1/D2, MYC, and Bcl-XL (65). In pre-clinical models, constitutive STAT3 activation alone results in malignant transformation (65) and inhibition of STAT3 activity results in arrest of tumor development and apoptosis (62).
In addition to its intrinsic role in promoting transformation and cancer progression, active STAT3 also contributes to cancer immune evasion and has been implicated in infectious (such as H. pylori and Epstein–Barr virus) and non-infectious (such as colitis) inflammation-induced carcinogenesis (63, 64, 66). STAT3 activation antagonizes expression of anti-tumor T helper 1 cytokines (such as IL-12 and IFN-γ), mediates T regulatory cell expansion in tumors, and is required for the immunosuppressive effects of MDSCs and tumor-associated macrophages, as well as for the development of tumor-promoting TH17 T cells (64). STAT3 activity has also been found to hamper the effect of locally administered TLR9 ligands (67). Ablating STAT3 through use of inhibitors or knock-out mice results in enhanced function of DCs, T cells, natural killer cells, and neutrophils in tumor-bearing mice and induces growth inhibition of established tumors (68).
Signal transducer and activator of transcription 3 may also promote leukemogenesis, and active STAT3 has been observed in primary leukemia blasts in a majority of AML patients (62). Furthermore, STAT3 activation is correlated with inferior outcome in AML. In a study of 63 patients with AML, STAT3 activation, as determined by STAT3 phosphorylation on Western Blot and DNA-binding activity on electrophoretic mobility shift assay, was present in 44% of patients, where it was associated with a significantly decreased disease-free survival (median 20.6 versus 8.7 months) (69). Thus, STAT3 has become an attractive therapeutic target in AML. However, until recently, it was unclear whether the adverse effects of STAT3 activation in leukemia were a result of its anti-apoptotic and proliferative effects, or also a result of its immune modulatory effects.
In a pre-clinical study of a mouse model of Cbfb-MYH11/Mpl-induced leukemia, which mimics inv(16) AML in humans, systemic administration of CpG-STAT3 small interfering RNA (siRNA) resulted in eradication of established AML in mice (70). STAT3 inhibition failed to restrain leukemia progression in immunodeficient mice, demonstrating that the anti-leukemia effects of STAT3 inhibition were likely immune-dependent. Furthermore, targeted STAT3 blocking/TLR9 activation resulted in differentiation of AML blasts to APCs with an activated DC phenotype, increased the ratio of tumor-infiltrating CD8+ T cells to regulatory T cells, and promoted CD8+ T cell dependent regression of leukemia (70).
Current methods of inhibiting STAT3 are indirect, including inhibition of upstream tyrosine phosphorylation through tyrosine kinase inhibitors, activating negative regulators of STAT (including SOCS and PIAS), disruption of STAT nucleocytoplasmic shuttling, and blockade of cytokine/growth factor receptor binding (62). However, small molecule inhibitors are being developed to block STAT3 dimerization and transcriptional activity (71). While further study and development of STAT3 inhibitors are needed, based on the promising pre-clinical results and evidence of activation in AML, STAT3 inhibition warrants further evaluation in clinical trials in leukemia.
While immune-based therapies, such as CTLA-4 and PD-1-blocking antibodies are highly effective in a subset of patients with melanoma and other solid cancers, monotherapy for most patients is not sufficient for complete or prolonged tumor eradication, likely due to compensatory immune evasion pathways activated in the cancer-bearing host (19, 20, 72, 73). Thus, it follows that targeting the innate immune system alone is unlikely to be a sufficient strategy to eliminate AML. In addition, as discussed below, the increased IFN-γ produced by innate immune activation leads to up-regulation of several compensatory immune evasion pathways, making combination therapy even more pertinent in this setting. We envision that the simultaneous manipulation of several non-redundant negative regulatory pathways, including negative costimulatory receptor (i.e., PD-1, CTLA-4, TIM-3, LAG-3) blockade, removal of suppressor cell populations, and targeting inhibitory enzymes (IDO, arginase), in combination with innate immune targeting may lead to synergistic effects. Although a detailed discussion of the other immune evasion pathways is beyond the scope of this review, a few promising targets will be discussed and are shown in Figure 1.
Programed cell death-1 (PD-1) is a negative regulatory receptor expressed on the surface of activated T cells, B cells, and NK cells and binds to PD-L1, which is expressed on various malignant cells, including AML cells. Zhang and colleagues have demonstrated that the ligand for PD-1, PD-L1 is up-regulated on a murine leukemia cell line in vivo and after in vitro exposure to IFN-γ (15). Other groups have also demonstrated that up-regulated expression of PD-L1 is dependent on exposure of tumor cells to IFN-γ produced by effector T cells (74). Because many of the therapies targeting innate immune activation result in T cell activation and IFN-γ production, targeting PD-L1 in combination with innate immune activation may be synergistic. Trials of PD-1 and PD-L1 inhibitors have demonstrated efficacy in solid tumors (21, 72), and clinical trials of PD-1 and PD-L1 inhibitors are currently underway in AML (Table 1). Future trials may consider combination of PD-1 or PD-L1 inhibitors with innate immune activators.
Inhibitory enzymes, such as indoleamine-2,3-dioxygenase (IDO), may also be beneficial synergistic targets with innate immune activators. IDO catalyzes tryptophan degradation, with tryptophan metabolites negatively regulating T cell activation and survival. Similar to PD-L1, IFN-γ production by CD8+ T cells has been shown to result in up-regulation of IDO (74). IDO inhibitors are currently being investigated in clinical trials in lymphoid malignancies and MDS (Table 1). IDO expression has been demonstrated in blasts of AML patients (75) and is correlated with decreased survival (76). Based on this data, trials in AML, including combination with innate immune activators are warranted.
Regulatory T cells (Tregs) are naturally occurring immunosuppressive CD4+ T cells, which are important in the maintenance of peripheral tolerance to self-antigens (77–79). Inhibition or depletion of Tregs results in enhanced anti-tumor T cell responses and control of tumor progression in transplantable cancer models (80, 81). In addition, Tregs accumulate in leukemia-bearing mice and depletion results in enhanced anti-leukemia T cell responses (16). CD8+ T cell infiltration is associated with recruitment of Tregs in the tumor microenvironment, making this an attractive immunotherapeutic target (74). In the clinical setting, Treg depletion has been demonstrated to be achievable in cancer patients, although responses have been conflicting (82–84). However, based on the evidence available, Treg depletion may be beneficial in combination with innate immune activation.
It is unlikely that targeting any single immune pathway will be sufficient for effective and prolonged treatment of AML. Because AML appears to rapidly induce tolerance of leukemia-specific T cells in a manner, which depends on the innate immune system, we envision that activation of the innate immune will be a key component of immune-based therapies and necessary in order for functional anti-leukemia T cell responses to be primed. However, it is likely that even functionally primed T cells will be subjected to additional negative regulation in the host and thus combination with other immune evasion targeted therapies will be important in order for clinically meaningful anti-leukemia immune responses to be raised in the host.
The innate immune system appears to play a key role in immune evasion by AML and thus, activating innate immunity will likely be critical in immunotherapeutic strategies for this disease. Further insight into the unique mechanisms of immune evasion in AML is needed, but early data suggest promising immunotherapeutic approaches with TLR agonists, the calreticulin-CD47 pathway, STING activation, and STAT3 inhibition. However, because of compensatory immune evasion pathways, combination with other immune targets, such as PD-1/PD-L1, IDO inhibition, and Treg depletion will likely be needed. Given these findings, future studies may benefit from incorporating innate immune activation strategies and will hopefully lead to the same exciting results already demonstrated in solid malignancies.
EC and JK reviewed relevant literature. EC drafted the manuscript. JK revised the manuscript and supervised EC. LC assisted with manuscript revisions and designed the figures. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by T32 CA009566-27 and NIH/NIGMS Clinical Therapeutics Training Grant T32GM007019 to EC and R01 CA16670 to JK.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin (2014) 64:9–29. doi:10.3322/caac.21208
2. Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol (2012) 30:4515–23. doi:10.1200/JCO.2012.43.4738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Gupta V, Tallman MS, Weisdorf DJ. Allogeneic hematopoietic cell transplantation for adults with acute myeloid leukemia: myths, controversies, and unknowns. Blood (2011) 117:2307–18. doi:10.1182/blood-2010-10-265603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med (1979) 300:1068–73. doi:10.1056/NEJM197905103001902
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Baron F, Maris MB, Sandmaier BM, Storer BE, Sorror M, Diaconescu R, et al. Graft-versus-tumor effects after allogeneic hematopoietic cell transplantation with nonmyeloablative conditioning. J Clin Oncol (2005) 23:1993–2003. doi:10.1200/JCO.2005.08.136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Gyurkocza B, Storb R, Storer BE, Chauncey TR, Lange T, Shizuru JA, et al. Nonmyeloablative allogeneic hematopoietic cell transplantation in patients with acute myeloid leukemia. J Clin Oncol (2010) 28:2859–67. doi:10.1200/JCO.2009.27.1460
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Scheibenbogen C, Letsch A, Thiel E, Schmittel A, Mailaender V, Baerwolf S, et al. CD8 T-cell responses to Wilms tumor gene product WT1 and proteinase 3 in patients with acute myeloid leukemia. Blood (2002) 100:2132–7. doi:10.1182/blood-2002-01-0163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Greiner J, Bullinger L, Guinn BA, Dohner H, Schmitt M. Leukemia-associated antigens are critical for the proliferation of acute myeloid leukemia cells. Clin Cancer Res (2008) 14:7161–6. doi:10.1158/1078-0432.CCR-08-1102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood (2000) 95:286–93.
10. Borrello IM, Levitsky HI, Stock W, Sher D, Qin L, Deangelo DJ, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML). Blood (2009) 114:1736–45. doi:10.1182/blood-2009-02-205278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med (2004) 10:909–15. doi:10.1038/nm1100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med (2009) 361:1058–66. doi:10.1056/NEJMoa0903840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Cancer Genome Atlas Research Network T. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368:2059–74. doi:10.1056/NEJMoa1301689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Wang X, Zheng J, Liu J, Yao J, He Y, Li X, et al. Increased population of CD4(+)CD25(high), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur J Haematol (2005) 75:468–76. doi:10.1111/j.1600-0609.2005.00537.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood (2009) 114:1545–52. doi:10.1182/blood-2009-03-206672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Zhou Q, Bucher C, Munger ME, Highfill SL, Tolar J, Munn DH, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood (2009) 114:3793–802. doi:10.1182/blood-2009-03-208181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117:4501–10. doi:10.1182/blood-2010-10-310425
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood (2013) 122:749–58. doi:10.1182/blood-2013-01-480129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Hodi FS, O’day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
20. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet (2014) 384(9948):1109–17. doi:10.1016/S0140-6736(14)60958-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Zhang L, Chen X, Liu X, Kline DE, Teague RM, Gajewski TF, et al. CD40 ligation reverses T cell tolerance in acute myeloid leukemia. J Clin Invest (2013) 123:1999–2010. doi:10.1172/JCI63980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Kline DE, Fosco D, Chen X, Kline J. CD8α+ dendritic cells dictate immune responses against murine AML. J ImmunoTherapy Cancer (2013) 1(Suppl 1):P158. doi:10.1186/2051-1426-1-S1-P158
24. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol (2001) 1:135–45. doi:10.1038/35100529
25. Uematsu S, Akira S. Toll-like receptors and type I interferons. J Biol Chem (2007) 282:15319–23. doi:10.1074/jbc.R700009200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, et al. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol (2003) 33:827–33. doi:10.1002/eji.200323797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Jelinek I, Leonard JN, Price GE, Brown KN, Meyer-Manlapat A, Goldsmith PK, et al. TLR3-specific double-stranded RNA oligonucleotide adjuvants induce dendritic cell cross-presentation, CTL responses, and antiviral protection. J Immunol (2011) 186:2422–9. doi:10.4049/jimmunol.1002845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Smits EL, Ponsaerts P, Van De Velde AL, Van Driessche A, Cools N, Lenjou M, et al. Proinflammatory response of human leukemic cells to dsRNA transfection linked to activation of dendritic cells. Leukemia (2007) 21:1691–9. doi:10.1038/sj.leu.2404763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Levine AS, Sivulich M, Wiernik PH, Levy HB. Initial clinical trials in cancer patients of polyriboinosinic-polyribocytidylic acid stabilized with poly-L-lysine, in carboxymethylcellulose [poly(ICLC)], a highly effective interferon inducer. Cancer Res (1979) 39:1645–50.
30. Lampkin BC, Levine AS, Levy H, Krivit W, Hammond D. Phase II trial of a complex polyriboinosinic-polyribocytidylic acid with poly-L-lysine and carboxymethyl cellulose in the treatment of children with acute leukemia and neuroblastoma: a report from the Children’s Cancer Study Group. Cancer Res (1985) 45:5904–9.
31. Hawkins MJ, Levin M, Borden EC. An eastern cooperative oncology group phase I-II pilot study of polyriboinosinic-polyribocytidylic acid poly-L-lysine complex in patients with metastatic malignant melanoma. J Biol Response Mod (1985) 4:664–8.
32. Krown SE, Kerr D, Stewart WE II, Field AK, Oettgen HF. Phase I trials of poly(I,C) complexes in advanced cancer. J Biol Response Mod (1985) 4:640–9.
33. Stevenson HC, Abrams PG, Schoenberger CS, Smalley RB, Herberman RB, Foon KA. A phase I evaluation of poly(I,C)-LC in cancer patients. J Biol Response Mod (1985) 4:650–5.
34. Salazar AM, Levy HB, Ondra S, Kende M, Scherokman B, Brown D, et al. Long-term treatment of malignant gliomas with intramuscularly administered polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose: an open pilot study. Neurosurgery (1996) 38:1096–103. doi:10.1227/00006123-199606000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Hafner AM, Corthesy B, Merkle HP. Particulate formulations for the delivery of poly(I:C) as vaccine adjuvant. Adv Drug Deliv Rev (2013) 65:1386–99. doi:10.1016/j.addr.2013.05.013
36. Ammi R, De Waele J, Willemen Y, Van Brussel I, Schrijvers DM, Lion E, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther (2015) 146:120–31. doi:10.1016/j.pharmthera.2014.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol (2011) 29:330–6. doi:10.1200/JCO.2010.30.7744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, Potter DM, et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J Clin Oncol (2014) 32:2050–8. doi:10.1200/JCO.2013.54.0526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Jiang W, Swiggard WJ, Heufler C, Peng M, Mirza A, Steinman RM, et al. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature (1995) 375:151–5. doi:10.1038/375151a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Almstedt M, Blagitko-Dorfs N, Duque-Afonso J, Karbach J, Pfeifer D, Jager E, et al. The DNA demethylating agent 5-aza-2’-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk Res (2010) 34:899–905. doi:10.1016/j.leukres.2010.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med (2014) 6:232ra251. doi:10.1126/scitranslmed.3008068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Golden EB, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC. The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol (2012) 2:88. doi:10.3389/fonc.2012.00088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123:321–34. doi:10.1016/j.cell.2005.08.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13:54–61. doi:10.1038/nm1523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med (2010) 2:63ra94. doi:10.1126/scitranslmed.3001375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Wemeau M, Kepp O, Tesniere A, Panaretakis T, Flament C, De Botton S, et al. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis (2010) 1:e104. doi:10.1038/cddis.2010.82
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Subramanian S, Boder ET, Discher DE. Phylogenetic divergence of CD47 interactions with human signal regulatory protein alpha reveals locus of species specificity. Implications for the binding site. J Biol Chem (2007) 282:1805–18. doi:10.1074/jbc.M603923200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell (2009) 138:286–99. doi:10.1016/j.cell.2009.05.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A (2012) 109:6662–7. doi:10.1073/pnas.1121623109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A (2013) 110:11103–8. doi:10.1073/pnas.1305569110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res (2011) 71:1374–84. doi:10.1158/0008-5472.CAN-10-2238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell (2010) 142:699–713. doi:10.1016/j.cell.2010.07.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol (2005) 6:722–9. doi:10.1038/ni1213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med (2011) 208:1989–2003. doi:10.1084/jem.20101158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med (2011) 208:2005–16. doi:10.1084/jem.20101159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity (2014) 41:830–42. doi:10.1016/j.immuni.2014.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi:10.1038/nature07317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461:788–92. doi:10.1038/nature08476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi:10.1126/science.1232458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339:826–30. doi:10.1126/science.1229963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Curran EK, Chen X, Corrales L, Kline J. Activation of the sting pathway enhances immunity and improves survival in a murine myeloid leukemia model. Blood (2014) 124(21):3759.
62. Benekli M, Baumann H, Wetzler M. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J Clin Oncol (2009) 27:4422–32. doi:10.1200/JCO.2008.21.3264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Rebe C, Vegran F, Berger H, Ghiringhelli F. STAT3 activation: a key factor in tumor immunoescape. JAKSTAT (2013) 2:e23010. doi:10.4161/jkst.23010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9:798–809. doi:10.1038/nrc2734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. STAT3 as an oncogene. Cell (1999) 98:295–303. doi:10.1016/S0092-8674(00)81959-5
66. Rezvani K, Barrett J. STAT3: the “Achilles” heel for AML? Blood (2014) 123:1–2. doi:10.1182/blood-2013-11-537092
67. Kortylewski M, Kujawski M, Herrmann A, Yang C, Wang L, Liu Y, et al. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res (2009) 69:2497–505. doi:10.1158/0008-5472.CAN-08-3031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting STAT3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med (2005) 11:1314–21. doi:10.1038/nm1325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Benekli M, Xia Z, Donohue KA, Ford LA, Pixley LA, Baer MR, et al. Constitutive activity of signal transducer and activator of transcription 3 protein in acute myeloid leukemia blasts is associated with short disease-free survival. Blood (2002) 99:252–7. doi:10.1182/blood.V99.1.252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Hossain DM, Dos Santos C, Zhang Q, Kozlowska A, Liu H, Gao C, et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood (2014) 123:15–25. doi:10.1182/blood-2013-07-517987
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Zhao M, Jiang B, Gao FH. Small molecule inhibitors of STAT3 for cancer therapy. Curr Med Chem (2011) 18:4012–8. doi:10.2174/092986711796957284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi:10.1056/NEJMoa1200694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res (2012) 18:2039–47. doi:10.1158/1078-0432.CCR-11-1823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med (2013) 5:200ra116. doi:10.1126/scitranslmed.3006504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Curti A, Trabanelli S, Salvestrini V, Baccarani M, Lemoli RM. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: focus on hematology. Blood (2009) 113:2394–401. doi:10.1182/blood-2008-07-144485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Chamuleau ME, Van De Loosdrecht AA, Hess CJ, Janssen JJ, Zevenbergen A, Delwel R, et al. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica (2008) 93:1894–8. doi:10.3324/haematol.13113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
78. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
79. Workman CJ, Szymczak-Workman AL, Collison LW, Pillai MR, Vignali DA. The development and function of regulatory T cells. Cell Mol Life Sci (2009) 66:2603–22. doi:10.1007/s00018-009-0026-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res (1999) 59:3128–33.
81. Li J, Hu P, Khawli LA, Epstein AL. Complete regression of experimental solid tumors by combination LEC/chTNT-3 immunotherapy and CD25(+) T-cell depletion. Cancer Res (2003) 63:8384–92.
82. Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res (2010) 16:5067–78. doi:10.1158/1078-0432.CCR-10-1757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med (2012) 4:134ra162. doi:10.1126/scitranslmed.3003330
84. Sampson JH, Schmittling RJ, Archer GE, Congdon KL, Nair SK, Reap EA, et al. A pilot study of IL-2Ralpha blockade during lymphopenia depletes regulatory T-cells and correlates with enhanced immunity in patients with glioblastoma. PLoS One (2012) 7:e31046. doi:10.1371/journal.pone.0031046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: acute myeloid leukemia, innate immune system, toll-like receptors, calreticulin, type I interferon, STAT3
Citation: Curran E, Corrales L and Kline J (2015) Targeting the innate immune system as immunotherapy for acute myeloid leukemia. Front. Oncol. 5:83. doi: 10.3389/fonc.2015.00083
Received: 06 November 2014; Accepted: 21 March 2015;
Published online: 09 April 2015.
Edited by:
William L. Redmond, Earle A. Chiles Research Institute, USAReviewed by:
Christian Capitini, University of Wisconsin, USACopyright: © 2015 Curran, Corrales and Kline. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justin Kline, University of Chicago, 5841 S. Maryland Avenue, MC 2115, Chicago, IL 60637, USA e-mail:amtsaW5lQG1lZGljaW5lLmJzZC51Y2hpY2Fnby5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.